

Abstract
Silent information regulator 1 (SIRT1) mediates many effects of caloric restriction (CR) on an organism’s lifespan and metabolic pathways. Recent reports have also emphasized its role in vascular function. The present study was designed to investigate the effects of SIRT1 on the properties of mouse spleen derived endothelial progenitor cells (EPCs). SIRT1 in EPCs was significantly increased by serum and by vascular endothelial growth factor (VEGF). Moreover, an adenovirus (Ad) vector expressing SIRT1 (Ad-SIRT1)-mediated overexpression of SIRT1 directly enhanced migration and proliferation of EPCs, whereas silencing of endogenous SIRT1 in EPCs inhibited cell functions. In addition, LY294002 (a PI3K inhibitor), sc-221226 (an Akt inhibitor), and L-NAME (an NOS inhibitor) abolished Ad-SIRT1-induced migration and proliferation of EPCs, and prevented nitric oxide (NO) production. Phosphorylation of Akt, PI3K, and endothelial nitricoxide synthase (eNOS) were up-regulated by Ad-SIRT1, which was attenuated by LY294002, sc-221226, and L-NAME. Together, the results suggested that through the PI3K/Akt/eNOS signaling pathway, SIRT1 plays an important role in the biological properties of EPCs.
Keywords: Silent information regulator 1, endothelial progenitor cells, migration, proliferation
Introduction
Enhancement of re-endothelialization plays an important role in the repair of injured blood vessels. Endothelial progenitor cells (EPCs) have the capacity to proliferate and differentiate into mature endothelial cells, which facilitate repair of injured blood vessels [1,2]. EPCs can migrate to sites of injury and differentiate into endothelial cells (ECs), to eventually participate in the re-endothelialization after vascular injury [3-5]. However, the regulatory mechanisms of the migration and proliferation of EPCs in re-endothelialization after vascular injury remain unclear. It was reported that one of the sirtuins, silent information regulator 1 (SIRT1), is responsible for maintenance of vascular endothelial cell homoeostasis [6-8], and was shown to exert anti-atherosclerotic effects against EPC dysfunction [9,10].
The sirtuins are a highly conserved family of NAD+-dependent histone deacetylases that help regulate the lifespan of diverse organisms. It has been reported that sirtuins are associated with age-related diseases, obesity-associated metabolic diseases, and cardiac aging [11]. The human genome encodes seven different sirtuins (SIRT1-7), which share a common catalytic core domain, but possess distinct N-terminal and C-terminal extensions. Of the seven mammalian sirtuin proteins, SIRT1 has been the most extensively characterized. SIRT1 regulates a variety of physiological functions, such as metabolism, senescence, and differentiation in multiple cell types. It is highly expressed in the vasculature during blood vessel growth and controls the angiogenic activity of EPCs. Recently, SIRT1 has been shown to be a key component of EPC dysfunction in metabolic syndrome and re-endothelialization after vascular injury [12], although, the signaling mechanisms responsible for these SIRT1-mediated EPC functions have not been determined.
Activations of the survival signal PI3K/Akt pathway and the endothelial specific eNOS/NO pathway are closely associated with vascular remodeling and angiogenesis [13-15]. PI3K/Akt activation induced by pro-angiogenic factors has been shown to participate in the proliferation and migration of EPCs [16]. NO plays critical roles in EPC migration and proliferation. It is mainly produced by endothelial nitricoxide synthase (eNOS), and endothelial dysfunction is characterized by a loss of NO bioavailability. Furthermore, VEGF increased EPC survival and angiogenesis by promoting Akt-dependent eNOS phosphorylation and NO production [14].
In this study, we identified the effects of SIRT1 on the migration and proliferation of EPCs, in cultured mouse spleen-derived EPCs. In addition, we provided evidence that the biological properties of EPCs are mediated through the PI3K/Akt/eNOS pathway.
Materials and methods
Ethics statement
All experimental procedures were approved by the Ethics Committee of the 305 Hospital of PLA (Beijing, P. R. China).
Isolation and characterization of EPCs
Culture and characterization of EPCs were done as previously described by Werner N et al [5]. Spleens were explanted from C57BL/6 mice (6 to 8 weeks of age, 20 to 25 g of weight, Beijing, P. R. China). Total spleen-derived mononuclear cells were isolated using a Ficoll gradient (Lympholite-M, Cedarlane). After three washing steps, 4 × 106 spleen-derived mononuclear cells were seeded on fibronectin-coated cell culture flasks and re-suspended in 6 ml endothelial basal medium (Cell Systems) supplemented with 1 μg/ml hydrocortisone, 3 μg/ml bovine brain extract, 30 μg/ml gentamicin, 50 μg/ml amphotericin B, 10 μg/ml human endothelial growth factor, and 20% fetal calf serum (FCS). The harvested cells were cultured at 37°C under an atmosphere of 5% CO2. Forty-eight hours later, non-adherent cells were removed, and the adherent cells were cultured continuously. Only adherent cells were used in further experiments. The medium was changed every second day. For characterization, after 4 days in culture, the cells were incubated with 10 mg/ml acetylated low-density lipoprotein/binding (Dil-Ac-LDL, Invitrogen, CA, USA) for 4 h, fixed with 4% paraformaldehyde and then incubated with 10 mg/ml fluorescein isothiocyanate-Ulex Europeaus lectin-1 (UEA-1, Sigma-Aldrich, St Louis, MO, USA) for 1 h. Finally, the cells were incubated with 1 μg/ml 4’,6-diamidino-2-phenylindole (DAPI, Life Technologies, NY, USA) for 5 min. Triple-stained cells positive for Dil-Ac-LDL, lectin, and DAPI, were identified as EPCs. Additionally, fluorescence activated cell sorting (FACS) analysis was performed using the following monoclonal antibodies: FITC conjugated anti-Sca-1 (Abcam, Cambridge, MA, USA), PE conjugated anti-VEGFR-2 (Biosciences, San Diego, CA, USA), or their corresponding isotype controls (Biosciences, San Diego, CA, USA).
Recombinant adenoviral vectors expressing SIRT1
In order to evaluate the role of SIRT1, adenovirus vector expressing SIRT1 was generated using the pAd-Easy system. Briefly, full-length murine SIRT1 cDNA was first TA-cloned into pMD19-T simple vector and then subcloned into pAdTrack-CMV, resulting in pAdTrack-SIRT1. The shuttle vector was used to generate recombinant adenovirus Ad-SIRT1 according to the manufacturer’s protocol. All PCR-amplified fragments and cloning junctions were verified by DNA sequencing (Sangon, Shanghai, China). An adenovirus encoding green fluorescent protein (GFP; Ad-GFP) was used as control. All adenoviruses were replication deficient and used at 20 multiplicity of infection (mois) for 24 h without apparent cytotoxicity.
