

Abstract
Peritoneal fibrosis resulting from long-term peritoneal dialysis is a major cause of failure of peritoneal ultrafiltration function and main reason of dropout from peritoneal dialysis. Epithelial-mesenchymal transition (EMT) of peritoneal mesochelial cells (HPMCs) is a major contributor of peritoneal fibrosis. Recently, the association between histone acetylation and kinds of fibrosis including liver, lung and kidney fibrosis is well established. Thus, in this study we tried to profile whether histone acetylation is also operates EMT process in HPMCs and what’s the regulatory mechanism. We established an EMT model of HPMCs through high glucose treatment. And hyperacetylation of H3 histone was found using western blot in EMT model. After treated with C646, a histone acetyltransferase (HAT) inhibitor, high glucose-induced EMT in HPMCs was counteracted. To further understand the molecular mechanism of C646 rescues high glucose-induced EMT, CHIP-qPCRwas used to examine the modulation of histone H3 acetylation at promoters of series signaling target genes. We found that the H3 acetylation level at TGF-β1 gene promoter was down-regulation by C646 treatment. Moreover, we also found that TGF-β1/Smad3 signaling was blocked. Hence, our results suggest that histone H3 acetylation activated TGF-β1/Smad3 signaling during EMT of HPMCs, and C646 can rescue the mesenchymal phenotype transition. These findings may provide a novel pathogenic mechanism and therapeutic target for peritoneal fibrosis.
Keywords: Peritoneal fibrosis, epithelial-mesenchymal transition, histone acetylation, histone acetyltransferase (HAT) inhibitor, TGF-β1/Smad3 signaling
Introduction
End-stage renal disease (ESRD) is an important public health problem all over the world. According to statistics, ESRD affects more than 100000 people in China, and increasing numbers of new cases is diagnosed each year [1]. Currently, peritoneal dialysis (PD) is the most widely used renal replacement therapy of ESRD. However, long-term PD often causes peritoneal fibrosis and subsequently leads to the failure of peritoneal ultrafiltration function. Ultimately, patients have to withdraw from the PD treatment. Therefore, researching the mechanism of peritoneal fibrosis is important for ESRD treatment.
Peritoneal fibrosis is characterized by loss of the mesothelial cell layer and enlargement of the submesothelial layer, which is accompanied by increased myfibroblasts, collagen deposition and changes in the structure of blood vessels [2,3]. A variety of mechanisms contribute to the progression of peritoneal fibrosis, such as chronic inflammation, oxidative stress and epithelial-mesenchymal transition (EMT) of peritoneal mesothelial cells (PMC) [4-6]. Previous studies believed that peritoneal fibrosis is mostly attributed to the EMT of PMC [7-9].
EMT is a dynamic process of degeneration of mature epithelial characteristics and acquisition of a mesenchymal phenotype [10]. During the development of EMT, epithelial cells usually lose their adhesion ability and rearrange cytoskeletal. Subsequently, this process further promotes the loss of peritoneal member function [11,12]. Many signaling pathways have been shown to contribute to EMT, such as ROS/MMP-9 pathways [13], PI3k/AKT pathways [14] and TGF-β signaling pathways [15,16]. Increasing evidence indicates transforming growth factor-β1 (TGF-β1) is a well-characterized inducer of EMT, and TGF-β1/Smad3 signaling seems to be a crucial element in EMT process [17,18]. Although extensive studies have been reported, the precise mechanisms leading to EMT are only partially understood.
Hyperacetylation of histones have been shown to associate with open chromatin structure and activation gene transcription, while deacetylation of histones is correlated with gene repression. The balance between acetylation and deacetylation often decide cell fate. In fact, this balance has been identified as an important role in many human diseases, such as cancers, Parkinson’s disease, inflammatory and immune diseases [19-21]. Previous studies reported that histones acetylation induces epithelial-to-mesenchymal cell transition in cancer and lens epithelial cells [22,23]. However, the relationship between histones acetylation and peritoneal fibrosis has none reported. Thus, we propose a hypothesis: whether acetylation of histones is also operates EMT process in peritoneal mesothelial cells?
