

Abstract
Background and Purpose
Hypoxia inducible factor-1 (HIF-1) promotes transitory neuronal survival suggesting that additional mechanisms such as the endoplasmic reticulum (ER) stress might be involved in determining neuronal survival or death. Here, we examined the involvement of ER stress in hypoxia-induced neuronal death and analysed the relationship between ER stress and the HIF-1 pathways.
Experimental Approach
Cultures of rat cortical neurons were exposed to chemical hypoxia induced by 200 μM CoCl2, and its effect on neuronal viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and counting apoptotic nuclei. Protein levels were determined by Western blot analysis. RT-PCR was performed to analyse the content and the t1/2 of HIF-1α mRNA.
Key Results
Chemical hypoxia induced neuronal apoptosis in a time-dependent manner and activated the ER stress PRK-like endoplasmic reticulum kinase (PERK)-dependent pathway. At later stages, chemical hypoxia increased the expression of the C/EBP homologous protein (CHOP) and caspase 12 activity. CoCl2 reduced HIF-1α mRNA t1/2 leading to a decrease in HIF-1α mRNA and protein content, simultaneously activating the ER stress PERK-dependent pathway. Salubrinal, a selective inhibitor of phospho-eIF2α phosphatase, protected neurons from chemical hypoxia by reducing CHOP levels and caspase 12 activity, and increasing the t1/2 of HIF-1α mRNA and the levels of HIF-1α protein. Knocking down HIF-1α blocked the neuroprotective effects of salubrinal.
Conclusions and Implications
Neuronal apoptosis induced by chemical hypoxia is a process regulated by HIF-1α stabilization early on and by ER stress activation at later stages. Our data also suggested that HIF-1α levels were regulated by ER stress.
Table of Links
| TARGETS |
|---|
| Enzymesa |
| Caspase 3 |
| Caspase 9 |
| PERK, endoplasmic reticulum kinase |
| Transportersb |
| GLUT1 (SLC2A1), glucose transporter 1 |
This Table lists key protein targets and ligands in this article which are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Pawson et al., 1999) and are permanently archived in the Concise Guide to PHARMACOLOGY 2013/14 (
Introduction
Hypoxia inducible factors (HIFs) are considered to be the master regulators of oxygen homeostasis. They play a central role during development in post-natal physiology as well as in disease pathogenesis (Ratan et al., 2007). Among the HIF isoforms (Semenza, 2008), HIF-1α is the best studied and most widely described (Semenza, 2000; Jaakkola et al., 2001). Under hypoxic conditions or in the presence of chemical compounds such as CoCl2 or deferoxamine (Triantafyllou et al., 2006), the HIF-1α subunit dimerizes with the constitutively expressed HIF-1β subunit (Jaakkola et al., 2001) forming HIF-1. In the nucleus, HIF-1 binds to the specific hypoxia response elements of target genes such as erythropoietin (EPO), LDH-A and glucose transporter 1 (GLUT1) (Semenza, 2000; Goda and Kanai, 2012), enhancing its expression and mediating the adaptive response to hypoxia (Ratcliffe et al., 1998; Semenza, 2000). In the CNS, HIF-1 is induced in neurons in response to hypoxia (Ruscher et al., 1998) and seems to promote neuronal survival (Baranova et al., 2007). However, this neuroprotective effect seems to be transitory. For instance, HIF-1α protects neurons from apoptosis during the early phase of chemical hypoxia or from mild hypoxia but, under prolonged hypoxia, the neuroprotective effect was lost and neurons eventually die from apoptosis (Posadas et al., 2009; Lopez-Hernandez et al., 2012). These findings suggest that additional mechanisms are involved in determining neuronal survival or death at the later stages of hypoxia.
The endoplasmic reticulum (ER) has been described as a potential sensor capable of triggering both adaptive and pathological signalling. The glucose-regulated protein 78 (GRP-78/Bip) is a chaperonin usually bound to the luminal side of the ER to the three ER stress transducers: the inositol-requiring 1 α (IRE1α), the PRK-like ER kinase (PERK) and the activating transcription factor 6 (ATF6). When ER function is altered, GRP-78/Bip is released, allowing aggregation of the transmembrane signalling proteins PERK, IRE1α and ATF6 and activating the ER stress response (Sommer and Jarosch, 2002).
Activation of the PERK pathway reduces the protein load on the ER through phosphorylation and inactivation of the eukaryotic initiation elongation factor 2α (eIF2α) (Boyce and Yuan, 2006). However, certain mRNAs such as that for ATF4 gain a selective advantage for translation under these conditions (Xu et al., 2005). Initially, ER stress leads to adaptations to the changing environment and restoration of normal ER function. However, when persistent ER stress cannot be corrected, it triggers cell apoptosis (Stefani et al., 2012).
In the CNS, ER stress has been associated with neuronal death in neurodegenerative diseases (Stefani et al., 2012; Roussel et al., 2013), and there is now increasing evidence that ER stress plays a crucial role in hypoxia/ischaemia-induced cell death in vitro (Chen et al., 2008; Roussel et al., 2013) and in vivo (Galehdar et al., 2010; Stefani et al., 2012). In the ischaemic penumbra, translational arrest induced by PERK-mediated phosphorylation of eIF2α is associated with cell survival (Liu et al., 2006), whereas dephosphorylation of eIF2α observed under prolonged ER stress seems to promote neuronal apoptosis through activation of C/EBP homologous protein (CHOP) and caspase 12 (Galehdar et al., 2010; Stefani et al., 2012). Paradoxically, it has also been reported that phosphorylation of eIF2α results in neuronal death (Halterman et al., 2008; Binet et al., 2013) whereas delayed CHOP expression protects neurons against hypoxic injury (Halterman et al., 2010). These discrepant data suggest that ER stress may be involved in the cell response to hypoxia/ischaemia but its role in neuronal viability remains unclear.
Because HIF-1 displays a neuroprotective role only during the initial phase of hypoxia, and prolonged ER stress has been related to neuronal toxicity, we hypothesized that there might be a causal relationship between the HIF-1α and ER stress pathways. Thus, the aim of this study was to clarify whether ER stress played a protective or detrimental role during hypoxia-mediated delayed neuronal death, and to explore whether this ER response was related to the HIF-1α loss of function that we previously observed during prolonged hypoxia (Posadas et al., 2009; Lopez-Hernandez et al., 2012).
Methods
Animals
All animal care and experimental studies complied with the guidelines of the European Union (2010/63/EU) and were approved by the Ethical Committee of Animal Experimentation at UCLM. Studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010). A total of 15 pregnant rats were used in the experiments described here.
Cell culture
The frontolateral cortical lobes were dissected out of Sprague–Dawley embryonic day 17 fetuses and were mechanically dissociated in Hanks' balanced solution. Sprague–Dawley (female, 3 months old, 350 g) pregnant rats were obtained from Janvier Labs (Saint-Berthevin, France). This method is fully described by Posadas et al., (2010). Cortical cells were maintained at 37°C in a saturated humidity atmosphere containing 95% air and 5% CO2 and were used or experiments after 7–12 days n vitro (DIV).
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
MTT assay was performed as previously described (Perez-Carrion et al., 2012). Cortical neurons were treated with vehicle (double distilled water) or 200 μM CoCl2 in the presence or absence of salubrinal for various times. Afterwards, MTT (5 mg·mL−1) was added and the insoluble formazan crystals were dissolved in 300 μL DMSO. Aliquots of 50 μL were transferred to a 96-well microplate and measured in a microplate spectrophotometer (Microplate Reader 550; Bio-Rad, Madrid, Spain) at 590 nm.
Hoechst 33342 staining
Hoechst 33342 staining was performed as previously described (Jordan et al., 2000). Cells were treated with vehicle (double distilled water) or 200 μM CoCl2 in the presence or absence of salubrinal for various time periods. Afterwards, neurons were loaded with 1 μM Hoechst 33342 for 5 min at 37°C. The fluorescence was observed using an excitation filter of 350 nm and an emission filter of 450 nm in a Nikon Diaphot inverted microscope equipped with a 75 W Xenon lamp and a Nikon 40×, 1.3 numerical aperture, epifluorescence oil immersion objective (Nikon Instruments, Barcelona, Spain). Images were acquired with a CCD camera (Hamamatsu Photonics, Barcelona, Spain).
Mitochondrial transmembrane potential
Mitochondrial transmembrane potential (Ψm) was determined as previously described (Lopez-Hernandez et al., 2012). Cortical neurons were treated with vehicle (double distilled water) or 200 μM CoCl2 in the presence or absence of salubrinal for various time periods. Briefly, cells were loaded with 10 μM tetramethylrhodamine methyl ester (TMRM; Molecular Probes, Carlsbad, CA, USA). The fluorescence was acquired every 15 s for 5 min, with a CCD camera (Hamamatsu Photonics) using an excitation filter of 535 nm and an emission filter of 590 nm in a Nikon Diaphot inverted microscope equipped with a 75 W Xenon lamp and a Nikon 40×, 1.3 numerical aperture, epifluorescence oil immersion objective (Nikon Instruments). Linear regression of fluorescence data was obtained and the slopes of the lines fitted by least squares were taken as the rate of loss of Ψm. The percentages of Ψm were calculated with respect to vehicle-treated cells.
