

Abstract
Generally accepted, inflammation and neuron apoptosis are two characterized pathological features of cerebral ischemia-reperfusion (IR) injury. Ginsenoside Rg1 was reported showing distinct neuroprotective effect in cerebral IR injury but the underlying mechanisms are still unclear. PPARγ/Heme oxygenase-1 (HO-1) signaling was proved effective in suppressing both apoptosis and inflammation. This study was aimed to investigate whether PPARγ/HO-1 signaling was involved in cerebral IR injury and ginsenoside Rg1’s neuroprotective effect in cerebral IR injury. Cerebral IR injury was induced by middle cerebral artery occlusion in rats. The PPARγ agonist rosiglitazone (ROZ) and the HO-1 inhibitor zinc protoporphyrin-IX (ZnPP) and ginsenoside Rg1 at various concentrations were used to treat the modeled rats. Neurological deficits, apoptosis and inflammation in hippocampus were evaluated. Furthermore, HO-1 enzymatic activity, expression levels of apoptosis-related and inflammation-related proteins, concentrations of inflammatory cytokines were also determined. The results showed that PPARγ activation by ROZ significantly attenuated neurological deficits, apoptosis and inflammation in hippocampus in cerebral IR rats. However, the neuroprotective effect of ROZ was then impaired by HO-1 inhibitor ZnPP. This effect was evidenced by changes of expression levels of PPARγ, bcl-2, cleaved caspase-3, cleaved caspase-9, IL-1β, TNF-α, HMGB1, and RAGE in hippocampus of modeled animals. Ginsenoside Rg1 showed similar effect to ROZ in activating PPARγ/HO-1 in protecting against apoptosis and inflammation but also impaired by ZnPP administration. In conclusion, PPARγ/HO-1 signaling was critical in mediating apoptosis and inflammation, which was also the therapeutic target of ginsenoside Rg1 in cerebral IR injury.
Keywords: Cerebral ischemia-reperfusion injury, PPARγ, apoptosis, inflammation
Introduction
Resulted from either permanent or transient cerebral blood flow reduction or pause, ischemic stroke becomes one of the leading causes of morbidity and mortality globally [1]. It was believed that ischemia- reperfusion (IR) injury was the underlying pathogenesis for neurological damage in stroke [2]. Generally accepted and frequently reported, cell apoptosis is the associated molecular mechanisms of IR injury. In the brain, pyramidal neurons in hippocampal CA1 region were considered the most vulnerable neurocytes to IR injury- induced apoptosis according to previous studies [3]. Moreover, loss and dysfunction of CA1 neurons was considered to lead to secondary neurological injuries such as long- term learning deficits [4].
Inflammatory responses right after occurrence of stroke were also believed to take responsibility in brain IR injury. The immune cells such as neutrophils were activated to participate in inflammatory response [5]. Several previous studies reported that inflammatory cytokines including interleukin (IL)-1 beta, IL-10, tumor necrosis factor (TNF)-alpha mediated inflammation reactions in cerebral IR injury by inducing microcirculation dysfunction, aggravating cell apoptosis and recruiting inflammatory cell accumulation [6]. Application of anti- inflammation therapies showed significant attenuating effects on the neurological injuries in experimental stroke animal models [7]. Thus, drugs targeting inflammation and neuron apoptosis are potential ideal candidates in neuroprotective treatment of ischemic stroke.
As a typical and representative drug in traditional medicine in Eastern Asian region, ginseng has been used to treat many kinds of organic and psychosomatic diseases from ancient times. Ginsenoside Rg1 (GRg1) is one of the bioactive components extracted from ginseng, which exerts anti-inflammation, antioxidant, anti-proliferative and anti-apoptosis pharmacological activities [8,9]. In several IR-injury related diseases, such as myocardial infarction and liver ischemia, GRg1 showed abundant protective and curative effects [10,11]. However, mechanisms of effects of GRg1’s protective role in cerebral IR injury are rare are still unclear.
Peroxisome proliferator-activated receptor γ (PPARγ) is a member of nuclear hormone receptor superfamily, which was believed mediating many signaling pathways in various pathological conditions [12]. Activation of PPARγ was considered protective against both apoptosis and inflammation [13,14]. After activated by specific ligands, PPARγ was translocated to further activate PPAR response elements (PPREs) to initiate target gene transcription. Heme oxygenase-1 (HO-1) is one of the down-stream effecter of PPARγ [15]. Notably, HO-1 was shown to suppress apoptosis and inflammation [16,17]. It was reported in a recent study that GRg1 attenuated neurological damage in hippocampus in rat model of Alzheimer’s disease by up-regulating PPARγ [18].
Thus, we speculated that whether PPARγ/HO-1 activation was involved in GRg1’s neuroprotective effects against cerebral ischemia-reperfusion injury. To testify our hypothesis, the neurological deficits were evaluated in rat model of cerebral IR injury treated by GRg1, PPARγ agonist and HO-1 inhibitor. Moreover, PPARγ expression, HO-1 activity, inflammatory cytokines and apoptosis in hippocampus were also assessed. We believe that the results in this study would not only enrich our knowledge concerning the pathogenesis, but also provide solid supporting evidence for further application of GRg1 in treatment of ischemic stroke.