Small interfering RNA-mediated silencing of SIRT1 expression
Transient silencing of SIRT1 was accomplished by transfection with small interfering RNAs (si-SIRT1). The selected siRNA duplex sequences specifically targeted mouse SIRT1 (GenBank accession number NM_019812.2), and showed no homology to any other sequences, as determined by a blast search. A non-silencing control (si-CON) sequence was designed to be used as a negative control. Transfection of si-SIRT1 used the Lipofectamine 2000 reagent with a molar ratio between DNA and lipid of approximately 1:3. Forty-eight hours after transfection, cells were collected and used for functional assays.
Reverse transcription-PCR (RT-PCR)
Total RNA was extracted from EPCs using TRIzol (Life Technologies, NY, USA), followed by cDNA synthesis using oligo (dT) and M-MLV reverse transcriptase (Takara, Dalian, China), according to the manufacturer’s instructions. For quantitative RT-PCR analyses, the ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) and SYBR Green PCR Master Mix (Takara, Dalian, China) were used with the following primers: SITR1 sense: 5’-ACTGCAGAAACTTTTAGCCTTTCAA-3’; SIRT1 antisense: 5’-GGCAATGTTCCAAAGAAGTCTGT-3’; GAPDH sense: 5’-TGAACGGGAAGCTCACTGG-3’; GAPDH antisense: 5’-GCTTCACCACCTTCTTGATGTC-3’. All primers were synthesized by Invitrogen (Shanghai, China) and were the highest available purity.
Western blot analysis
After treatment, cells were lysed in lysis buffer. The protein concentration of cell lysates was determined using the Bradford method. The same amounts of protein were separated by SDS-PAGE and electrophoretically transferred onto a polyvinylidene fluoride membrane. Membranes were blocked with 5% non-fat milk solution in TBS, with 0.5% Tween-20. Membrane-bound proteins were probed with primary antibodies against SIRT1 (1:200) and GAPDH (1:500), followed by probing with secondary horseradish peroxidase-conjugated antibodies. Protein bands were visualized by chemiluminescent detection (Amersham Pharmacia Biotech, UK), and quantified using Quantity One software (Bio-Rad, USA). Anti-GAPDH monoclonal antibody was used to test for equal protein loading.
EPC migration assay
The migration of EPCs was assayed using a Transwell system (Corning Costar, USA) containing 8 μm polycarbonate filter inserts in 24-well plates. EPCs (2 × 105) in 100 μl of serum-free DMEM were placed in the upper chamber. DMEM containing 10% FCS (500 μl) was placed in the lower chamber. After 6 h in culture, cells on the bottom of the Transwell membrane were fixed with 4% paraformaldehyde at 37°C for 20 min and stained with 1% crystal violet at 37°C for 5 min. Migration activity was determined as the mean number of migrated cells in six random high-power fields (× 200) per chamber.
EPC proliferation assay
The EPCs were harvested from the cultures and placed, in triplicate, into fibronectin-coated 96-well plates (2 × 106 cells/ml). Cell proliferation was measured using the MTS assay (Cell Titer 96 Aqueous, Promega, USA) according to the manufacturer’s protocol. Before reading the optical density at 490 nm, 20 μl of MTS solution was added to each well. All groups of experiments were performed in triplicate.
Concentration of NO in media
The concentration of NO released from EPCs was determined using a NO assay kit (Nanjing Jiancheng Institute of Biological Engineering, China) according to the manufacturer’s instructions. EPCs were pretreated with LY294002 (30 μM), sc-221226 (30 μM), or L-NAM (200 μM) for 1 h, then treated with Ad-SIRT1 or Ad-GFP for 24 h. The concentration of NO in 100μl of supernatant in media from different groups was detected at 550 nm. The total protein in every group was quantified using the BCA method. The NO-releasing ability of EPCs was calculated as the ratio of NO concentration and total protein.
Statistical analysis
Data from at least three independent experiments were expressed as the mean ± S.D. SPSS 18.0 software was used for statistical analysis. Comparisons between multiple groups were performed using Multi-Way ANOVA or One-Way ANOVA. Comparisons between groups were performed using Fisher’s LSD test. P values < 0.05 were considered to be statistically significant.
Results
Characterization of spleen-derived EPCs
After 4-7 days of culture (typical culture period before coculture and further experiments), adherent EPCs were characterized by immunofluorescence and fluorescence activated cell sorting (FACS) analysis. The majority of cells (>90%) stained positively for Dil-Ac-LDL, lectin, and DAPI (Figure 1A). In addition, 81.53 ± 3.97% of these cells expressed mouse stem-cell marker Sca-1, and 56.32 ± 2.18% expressed endothelial cell marker VEGFR-2 (Figure 1B).
Figure 1.

The isolation and characterization of EPCs. A. EPCs stained with Dil-Ac-LDL, lectin, and DAPI. B. FACS analysis of primary EPCs cultured for 5-7 days. FACS analysis of cultured EPCs for FITC-Sca-1 and VEGFR2, representing a stem/progenitor cell marker and endothelial cell marker, respectively. Positive cells were 81.53 ± 3.96% (n = 3) for Sca-1 expression and 49.72 ± 2.58% (n = 3) for VEGFR2 expression. The left peak in each box denotes corresponding negative isotype control labeling, and the positive gate M1 is shown.
SIRT1 expression and localization in EPCs
SITR1 was present at low levels in quiescent EPCs, but was up-regulated upon stimulation with serum and VEGF, as determined using either mRNA levels as determined by RT-PCR, or by protein levels as determined by western blotting (Figure 2).
Figure 2.
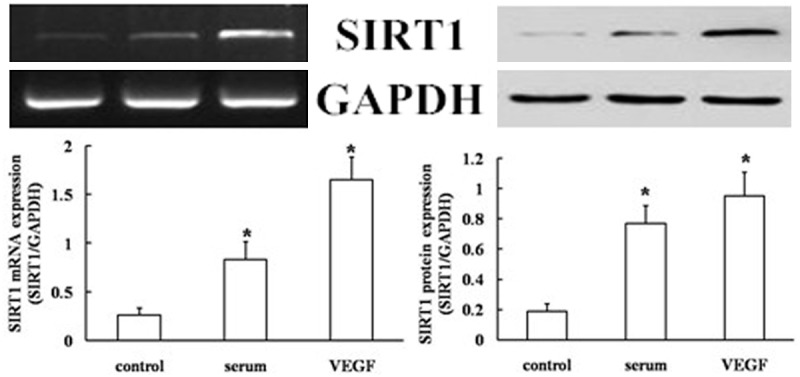
SIRT1 expression in EPCs. Protein and mRNA levels of SIRT1 in EPCs using western blotting and RT-PCR analysis. Values are the percentage of GAPDH (serum- and VEGF-free, null treatment). Representative images from semi-quantitative RT-PCR and western blots. Data are expressed as mean ± SD of three independent experiments done in triplicate, with *P < 0.05 compared with the controls.