In the present study, we examined the acetylation of histones (H3 and H4) during high glucose induced EMT of human peritoneal mesothelial cells (HPMCs). Then, chromatin immunoprecipitation followed by qPCR (CHIP-qPCR) was used to compare acetylation at the promoters of target genes in different signaling pathways. Finally, we also investigated whether C646, a histone acetyltransferase (HAT) inhibitor, can reverse the mesenchymal phenotype. Here, we will firstly demonstrate the role and mechanism of histones acetylation in the EMT of HPMCs.
Materials and methods
Reagents
Antibodies against acety-H3, Acety-H4 and β-actin were purchased from Cell Signaling Technologies (Danvers, MA). Whereas, antibodies against E-cadherin, α-SMA, collagen I, fibronection, Smad3, p-Smad3 and TGF-β1 were purchased from Abcam (Cambridge, MA, USA). Rabbit antibodies conjugated with horseradish peroxidase (HRP) and sheep anti-mouse-HRP were purchased from Zhongsan Jinqiao (Beijing, China). D-glucose purchased from Sigma (Saint Louis, MO, USA). Histone acetyltransferase inhibitor C646 purchased from Selleckchem (Houston, TX). All others chemical reagents were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Cell culture
Human peritoneal mesothelial cell lines HMrSV5 were purchased from Focusbio (Guangzhou, China). Cells were cultured in Dulbecco’s modified Eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco), 100U/ml penicillin and 100 μg/ml streptomycin. HPMCs were cultured at 37°C under 5% CO2.
Cell proliferation assay
We used cell counting Kit-8 (Beyotime, China) to evaluate the proliferation of HPMCs treated with different concentration of D-glucose. Briefly, HPMCs were transferred into a 96-well cell culture plates with 200 Μl suspension per well, and grown overnight. Then, cells were treated with different concentration of D-glucose (30 Mm, 40 Mm, 50 Mm, 60 Mm and 120 Mm). All groups were performed in triplicate. At 0 h, 6 h, 12 h, 24 h, 36 h and 48 h, 20 Μl CCK-8 was added to each well, and then the plates were incubated for 2 h. Finally, absorbance was measured at 490 nm with a microplate reader (BioRad).
Western blotting analysis
Cells were harvested and homogenized with cell lysis buffer (Beyotime, China). Then, the homogenates were centrifuged for 30 min at 4°C, 12000 rpm, and the supernatants were collected as protein samples. Protein amounts were measured using BCA Protein Assay Kit (Beyotime, China). Equal amounts of protein samples were separated by denaturing 10% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. Membranes were incubated in a 5% skim milk TBST blocking solution at room temperature (RT) for 1 h. Then, membranes were incubated with rotation at 4°C overnight with specific primary antibodies aganist E-cadherin (1:1000), α-SMA (1:1000), Collagen I (1:1500), fibronection (1:2000), Smad3 (1:1000), p-Smad3 (1:1000), TGF-β1 (1:1500), Acety-H3 (1:1500), Acety-H4 (1:1500) and β-Actin (1:1000). After that, membranes incubated by secondary antibodies (1:1000) conjugated with horseradish peroxidase (HRP) at RT for 50 min. Finally, protein bands were visualized using an enhanced chemiluminescence (ECL) western blotting detection system (GE Healthcare, Amersham, UK) according to operation standard.
Histone acetyltransferase (HAT) activity assay
HAT activity was determined using HAT activity colorimetric assay kit (BioVision, Milpitas, USA) according to the manufacturer’s protocol. Briefly, nuclear extract was obtained by incubating cells with extraction buffer and centrifugation at 15,000 rpm for 20 min under 4°C. Prepare test samples (50 μg of nuclear extract) in 40 Μl water for each assay in a 96-well plate. Then, add 68 Μl of Assay Mix to each well, and incubate plates at 37°C for 1-4 hours. Finally, read absorbance at 490 nm with a microplate reader (BioRad).