Material and methods
Animals, cerebral IR injury modeling and treatment
108 male Sprague-Dawley (SD) rats were randomly and evenly divided into 8 groups: namely Sham group (Sham), ischemia-reperfusion group (IR), rosiglitazone treated ischemia-reperfusion group (ROZ + IR), rosiglitazone and zinc protoporphyrin-IX treated ischemia-reperfusion group (ROZ + ZnPP + IR), high-dose GRg1 treated ischemia-reperfusion group (HGRg1 + IR), medium-dose GRg1 treated ischemia-reperfusion group (MGRg1 + IR), low-dose GRg1 treated ischemia-reperfusion group (LGRg1 + IR), GRg1 and ZnPP treated ischemia-reperfusion group (ZnPP + GRg1 + IR). This study was approved by the Institutional Research Committee of Xi’an Jiaotong University and performed at the Surgical Dream Works Laboratory. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health.
The IR injury was modeled by middle cerebral artery occlusion (MCAO) according to protocol described previously [6]. The internal and external carotid artery were exposed and isolated. Then the distal part of external carotid artery was dissected. Monofilament nylon (4-0) was introduced into internal carotid artery through external carotid artery to perform MCAO. After 2 hours, MCAO was withdrawn to introduce reperfusion.
Rosiglitazone (GSK, 5 mg/kg) was administrated orally 6 hours prior to MCAO; ZnPP (Sigma-Aldrich, 10 mg/kg) was administrated by intraperitoneal injections with every rosiglitazone treatment; high-dose (60 mg/kg) GRg1 (Sigma-Aldrich), medium-dose (40 mg/kg) GRg1 or low-dose GRg1 (20 mg/kg) which was showed effective neuroprotective effect according to previous studies was dissolved in physiological saline and administrated by intravenous injection 1 hour before MCAO.
Neurological deficits determination
As described in previous studies [19], a five-point scale was employed to evaluate the neurological defects 24 hours after reperfusion. All investigators were blinded to animal groups. The scale was divided into five grades from 0 to 4. Grade 0 referred to no neurologic deficit (normal), grade 1 referred to flexion of forelimbs and contralateral torso when lifted by tail (mild); grade 2 referred to circling to contralateral side but maintaining normal posture at rest (moderate); grade 3 referred to leaning to the contralateral side at rest (severe); grade 4 referred to no spontaneous motor activities (very severe).
In situ apoptosis assay
The cell apoptosis in hippocampus was assessed by TUNEL fluorescent staining according to previous descriptions. Briefly, the rats were scarified by over-dose anesthesia by chloral hydrate (10%) intraperitoneal injection. The brain tissue was harvested and further embedded by OCT (Sakura) and then cut to frozen sections of 10 μm at -20°C. 0.1% Triton X-100 buffer was used to treat the sections. After washing by cold PBS buffer, TUNEL stain mixture (Roche) was added to the tissue sections to incubate at 37°C for 1 hour in a dark chamber. Excitation at 450 nm and observed at 550 nm, a fluorescence microscopy (Nikon) was employed to observe and quantify the apoptotic cells.
Enzyme-linked immunosorbent (ELISA) assay
The modeled rats were scarified by over-dose anesthesia by chloral hydrate (10%) intraperitoneal injection. The brain tissue was harvested and the hippocampus was isolated from ischemic cortex. Homogenates from the fresh hippocampus tissue were used for ELISA assay. Rat ELISA kits (R & D) were used to detect the concentrations of IL-1β, TNFα and high-mobility group box-1 (HMGB1) in hippocampus homogenates. All experimental protocols were in accordance with the instructions from the manufacturer.
HO-1 enzymatic activity evaluation
The testing sample was acquired as the supernatant which was resulted from centrifugation of hippocampus tissue homogenates at 10000 g for 15 minute at 4°C. A HO-1 enzymatic activity assay kit (GenMed) was used to evaluate the HO-1 enzymatic activity per manufacturer’s instructions.
Western blotting
Western blotting assay was implemented according to protocol described previously. Harvested hippocampus tissue were homogenized by RIPA lysis buffer system (Santa Cruz) supplemented with protease inhibitor cocktail (Santa Cruz) on ice. The total protein was collected after the homogenates were centrifuged at 14000 g for 20 minutes at 4°C. The protein concentration was detected by BCA method with a BCA Protein Assay kit (Thermo Scientific). 30 μg protein from each sample was separated by electrophoresis on SDS-PAGE gel and then transferred to PVDF membranes electronically. 5% defatted milk was used to block the possible unspecific binding. Specific antibodies against PPARγ (Cell Signaling Tech), Bcl-2 (Abcam), cleaved caspase-3 (Cell Signalling Tech), cleaved caspase-9 (Cell Signalling Tech), receptor for advanced glycation end product (RAGE) (Abcam) and GAPDH (Santa Cruz) were used to incubate the membranes at 4°C for 12 hours. After the horseradish peroxidase-conjungated secondary antibodies incubation, the immunoblots were detected by using Enzymatic Chemiluminescence (ECL) kit (Bio-Rad). The intensities of the immunoblots were analyzed by software ImageJ2x.
Statistical considerations
Data acquired in this study were expressed in a (mean ± SD) manner. One-way analysis of variance (ANOVA) was used to analyze the differences between groups. A LSD analysis was followed. P < 0.05 was considered statistically significant.
Results
Effects of rosiglitazone, ZnPP and RGg1 treatments on neurological impairments in IR rats
We first evaluated the effects of rosiglitazone and ZnPP on neurological deficits. 24 hours after reperfusion, neurologic deficits were scored in rats. As shown in Figure 1A, no rats showed neurological deficit in Sham; neurologic score reduced significantly in ROZ + IR compared with IR. However, neurologic score increased in ROZ + ZnPP + IR compared with ROZ + IR significantly. Wheather RGg1 attenuated neurological impairment in IR rats? As shown in Figure 1B, neurological score decreased significantly in GRg1 treated rats than IR in a concentration-dependent manner. However, this effect was reversed by ZnPP treatment in ZnPP + GRg1 + IR.
Figure 1.