Overexpression of SIRT1 enhances migration and proliferation of EPCs
To determine if SIRT1 was involved in the regulation of migration and proliferation of EPCs, an adenoviral vector was constructed that expressed SIRT1 exogenously using the pAd-Easy system. The transfection efficacy was approximately 60-70% as assessed by western blot analysis. Increased levels of SIRT1 after adenovirus-mediated overexpression of SIRT1 was confirmed by RT-PCR and western blot analysis (Figure 3). The parental adenoviral vector or Ad-GFP was used as a transfection control.
Figure 3.
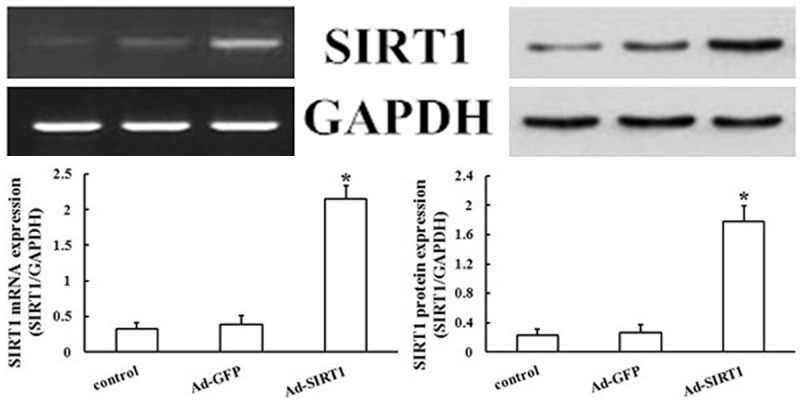
Effect of SIRT1 overexpression on levels of SIRT1. Changes in levels of protein and mRNA of SIRT1 were detected by overexpression of SIRT1 in EPCs using western blotting and RT-PCR analysis. Values are the percentage of GAPDH. SIRT1 gene and protein levels were increased by SIRT1 overexpression, whereas Ad-GFP levels were not affected (*P < 0.05 vs. Ad-GFP).
EPCs transfected with Ad-SIRT1 or control Ad-GFP were subsequently subjected to separate assays to examine their migration and proliferation. The Transwell system was used to examine the effects of SIRT1 overexpression on EPC migration. As shown in Figure 4A and 4C, transfection of EPCs with Ad-SIRT1 increased the number of migrating cells compared with Ad-GFP cells (P < 0.05). The MTS assay was used to examine how SIRT1 overexpression affected EPC proliferation. The proliferation of EPCs transfected with Ad-SIRT1 was enhanced approximately 300% compared with Ad-GFP transfected cells and control cells (P < 0.05) (Figure 5A). Taken together, and as expected, the results showed that Ad-SIRT1 promoted EPCs migration and proliferation in vitro. SIRT1 therefore is an important component in the regulation of migration and proliferation of EPCs.
Figure 4.
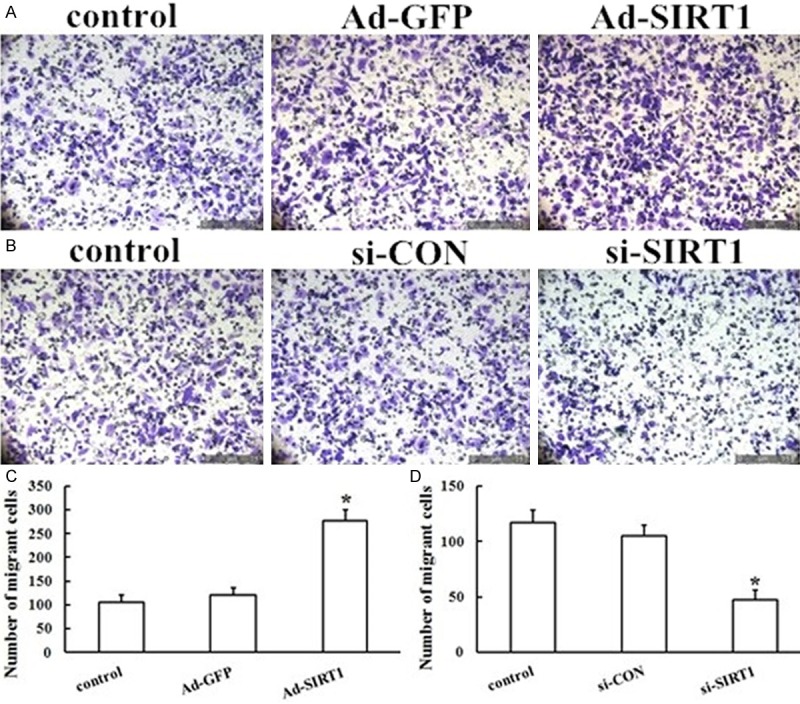
Effect of SIRT1 overexpression and silencing on the migration of EPCs. A, B. Representative photographs of SIRT1 overexpression and silencing on the migration of EPCs. EPC migration in response to Ad-SIRT1 and si-SIRT1 was detected using the Transwell system. C, D. The migration of EPCs transfected with Ad-SIRT1 was enhanced as compared with that of Ad-GFP-transfected EPCs, but knockdown of endogenous SIRT1 significantly reduced the migration of EPCs compared with the Ad-GFP group. The results are expressed as the mean ± SD (*P < 0.05 vs. Ad-GFP).
Figure 5.
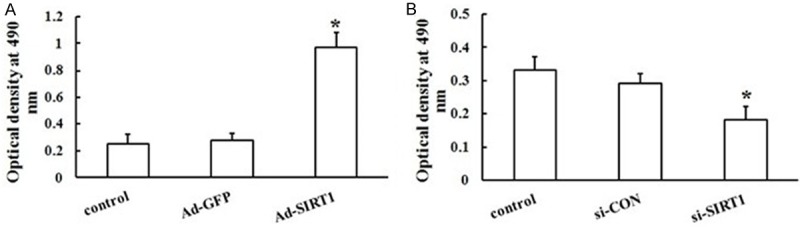
Effects of SIRT1 overexpression and silencing on the proliferation of EPCs. Proliferation of EPCs was examined using the MTS assay. A. EPCs were transfected with or without Ad-GFP, and Ad-SIRT1. Three separate experiments were done in triplicate. B. EPCs were transfected with or without negative control of siRNA or si-SIRT1. The effect was decreased in the presence of si-SIRT1. The results are expressed as the mean ± SD (*P < 0.05 vs. Ad-GFP or si-CON).