Quantitative real-time RT-PCR
Total RNA was extracted using Trizol reagents (Invitrogen) according to the manufacturer’s instructions and diluted to 200 ng/Μl. Then, quantitative real-time RT-PCR (Qrt-PCR) was performed using One Step SYBR® PrimeScript™ RT-PCR Kit II (TaKaRa, China) according to standard protocol. GAPDH gene was used as an internal control. The Qrt-PCR amplification was performed as follows: 42°C for 5 min, 95°C for 10 s, followed by 40 cycles of 95°C for 5 s, 60°C for 20 s, and 72°C for 15 s. PCR was followed by a melt curve analysis to determine the reaction specificity. The relative gene expression was calculated using 2-ΔΔCt method. Primers used in Qrt-PCR were as follows: p300: 5’-agattcagagggcagcagcagagac-3’ (forward probe), 5’-gccataggaggtgggttcatac-3’ (reverse probe); GCN5: 5’-ggaaaggagaagggcaaggag-3’ (forward probe), 5’-gtcaatggggaagcggataac-3’ (reverse probe); GAPDH: 5’-ggaccaatacgaccaaatccg-3’ (forward probe), 5’-agccacatcgctcagacac-3’ (reverse probe).
Chromatin immunoprecipitation (CHIP) assay and quantitative real-time polymerase chain reaction (QPCR)
CHIP assay was performed using the commercially available CHIP assay kit from Upstate Biotechnology (NY, USA) according to the manufacturer’s protocol. In brief, after cells were cross-liked with 1% formaldehyde for 10 min, cells were neutralized in glycine and collected by centrifugation. CHIP lysis buffer (1% SDS, 10 Mm EDTA, 50 mM Tris-HCl pH 8.0) was then added and incubated 10 minutes on ice. Cells were sonicated to shear DNA to lengths between 200-1000 bp. Then, sample was divided into 2 equal portions. One sample was removed and used for input control, whereas another was incubated with antibodies against Acety-H3 or non-immune IgG for 2 h at 4°C. The immunoprecipitated protein-DNA complexes were collected using protein A agarose beads. Subsequently, beads were washed for 5 minutes with 1 Ml of the buffers listed in the order as given below: low salt wash buffer, high salt wash buffer, LiCl buffer, and then TE buffer. After that, protein-DNA complexes were subjected with reversed cross-links, proteinase K digestion to remove histones, and DNA purification. The QPCR then performed using primers specific for TGF-β1, MMP-9 and PI3K promoters. We used γ-satellite as a constitutive heterochromatin according to a previous study [24]. Primers were as follows: TGF-β1: 5’-aggctgcttagccacatg-3’ (forward probe), 5’-gtgggaggagggggcaa-3’(reverse probe); MMP-9:5’-agagcctgctcccagagggc-3’ (forward probe), 5’-gccaagtcaggcaggacccc-3’ (reverse probe); PI3K: 5’-aaagtagaacgaccaataag-3’ (forward probe), 5’-gagatgagggaagaggag-3’ (reverse probe); γ-satellite: 5’-tatggcgaggaaaactgaaa-3’ (forward probe), 5’-ttcacgtcctaaagtgtgtat -3’ (reverse probe). QPCR was performed with the following conditions: 95°C for 5 min, followed by 40 cycles at 95 °C for 20 s, 58°C for 20 s, and 72°C for 20 s. Each QPCR reaction was repeated in triplicate. QPCR was followed by a melt curve analysis to determine the reaction specificity. The relative gene expression was calculated using 2-ΔΔCt method.
Statistical analysis
Data are reported as mean ± standard deviation (SD). Statistical significance was determined using Double-sided Student’s t test. Multiple groups were analyzed using ANOVA. A P value of less than 0.05 was considered to be significant.
Results
High glucose induced epithelial-to-mesenchymal transition (EMT) in human peritoneal mesothelial cells (HPMCs)
To establish an EMT model of HPMCs, cell lines HMrSV5 were treated with different concentrations of D-glucose. Cells proliferation was measured by a CCK-8 assay. As shown in Figure 1A and 1B, we found that cells be treated with 60 mmol/LD-glucose for 24 h without significant cell toxicity. After treated with 60 mmol/LD-glucose, variable morphological features of cells were observed. Cells changed from a cobblestone-like appearance to fibroblast-like cells (Figure 1C). After that, we monitored expressions of E-cadherin (epithelial marker), α-SMA, Collagen I and fibronection (mesenchymal markers), using western blot assay. As shown in Figure 1D, the expression of E-cadherin was significantly decreased and the expression of α-SMA, Collagen I and fibronection were significantly increased in D-glucose treated cells. These data indicated that HMrSV5 cells have occurred EMT after D-glucose treated. Therefore, we used 60 mmol/L D-glucose for the rest of the experiment of EMT model of HMrSV5.