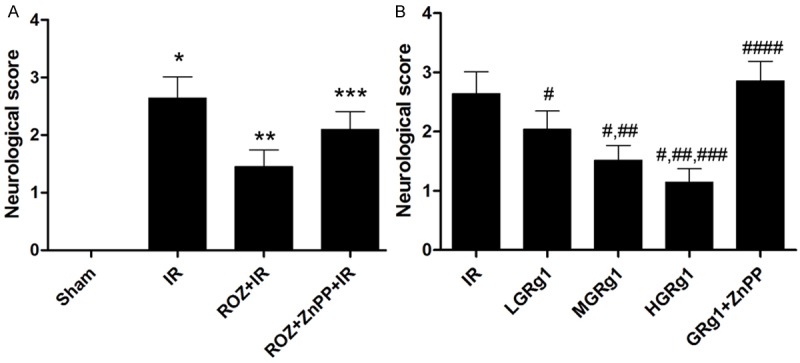
Effects of rosiglitazone, ZnPP and RGg1 treatments on neurological impairments on IR rats. Columns on this figure indicated the neurological score in IR rats received different treatments. The neurological score was determined by a 5-point scale. A. Neurological deficits score in sham, IR, ROZ + IR and ROZ + ZnPP + IR respectively. Differences were significant when compared with *Sham, **IR, ***ROZ + IR. B. Neurological deficits score in IR, LGRg1, MGRg1, HGRg1 and GRg1 + ZnPP respectively. Differences were significant when compared with #IR, ##LGRg1, ###MGRg1, ####HGRg1.
Effects of rosiglitazone, ZnPP and RGg1 treatments on apoptosis in hippocampus in IR rats
TUNEL fluorescent staining was employed to detect the cell apoptosis in hippocampus region. As demonstrated in Figure 2, apoptotic rate in hippocampus region elevated significantly in IR compared with Sham. The apoptotic rate decreased significantly by rosiglitazone administration in ROZ + IR than IR. Nevertheless, ZnPP reversed rosiglitazon’s anti-apoptotic effect in ROZ + ZnPP + IR. In addition, GRg1 administration dramatically alleviated apoptosis in hippocampus in a concentration-dependent manner (Figure 2). However, apoptotic rate in ZnPP + GRg1 + IR was significantly higher than IR rats treated with GRg1 (Figure 2).
Figure 2.
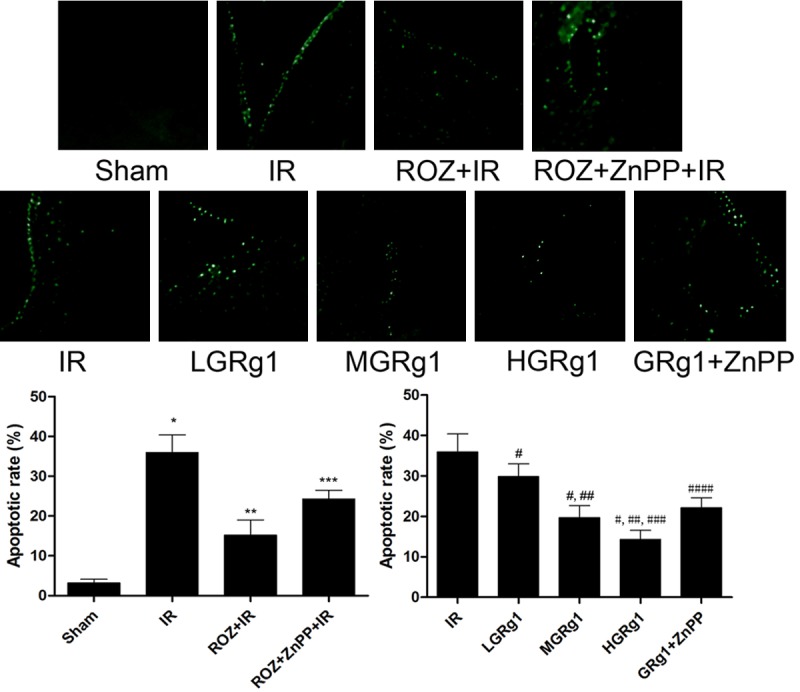
Effects of rosiglitazone, ZnPP and RGg1 treatments on apoptosis in hippocampus in IR rats. The upper panel in this figure showed the captured images of TUNEL fluorescent stain in hippocampus in different groups. The positive staining was shown as green fluorescence in these images. The lower panel demonstrated the calculated apoptotic rate in different groups. Differences were significant when compared with *Sham, **IR, ***ROZ + IR; Differences were significant when compared with #IR, ##LGRg1, ###MGRg1, ####HGRg1.
Rosiglitazone and RGg1 exerted anti-inflammation effect which was impaired by ZnPP in hippocampus
Figure 3 demonstrated the expression levels of several inflammatory cytokines including IL-1β, TNFα and HMGB1 in hippocampus homogenates from IR rats. We found that the expression levels of IL-1β, TNFα and HMGB1 in IR were increased significantly but inhibited in ROZ + IR. We also found that ZnPP treatment in ZnPP + ROZ + IR elevated the expression level of these cytokines compared with ROZ + IR. Moreover, After RGg1 treatment, the IL-1β, TNFα and HMGB1 concentrations decreased in IR rats. However, ZnPP treatment significantly impaired RGg1’s anti-inflammatory effect (Figure 3).
Figure 3.