EPC migration and proliferation are inhibited by siRNA-mediated knockdown of SIRT1
Although overexpression of exogenous SIRT1 directly enhanced the migration and proliferation of EPCs, the role of endogenous SIRT1 was not determined. To determine whether endogenous SIRT1 affected the migration and proliferation of EPCs, siRNA fragments were used to knockdown SIRT1 protein levels. Forty-eight hours after transfection, si-SIRT1 caused a significant loss of SIRT1 in EPCs as measured by RT-PCR and western blot (Figure 6) (all, P < 0.05). Importantly, EPCs exhibited a decrease in cell migration (Figure 4B, 4D) and proliferation (Figure 5B) compared to si-CON cells (all P < 0.05). The results were reproducible in at least three independent experiments. Thus, knockdown of endogenous SIRT1 significantly reduced the migration and proliferation formation of EPCs, suggesting an important role of endogenous SIRT1 in EPCs.
Figure 6.
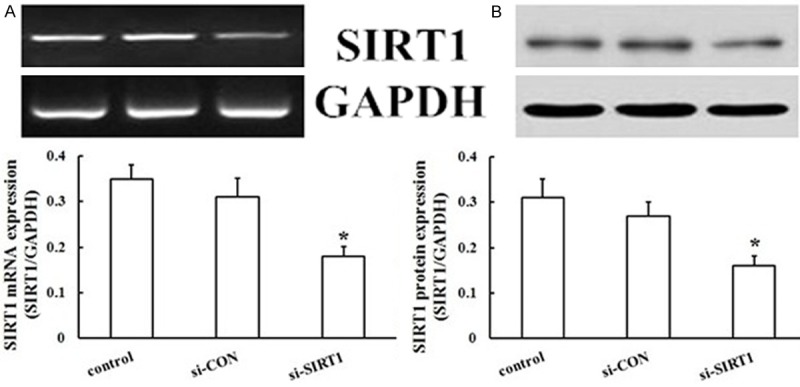
Influence of SIRT1 silencing on changes in levels of SIRT1. Changes in levels of protein and mRNA of SIRT1 were detected using western blotting and RT-PCR analysis after silencing of SIRT1 in EPCs. Values are the percentage of GAPDH. SIRT1 gene and protein levels were decreased by SIRT1 silencing, but si-CON was not affected (*P < 0.05 vs. control).
The role of the PI3K/Akt/eNOS pathway in SIRT1-induced migration and proliferation of EPCs
To evaluate the functional roles of the PI3K/Akt/eNOS signaling pathway in SIRT1-induced migration and proliferation, EPCs approximately 7 days old were pretreated with LY294002 (a PI3K inhibitor, 30 μM), sc-221226 (an Akt inhibitor, 30 μM), and L-NAME (an NOS inhibitor, 200 μM ) for 1 h, before migration and proliferation assays. Pretreatment of EPCs with LY294002 almost completely blocked Ad-SIRT1-induced EPC migration (Figures 7A and 8) and proliferation (Figure 7B). Simultaneously, Ad-SIRT1-induced migration (Figures 7A and 8) and proliferation (Figure 7B) were all significantly inhibited by pretreatment with sc-221226. To further study the molecular mechanisms of Ad-SIRT1-mediated migration and proliferation, EPCs at 7 days were pretreated with eNOS inhibitor L-NAME. As shown in Figures 7 and 8, L-NAME significantly attenuated Ad-SIRT1-induced EPC migration (Figures 7A and 8) and proliferation (Figure 7B). Taken together, these results demonstrated that the PI3K/Akt/eNOS signal transduction pathway plays an important role in SIRT1-induced EPCs migration and proliferation.
Figure 7.
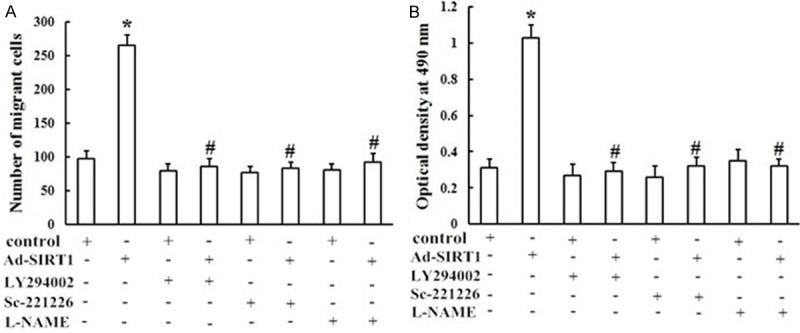
Role of the PI3K/Akt/eNOS signaling pathway in SIRT1-induced migration and proliferation of EPCs. Cells in the control group and the Ad-SIRT1 group with or without pretreatment with LY294002 (30 μM), sc-221226 (30 μM), or L-NAM (200 μM). Ad-SIRT1-induced migration (A) and proliferation (B) of EPCs were significantly inhibited by pretreatment with LY294002, sc-221226, or L-NAME (*P < 0.05 vs. control, #P < 0.05 vs. Ad-SIRT1).
Figure 8.
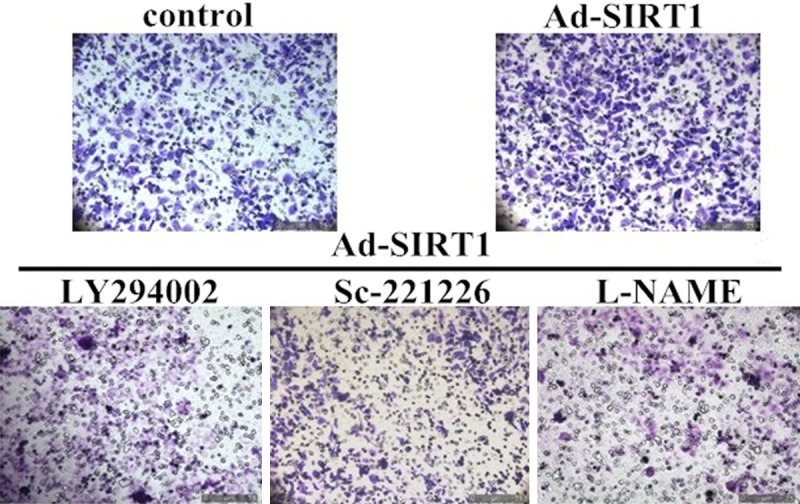
The role of the PI3K/Akt/eNOS signaling pathway in SIRT1-induced migration of EPCs. Representative photographs of EPC migration.