Caspase activity
Caspase 3 and caspase 9 activities were determined in total lysates as previously described (Posadas et al., 2007; Tornero et al., 2011). For caspase 12 activity, lysates (40 μg of protein) were incubated in reaction buffer (10 mM Tris-HCl, 2 mM EDTA, 0.1% CHAPS, 10 mM DTT, 50 μM PMSF, 10 μg·mL−1 aprotinin, 10 μg·mL−1 leupeptin, pH 7.4) containing 50 μM fluorescence substrate Ac-ATAD-AFC at 37°C for 1 h following the manufacturer's instructions (BioVision, Milpitas, CA, USA). Cleavage of the AFC fluorophore was determined in a spectrofluorometer (Victor3; Perkin Elmer, Madrid, Spain) at an excitation wavelength of 400 nm and fluorescence was detected at an emission wavelength of 505 nm. Caspase activities were calculated as units of fluorescence (mg of protein × h)−1.
Western blot analysis
An immunoblot analysis was performed on total lysates as previously described (Posadas et al., 2012). The following primary antibodies were used: goat monoclonal anti-HIF-1α antibody (1:1000), rabbit polyclonal LDH-A antibody (1:1000), goat polyclonal EPO antibody (1:500), rabbit polyclonal GLUT1 antibody (1:1000), mouse monoclonal anti-eIF2α antibody (1:1000), rabbit monoclonal anti-peIF2α antibody (1:1000), rabbit monoclonal anti-Bip antibody (1:1000), mouse monoclonal anti-CHOP antibody, rabbit monoclonal anti-ATF4 antibody (1:1000) or mouse polyclonal α-tubulin antibody (1:2000). The following secondary antibodies were used: HRP anti-rabbit IgG (1:10 000), HRP anti-mouse IgG (1:10 000), HRP anti-goat IgG (1:10 000). Immunoreactive bands were visualized using an enhanced chemiluminescence system (ECL).
Real-time reverse transcription PCR (RT-PCR) analysis
RT-PCR analysis was performed as previously described (Perez-Martinez et al., 2012). To determine the mRNA t1/2 24 h after treatment, cells were incubated with 0.1 μM actinomycin D for various time periods. Total RNA was isolated using a commercially available reagent (Tripure; Sigma-Aldrich, Madrid, Spain) according to the manufacturer's instructions. cDNA was amplified using SYBR Green PCR Master mix with the StepOne real-time PCR system and StepOne v2.0 software (Applied Biosystems, Foster City, CA, USA). The primers used to amplify the HIF-1α gene were 5′- GCTGCCTCTTCGACAAGCTT-3′ (forward) and 5′- CGCTGGAGCTAGCAGAGTCA-3′ (reverse). To normalize the data, the β-actin RNA expression level was used as a housekeeping gene. The primers used to amplify the β-actin gene were 5′-TTCAACACCCCAGCCATGT-3′ (forward) and 5′-CAGAGGCATACAGGGACAACAC-3′ (reverse). The real-time RT-PCR reaction was maintained at 95°C for 10 min followed by 40 cycles at 95°C for 15 s and 60°C for 1 min. The dissociation curves were analysed to guarantee the amplification of a single PCR product. All samples were processed three times to ensure the reliability of the results obtained. The quantification was performed by the comparative cycle threshold (Ct (2−ΔΔCt)) method (Livak and Schmittgen, 2001).
Transduction of cortical neurons with lentiviral vectors
Lentiviral vectors were prepared as previously described (Lopez-Hernandez et al., 2012). For transfection experiments, cortical neurons were plated and, immediately after plating, various volumes of viral stocks were added. After 24 h, the medium was replaced and cells were maintained at 37°C until used at 7–12 DIV.
Data analysis
Data are expressed as mean ± SEM. Statistical analyses were carried out using anova and the a posteriori Bonferroni's test for multiple comparisons using GraphPad Software, Inc. (La Jolla, CA, USA). P-values less than 0.05 were considered significant. Statistical results are reported in the Figure legends.
Materials
The BCA protein assay kit was from Pierce Biotechnology, Inc. (Rockford, IL, USA). The caspase substrates Z-DEVD-AFC and Ac-LEHD-AFC were from Calbiochem (Madrid, Spain). The fluorescent probe TMRM was obtained from Molecular Probes. The caspase 12 substrate Ac-ATAD-AFC was from BioVision. The antibodies against HIF-1α and EPO were from R&D System (Abingdon, UK); the antibody against GLUT1 was from Abcam (Cambridge, UK); the antibodies against LDH-A, eIF2α, peIF2α, ATF4, Bip and CHOP were from Cell Signaling (Danvers, MA, USA); and the antibody against α-tubulin was from Calbiochem. The HRP-conjugated IgG antibodies were purchased from DakoCytomation S.A. (Barcelona, Spain). The ECL reagent was from GE Healthcare (Barcelona, Spain). The primers used to amplify the HIF-1α and the β-actin genes were from Eurofins MWG Operon (Madrid, Spain). The cDNA reverse transcription kit and the SYBR Green PCR Master mix were purchased from Applied Biosystems. All other reagents were obtained from Sigma-Aldrich.
Results
Characterization of prolonged chemical hypoxia-induced neuronal death
Cultures of cortical neurons were treated with 200 μM CoCl2. Mitochondrial function, determined as the percentage of transformed MTT and by measuring the Ψm, and apoptotic cell death, quantified through Hoechst 33342 staining, were measured. CoCl2 induced loss of Ψm in a time-dependent manner (Figure 1A). The loss of Ψm was apparent 6 h after CoCl2 incubation, and at 18–24 h the Ψm was halved (Figure 1A). Similarly, the percentage of MTT transformed was significantly reduced at 18 h and was till low at 96 h. Quantification of apoptotic nucleus stained with Hoechst 33342 showed that CoCl2 increased the number of apoptotic cells over time (Figure 1B). At 96 h about 50% of the cells showed chromatin condensation and/or fragmentation (Figure 1C).
Figure 1.
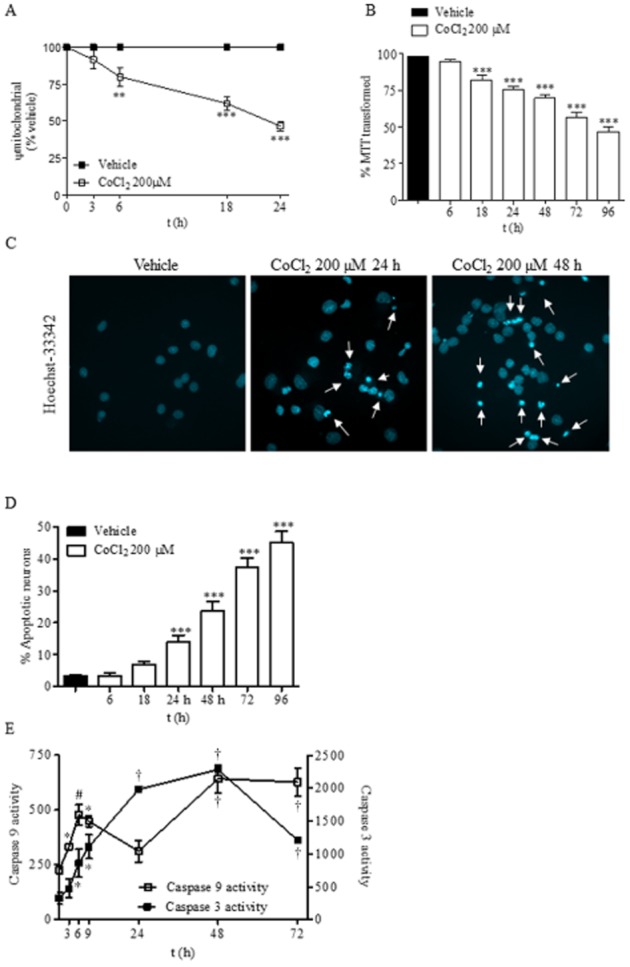
Chemical hypoxia induces apoptosis in rat cortical neurons. (A) Time course of the loss of mitochondrial membrane potential (Ψm) induced by 200 μM CoCl2. Ψm values are expressed as the percentage of Ψm of the vehicle-treated neurons. (B) Time course of neuronal toxicity induced by 200 μM CoCl2. Neurons were exposed to CoCl2 for the indicated times and the mitochondrial function was determined as the percentage of MTT transformed. (C) CoCl2 induces an increase of apoptotic nuclei in a time-dependent manner. White arrows indicate cells with fragmented or condensed chromatin. Images are representative of three different experiments from, at least, six different cell cultures. (D) Quantification of apoptotic nuclei in neurons treated with vehicle or 200 μM CoCl2 for different times. (E) Time course of caspase 9 and caspase 3 activity induced by 200 μM CoCl2. Data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures. In figures (A), (B) and (D) **P < 0.01; ***P < 0.001; in figure (E) *P < 0.05; #P < 0.01; †P < 0.001, significantly different from vehicle-treated cells.