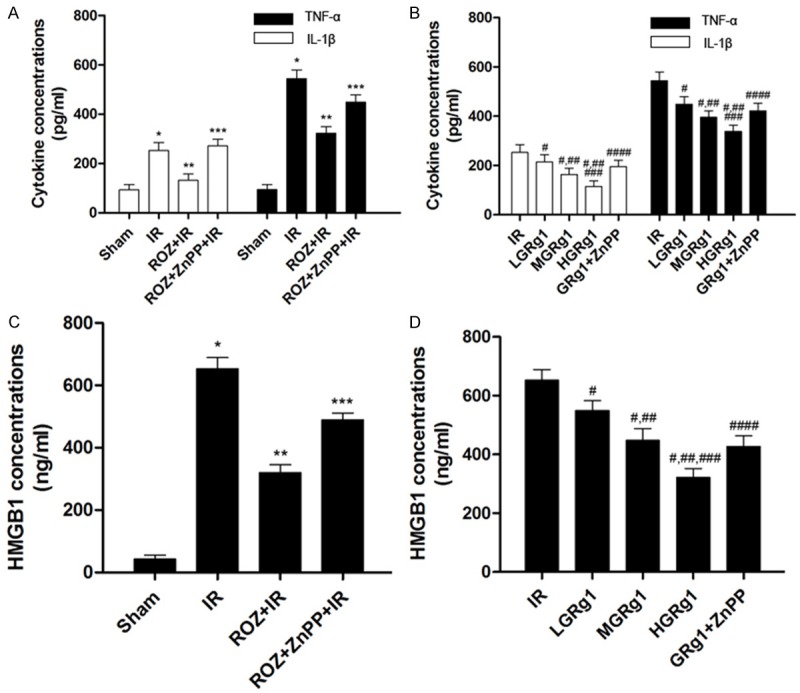
Effects of rosiglitazone, ZnPP and RGg1 treatments on inflammation in hippocampus in IR rats. The concentrations of inflammatory cytokines were determined by ELISA in this study. A and C showed the concentrations of IL-1β, TNF-α and HMGB1 in sham, IR, ROZ + IR and ROZ + ZnPP + IR respectively. B and D demonstrated the concentrations of IL-1β, TNF-α and HMGB1 in IR, LGRg1, MGRg1, HGRg1 and GRg1 + ZnPP respectively. Differences were significant when compared with *Sham, **IR, ***ROZ + IR; Differences were significant when compared with #IR, ##LGRg1, ###MGRg1, ####HGRg1.
Effects of rosiglitazone, ZnPP and GRg1 on PPARγ expression and HO-1 activity in hippocampus from IR rats
Firstly, we found that PPARγ expression and HO-1 activity was suppressed in IR rats compared with Sham. Rosiglitazone administration elevated PPARγ expression and HO-1 activity but HO-1 activity was suppressed by ZnPP treatment. Secondly, GRg1 treatment showed similar effect on PPARγ expression and HO-1 activity as rosiglitazone but HO-1 activity elevation was also reversed by ZnPP (Figure 4).
Figure 4.
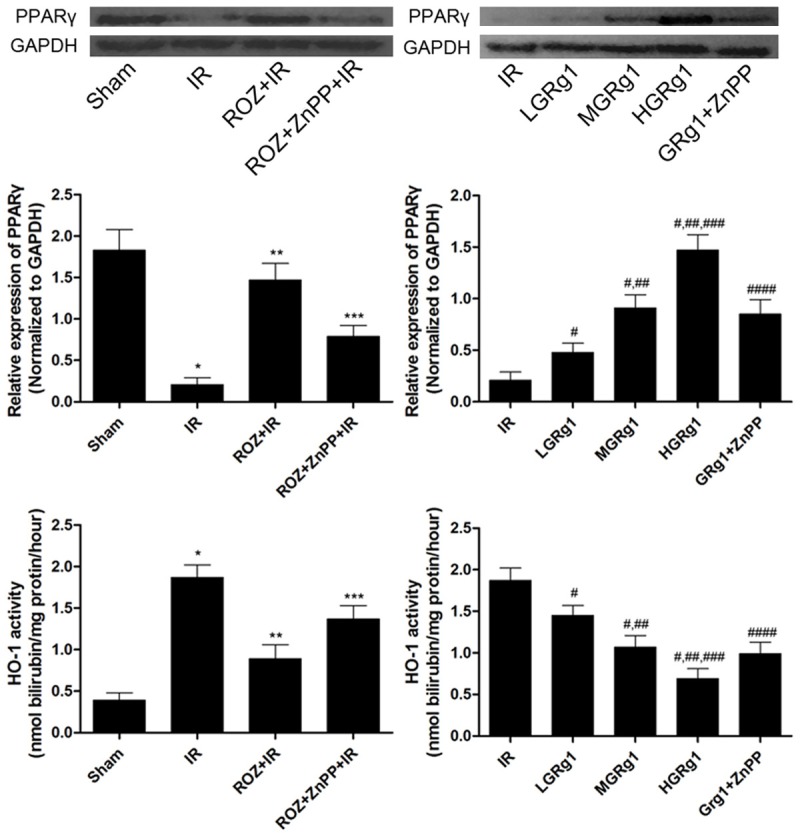
Effects of rosiglitazone, ZnPP and GRg1 treatments on PPARγ/HO-1 activation in IR rats. The upper panel demonstrated the immunoblots of PPARγ and GAPDH in different groups. The columns in the middle panel indicated the relative protein expression of PPARγ (normalized to GAPDH) in different groups. The columns in at the bottom of this figure indicated the HO-1 activity in different groups. Differences were significant when compared with *Sham, **IR, ***ROZ + IR; Differences were significant when compared with #IR, ##LGRg1, ###MGRg1, ####HGRg1.