Figure 1.
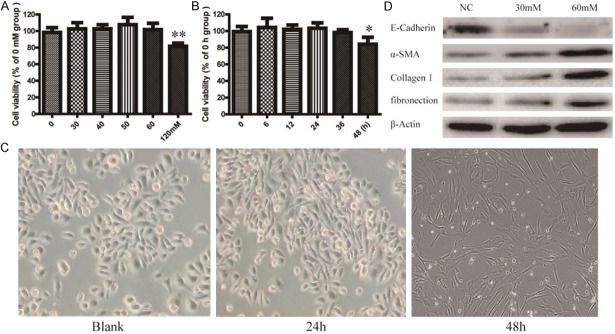
Biomarker alteration and cell morphology transform occur in epithelial-mesenchymal transition (EMT) of human peritoneal mesothelial cells treated with high glucose (HG). A. HMrSV5 cells were treated with 0-120 mM HG for 24 h, and the cell viability was measured using CCK-8 assay. Values represent mean ± SD of three independent experiments. B. HMrSV5 cells were treated with 60 mM HG for 0, 6, 12, 24, 36 and 48 h, and the cell viability was analyzed by CCK-8. Values represent mean ± SD of three independent experiments. C. Phase contrast microscopy shows different morphological characteristics of HMrSV5 treated with 60 mM HG at different time. D. HMrSV5 cells were treated with 60 mM HG for 36 h, and the expression of EMT markers E-cadherin, α-SMA, Collagen I and fibronection were detected by Western blot. β-Actin was used as an internal control.
Increase of histone H3 acetylation in high glucose-induced EMT in HPMCs
To investigate the relationship between histones acetylation and the EMT of HPMCs, we detected the acetylation of histones (H3, H4) using western blot. As shown in Figure 2A, an increase in histone H3 acetylation was found at high glucose treated groups in a dose-dependent manner. However, the histone H4 acetylation was not noticed. Then, HAT activity was determined by HAT activity colorimetric assay kit (BioVision, Milpitas, USA). We found HAT activity was increased in the presence of high glucose in cells. And cells treated with 60 mM high glucose increased HAT activity by about 130% (Figure 2B). We further detected the expression levels of the HAT subtypes (p300, GCN5) using qRT-PCR. The results show that the expression levels of p300 were significantly increased in high glucose treated cells. Moreover, the expression levels of GCN5 were not significantly altered in treated groups (Figure 2C).
Figure 2.

Increase of histone acetylation and histone acetylase(HAT) activities in HG-induced EMT of HPMCs. A. Expression of acetylated histone H3 and H4 in HG-induced EMT were detected by western blot assay. β-Actin was used as an internal control. B. HAT activities were determined using the commercially available HAT Activity Colorimetric Assay kit according to the manufacture’s protocol. C. The expression level of HAT subtypes p300 and GCN5 mRNA were detected using qRT-PCR assay. Data are expressed as means ± SD from three independent experiments and analyzed by one-way ANOVA. *P < 0.05, **P < 0.01 vs. without HG group.
Histone acetyltransferase (HAT) inhibitor C646 counteracts high glucose-induced EMT in HPMCs
To further determine whether histone acetylation is functionally linked to the process of EMT, we treated high glucose-induced cells with C646, a HAT inhibitor for 36 h in culture. In high glucose-induced HPMCs, C646 treatment resulted in down-regulation of histone H3 acetylation (Figure 3A). Meanwhile, gene expression of E-cadherin was significantly up-regulation and the expression of α-SMA, Collagen I and fibronection were significantly down-regulation in HPMCs cells with C646 treatment (Figure 3B). In addition, the effect of C646 treatment rescues high glucose-induced morphological features changing was also observed (Figure 3C). Therefore, these data indicates that C646 counteracts high glucose-induced EMT in HPMCs.
Figure 3.