Next, we determined two characteristic features of the intrinsic apoptotic pathway, that is, activation of caspases 9 and 3. CoCl2 induced an activation of caspase 3 activity that was preceded temporarily by the activation of caspase 9 (Figure 1D). It is interesting to note that caspase 9 activity increased significantly at initial times of chemical hypoxia but decreased at 24 h and showing a reactivation by 48 h (Figure 1C).
Activation of both caspases preceded the detection of apoptotic nuclei, suggesting that chemical hypoxia induced neuronal death through activation of the intrinsic apoptotic pathway.
Chemical hypoxia induces ER stress
ER stress has been observed in response to a number of stimuli including ischaemia and hypoxia. Thus, we examined whether chemical hypoxia induced an ER stress response by studying several ER stress markers such as GRP-78/Bip, eIF2α2, peIF2α, ATF4, CHOP and caspase 12.
Under our experimental conditions, CoCl2 induced ER stress and activation of the PERK-dependent pathway at early times that lasted throughout the study (Figure 2A). A significant increase in GRP-78/Bip protein expression was observed 6 h after CoCl2 treatment and lasted for 48 h (Figure 2A; Table 1). In addition, although the levels of eIF2α expression were not altered by chemical hypoxia, incubation with CoCl2 increased phosphorylation of this elongation factor (Figure 2A). Densitometric analysis showed an early (3 h) increase of phosphorylated eIF2α (peIF2α), reaching maximal levels at 24 h and decreasing thereafter (Table 1). In addition, a weak but significant increase in ATF4 levels was detected early while there was a strong increase in this transcription factor at later stages (Figure 2A; Table 1), suggesting that prolonged hypoxia strongly activates the ER stress, PERK-dependent, pathway.
Figure 2.
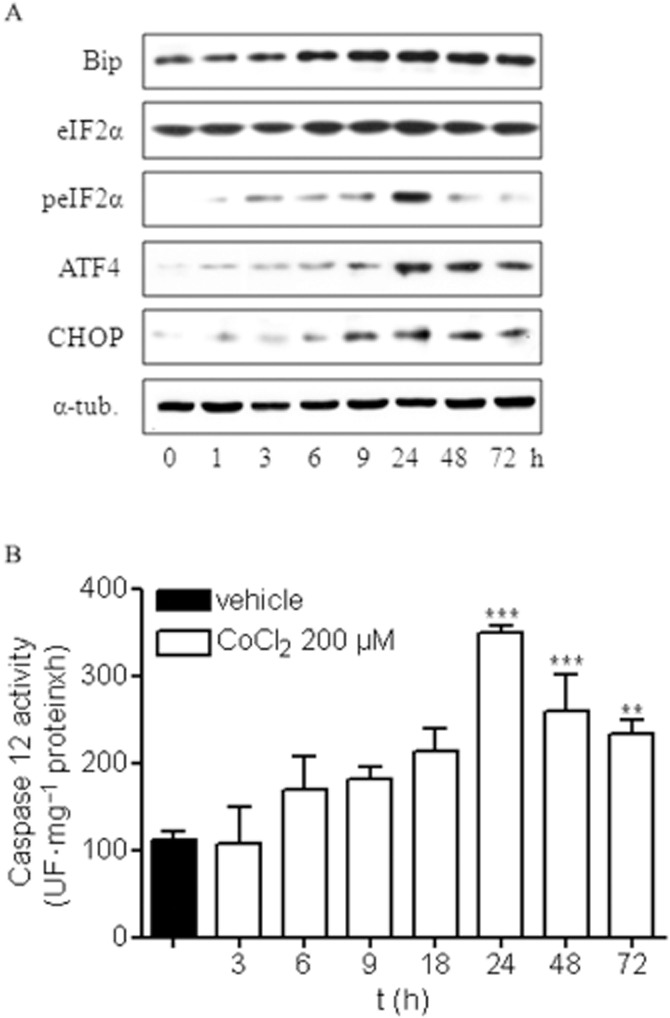
Chemical hypoxia induces ER stress activation in rat cortical neurons. (A) Cortical neurons were treated with 200 μM CoCl2 for different times, and the expression of different ER stress proteins was analysed. Images are representative of three different experiments from, at least, six different cell cultures. (B) Quantification of caspase 12 activity in rat cortical neurons treated with 200 μM CoCl2 for different times showed that CoCl2 induces caspase 12 activation in a time-dependent manner. Data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures. ***P < 0.001, significantly different from vehicle-treated cells.
Table 1.
Densitometric analysis of the expression of different ER stress proteins after exposure to vehicle (V) or CoCl2 200 μM (Co) for various periods of time
| V | Co 1 h | Co 3 h | Co 6 h | Co 9 h | Co 24 h | Co 48 h | Co 72 h | |
|---|---|---|---|---|---|---|---|---|
| Bip | 0.68 ± 0.10 | 0.57 ± 0.12 | 0.80 ± 0.12 | 1.07 ± 0.11** | 1.09 ± 0.10** | 1.30 ± 0.16*** | 1.10 ± 0.15*** | 0.85 ± 0.09 |
| eIF2α | 0.94 ± 0.09 | 0.91 ± 0.09 | 1.11 ± 0.20 | 1.25 ± 0.24 | 1.12 ± 0.10 | 1.33 ± 0.27 | 1.06 ± 0.09 | 0.99 ± 0.10 |
| peIF2α | 0.02 ± 0.01 | 0.03 ± 0.01 | 0.34 ± 0.01*** | 0.26 ± 0.01*** | 0.39 ± 0.02*** | 1.05 ± 0.08*** | 0.22 ± 0.02*** | 0.10 ± 0.01** |
| peIF2α/eIF2α | 0.03 ± 0.01 | 0.04 ± 0.01 | 0.30 ± 0.02*** | 0.15 ± 0.01*** | 0.27 ± 0.02*** | 0.78 ± 0.06*** | 0.16 ± 0.02*** | 0.08 ± 0.01** |
| ATF4 | 0.06 ± 0.01 | 0.12 ± 0.01 | 0.22 ± 0.01* | 0.28 ± 0.01** | 0.32 ± 0.02*** | 1.03 ± 0.09*** | 0.76 ± 0.03*** | 0.49 ± 0.02*** |
| CHOP | 0.05 ± 0.01 | 0.14 ± 0.01 | 0.15 ± 0.01 | 0.19 ± 0.01 | 0.57 ± 0.02*** | 0.81 ± 0.03*** | 0.56 ± 0.02*** | 0.40 ± 0.01*** |
Values are expressed as the ratio between the OD of the protein studied and that of α-tubulin (α-tub; loading control). Data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures.
*P < 0.05; **P < 0.01; ***P < 0.001, significantly different from vehicle-treated cells at each time point.
Chemical hypoxia-induced ER stress also triggered an increase in the expression of the transcription factor CHOP that became apparent 9 h after the stress, reaching a plateau thereafter (Figure 2A; Table 1). Similarly, the late phase of chemical hypoxia induced the activation of caspase 12 (Figure 2B), suggesting a possible participation of both CHOP and caspase 12 in the neuronal death observed in the later phase of hypoxia.
Taken together, our results reveal an activation of ER stress, PERK-dependent, pathway induced by chemical hypoxia in cortical neurons that, initially, might contribute to the adaptive response to hypoxia. However, under prolonged hypoxia, ER stress might contribute to neuronal apoptosis as CHOP expression and caspase 12 activity correlated with neuronal apoptosis.
Salubrinal prevents chemical hypoxia-induced neuronal death
To test whether ER stress contributed to chemical hypoxia-induced neuronal death, we explored the effect of salubrinal, a selective inhibitor of eIF2α dephosphorylation, on CoCl2-treated neurons. Salubrinal prevented the loss of mitochondrial function induced by chemical hypoxia in a concentration-dependent manner and, at 10 μM, salubrinal completely prevented the deleterious effect of chemical hypoxia on cortical neurons. Thus, we selected the concentration of 10 μM for our subsequent experiments (Figure 3A). In agreement with this finding, 10 μM salubrinal strongly reduced the loss of Ψm (Figure 3B) and the number of apoptotic cells at 24 and 48 h after exposure to 200 μM CoCl2 (Figure 3B). Salubrinal also prevented activation of caspases 3, 9 and 12, supporting the neuroprotective effect of this drug on chemical hypoxia-induced neurotoxicity (Figure 3C).
Figure 3.