Ad-SIRT1 treatment activated the PI3K/Akt/eNOS pathway
Because SIRT1-induced migration and proliferation of EPCs were regulated by the PI3K/Akt/eNOS signaling pathway, we examined the effect of Ad-SIRT1 on PI3K, Akt, and eNOS phosphorylation in EPCs. Exogenous stimulation with Ad-SIRT1 significantly up-regulated the phosphotyrosine levels of PI3K (Figure 9B), Akt (Figure 9C), and eNOS (Figure 9D) (all P < 0.05), confirming the activation of the PI3K/Akt/eNOS signaling pathway.LY294002 (30 μM), sc-221226 (30 μM), and L-NAM (200 μM) were further used to determine the effects of Ad-SIRT1 on PI3K, Akt, and eNOS phosphorylation in EPCs. LY294002, a highly selective inhibitor of PI3K, prevented the Ad-SIRT1-induced phosphorylation of PI3K (Figure 9B), Akt (Figure 9C), and eNOS (Figure 9D). As shown in Figure 9, treatment with sc-221226 attenuated levels of SIRT1-induced p-Akt (Figure 9C) and p-eNOS (Figure 9D) expression in EPCs transfected with Ad-SIRT1, but not p-PI3K (Figure 9B). SIRT1-induced p-eNOS expression was abrogated by L-NAME as determined by western blot analysis (Figure 9A, 9D). Together, the results suggested that eNOS activation was mediated through the PI3K/Akt signaling pathway.
Figure 9.
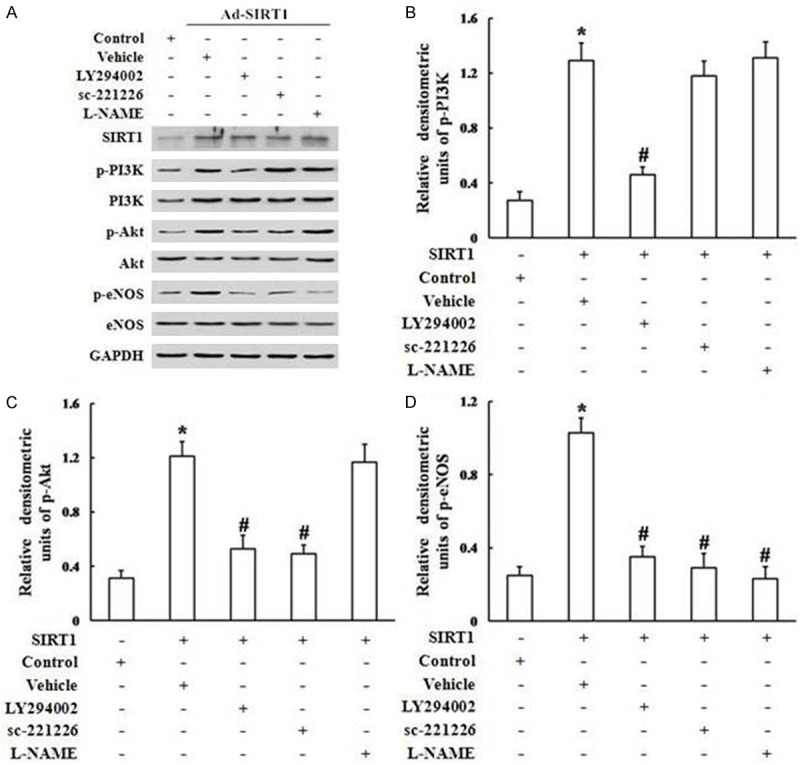
Blockage of the PI3K/Akt/eNOS signaling pathway abrogated SIRT1-induced phosphorylation of PI3K, Akt, and eNOS. (A) Western blot demonstrating that blockade by the PI3K inhibitor LY294002, the Akt-specific inhibitor sc-221226, and the NOS inhibitor L-NAME, reduced p-PI3K, p-Akt, and p-eNOS in EPCs transfected with Ad-SIRT1. Ad-SIRT1-induced phospho-PI3K (B), phospho-Akt (C), and phospho-eNOS (D) were normalized to total PI3K, Akt, or eNOS, respectively (*P < 0.05 vs. control, #P < 0.05 vs. Ad-SIRT1).
Ad-SIRT1 regulated intracellular NO levels via the PI3K/Akt/eNOS signaling pathway in EPCs
The eNOS/NO signaling pathway is recognized as an important mediator of the angiogenic processes. However, the effect of SIRT1 on NO production is unknown. As shown in Figure 10, the intracellular NO levels were elevated after treatment with Ad-SIRT1. However, in comparison with controls, pretreatment of EPCs with LY294002 (30 μM), sc-221226 (30 μM), and L-NAM (200 μM) downregulated Ad-SIRT1-induced NO production in EPCs (Figure 10). Taken together, the results provided direct evidence that Ad-SIRT1 enhanced NO production via the PI3K/Akt/eNOS pathway.
Figure 10.
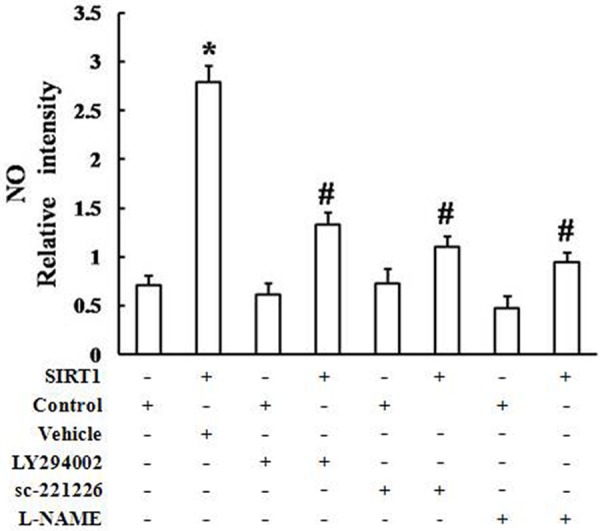
Ad-SIRT1 induced NO production via the PI3K/Akt/eNOS signaling pathway. The intracellular NO level was elevated after treatment with Ad-SIRT1. Pretreatment of EPCs with LY294002 (30 μM), sc-221226 (30 μM), or L-NAM (200 μM) downregulated Ad-SIRT1-induced NO production in EPCs, in comparison with the controls (*P < 0.05 vs. control, #P < 0.05 vs. Ad-SIRT1).
Discussion
Damage of endothelial cells following percutaneous coronary interventions such as angioplasty and stenting is an important pathophysiological event during atherosclerosis and restenosis. EPCs have been found to be the main endogenous repair mechanism that responds to endothelium repair, and contributes to re-endothelialization by reducing neointima formation after vascular injury [17]. This mechanism is regulated by various processes and signals [18-23]. However, the regulatory mechanisms for the biological properties of EPCs remain unclear. Recent studies demonstrated that endothelial SIRT1 may serve as an anti-atherosclerosis factor [9,10], which may be a key component of EPC dysfunction.