Effects of rosiglitazone, ZnPP and GRg1 on inflammation-and apoptosis-related proteins
Bcl-2 expression was down-regulated, while expressions of cleaved caspase-3, cleaved caspase-9 and RAGE were up-regulated in IR than Sham. In ROZ + IR, Bcl-2 expression was up-regulated while expressions of cleaved caspase-3, cleaved caspase-9 and RAGE were suppressed but then reversed by ZnPP. Similar to the effect of rosiglitazone, GRg1 also up-regulated Bcl-2 expression but down-regulated expressions of cleaved caspase-3, cleaved caspase-9 and RAGE in IR rats. However, this effect was attenuated by ZnPP administration (Figure 5).
Figure 5.
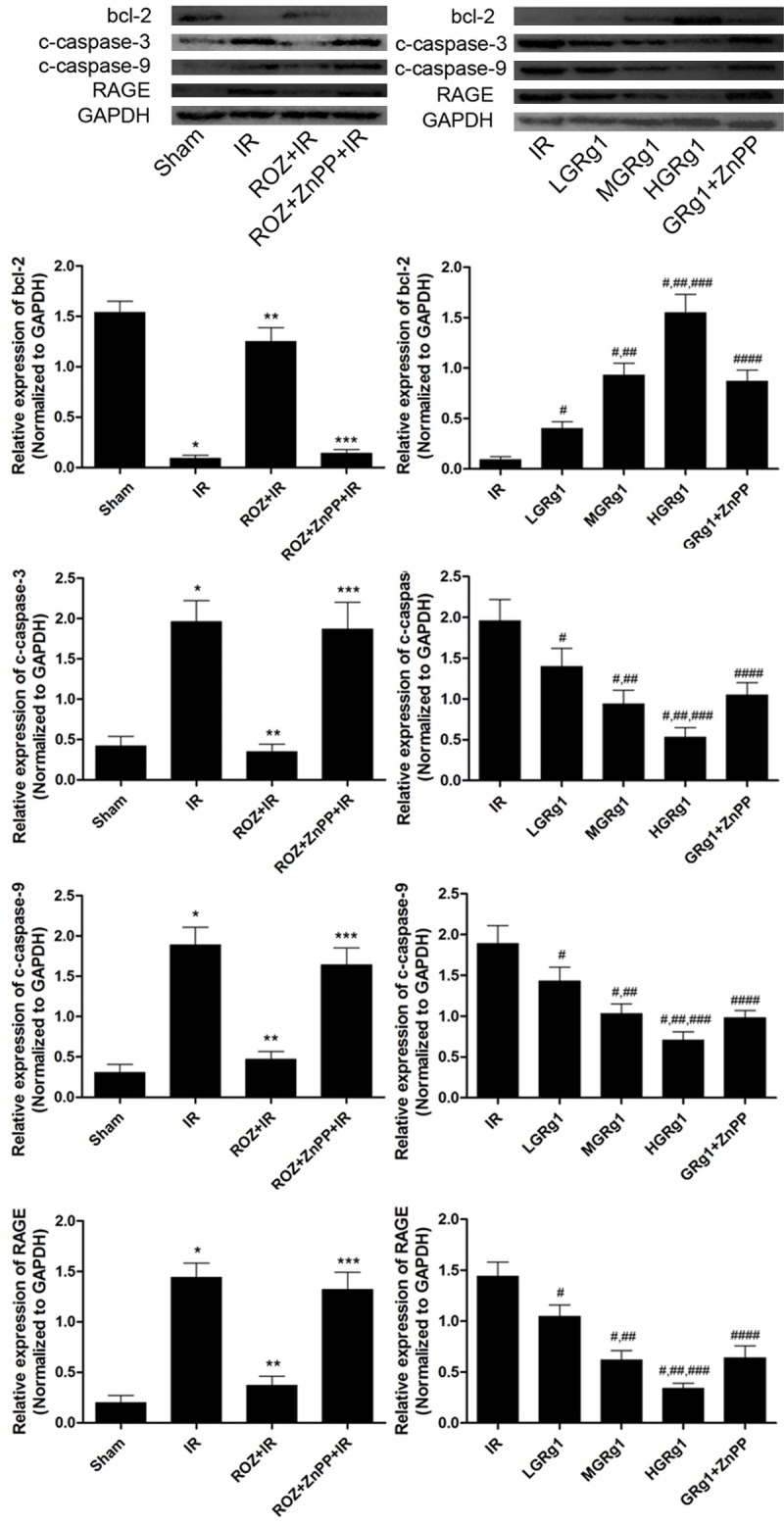
Effects of rosiglitazone ZnPP and GRg1 treatments on expression levels of bcl-2, cleaved caspase-3, cleaved caspase-9 and RAGE in IR rats. The upper panels showed the immunoblots of bcl-2, cleaved caspase-3 (c-caspase-3), cleaved caspase-9 (c-caspase-9), RAGE and GAPDH in different groups. The columns on the lower panels demonstrated the relative expression levels of bcl-2, c-caspase-3, c-caspase-9 and RAGE in different groups. Differences were significant when compared with *Sham, **IR, ***ROZ + IR; Differences were significant when compared with #IR, ##LGRg1, ###MGRg1, ####HGRg1.
Discussion
In this present study, we firstly investigated the role of PPARγ in cerebral IR injury in hippocampus. The cerebral IR model was introduced by MCAO, which resulted in neurological deficits deterioration. The administration of rosiglitazone, the PPARγ agonist, significantly reduced the apoptosis and inflammation in hippocampus and thus neurological deficits and thus attenuated neurological deficits in IR rats. However, the HO-1 inhibitor ZnPP reversed PPARγ activation’s neuroprotective effect. Secondly, we also investigated the neuroprotective effect of GRg1 in cerebral IR rats. The results suggested that PPARγ mediated signaling was involved in the anti-apoptosis and anti-inflammation effects of GRg1 in hippocampus cerebral IR injury.