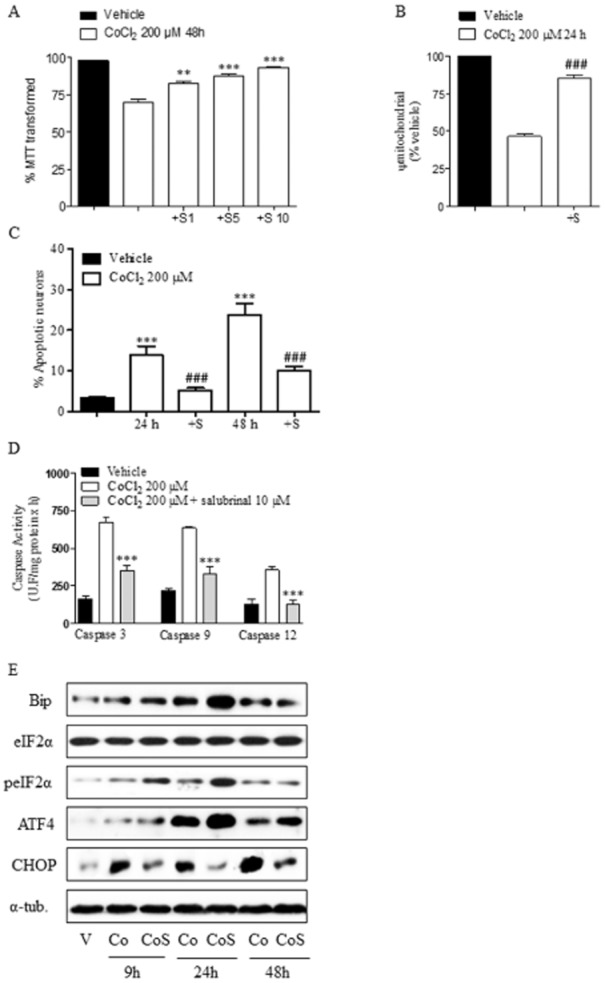
Salubrinal prevents CoCl2 toxicity and ER stress activation. (A) Salubrinal prevents CoCl2-induced neuronal death, determined as the percentage of MTT transformed, in a concentration-dependent manner (S1, S5, S10: salubrinal 1, 5 10 μM). **P < 0.01; ***P < 0.001, significantly different from CoCl2-treated cells for 48 h. (B) Salubrinal (S; 10μM) prevents the loss of Ψm induced by 200 μM CoCl2 for 24 h. Ψm values are expressed as the percentage of Ψm of the vehicle-treated neurons. ###P < 0.001, significant effect of salubrinal. (C) Salubrinal (S; 10μM) prevents CoCl2-induced apoptotic neuronal death. Cells were treated with 200 μM CoCl2 for 24 and 48 h and then stained with Hoechst 33342. Total cells and cells showing fragmented or condensed chromatin were counted and the percentage of apoptotic neurons was calculated. ***P < 0.001, significantly different from vehicle-treated cells; ###P < 0.001, significant effect of salubrinal. (D) Salubrinal reduces activation of caspases 3, 9 and 12 induced by CoCl2 treatment. ***P < 0.001, significantly different from CoCl2-treated cells. (E) Effect of salubrinal (S; 10μM) on CoCl2 (Co)-induced ER stress activation. Cells were treated with 200 μM CoCl2 for 9, 24 and 48 h and the expression of different ER stress proteins was analysed. Images are representative of three different experiments from, at least, six different cell cultures. Data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures.
Once we had confirmed that salubrinal was able to protect cells from hypoxia-induced death, we determined the effect of this pharmacological inhibitor on ER stress under our experimental conditions. To this end, cortical neurons were treated with 200 μM CoCl2 in the presence or absence of 10 μM salubrinal for 9, 24 and 48 h and the expression of the ER stress markers previously studied was examined. As expected, salubrinal induced a marked increase in the phosphorylated status of eIF2α in relation to that induced by CoCl2 alone (Figure 3D; Table 2). In addition, densitometric analysis showed that salubrinal significantly increased GRP-78/Bip levels at 9 and 24 h (Table 2). Interestingly, salubrinal strongly reduced CHOP expression induced by CoCl2 and increased ATF4 expression at all times studied (Figure 3D; Table 2).
Table 2.
Densitometric analysis of the expression of different ER stress proteins after exposure to vehicle (V) or CoCl2 200 μM (Co) in the absence or presence of salubrinal 10 μM (CoS) for 9, 24 and 48 h
| V | Co 9 h | CoS 9 h | Co 24 h | CoS 24 h | Co 48 h | CoS 48 h | |
|---|---|---|---|---|---|---|---|
| Bip | 0.64 ± 0.02 | 1.18 ± 0.15 | 1.49 ± 0.12* | 1.75 ± 0.11 | 2.52 ± 0.19*** | 1.50 ± 0.12 | 10.9 ± 0.2 |
| eIF2α | 1.03 ± 0.11 | 0.97 ± 0.09 | 1.04 ± 0.11 | 1.00 ± 0.10 | 1.04 ± 0.12 | 1.08 ± 0.10 | 0.97 ± 0.11 |
| peIF2α | 0.09 ± 0.01 | 0.44 ± 0.03 | 1.12 ± 0.12*** | 0.68 ± 0.03 | 1.50 ± 0.13*** | 0.30 ± 0.02 | 0.29 ± 0.01 |
| peIF2α/eIF2α | 0.10 ± 0.01 | 0.49 ± 0.06 | 1.08 ± 0.16*** | 0.68 ± 0.01 | 1.43 ± 0.12*** | 0.27 ± 0.02 | 0.26 ± 0.05 |
| ATF4 | 0.10 ± 0.01 | 0.22 ± 0.02 | 0.45 ± 0.03** | 0.96 ± 0.03 | 1.26 ± 0.09*** | 0.50 ± 0.01 | 0.62 ± 0.03* |
| CHOP | 0.20 ± 0.01 | 0.97 ± 0.09 | 0.50 ± 0.04*** | 0.87 ± 0.08 | 0.25 ± 0.02*** | 1.24 ± 0.17 | 0.58 ± 0.03*** |
Values are expressed as the ratio between the OD of the protein studied and that of α-tubulin (α-tub; loading control). Data are expressed as mean ± SEM from 6 to 12 determinations from at least three different cell cultures.
*P < 0.05; **P < 0.01; ***P < 0.001, significantly different from the corresponding CoCl2-treated cells at each time point.
It is noteworthy that the neuroprotective effect of salubrinal correlated with a sustained increase in ATF4 expression and a marked reduction of CHOP expression. Thus, the results suggest that ER stress might be responsible, at least in part, for the neuronal death detected after prolonged exposure to chemical hypoxia.
Prolonged exposure to chemical hypoxia reduces HIF-1α levels
We previously reported that chemical hypoxia stabilizes and induces HIF-1α translocation from cytosol to the nucleus in the early stages. This event seems to confer neuroprotection against chemical hypoxia-mediated toxicity at early, but not at later, times (Posadas et al., 2009), so we explored the time course of the mRNA and protein levels of HIF-1α for up to 72 h in response to CoCl2.
Quantification of HIF-1α mRNA showed that chemical hypoxia induced a reduction in total mRNA starting at 9 h, reaching minimal levels from 24 to 48 h and recovering thereafter (Figure 4A). At 24 h, HIF-1α mRNA levels were reduced to approximately 25% of those detected in vehicle-treated neurons. In agreement with previously reported data, CoCl2 treatment induced a stabilization of HIF-1α protein levels early on (Figure 4B). Interestingly, HIF-1α protein levels drastically diminished after 24 h of treatment, with the minimum levels of HIF-1α mRNA detected at 24–48 h (Figure 4A and B).
Figure 4.
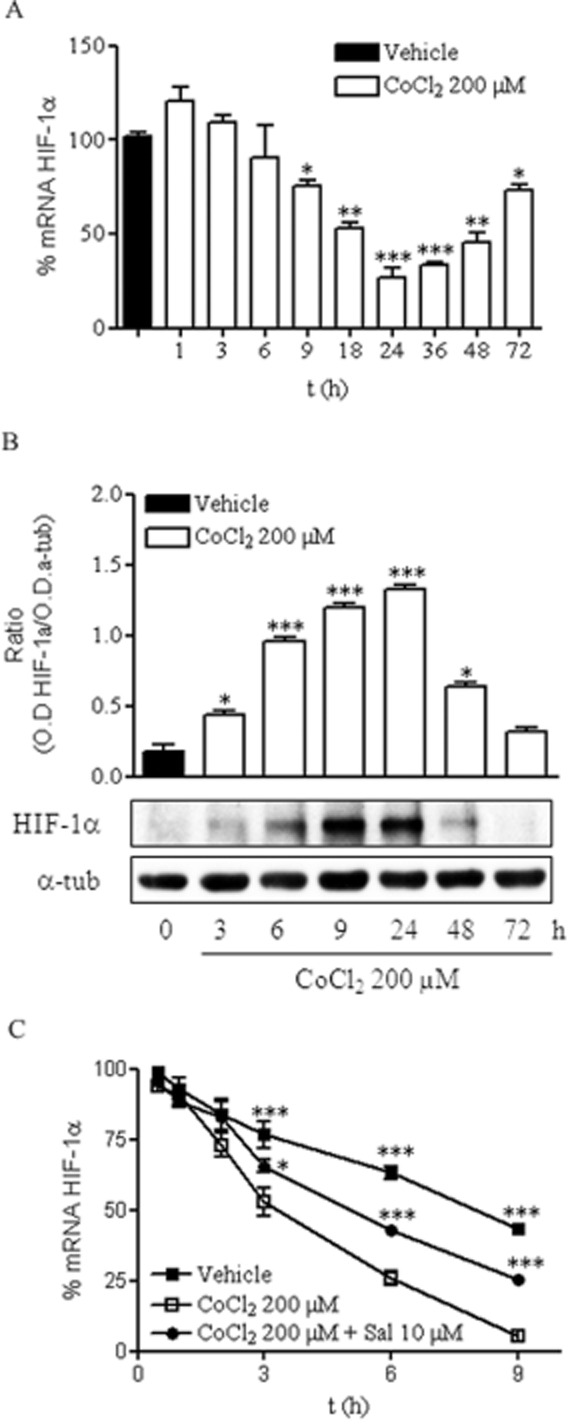
Prolonged chemical hypoxia reduces HIF-1α levels. (A) Time course of mRNA HIF-1α levels, expressed relative to the values in the vehicle-treated group (100%), in the presence of 200 μM CoCl2. (B) Time course of HIF-1α protein expression in the presence of 200 μM CoCl2, shown as the ratio of OD of HIF-1α to that of α-tubulin (α-tub), used as the loading control. Images are representative of three different experiments from at least six different cell cultures. (C) t1/2 of mRNA HIF-1α determined in vehicle- and CoCl2-treated cortical neurons in the absence and presence of 10 μM salubrinal. Data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures. *P < 0.05; **P < 0.01; ***P < 0.001, significantly different from CoCl2-treated cells at the same time.