The present study offers novel insights into the relationship between the SIRT1 pathway and PI3K/Akt/eNOS signaling in EPCs during migration and proliferation. The results demonstrated that SIRT1 stimulated EPCs migration and proliferation; upregulated the phosphotyrosine levels of PI3K, Akt, and eNOS; and enhanced NO production via the PI3K/Akt/eNOS pathway. These effects were abrogated in the presence of the PI3K-specific inhibitor LY294002, the Akt inhibitor sc-221226, and the NOS inhibitor L-NAME. In addition, knockdown of endogenous SIRT1 significantly reduced the migration and proliferation of EPCs. These results demonstrated that the migration and proliferation of EPCs are all mediated through the SIRT1/PI3K/Akt/eNOS signaling pathway.
SIRT1 is a member of a protein class known as sirtuins, belonging to the Sir2 family, which has been identified as NAD+-dependent deacetylases [24,25]. SIRT1 affects the activity of many proteins, resulting in the regulation of a number of proteins and their translation, which have important roles in biological processes such as metabolism, oxidative stress, and cell proliferation [26-29]. SIRT1 has been implicated in cancer, aging, metabolic diseases, and cardiovascular dysfunctions [30-33]. Direct application of the SIRT1 activator resveratrol has been shown to protect cardiomyocytes against H2O2- and hypoxia-induced apoptosis [34-37]. The antioxidant ability of resveratrol is SIRT1-dependent, because knockdown of SIRT1 resulted in the loss of resveratrol-mediated reduction of reactive oxygen species and cell protection [37,38]. Endothelial SIRT1 may also serve as an anti-atherosclerosis factor. SIRT1 can protect endothelial cells from oxidative stress, and oxidative low-density lipoprotein-induced apoptosis [39,40]. In endothelial cell-specific SIRT1 transgenic mice, high fat-induced impairment in endothelium-dependent vasorelaxation decreased, accompanied by less atherosclerotic lesions [41], suggesting that SIRT1 improved endothelial function to prevent atherosclerosis. SIRT1 is highly expressed in endothelial cells and controls their angiogenic function. It is involved in vascular growth of cultured endothelium, in the formation of the vascular network of the developing zebrafish, and even in ischemia-induced neovascularization of the adult mouse [42-44].
The PI3K/Akt pathway provides essential signaling for cell survival and proliferation. Signaling from different eNOS agonists, such as VEGF, insulin, estrogen, and platelet-derived lipid mediators, can affect eNOS activity through the PI3K/AKT pathway. It has been reported that activations of the survival signal PI3K/Akt pathway and the endothelial specific eNOS/NO pathway were closely associated with vascular remodeling and angiogenesis [13,14,45]. Recent studies also reported that activation of SIRT1 improved endothelium relaxation through up-regulating endothelial nitric oxide synthase (eNOS) expression and production of nitric oxide [46,47]. However, whether SIRT1 affects the biological properties of EPCs, and the role of PI3K/Akt/eNOS signaling pathway in SIRT1-induced migration and proliferation, have remained poorly understood.
In the present study, we determined the effects of the SIRT1/PI3K/Akt/eNOS signaling pathway on EPC migration and proliferation. The results showed that SIRT1 induced the activation of the PI3K/Akt/eNOS signaling pathway during these processes. In vitro transfection of EPCs with Ad-SIRT1 induced phosphorylation of Akt via PI3K, the phosphorylation of eNOS via AKt, and increased the expression of NO via eNOS. In addition, knockdown of endogenous SIRT1 reduced migration and proliferation of EPCs. Together, the results suggested that the migration and proliferation of EPCs are attributable to the up-regulation of SIRT1, p-PI3K, p-Akt, and p-eNOS, as well as the production of NO. In addition, blockage of the PI3K/Akt/eNOS signaling pathway by the PI3K inhibitor LY294002, the Akt-specific inhibitor sc-221226, and the NOS inhibitor L-NAME, abrogated SIRT1-induced EPC migration and proliferation. Furthermore, treatment with the NOS inhibitor L-NAME decreased phosphorylation of eNOS, but did not affect PI3K/Akt activity, suggesting that PI3K/Akt is upstream of eNOS. These findings, therefore demonstrated the existence of a SIRT1/PI3K/Akt/eNOS signaling pathway during EPC migration and proliferation.
In conclusion, by mediating EPC proliferation and recruitment, the present study demonstrated important roles of SIRT1 in neovascularization and re-endothelialization; In addition, the SIRT1/PI3K/Akt/eNOS signaling pathway may also play an important role during these same processes. Future studies should therefore be directed towards characterization of the complex mechanisms and therapeutic potentials ofSIRT1, PI3K, Akt, and eNOS in the angiogenesis and tissue regeneration process mediated by EPCs.
Acknowledgements
Appreciation goes to Huali Kang (technician at the Institute of Cardiovascular Science of PLA) for excellent technical assistance.
Disclosure of conflict of interest
None.
References
- 1.Napoli C, Hayashi T, Cacciatore F, Casamassimi A, Casini C, Al-Omran M, Ignarro LJ. Endothelial progenitor cells as therapeutic agents in the microcirculation: an update. Atherosclerosis. 2011;215:9–22. doi: 10.1016/j.atherosclerosis.2010.10.039. [DOI] [PubMed] [Google Scholar]
- 2.Walter DH, Rittig K, Bahlmann FH, Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara T, Isner JM. Statin therapy accelerates reendothelialization: a novel effect involving mobilization and incorporation of bone marrow-derived endothelial progenitor cells. Circulation. 2002;105:3017–3024. doi: 10.1161/01.cir.0000018166.84319.55. [DOI] [PubMed] [Google Scholar]
- 3.Cho HJ, Kim HS, Lee MM, Kim DH, Yang HJ, Hur J, Hwang KK, Oh S, Choi YJ, Chae IH, Oh BH, Choi YS, Walsh K, Park YB. Mobilized endothelial progenitor cells by granulocyte-macrophage colony-stimulating factor accelerate reendothelialization and reduce vascular inflammation after intravascular radiation. Circulation. 2003;108:2918–2925. doi: 10.1161/01.CIR.0000097001.79750.78. [DOI] [PubMed] [Google Scholar]