IR injury refers to the tissue and cellular damage induced by oxygen supply was restored to tissue suffered a prolonged ischemia. Cell damage were obvious in both ischemia and reperfusion phases [20]. It was believed that cell apoptosis and inflammation were characterized pathological changes during cerebral IR injury, leading to neurological dysfunctions [7]. In this study, we applied MCAO to induce cerebral IR injury in rats and the neurological deficits were found deteriorated. Furthermore, in hippocampus of cerebral IR rats, the cell apoptosis rate was found increased by TUNEL staining and the inflammation was found stimulated evidenced by inflammatory cytokines level elevation.
Indeed, the notion that PPARγ activation protected against apoptosis and inflammation was well established in many previous studies [21,22]. Furthermore, it has been also shown that some of these protective effects of PPARγ were introduced by one of its down-stream effectors HO-1 [23]. HO-1 which could be categorized into stress-responsive proteins degrades pro-oxidant heme into free iron, biliverdin (bilirubin) and carbon monoxide. HO-1 exerts its various biological activities by these enzymatic reactions. Previous studies reported that lack of HO-1 resulted in significant increase in apoptotic cell rate [24]. Other studies indicated that caspase-3 cleavage reduction was involved in HO-1 mediated apoptosis inhibitory effect [25]. Besides, by activating anti-apoptotic factors such as bcl-2, HO-1 was proved to prevent apoptosis via stabilizing mitochondrial transport carriers and maintaining mitochondrial functions [26]. Elevated HO-1 activity was also found to inhibit cytochrome C release to improve cell survival [27]. In the present study, we found that the HO-1 activity was increased after administration of rosiglitazone in cerebral IR rats; as a result, the apoptosis was reduced in hippocampus. Expression changes of corresponding proteins, including bcl-2, cleaved caspase-3 and cleaved caspase-9 were also observed. ZnPP treatment reversed this anti-apoptotic effect, which further indicated the anti-apoptotic role of PPARγ was induced via HO-1 in cerebral IR injury.
During the onset of cerebral IR injury, complex cellular biological processes were triggered. It was reported that within several hours after cerebral IR injury, the resident glial cells were activated; circulating inflammatory cells such as neutrophils were recruited and infiltrated into brain [28]. Thus, inflammatory responses were considered playing an important role in cerebral IR injury [29]. The infiltrated inflammatory cells would secrete inflammatory cytokines. These cytokines then further activate and recruit more inflammatory cells which mediate neurological damage by releasing proteases and reactive oxygen spices (ROS) [30]. In some recent studies, HMGB1 which was also considered as an inflammatory take part in several inflammatory pathological conditions such as acute lung injury, asthma and acute myocardial infarction [31,32]. After binding to its receptor, namely RAGE, many signaling cascades were activated by HMGB1-RAGE axis, such as NF-κB and MAPK which eventually initiated the transcription of genes of various inflammatory mediators including IL-1βand TNF-α [33,34] we detected by ELISA in the present study. Expression levels of IL-1β, TNF-α and HMGB1 in hippocampus from cerebral IR rats were reduced after PPARγ was activated by rosiglitazone which was reversed by ZnPP administration. Considering the conclusion that HO-1 induction was coupled with subsequent reduction of HMGB1-RAGE axis, the above results in our study concerning apoptosis and inflammation indicated that PPARγ/HO-1 was located on the central position in the network regulating apoptosis and inflammation in cerebral IR injury.
GRg1 is one of the major active ingredients of ginseng, which shows a brand spectrum of biological activities. Previous studies described the neuroprotective effects of GRg1 in treating neurodegenerative diseases [35]. However, the exact mechanisms of GRg1 in cerebral IR injury treatment are still unclear. In this study, we found that GRg1 showed dramatic curative effect in cerebral IR rats which was evidenced by improvement of neurological deficits score. In a recent study in regard of Alzheimer’s disease, GRg1 was proposed to attenuate learning and memory by activating PPARγ in hippocampus in rat [18]. Similar to this finding, we detected dramatically increased PPARγ in hippocampus in cerebral IR rats. Furthermore, both of the apoptosis and inflammation were attenuated by GRg1 treatment. Concurrently, the HO-1 activity was also found increased in GRg1 treated rats. However, GRg1’s neuroprotective effect was impaired by ZnPP administration, which was proved by neurological dysfunction, increased apoptosis and inflammation in hippocampus. Mechanically, compared with GRg1-treated cerebral IR rats, the expressions of molecules in PPARγ/HO-1 down-stream pathways were also reversed by ZnPP. These results suggested that the neuroprotective effect of GRg1 was similar to the PPARγ agonist rosiglitazone in cerebral IR injury by attenuating apoptosis and inflammation in hippocampus through PPARγ/HO-1 governed signaling pathways.
Disclosure of conflict of interest
None.