It seemed that prolonged exposure to chemical hypoxia triggered a reduction in HIF-1α mRNA that might have contributed to a reduction in HIF-1α expression. To further determine if the reduction in HIF-1α mRNA was due to a reduction in mRNA stability, we sought to determine if chemical hypoxia modified the stability of HIF-1α mRNA. To this end, we performed actinomycin D chase experiments to measure the t1/2 of HIF-1α mRNA in cortical neurons treated with vehicle or exposed to chemical hypoxia for various time periods. Chemical hypoxia dramatically enhanced the degradation of HIF-1α mRNA in cortical neurons. Whereas the t1/2 of HIF-1α mRNA in vehicle-treated cells was approximately 9 h, the t1/2 of HIF-1α mRNA in CoCl2-treated cells was reduced to about 3 h (Figure 4C). To rule out a non-specific and generalized effect of CoCl2 on mRNA translation, the t1/2 of actin mRNA was evaluated in vehicle- and CoCl2-treated neurons. No differences were found between the t1/2 of actin mRNA of both groups analysed (data not shown), indicating that the effect of chemical hypoxia on the t1/2 of HIF-1α mRNA was specific.
Thus, reduction in the t1/2 of HIF-1α mRNA induced by CoCl2 might be responsible for the reduction in HIF-1α mRNA and HIF-1α expression observed after prolonged exposure to chemical hypoxia.
Inhibition of ER stress increases HIF-1α levels
Chemical hypoxia activated ER stress. This activation coincided with the reduction in HIF-1α mRNA levels, which might have contributed to a drastic reduction in HIF-1α protein expression. Thus, we next studied whether both events were related. To this end, cortical neurons were treated with 200 μM CoCl2 for various time periods in the presence of 10 μM salubrinal and HIF-1α mRNA content, and HIF-1α protein levels as well as the t1/2 of HIF-1α mRNA were determined. Salubrinal, at 10 μM, prevented CoCl2-induced reduction in HIF-1α mRNA levels (Figure 5A) and HIF-1α protein expression at all time points studied, with the maximal effect observed at 24 h (Figure 5B). Interestingly, in vehicle-treated cells salubrinal did not modify the HIF-1α mRNA levels nor the HIF-1α protein expression, indicating that the effect of salubrinal was not due to a direct interaction of salubrinal with HIF-1α mRNA or with the stabilization of HIF-1α protein (Figure 5A and B). In addition, we observed that the t1/2 of HIF-1α mRNA in the presence of salubrinal under chemical hypoxia was approximately 5.8 h (Figure 4C). Hence, salubrinal partly prevented the reduction in HIF-1α mRNA levels, leading to an increase in HIF-1α protein levels. To determine whether the effect of salubrinal on HIF-1α protein levels led to an increase in the transcriptional activity of HIF-1α, the expression of LDH-A, GLUT1 and EPO was determined. CoCl2 treatment significantly increased the expression of LDH-A and GLUT1 at initial times, reaching maximal levels at 24 h and decreasing thereafter, whereas increased EPO expression was only detected at 24 h (Figure 5C; Table 3). Interestingly, salubrinal induced a sustained increase in LDH-A and EPO expression over time, and an increased GLUT1 expression at the later time point studied (Figure 5C; Table 3).
Figure 5.
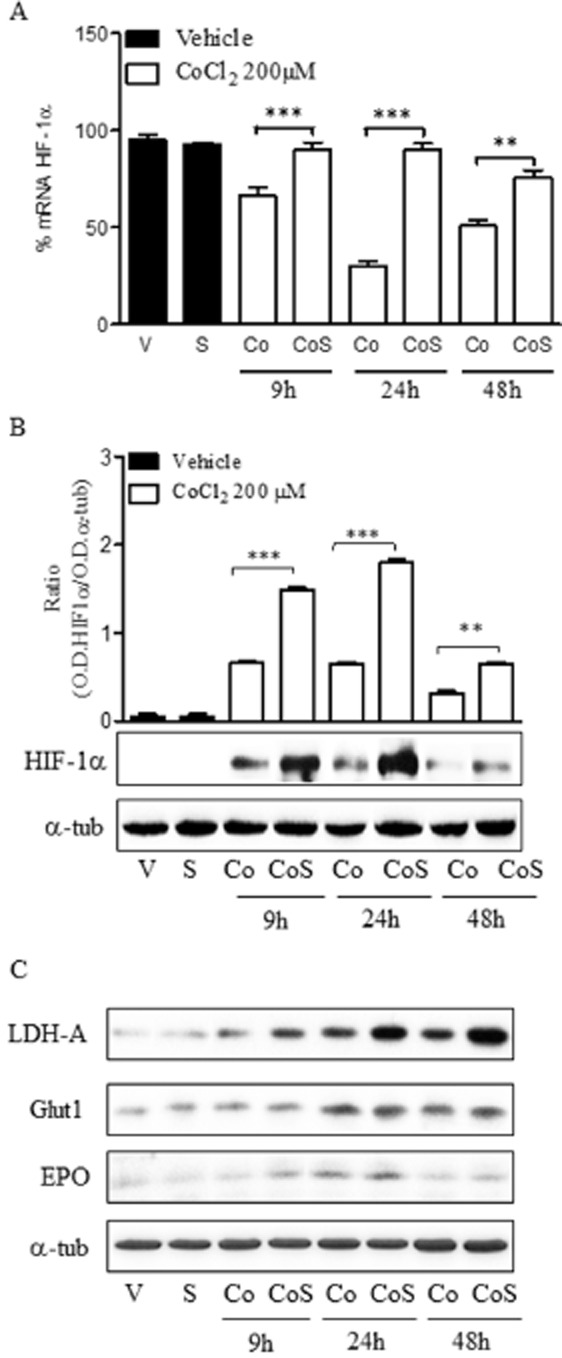
Salubrinal prevents the reduction of HIF-1α levels by CoCl2. (A) Effect of salubrinal (S) on HIF-1α mRNA levels in the absence [Vehicle (V), S] or the presence of 200 μM CoCl2 (Co, CoS) at different times. (B) Effect of salubrinal (S) on HIF-1α protein expression in the absence [Vehicle (V), S] or the presence of 200 μM CoCl2 (Co, CoS) at different times. Densitometric analysis represents the ratio between the OD of HIF-1α and that of α-tubulin (α-tub; loading control). (C) Effect of salubrinal (S) on LDH-A, GLUT1 and EPO expression in the absence [Vehicle (V), S] or the presence of 200 μM CoCl2 (Co, CoS) at different times. In B and C, the images are representative of three different experiments from at least six different cell cultures. In A and B, data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures.**P < 0.01; ***P < 0.001, significantly different from CoCl2-treated cells.
Table 3.
Densitometric analysis of the expression of different HIF-1α target proteins after exposure to vehicle (V) or CoCl2 200 μM (Co) in the absence or presence of salubrinal 10 μM (CoS) for 9, 24 and 48 h
| V | Co 9 h | CoS 9 h | Co 24 h | CoS 24 h | Co 48 h | CoS 48 h | |
|---|---|---|---|---|---|---|---|
| LDH-A | 0.18 ± 0.01 | 0.35 ± 0.02*** | 0.64 ± 0.05 ### | 0.65 ± 0.05*** | 1.25 ± 0.08### | 0.50 ± 0.12*** | 1.24 ± 0.11### |
| GLUT1 | 0.29 ± 0.02 | 0.36 ± 0.01** | 0.41 ± 0.03 | 0.79 ± 0.06*** | 0.83 ± 0.05 | 0.54 ± 0.05*** | 0.63 ± 0.04### |
| EPO | 0.22 ± 0.02 | 0.19 ± 0.02 | 0.34 ± 0.02### | 0.40 ± 0.03*** | 0.63 ± 0.04### | 0.19 ± 0.01 | 0.24 ± 0.02# |
Values are expressed as the ratio between the OD of the protein studied and that of α-tubulin (α-tub; loading control). Data are expressed as mean ± SEM from 6 to 12 determinations from at least three different cell cultures.
**P < 0.01; ***P < 0.001, significantly different from vehicle-treated neurons; #P < 0.05; ###P < 0.001, significantly different from the corresponding CoCl2-treated cells at each time point.
Therefore, our data suggest that ER stress activation plays a role in neuronal death under prolonged chemical hypoxia through a mechanism that seems to be related, at least in part, to the loss of HIF-1α function.