- 4.Iwakura A, Luedemann C, Shastry S, Hanley A, Kearney M, Aikawa R, Isner JM, Asahara T, Losordo DW. Estrogen-mediated, endothelial nitric oxide synthase-dependent mobilization of bone marrow-derived endothelial progenitor cells contributes to reendothelialization after arterial injury. Circulation. 2003;108:3115–3121. doi: 10.1161/01.CIR.0000106906.56972.83. [DOI] [PubMed] [Google Scholar]
- 5.Werner N, Junk S, Laufs U, Link A, Walenta K, Bohm M, Nickenig G. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003;93:e17–24. doi: 10.1161/01.RES.0000083812.30141.74. [DOI] [PubMed] [Google Scholar]
- 6.Chen Z, Peng IC, Cui X, Li YS, Chien S, Shyy JY. Shear stress, SIRT1, and vascular homeostasis. Proc Natl Acad Sci U S A. 2010;107:10268–10273. doi: 10.1073/pnas.1003833107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Breitenstein A, Wyss CA, Spescha RD, Franzeck FC, Hof D, Riwanto M, Hasun M, Akhmedov A, von Eckardstein A, Maier W, Landmesser U, Luscher TF, Camici GG. Peripheral blood monocyte Sirt1 expression is reduced in patients with coronary artery disease. PLoS One. 2013;8:e53106. doi: 10.1371/journal.pone.0053106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Potente M, Dimmeler S. Emerging roles of SIRT1 in vascular endothelial homeostasis. Cell Cycle. 2008;7:2117–2122. doi: 10.4161/cc.7.14.6267. [DOI] [PubMed] [Google Scholar]
- 9.Stein S, Matter CM. Protective roles of SIRT1 in atherosclerosis. Cell Cycle. 2011;10:640–647. doi: 10.4161/cc.10.4.14863. [DOI] [PubMed] [Google Scholar]
- 10.Yu W, Fu YC, Chen CJ, Wang X, Wang W. SIRT1: a novel target to prevent atherosclerosis. J Cell Biochem. 2009;108:10–13. doi: 10.1002/jcb.22240. [DOI] [PubMed] [Google Scholar]
- 11.Donmez G, Guarente L. Aging and disease: connections to sirtuins. Aging Cell. 2010;9:285–290. doi: 10.1111/j.1474-9726.2010.00548.x. [DOI] [PubMed] [Google Scholar]
- 12.Li L, Zhang HN, Chen HZ, Gao P, Zhu LH, Li HL, Lv X, Zhang QJ, Zhang R, Wang Z, She ZG, Wei YS, Du GH, Liu DP, Liang CC. SIRT1 acts as a modulator of neointima formation following vascular injury in mice. Circ Res. 2011;108:1180–1189. doi: 10.1161/CIRCRESAHA.110.237875. [DOI] [PubMed] [Google Scholar]
- 13.Namkoong S, Kim CK, Cho YL, Kim JH, Lee H, Ha KS, Choe J, Kim PH, Won MH, Kwon YG, Shim EB, Kim YM. Forskolin increases angiogenesis through the coordinated cross-talk of PKA-dependent VEGF expression and Epac-mediated PI3K/Akt/eNOS signaling. Cell Signal. 2009;21:906–915. doi: 10.1016/j.cellsig.2009.01.038. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Yan W, Lu X, Qian C, Zhang J, Li P, Shi L, Zhao P, Fu Z, Pu P, Kang C, Jiang T, Liu N, You Y. Overexpression of osteopontin induces angiogenesis of endothelial progenitor cells via the avbeta3/PI3K/AKT/eNOS/NO signaling pathway in glioma cells. Eur J Cell Biol. 2011;90:642–648. doi: 10.1016/j.ejcb.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 15.Kang Z, Jiang W, Luan H, Zhao F, Zhang S. Cornin induces angiogenesis through PI3K-Akt-eNOS-VEGF signaling pathway. Food Chem Toxicol. 2013;58:340–346. doi: 10.1016/j.fct.2013.05.017. [DOI] [PubMed] [Google Scholar]
- 16.Wang H, Yin Y, Li W, Zhao X, Yu Y, Zhu J, Qin Z, Wang Q, Wang K, Lu W, Liu J, Huang L. Over-expression of PDGFR-beta promotes PDGF-induced proliferation, migration, and angiogenesis of EPCs through PI3K/Akt signaling pathway. PLoS One. 2012;7:e30503. doi: 10.1371/journal.pone.0030503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu Y, Gao Y, Qin J, Kuang CY, Song MB, Yu SY, Cui B, Chen JF, Huang L. CCN1 promotes the differentiation of endothelial progenitor cells and reendothelialization in the early phase after vascular injury. Basic Res Cardiol. 2010;105:713–724. doi: 10.1007/s00395-010-0117-0. [DOI] [PubMed] [Google Scholar]
- 18.Lim WH, Seo WW, Choe W, Kang CK, Park J, Cho HJ, Kyeong S, Hur J, Yang HM, Lee YS, Kim HS. Stent coated with antibody against vascular endothelial-cadherin captures endothelial progenitor cells, accelerates re-endothelialization, and reduces neointimal formation. Arterioscler Thromb Vasc Biol. 2011;31:2798–2805. doi: 10.1161/ATVBAHA.111.226134. [DOI] [PubMed] [Google Scholar]
- 19.Takamiya M, Okigaki M, Jin D, Takai S, Nozawa Y, Adachi Y, Urao N, Tateishi K, Nomura T, Zen K, Ashihara E, Miyazaki M, Tatsumi T, Takahashi T, Matsubara H. Granulocyte colony-stimulating factor-mobilized circulating c-Kit+/Flk-1+ progenitor cells regenerate endothelium and inhibit neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2006;26:751–757. doi: 10.1161/01.ATV.0000205607.98538.9a. [DOI] [PubMed] [Google Scholar]
- 20.Thyberg J. Re-endothelialization via bone marrow-derived progenitor cells: still another target of statins in vascular disease. Arterioscler Thromb Vasc Biol. 2002;22:1509–1511. doi: 10.1161/01.atv.0000036415.04486.01. [DOI] [PubMed] [Google Scholar]
- 21.Hibbert B, Ma X, Pourdjabbar A, Holm E, Rayner K, Chen YX, Sun J, Filion L, O’Brien ER. Inhibition of endothelial progenitor cell glycogen synthase kinase-3beta results in attenuated neointima formation and enhanced re-endothelialization after arterial injury. Cardiovasc Res. 2009;83:16–23. doi: 10.1093/cvr/cvp156. [DOI] [PubMed] [Google Scholar]
- 22.Padfield GJ, Newby DE, Mills NL. Understanding the role of endothelial progenitor cells in percutaneous coronary intervention. J Am Coll Cardiol. 2010;55:1553–1565. doi: 10.1016/j.jacc.2009.10.070. [DOI] [PubMed] [Google Scholar]