References
- 1.Livesay SL. Clinical review and implications of the guideline for the early management of patients with acute ischemic stroke. AACN Adv Crit Care. 2014;25:130–141. doi: 10.1097/NCI.0000000000000017. [DOI] [PubMed] [Google Scholar]
- 2.Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab. 2006;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]
- 3.Zhou JG, Liu JC, Fang YQ. [Study on the effect of hyperbaric oxygen on apoptosis in the CA1 region of hippocampus following forebrain ischemia reperfusion] . Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2001;17:82–84. [PubMed] [Google Scholar]
- 4.Zamani M, Hassanshahi J, Soleimani M, Zamani F. Neuroprotective effect of olive oil in the hippocampus CA1 neurons following ischemia: Reperfusion in mice. J Neurosci Rural Pract. 2013;4:164–170. doi: 10.4103/0976-3147.112753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Justicia C, Panes J, Sole S, Cervera A, Deulofeu R, Chamorro A, Planas AM. Neutrophil infiltration increases matrix metalloproteinase-9 in the ischemic brain after occlusion/reperfusion of the middle cerebral artery in rats. J Cereb Blood Flow Metab. 2003;23:1430–1440. doi: 10.1097/01.WCB.0000090680.07515.C8. [DOI] [PubMed] [Google Scholar]
- 6.Ye XH, Wu Y, Guo PP, Wang J, Yuan SY, Shang Y, Yao SL. Lipoxin A4 analogue protects brain and reduces inflammation in a rat model of focal cerebral ischemia reperfusion. Brain Res. 2010;1323:174–183. doi: 10.1016/j.brainres.2010.01.079. [DOI] [PubMed] [Google Scholar]
- 7.Zhang S, Qi Y, Xu Y, Han X, Peng J, Liu K, Sun CK. Protective effect of flavonoid-rich extract from Rosa laevigata Michx on cerebral ischemia-reperfusion injury through suppression of apoptosis and inflammation. Neurochem Int. 2013;63:522–532. doi: 10.1016/j.neuint.2013.08.008. [DOI] [PubMed] [Google Scholar]
- 8.Wei CB, Jia JP, Liang P, Guan YQ. [Ginsenoside-Rg1 inhibits cell apoptosis induced by beta amyloid] . Zhonghua Yi Xue Za Zhi. 2008;88:1763–1766. [PubMed] [Google Scholar]
- 9.Ma ZC, Gao Y, Wang YG, Tan HL, Xiao CR, Wang SQ. Ginsenoside Rg1 inhibits proliferation of vascular smooth muscle cells stimulated by tumor necrosis factor-alpha. Acta Pharmacol Sin. 2006;27:1000–1006. doi: 10.1111/j.1745-7254.2006.00331.x. [DOI] [PubMed] [Google Scholar]
- 10.Tao T, Chen F, Bo L, Xie Q, Yi W, Zou Y, Hu B, Li J, Deng X. Ginsenoside Rg1 protects mouse liver against ischemia-reperfusion injury through anti-inflammatory and anti-apoptosis properties. J Surg Res. 2014;191:231–238. doi: 10.1016/j.jss.2014.03.067. [DOI] [PubMed] [Google Scholar]
- 11.Yin H, Liu Z, Li F, Ni M, Wang B, Qiao Y, Xu X, Zhang M, Zhang J, Lu H, Zhang Y. Ginsenoside-Rg1 enhances angiogenesis and ameliorates ventricular remodeling in a rat model of myocardial infarction. J Mol Med (Berl) 2011;89:363–375. doi: 10.1007/s00109-011-0723-9. [DOI] [PubMed] [Google Scholar]
- 12.Aleshin S, Strokin M, Sergeeva M, Reiser G. Peroxisome proliferator-activated receptor (PPAR)beta/delta, a possible nexus of PPARalpha- and PPARgamma-dependent molecular pathways in neurodegenerative diseases: Review and novel hypotheses. Neurochem Int. 2013;63:322–330. doi: 10.1016/j.neuint.2013.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Klotz L, Diehl L, Dani I, Neumann H, von Oppen N, Dolf A, Endl E, Klockgether T, Engelhardt B, Knolle P. Brain endothelial PPARgamma controls inflammation-induced CD4+ T cell adhesion and transmigration in vitro. J Neuroimmunol. 2007;190:34–43. doi: 10.1016/j.jneuroim.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 14.Ishibashi Y, Matsui T, Ohta K, Tanoue R, Takeuchi M, Asanuma K, Fukami K, Okuda S, Nakamura K, Yamagishi S. PEDF inhibits AGE-induced podocyte apoptosis via PPAR-gamma activation. Microvasc Res. 2013;85:54–58. doi: 10.1016/j.mvr.2012.10.007. [DOI] [PubMed] [Google Scholar]
- 15.Park SY, Bae JU, Hong KW, Kim CD. HO-1 Induced by Cilostazol Protects Against TNF-alpha-associated Cytotoxicity via a PPAR-gamma-dependent Pathway in Human Endothelial Cells. Korean J Physiol Pharmacol. 2011;15:83–88. doi: 10.4196/kjpp.2011.15.2.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li YX, Li G, Dong WP, Lu DR, Tan JM. Protection of human islets from induction of apoptosis and improved islet function with HO-1 gene transduction. Chin Med J (Engl) 2006;119:1639–1645. [PubMed] [Google Scholar]
- 17.Carter EP, Garat C, Imamura M. Continual emerging roles of HO-1: protection against airway inflammation. Am J Physiol Lung Cell Mol Physiol. 2004;287:L24–25. doi: 10.1152/ajplung.00097.2004. [DOI] [PubMed] [Google Scholar]