Neuroprotective effect of salubrinal is dependent on HIF-1α
If the loss of function of HIF-1α observed during the late phase of chemical hypoxia was related to the activation of ER stress, and the neuroprotective effect of salubrinal was related to the recovery of HIF-1α levels and the increase of its transcriptional activity, blockade of HIF-1α expression would reduce the neuroprotective effect of this selective inhibitor of p-eIF2α dephosphorylation. To test this hypothesis, we silenced HIF-1α expression using a lentivirus-mediated transduction of a shRNAi against HIF-1α mRNA (pLVTHM-shHIF-1α). Cortical neurons were transduced with pLVTHM-shHIF-1α (shH), which selectively knocks-down HIF-1α expression, or with pLVTHM-sh-Random (shR), which encodes a random sequence, as control. After transduction, neurons were treated with vehicle or 200 μM CoCl2 in the presence or absence of 10 μM salubrinal for 48 h and the neuroprotective effect of salubrinal was studied.
In agreement with previously reported data, HIF-1α knockdown did not modify mitochondrial function in vehicle- or CoCl2-treated neurons, confirming the loss of function of HIF-1α under prolonged hypoxia. Interestingly, HIF-1α knockdown significantly prevented the neuroprotective effect of salubrinal, whereas transduction of cortical neurons with shR did not modify the effect of the pharmacological inhibitor (Figure 6B). These data suggest that the neuroprotective action of salubrinal requires the presence of HIF-1α, because of its effects on the t1/2 of HIF-1α mRNA. Such conclusions would support a causal connection between neuronal death induced by ER stress and the loss of function of HIF-1α.
Figure 6.
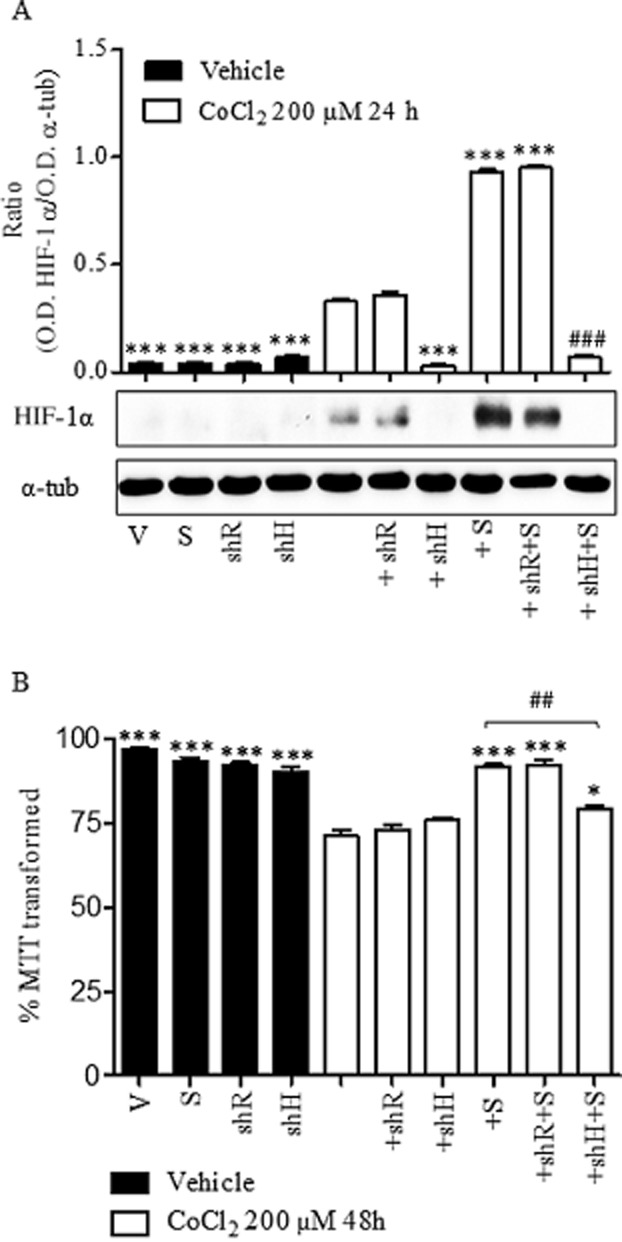
Effect of HIF-1α knock-down on CoCl2-mediated neuronal toxicity. Rat cortical neurons were transfected with pLVTHM-shR (shR) or pLVTHM-shH (shH) and then incubated with vehicle (V) or 200 μM CoCl2 for 48 h in the absence or presence of 10 μM salubrinal (S). (A) Expression of HIF-1α determined in total lysates obtained from cortical neurons after treatment with vehicle (V), 10 μM salubrinal (S) or 200 μM CoCl2 alone or in the presence of 10 μM salubrinal (+S) previously transfected with shR or shH. Densitometric analysis represents the ratio between the OD of HIF-1α and that of α-tubulin (α-tub; loading control). Images are representative of three different experiments from at least six different cell cultures. (B) Effect of HIF-1α silencing on cellular viability, determined as the percentage of MTT transformed, of cortical neurons after treatment with vehicle (V), 10 μM salubrinal (S) or 200 μM CoCl2 alone or in the presence of 10 μM salubrinal ( +S). Data are expressed as mean ± SEM from 6 to 12 determinations from at least six different cell cultures. *P < 0.05, ***P < 0.001 as compared with CoCl2-treated cells. ##P < 0.01, ###P <,0.001; significantly different from 200 μM CoCl2 + 10 μM salubrinal (+S) group.
Discussion and conclusions
Hypoxia is a process relevant to several CNS disorders including stroke and neurodegenerative diseases (Ratan et al., 2007). In the CNS, HIF-1α stabilization after a hypoxic or ischaemic injury has been reported to prevent neuronal death (Baranova et al., 2007). In addition, we have recently reported that HIF-1α was neuroprotective in the initial phases of both chemical and moderate hypoxia (Posadas et al., 2009; Lopez-Hernandez et al., 2012). However, the lack of involvement of HIF-1α in the mechanism activated during the late stages of hypoxia suggested that other mechanisms were involved in neuronal death following prolonged hypoxia (Posadas et al., 2009; Lopez-Hernandez et al., 2012).
Chemical hypoxia induces neuronal apoptosis
In the present study, we have selected an experimental model of chemical hypoxia by exposing cortical neurons to CoCl2, to explore a possible relationship between ER stress and the HIF-1 signalling pathway. CoCl2 is a well-known and well-characterized method of inducing a hypoxia-mimicking response in vitro and in vitro. In brain cells, CoCl2 induces hypoxic conditions characterized not only by HIF-1α stabilization but also by mitochondrial damage, ATP depletion, ROS generation and apoptotic cell death (Yuan et al., 2003; Zhang et al., 2007; Millonig et al., 2009; Posadas et al., 2009; Jones et al., 2013; Sokolowska et al., 2014).
Under our experimental conditions, neuronal death seems to be mediated by mitochondrial dysfunction, probably related to the loss of Ψm, and caspases 9 and 3 activation, which may trigger DNA fragmentation and apoptotic neuronal death. The activation of caspase 9 can be triggered through activation of the canonical intrinsic apoptotic pathway (Zou et al., 1999; Wurstle et al., 2012), but it can be also activated by caspase 12 through a mechanism that is independent of the intrinsic apoptotic pathway (Morishima et al., 2002; Rao et al., 2002). Caspase 12 is an ER-resident caspase that translocates to the cytosol and is activated under ER stress conditions (Nakagawa and Yuan, 2000; Szegezdi et al., 2003; Badiola et al., 2011). Our results showed that chemical hypoxia activated caspase 12 in the later stages of hypoxia suggesting that the ER stress response could be involved in chemical hypoxia-induced neuronal death. In the CNS, the activation of ER stress has been observed after hypoxic and ischaemic injury (Halterman et al., 2010; Gharibani et al., 2013).
The ER stress is involved in neuronal damage during prolonged chemical hypoxia
In the present study we found that in rat cortical neurons, chemical hypoxia induced a rapid increase in the phosphorylation of eIF2α, which was followed by up-regulation of the chaperonin GRP-78/Bip and later up-regulation of ATF4 and CHOP. It is noteworthy that up-regulation of GRP-78/Bip, which has been proposed to protect against ischaemic injury (Boyce and Yuan, 2006; Ouyang et al., 2011), takes place in the initial phases of hypoxia when no neuronal death was observed. However, maximal levels of ATF4 and its downstream target CHOP, a transcription factor involved in ER stress-induced apoptosis (Galehdar et al., 2010), were observed at later times, accompanying neuronal death. Thus, our results reinforce the hypothesis that ER stress may initially act to restore normal ER function and is therefore cytoprotective. However, when the stimulus causing ER stress persists, apoptosis occurs through a mechanism that involves CHOP and caspase 12 activation (Pan et al., 2012).