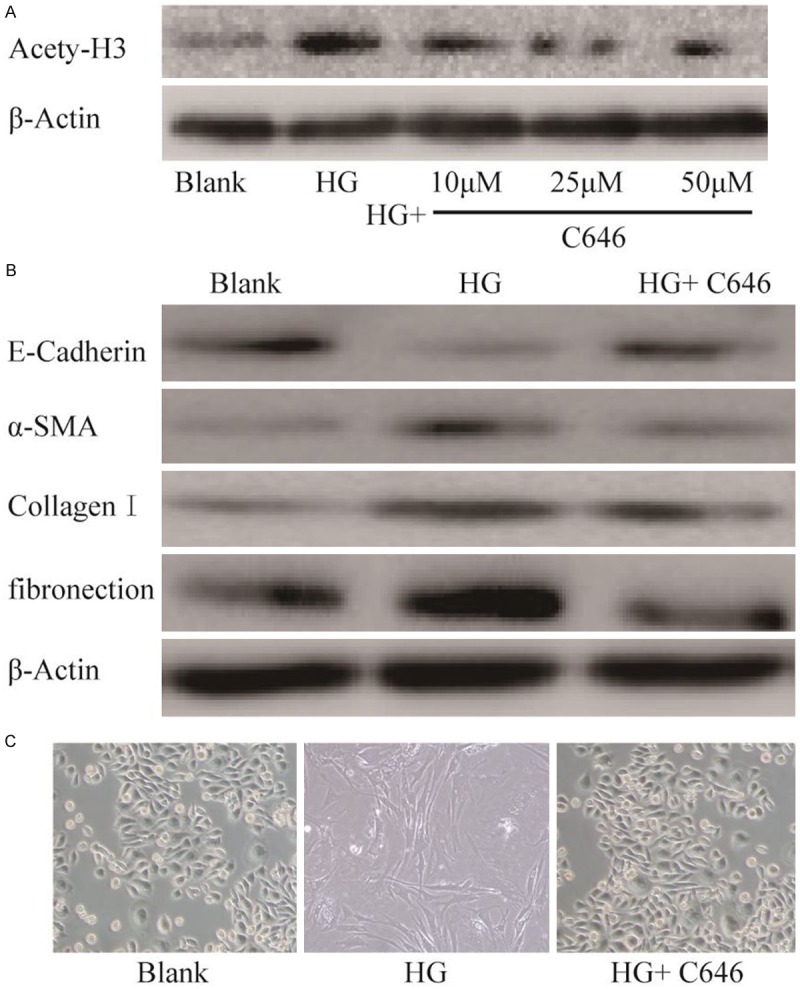
HAT inhibitor C646 counteracts HG-induced EMT in HPMCs. A. Western blot analysis of expression of acetylated histone H3 in HPMCs which were treated with different concentrations of C646 for 24 h and then exposed with HG for 36 h. B. Cells were treated as described above, and the expression of the EMT markers E-cadherin, α-SMA, Collagen I and fibronection were detected by Western blot. C. The effect of C646 treatment rescues HG-induced morphological features changing was observed. Magnification is 200 ×.
HAT inhibitor C646 blocks TGF-β1/Smad3 signaling in HG-induced HPMCs
In order to further understand the involvement of C646 counteracts high glucose-induced EMT in HPMCs, the modulation of histone H3 acetylation at promoters of series signaling target genes were examined. As shown in Figure 4A, CHIP-qPCR experiment indicated that the acetylation level of TGF-β1 gene promoter was significantly increased in high glucose-treated cells. However, the levels of MMP-9 and PI3K gene promoter have no obvious change. This data indicated that TGF-β1/ Smad3 signaling may play an important role in EMT.
Figure 4.
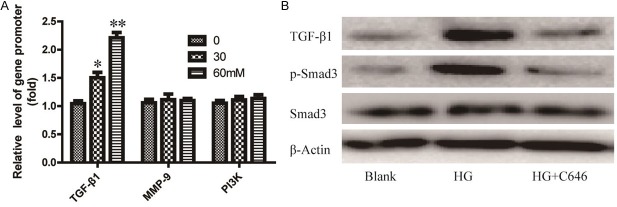
HAT inhibitor C646 counteracts EMT via regulates TGF-β1/Smad3 signaling in HG-induced HPMCs. A. Cells treated with 60 mM of C646 and then treated with HG for 36 h. Chromatin immunoprecipitation (CHIP) assays followed by real time PCR assessed acetylation levels in series signaling target genes promoter. Anti-rabbit IgG was used to determine non-specific binding. Data are expressed as means ± SD from three independent experiments and analyzed by one-way ANOVA.*P < 0.05, **P < 0.01 vs. without HG group. B. Cells were treated as described above, and the expression of TGF-β1 and phosphorylated Smad3 were detected by Western bolt.