- 23.Kawabe-Yako R, Ii M, Masuo O, Asahara T, Itakura T. Cilostazol activates function of bone marrow-derived endothelial progenitor cell for re-endothelialization in a carotid balloon injury model. PLoS One. 2011;6:e24646. doi: 10.1371/journal.pone.0024646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guarani V, Deflorian G, Franco CA, Kruger M, Phng LK, Bentley K, Toussaint L, Dequiedt F, Mostoslavsky R, Schmidt MH, Zimmermann B, Brandes RP, Mione M, Westphal CH, Braun T, Zeiher AM, Gerhardt H, Dimmeler S, Potente M. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234–238. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 27.Vinciguerra M, Santini MP, Martinez C, Pazienza V, Claycomb WC, Giuliani A, Rosenthal N. mIGF-1/JNK1/SirT1 signaling confers protection against oxidative stress in the heart. Aging Cell. 2012;11:139–149. doi: 10.1111/j.1474-9726.2011.00766.x. [DOI] [PubMed] [Google Scholar]
- 28.Wang Y, Shi X, Qi J, Li X, Uray K, Guan X. SIRT1 inhibits the mouse intestinal motility and epithelial proliferation. Am J Physiol Gastrointest Liver Physiol. 2012;302:G207–217. doi: 10.1152/ajpgi.00302.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ota H, Eto M, Kano MR, Ogawa S, Iijima K, Akishita M, Ouchi Y. Cilostazol inhibits oxidative stress-induced premature senescence via upregulation of Sirt1 in human endothelial cells. Arterioscler Thromb Vasc Biol. 2008;28:1634–1639. doi: 10.1161/ATVBAHA.108.164368. [DOI] [PubMed] [Google Scholar]
- 30.Nadtochiy SM, Yao H, McBurney MW, Gu W, Guarente L, Rahman I, Brookes PS. SIRT1-mediated acute cardioprotection. Am J Physiol Heart Circ Physiol. 2011;301:H1506–1512. doi: 10.1152/ajpheart.00587.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen HC, Jeng YM, Yuan RH, Hsu HC, Chen YL. SIRT1 promotes tumorigenesis and resistance to chemotherapy in hepatocellular carcinoma and its expression predicts poor prognosis. Ann Surg Oncol. 2012;19:2011–2019. doi: 10.1245/s10434-011-2159-4. [DOI] [PubMed] [Google Scholar]
- 32.Yang J, Wang N, Zhu Y, Feng P. Roles of SIRT1 in high glucose-induced endothelial impairment: association with diabetic atherosclerosis. Arch Med Res. 2011;42:354–360. doi: 10.1016/j.arcmed.2011.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Orimo M, Minamino T, Miyauchi H, Tateno K, Okada S, Moriya J, Komuro I. Protective role of SIRT1 in diabetic vascular dysfunction. Arterioscler Thromb Vasc Biol. 2009;29:889–894. doi: 10.1161/ATVBAHA.109.185694. [DOI] [PubMed] [Google Scholar]
- 34.Arunachalam G, Yao H, Sundar IK, Caito S, Rahman I. SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: Role of resveratrol. Biochem Biophys Res Commun. 2010;393:66–72. doi: 10.1016/j.bbrc.2010.01.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu YX, Cui H, Fan L, Pan XJ, Wu JH, Shi SZ, Cui SY, Wei ZM, Liu L. Resveratrol attenuates left ventricular remodeling in old rats with COPD induced by cigarette smoke exposure and LPS instillation. Can J Physiol Pharmacol. 2013;91:1044–1054. doi: 10.1139/cjpp-2012-0464. [DOI] [PubMed] [Google Scholar]
- 36.Li YG, Zhu W, Tao JP, Xin P, Liu MY, Li JB, Wei M. Resveratrol protects cardiomyocytes from oxidative stress through SIRT1 and mitochondrial biogenesis signaling pathways. Biochem Biophys Res Commun. 2013;438:270–276. doi: 10.1016/j.bbrc.2013.07.042. [DOI] [PubMed] [Google Scholar]
- 37.Chen CJ, Yu W, Fu YC, Wang X, Li JL, Wang W. Resveratrol protects cardiomyocytes from hypoxia-induced apoptosis through the SIRT1-FoxO1 pathway. Biochem Biophys Res Commun. 2009;378:389–393. doi: 10.1016/j.bbrc.2008.11.110. [DOI] [PubMed] [Google Scholar]
- 38.Sundaresan NR, Pillai VB, Gupta MP. Emerging roles of SIRT1 deacetylase in regulating cardiomyocyte survival and hypertrophy. J Mol Cell Cardiol. 2011;51:614–618. doi: 10.1016/j.yjmcc.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shimada T, Furuta H, Doi A, Ariyasu H, Kawashima H, Wakasaki H, Nishi M, Sasaki H, Akamizu T. Des-acyl ghrelin protects microvascular endothelial cells from oxidative stress-induced apoptosis through sirtuin 1 signaling pathway. Metabolism. 2014;63:469–474. doi: 10.1016/j.metabol.2013.12.011. [DOI] [PubMed] [Google Scholar]
- 40.Guo H, Chen Y, Liao L, Wu W. Resveratrol protects HUVECs from oxidized-LDL induced oxidative damage by autophagy upregulation via the AMPK/SIRT1 pathway. Cardiovasc Drugs Ther. 2013;27:189–198. doi: 10.1007/s10557-013-6442-4. [DOI] [PubMed] [Google Scholar]
- 41.Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng W, Liu G, Wei YS, Cai H, Liu DP, Liang CC. Endothelium-specific overexpression of class III deacetylase SIRT1 decreases atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res. 2008;80:191–199. doi: 10.1093/cvr/cvn224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Borradaile NM, Pickering JG. Nicotinamide phosphoribosyltransferase imparts human endothelial cells with extended replicative lifespan and enhanced angiogenic capacity in a high glucose environment. Aging Cell. 2009;8:100–112. doi: 10.1111/j.1474-9726.2009.00453.x. [DOI] [PubMed] [Google Scholar]
- 43.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Volkmann I, Kumarswamy R, Pfaff N, Fiedler J, Dangwal S, Holzmann A, Batkai S, Geffers R, Lother A, Hein L, Thum T. MicroRNA-mediated epigenetic silencing of sirtuin1 contributes to impaired angiogenic responses. Circ Res. 2013;113:997–1003. doi: 10.1161/CIRCRESAHA.113.301702. [DOI] [PubMed] [Google Scholar]
- 45.Lee SJ, Namkoong S, Kim YM, Kim CK, Lee H, Ha KS, Chung HT, Kwon YG. Fractalkine stimulates angiogenesis by activating the Raf-1/MEK/ERK- and PI3K/Akt/eNOS-dependent signal pathways. Am J Physiol Heart Circ Physiol. 2006;291:H2836–2846. doi: 10.1152/ajpheart.00113.2006. [DOI] [PubMed] [Google Scholar]
- 46.Davis PA, Pagnin E, Dal Maso L, Caielli P, Maiolino G, Fusaro M, Paolo Rossi G, Calo LA. SIRT1, heme oxygenase-1 and NO-mediated vasodilation in a human model of endogenous angiotensin II type 1 receptor antagonism: implications for hypertension. Hypertens Res. 2013;36:873–878. doi: 10.1038/hr.2013.48. [DOI] [PubMed] [Google Scholar]
- 47.Mattagajasingh I, Kim CS, Naqvi A, Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K, Irani K. SIRT1 promotes endothelium-dependent vascular relaxation by activating endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:14855–14860. doi: 10.1073/pnas.0704329104. [DOI] [PMC free article] [PubMed] [Google Scholar]