Salubrinal, an ER stress inhibitor, is a selective pharmacological inhibitor of phospho-eIF2α protein phosphatase 1 and prolongs translational arrest and promotes the stabilization of phospho-eIF2α (Boyce et al., 2005). As a result, salubrinal enhances the PERK-eIF2α signalling during ER stress and has been reported to protect a wide range of cells against ER stress-induced apoptosis. (Boyce and Yuan, 2006; Hosoi et al., 2010; Gong et al., 2012; Ohri et al., 2013). Under our experimental conditions, salubrinal prevented chemical hypoxia-induced neuronal death, reinforcing the role of ER stress in chemical hypoxia-induced delayed neuronal death. Interestingly, we have shown that under chemical hypoxia, salubrinal increased eIF2α phosphorylation, GRP-78/Bip and ATF4 expression but strongly reduced CHOP levels and caspase 12 activity, supporting the hypothesis that GRP-78/Bip and ATF4 play a neuroprotective role and that CHOP and caspase 12 mediate apoptosis in neurons (Halterman et al., 2010; Pan et al., 2012). Similarly, Ohri et al. (2013) have recently described that salubrinal protected oligodendrocytes from ER stress-mediated apoptosis through increasing eIF2α phosphorylation and reducing CHOP expression.
The ER stress is involved in HIF-1α loss of function during prolonged chemical hypoxia
ER stress activation is characterized by arrest of mRNA translation and activation of proteasomes. On the other hand, HIF-1 is degraded under prolonged hypoxia (Millonig et al., 2009). We found that chemical hypoxia decreased HIF-1α mRNA, in agreement with data previously reported for other cell types (Uchida et al., 2004; Bett et al., 2013). We also observed that chemical hypoxia induced a rapid stabilization of HIF-1α protein and a weak increase in HIF-1α mRNA levels at very early times, after the addition of CoCl2. However, under prolonged chemical hypoxia, both HIF-1α mRNA and HIF-1α protein levels were reduced. The reduction in mRNA content might be related to the shorter HIF-1α mRNA t1/2 induced by the chemical hypoxia. However, hypoxia-mediated HIF-1α protein degradation in a prolylhydroxylase-dependent (Millonig et al., 2009) or -independent (Luo et al., 2010) manner cannot not be ruled out.
Interestingly, we observed that in cortical neurons, salubrinal not only protected neurons from apoptosis but also prevented the reduction in HIF-1α mRNA and protein content, mediated by chemical hypoxia. The effect of salubrinal on HIF-1α mRNA stability seemed to be related to an increase in HIF-1α mRNA t1/2.
In addition, chemical hypoxia initially increased the expression of HIF-1 target genes but at later stage the expression of EPO, GLUT1 and LDH-A diminished. Intriguingly, salubrinal induced a sustained expression of LDH-A, EPO and GLUT1 even at the late stages, probably contributing to the switch from mitochondrial oxidative phosphorylation to anaerobic glycolysis (Goda and Kanai, 2012) and preserving neuron integrity.
Taken together, our data suggested that the neuroprotective effect of salubrinal might be mediated, at least in part, by HIF-1α stabilization and the enhancement of HIF-1 translational activity. In agreement, the neuroprotective effect of salubrinal was markedly reduced when the expression of HIF-1α is knocked down.
In the present work, we have described for the first time that activation of the ER stress pathway could induce degradation of the mRNA for HIF-1α, with a concomitant reduction in HIF-1α protein expression. This effect might be related to the loss of the neuroprotective effects of HIF-1α, already observed under prolonged chemical hypoxia (Posadas et al., 2009). In addition, we have also described for the first time that HIF-1α is involved in the mechanism of action of salubrinal. Thus, the present data provide the basis for designing a new class of drugs that would target ER stress and HIF-1α signalling, taking advantage of both molecular pathways.
Acknowledgments
We thank Elena Galera for her technical assistance. This work has been supported, in part, by the grants PI-2007/47 and PPII10-0303-6157 from Consejería de Educación JCCM to I. P. and grants BFU2011-30161-C02-01 and BFU2014-59009-P from Ministerio de Economía y Competitividad, and PII1I09-0163-4002 and POII10-0274-3182 from Consejería de Educación, JCCM to V. C.
Glossary
- ATF
activating transcription factor
- CHOP
C/EBP homologous protein
- ddH2O
double distillate water
- DIV
days in vitro
- eIF2α
initiation elongation factor 2α
- EPO
erythropoietin
- ER
endoplasmic reticulum
- GLUT1
glucose transporter 1
- GRP-78/Bip
glucose-regulated protein 78
- HIF-1
hypoxia inducible factor-1
- IRE1α
inositol-requiring 1 α
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PERK
PRK-like endoplasmic reticulum kinase
- TMRM
tetramethylrhodamine methyl ester
Author contributions
I. P. designed the research study. B. L.-H. and I. P. performed the research and analysed the data. V. C. contributed essential reagents and tools. I. P. wrote the manuscript. V. C. revised the manuscript critically. B. L.-H., V. C. and I. P. agree to be accountable for accuracy and integrity of any part of the manuscript.
Conflict of interest
The authors declare that they do not have any conflict of interest.
References
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL, Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Enzymes. Br J Pharmacol. 2013a;170:1797–1867. doi: 10.1111/bph.12451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Benson HE, Faccenda E, Pawson AJ, Sharman JL. Spedding M, et al. The Concise Guide to PHARMACOLOGY 2013/14: Transporters. Br J Pharmacol. 2013b;170:1706–1796. doi: 10.1111/bph.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiola N, Penas C, Minano-Molina A, Barneda-Zahonero B, Fado R, Sanchez-Opazo G, et al. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. doi: 10.1038/cddis.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1 alpha increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bett JS, Ibrahim AF, Garg AK, Kelly V, Pedrioli P, Rocha S, et al. The P-body component USP52/PAN2 is a novel regulator of HIF1A mRNA stability. Biochem J. 2013;451:185–194. doi: 10.1042/BJ20130026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binet F, Mawambo G, Sitaras N, Tetreault N, Lapalme E, Favret S, et al. Neuronal ER stress impedes myeloid-cell-induced vascular regeneration through IRE1alpha degradation of netrin-1. Cell Metab. 2013;17:353–371. doi: 10.1016/j.cmet.2013.02.003. [DOI] [PubMed] [Google Scholar]
- Boyce M, Yuan J. Cellular response to endoplasmic reticulum stress: a matter of life or death. Cell Death Differ. 2006;13:363–373. doi: 10.1038/sj.cdd.4401817. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2α dephosphorylation protects cell from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Chen X, Kintner DB, Luo J, Baba A, Matsuda T, Sun D. Endoplasmic reticulum Ca2+ dysregulation and endoplasmic reticulum stress following in vitro neuronal ischemia: role of Na+-K+-Cl− cotransporter. J Neurochem. 2008;106:1563–1576. doi: 10.1111/j.1471-4159.2008.05501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galehdar Z, Swan P, Fuerth B, Callaghan SM, Park DS, Cregan SP. Neuronal apoptosis induced by endoplasmic reticulum stress is regulated by ATF4-CHOP-mediated induction of the Bcl-2 homology 3-only member PUMA. J Neurosci. 2010;30:16938–16948. doi: 10.1523/JNEUROSCI.1598-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharibani PM, Modi J, Pan C, Menzie J, Ma Z, Chen PC, et al. The mechanism of taurine protection against endoplasmic reticulum stress in an animal stroke model of cerebral artery occlusion and stroke-related conditions in primary neuronal cell culture. Adv Exp Med Biol. 2013;776:241–258. doi: 10.1007/978-1-4614-6093-0_23. [DOI] [PubMed] [Google Scholar]
- Goda N, Kanai M. Hypoxia-inducible factors and their roles in energy metabolism. Int J Hematol. 2012;95:457–463. doi: 10.1007/s12185-012-1069-y. [DOI] [PubMed] [Google Scholar]
- Gong T, Wang Q, Lin Z, Chen ML, Sun GZ. Endoplasmic reticulum (ER) stress inhibitor salubrinal protects against ceramide-induced SH-SY5Y cell death. Biochem Biophys Res Commun. 2012;427:461–465. doi: 10.1016/j.bbrc.2012.08.068. [DOI] [PubMed] [Google Scholar]
- Halterman MW, De JC, Rempe DA, Schor NF, Federoff HJ. Loss of c/EBP-beta activity promotes the adaptive to apoptotic switch in hypoxic cortical neurons. Mol Cell Neurosci. 2008;38:125–137. doi: 10.1016/j.mcn.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halterman MW, Gill M, DeJesus C, Ogihara M, Schor NF, Federoff HJ. The endoplasmic reticulum stress response factor CHOP-10 protects against hypoxia-induced neuronal death. J Biol Chem. 2010;285:21329–21340. doi: 10.1074/jbc.M109.095299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T, Kume A, Otani K, Oba T, Ozawa K. A unique modulator of endoplasmic reticulum stress-signalling pathways: the novel pharmacological properties of amiloride in glial cells. Br J Pharmacol. 2010;159:428–437. doi: 10.1111/j.1476-5381.2009.00544.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, et al. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- Jones SM, Novak AE, Elliott JP. The role of HIF in cobalt-induced ischemic tolerance. Neuroscience. 2013;252:420–430. doi: 10.1016/j.neuroscience.2013.07.060. [DOI] [PubMed] [Google Scholar]