Then, we detected the expression of genes in TGF-β1/Smad3 signaling in C646 treated cells using western blot assay. As shown in Figure 4B, when cells treated with high glucose along, the expression of TGF-β1 and p-Smad3 were significantly increased. However, after treatment with C646 in high glucose-treated cells, the over-expression of TGF-β1 and p-Smad3 were inhibited.
Discussion
In the present study, we report a novel finding: histone H3 acetylation stimulates TGF-β1 gene transcription during EMT of HPMCs, and HAT inhibitor can rescue the mesenchymal phenotype.
It is widely accepted that EMT in mesothelial cells is an important step in the process of peritoneal fibrosis. Previous studies reported that EMT is characterized by loss of the expression of the epithelial marker E-cadherin and up-regulation of mesenchymal markers α-SMA, collagen I, fibronectin and vimentin [25,26]. In this study, using high glucose (HG), reduced expression of E-cadherin, increased expression of α-SMA and switched morphological characteristic were observed in HPMCs. Our results indicated that we successfully constructed a peritoneal EMT model using HG in vitro.
Emerging evidence suggests that TGF-β1/Smad3 signaling plays a pivotal role in EMT. TGF-β1 binds to its receptors to phosphorylate receptor-regulated Smad3. Then, phosphorylation Smad3 complexes translocate into nucleus to regulate expression of target genes, such as snail, α-SMA and collagen I [27]. Peter et al [7] reported overexpression of TGF-β1 induces EMT in the rodent peritoneum. It has been reported mice lacking Smad3 are protected against tubulointerstitial fibrosis [28], skin fibrosis [29] and pulmonary fibrosis [30]. All findings support a critical role of TGF-β1/Smad3 signaling mediated EMT in fibrosis. However, little is known about the mechanism of activation of TGF-β1/Smad3 signaling in EMT of peritoneal fibrosis.
A large number of evidence indicated that aberrant histone acetylation associated with many types of cancers, both epithelial and hematological. Tumor suppressor gene silencing by histone lysine deacetylases (HDAC) is an important mechanism in many tumor occurrences [31]. Several HDAC inhibitors have been proved for their ability to re-express tumor suppressor genes leading to anti-cancer function [32-34]. At present, several specific HDAC inhibitors are in early phase clinical experiment as anticancer drugs [35-37]. Thus, researching histones acetylation is linked to finding treatment strategies for diseases.
In recent years, the association between histone acetylation and the progression of fibrosis is well established. A report showed that Sirt1, a histone deacetylase, controls fibroblast activation and tissue fibrosis via regulating TGF-β signaling in systemic sclerosis [38]. Pang et al [39] demonstrated that HDAC level is related to the progression of many tissue fibrosis including liver, lung and kidney. In addition, it has been reported that HDAC inhibitors can retard and even reverse fibrotic disorders in many fibrotic disease [40-42]. Thus, we speculated that anomalous histone acetylation may play an important role in peritoneal fibrosis.
In this study, hyperacetylation of histone H3 have been detected in EMT model of HPMCs. Then, analysis of chromatin-protein association by CHIP-qPCR demonstrated TGF-β1 gene promoter was hyper acetylated and promoted TGF-β1 gene expression. This promotes phosphorylation of Smad-3. Thus, TGF-β1/Smad3 signaling is activated. Subsequently, C646, a histone acetyltransferase (HAT) inhibitor, was used to further investigate the relationship between acetylation and process of EMT. Our data showed that C646 can reverse the mesenchymal phenotype via blocking TGF-β1/Smad3 signaling pathway.
In summary, our present results show that highly acetylated H3 histone induces EMT of peritoneal fibrosis through activating TGF-β1/Smad3 signaling. In addition, HAT inhibitor can reverse this transition. These findings provide a novel concept for the treatment of peritoneal fibrosis: HAT perhaps is a promising target for peritoneal fibrosis therapy.