- 18.Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T. Efficacy of PPAR-gamma agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging. 2011;32:1626–1633. doi: 10.1016/j.neurobiolaging.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 19.Luan H, Kan Z, Xu Y, Lv C, Jiang W. Rosmarinic acid protects against experimental diabetes with cerebral ischemia: relation to inflammation response. J Neuroinflammation. 2013;10:28. doi: 10.1186/1742-2094-10-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren M, Senatorov VV, Chen RW, Chuang DM. Postinsult treatment with lithium reduces brain damage and facilitates neurological recovery in a rat ischemia/reperfusion model. Proc Natl Acad Sci U S A. 2003;100:6210–6215. doi: 10.1073/pnas.0937423100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Efrati S, Berman S, Ilgiyeav E, Averbukh Z, Weissgarten J. PPAR-gamma activation inhibits angiotensin II synthesis, apoptosis, and proliferation of mesangial cells from spontaneously hypertensive rats. Nephron Exp Nephrol. 2007;106:e107–112. doi: 10.1159/000104834. [DOI] [PubMed] [Google Scholar]
- 22.Bernardo A, Minghetti L. PPAR-gamma agonists as regulators of microglial activation and brain inflammation. Curr Pharm Des. 2006;12:93–109. doi: 10.2174/138161206780574579. [DOI] [PubMed] [Google Scholar]
- 23.Kronke G, Kadl A, Ikonomu E, Bluml S, Furnkranz A, Sarembock IJ, Bochkov VN, Exner M, Binder BR, Leitinger N. Expression of heme oxygenase-1 in human vascular cells is regulated by peroxisome proliferator-activated receptors. Arterioscler Thromb Vasc Biol. 2007;27:1276–1282. doi: 10.1161/ATVBAHA.107.142638. [DOI] [PubMed] [Google Scholar]
- 24.Cai C, Teng L, Vu D, He JQ, Guo Y, Li Q, Tang XL, Rokosh G, Bhatnagar A, Bolli R. The heme oxygenase 1 inducer (CoPP) protects human cardiac stem cells against apoptosis through activation of the extracellular signal-regulated kinase (ERK)/NRF2 signaling pathway and cytokine release. J Biol Chem. 2012;287:33720–33732. doi: 10.1074/jbc.M112.385542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lang D, Reuter S, Buzescu T, August C, Heidenreich S. Heme-induced heme oxygenase-1 (HO-1) in human monocytes inhibits apoptosis despite caspase-3 up-regulation. Int Immunol. 2005;17:155–165. doi: 10.1093/intimm/dxh196. [DOI] [PubMed] [Google Scholar]
- 26.Lin H, Yu CH, Jen CY, Cheng CF, Chou Y, Chang CC, Juan SH. Adiponectin-mediated heme oxygenase-1 induction protects against iron-induced liver injury via a PPARalpha dependent mechanism. Am J Pathol. 2010;177:1697–1709. doi: 10.2353/ajpath.2010.090789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bindu S, Pal C, Dey S, Goyal M, Alam A, Iqbal MS, Dutta S, Sarkar S, Kumar R, Maity P, Bandyopadhyay U. Translocation of heme oxygenase-1 to mitochondria is a novel cytoprotective mechanism against non-steroidal anti-inflammatory drug-induced mitochondrial oxidative stress, apoptosis, and gastric mucosal injury. J Biol Chem. 2011;286:39387–39402. doi: 10.1074/jbc.M111.279893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liou KT, Shen YC, Chen CF, Tsao CM, Tsai SK. Honokiol protects rat brain from focal cerebral ischemia-reperfusion injury by inhibiting neutrophil infiltration and reactive oxygen species production. Brain Res. 2003;992:159–166. doi: 10.1016/j.brainres.2003.08.026. [DOI] [PubMed] [Google Scholar]
- 29.Zeng L, Wang Y, Liu J, Wang L, Weng S, Chen K, Domino EF, Yang GY. Pro-inflammatory cytokine network in peripheral inflammation response to cerebral ischemia. Neurosci Lett. 2013;548:4–9. doi: 10.1016/j.neulet.2013.04.037. [DOI] [PubMed] [Google Scholar]
- 30.Kaundal RK, Sharma SS. GW1929: a nonthiazolidinedione PPARgamma agonist, ameliorates neurological damage in global cerebral ischemic-reperfusion injury through reduction in inflammation and DNA fragmentation. Behav Brain Res. 2011;216:606–612. doi: 10.1016/j.bbr.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Asavarut P, Zhao H, Gu J, Ma D. The role of HMGB1 in inflammation-mediated organ injury. Acta Anaesthesiol Taiwan. 2013;51:28–33. doi: 10.1016/j.aat.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 32.Buhimschi CS, Baumbusch MA, Dulay AT, Oliver EA, Lee S, Zhao G, Bhandari V, Ehrenkranz RA, Weiner CP, Madri JA, Buhimschi IA. Characterization of RAGE, HMGB1, and S100beta in inflammation-induced preterm birth and fetal tissue injury. Am J Pathol. 2009;175:958–975. doi: 10.2353/ajpath.2009.090156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ito Y, Bhawal UK, Sasahira T, Toyama T, Sato T, Matsuda D, Nishikiori H, Kobayashi M, Sugiyama M, Hamada N, Arakawa H, Kuniyasu H. Involvement of HMGB1 and RAGE in IL-1beta-induced gingival inflammation. Arch Oral Biol. 2012;57:73–80. doi: 10.1016/j.archoralbio.2011.08.001. [DOI] [PubMed] [Google Scholar]
- 34.Wu X, Mi Y, Yang H, Hu A, Zhang Q, Shang C. The activation of HMGB1 as a progression factor on inflammation response in normal human bronchial epithelial cells through RAGE/JNK/NF-kappaB pathway. Mol Cell Biochem. 2013;380:249–257. doi: 10.1007/s11010-013-1680-0. [DOI] [PubMed] [Google Scholar]
- 35.Hu JF, Song XY, Chu SF, Chen J, Ji HJ, Chen XY, Yuan YH, Han N, Zhang JT, Chen NH. Inhibitory effect of ginsenoside Rg1 on lipopolysaccharide-induced microglial activation in mice. Brain Res. 2011;1374:8–14. doi: 10.1016/j.brainres.2010.11.069. [DOI] [PubMed] [Google Scholar]