- Jordan J, Galindo MF, Calvo S, Gonzalez-Garcia C, Cena V. Veratridine induces apoptotic death in bovine chromaffin cells through superoxide production. Br J Pharmacol. 2000;130:1496–1504. doi: 10.1038/sj.bjp.0703451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Liu L, Cash TP, Jones RG, Keith B, Thompson CB, Simon MC. Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol Cell. 2006;21:521–531. doi: 10.1016/j.molcel.2006.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Hernandez B, Posadas I, Podlesniy P, Abad MA, Trullas R, Ceña V. HIF-1alpha is neuroprotective during the early phases of mild hypoxia in rat cortical neurons. Exp Neurol. 2012;233:543–554. doi: 10.1016/j.expneurol.2011.11.040. [DOI] [PubMed] [Google Scholar]
- Luo W, Zhong J, Chang R, Hu H, Pandey A, Semenza GL. Hsp70 and CHIP selectively mediate ubiquitination and degradation of hypoxia-inducible factor (HIF)-1alpha but Not HIF-2alpha. J Biol Chem. 2010;285:3651–3663. doi: 10.1074/jbc.M109.068577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Drummond GB, McLachlan EM, Kilkenny C, Wainwright CL. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millonig G, Hegedusch S, Becker L, Seitz HK, Schuppan D, Mueller S. Hypoxia-inducible factor 1 alpha under rapid enzymatic hypoxia: cells sense decrements of oxygen but not hypoxia per se. Free Radic Biol Med. 2009;46:182–191. doi: 10.1016/j.freeradbiomed.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Morishima N, Nakanishi K, Takenouchi H, Shibata T, Yasuhiko Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis. Cytochrome c-independent activation of caspase-9 by caspase-12. J Biol Chem. 2002;277:34287–34294. doi: 10.1074/jbc.M204973200. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohri SS, Hetman M, Whittemore SR. Restoring endoplasmic reticulum homeostasis improves functional recovery after spinal cord injury. Neurobiol Dis. 2013;58:29–37. doi: 10.1016/j.nbd.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu LJ, Emery JF, Lee AS, Giffard RG. Overexpressing GRP78 influences Ca2+ handling and function of mitochondria in astrocytes after ischemia-like stress. Mitochondrion. 2011;11:279–286. doi: 10.1016/j.mito.2010.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan C, Prentice H, Price AL, Wu JY. Beneficial effect of taurine on hypoxia- and glutamate-induced endoplasmic reticulum stress pathways in primary neuronal culture. Amino Acids. 2012;43:845–855. doi: 10.1007/s00726-011-1141-6. [DOI] [PubMed] [Google Scholar]
- Pawson AJ, Sharman JL, Benson HE, Faccenda E, Alexander SP, Buneman OP, et al. NC-IUPHAR. The IUPHAR/BPS Guide to PHARMACOLOGY: an expert-driven knowledge base of drug targets and their ligands. Nucl Acids Res. 2014;42(Database Issue):D1098–D1106. doi: 10.1093/nar/gkt1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Carrion MD, Perez-Martinez FC, Merino S, Sanchez-Verdu P, Martinez-Hernandez J, Lujan R, et al. Dendrimer-mediated siRNA delivery knocks down Beclin 1 and potentiates NMDA-mediated toxicity in rat cortical neurons. J Neurochem. 2012;120:259–268. doi: 10.1111/j.1471-4159.2011.07556.x. [DOI] [PubMed] [Google Scholar]
- Perez-Martinez FC, Carrion B, Lucio MI, Rubio N, Herrero MA, Vazquez E, et al. Enhanced docetaxel-mediated cytotoxicity in human prostate cancer cells through knockdown of cofilin-1 by carbon nanohorn delivered siRNA. Biomaterials. 2012;33:8152–8159. doi: 10.1016/j.biomaterials.2012.07.038. [DOI] [PubMed] [Google Scholar]
- Posadas I, Vellecco V, Santos P, Prieto-Lloret J, Ceña V. Acetaminophen potentiates staurosporine-induced death in a human neuroblastoma cell line. Br J Pharmacol. 2007;150:577–585. doi: 10.1038/sj.bjp.0706993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posadas I, Lopez-Hernandez B, Clemente MI, Jimenez JL, Ortega P, de la Mata J, et al. Highly efficient transfection of rat cortical neurons using carbosilane dendrimers unveils a neuroprotective role for HIF-1alpha in early chemical hypoxia-mediated neurotoxicity. Pharm Res. 2009;26:1181–1191. doi: 10.1007/s11095-009-9839-9. [DOI] [PubMed] [Google Scholar]
- Posadas I, Santos P, Blanco A, Munoz-Fernandez M, Ceña V. Acetaminophen induces apoptosis in rat cortical neurons. PLoS ONE. 2010;5:e15360. doi: 10.1371/journal.pone.0015360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posadas I, Perez-Martinez FC, Guerra J, Sanchez-Verdu P, Ceña V. Cofilin activation mediates Bax translocation to mitochondria during excitotoxic neuronal death. J Neurochem. 2012;120:515–527. doi: 10.1111/j.1471-4159.2011.07599.x. [DOI] [PubMed] [Google Scholar]
- Rao RV, Castro-Obregon S, Frankowski H, Schuler M, Stoka V, del Rio G, et al. Coupling endoplasmic reticulum stress to the cell death program. An Apaf-1-independent intrinsic pathway. J Biol Chem. 2002;277:21836–21842. doi: 10.1074/jbc.M202726200. [DOI] [PubMed] [Google Scholar]
- Ratan RR, Siddiq A, Smirnova N, Karpisheva K, Haskew-Layton R, McConoughey S, et al. Harnessing hypoxic adaptation to prevent, treat, and repair stroke. J Mol Med (Berl) 2007;85:1331–1338. doi: 10.1007/s00109-007-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliffe PJ, O'Rourke JF, Maxwell PH, Pugh CW. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J Exp Biol. 1998;201:1153–1162. doi: 10.1242/jeb.201.8.1153. [DOI] [PubMed] [Google Scholar]
- Roussel BD, Kruppa AJ, Miranda E, Crowther DC, Lomas DA, Marciniak SJ. Endoplasmic reticulum dysfunction in neurological disease. Lancet Neurol. 2013;12:105–118. doi: 10.1016/S1474-4422(12)70238-7. [DOI] [PubMed] [Google Scholar]
- Ruscher K, Isaev N, Trendelenburg G, Weih M, Iurato L, Meisel A, et al. Induction of hypoxia inducible factor 1 by oxygen glucose deprivation is attenuated by hypoxic preconditioning in rat cultured neurons. Neurosci Lett. 1998;254:117–120. doi: 10.1016/s0304-3940(98)00688-0. [DOI] [PubMed] [Google Scholar]
- Semenza GL. HIF-1: mediator of physiological and pathophysiological responses to hypoxia. J Appl Physiol. 2000;88:1474–1480. doi: 10.1152/jappl.2000.88.4.1474. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1 and cancer pathogenesis. IUBMB Life. 2008;60:591–597. doi: 10.1002/iub.93. [DOI] [PubMed] [Google Scholar]
- Sokolowska P, Urbanska A, Bieganska K, Wagner W, Ciszewski W, Namiecinska M, et al. Orexins protect neuronal cell cultures against hypoxic stress: an involvement of Akt signaling. J Mol Neurosci. 2014;52:48–55. doi: 10.1007/s12031-013-0165-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer T, Jarosch E. BiP binding keeps ATF6 at bay. Dev Cell. 2002;3:1–2. doi: 10.1016/s1534-5807(02)00210-1. [DOI] [PubMed] [Google Scholar]
- Stefani IC, Wright D, Polizzi KM, Kontoravdi C. The role of ER stress-induced apoptosis in neurodegeneration. Curr Alzheimer Res. 2012;9:373–387. doi: 10.2174/156720512800107618. [DOI] [PubMed] [Google Scholar]
- Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–194. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- Tornero D, Posadas I, Ceña V. Bcl-x(L) blocks a mitochondrial inner membrane channel and prevents Ca2+ overload-mediated cell death. PLoS ONE. 2011;6:e20423. doi: 10.1371/journal.pone.0020423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triantafyllou A, Liakos P, Tsakalof A, Georgatsou E, Simos G, Bonanou S. Cobalt induces hypoxia-inducible factor-1alpha (HIF-1alpha) in HeLa cells by an iron-independent, but ROS-, PI-3K- and MAPK-dependent mechanism. Free Radic Res. 2006;40:847–856. doi: 10.1080/10715760600730810. [DOI] [PubMed] [Google Scholar]
- Uchida T, Rossignol F, Matthay MA, Mounier R, Couette S, Clottes E, et al. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: implication of natural antisense HIF-1alpha. J Biol Chem. 2004;279:14871–14878. doi: 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- Wurstle ML, Laussmann MA, Rehm M. The central role of initiator caspase-9 in apoptosis signal transduction and the regulation of its activation and activity on the apoptosome. Exp Cell Res. 2012;318:1213–1220. doi: 10.1016/j.yexcr.2012.02.013. [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Hilliard G, Ferguson T, Millhorn DE. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- Zou H, Li Y, Liu X, Wang X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem. 1999;274:11549–11556. doi: 10.1074/jbc.274.17.11549. [DOI] [PubMed] [Google Scholar]