Acknowledgements
This study is funded by Science and Technology Plan of Hunan Province (2013FJ6058) and Science and Technology Plan of Changsha City (K1303034-31).
Disclosure of conflict of interest
None.
References
- 1.Zuo L, Wang M. Current burden and probable increasing incidence of ESRD in China. Clin Nephrol. 2010;74(Suppl 1):S20–2. [PubMed] [Google Scholar]
- 2.Plum J, Hermann S, Fusshöller A, Schoenicke G, Donner A, Röhrborn A, Grabensee B. Peritoneal sclerosis in peritoneal dialysis patients related to dialysis settings and peritoneal transport properties. Kidney Int Suppl. 2001;78:S42–7. doi: 10.1046/j.1523-1755.2001.59780042.x. [DOI] [PubMed] [Google Scholar]
- 3.Tomino Y. Mechanisms and interventions in peritoneal fibrosis. Clin Exp Nephrol. 2012;16:109–114. doi: 10.1007/s10157-011-0533-y. [DOI] [PubMed] [Google Scholar]
- 4.de Lima SM, Otoni A, Sabino Ade P, Dusse LM, Gomes KB, Pinto SW, Marinho MA, Rios DR. Inflammation, neoangiogenesis and fibrosis in peritoneal dialysis. Clin Chim Acta. 2013;421:46–50. doi: 10.1016/j.cca.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 5.Kitamoto M, Kato K, Sugimoto A, Kitamura H, Uemura K, Takeda T, Wu C, Nogaki F, Morimoto T, Ono T. Sairei-to ameliorates rat peritoneal fibrosis partly through suppression of oxidative stress. Nephron Exp Nephrol. 2010;117:e71–e81. doi: 10.1159/000321147. [DOI] [PubMed] [Google Scholar]
- 6.Yáñez-Mó M, Lara-Pezzi E, Selgas R, Ramírez-Huesca M, Domínguez-Jiménez C, Jiménez-Heffernan JA, Aguilera A, Sánchez-Tomero JA, Bajo MA, Álvarez V. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. New Engl J Med. 2003;348:403–413. doi: 10.1056/NEJMoa020809. [DOI] [PubMed] [Google Scholar]
- 7.Margetts PJ, Bonniaud P, Liu L, Hoff CM, Holmes CJ, West-Mays JA, Kelly MM. Transient overexpression of TGF-β1 induces epithelial mesenchymal transition in the rodent peritoneum. J Am Soc Nephrol. 2005;16:425–436. doi: 10.1681/ASN.2004060436. [DOI] [PubMed] [Google Scholar]
- 8.Devuyst O, Margetts PJ, Topley N. The pathophysiology of the peritoneal membrane. J Am Soc Nephrol. 2010;21:1077–1085. doi: 10.1681/ASN.2009070694. [DOI] [PubMed] [Google Scholar]
- 9.Yang AH, Chen JY, Lin JK. Myofibroblastic conversion of mesothelial cells. Kidney Int. 2003;63:1530–1539. doi: 10.1046/j.1523-1755.2003.00861.x. [DOI] [PubMed] [Google Scholar]
- 10.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–1784. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyer B, Vallés AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochem Pharmacol. 2000;60:1091–1099. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tobar N, Villar V, Santibanez JF. ROS-NFκΒ mediates TGF-β1-induced expression of urokinase-type plasminogen activator, matrix metalloproteinase-9 and cell invasion. Mol Cell Biochem. 2010;340:195–202. doi: 10.1007/s11010-010-0418-5. [DOI] [PubMed] [Google Scholar]
- 14.Boca M, D’Amato L, Distefano G, Polishchuk RS, Germino GG, Boletta A. Polycystin-1 Induces Cell Migration by Regulating Phosphatidylinositol 3-kinase-dependent Cytoskeletal Rearrangements and GSK3β-dependent Cell-Cell Mechanical Adhesion. Mol Bio Cell. 2007;18:4050–4061. doi: 10.1091/mbc.E07-02-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee CJ, Subeq YM, Lee RP, Liou HH, Hsu BG. Calcitriol decreases TGF-β1 and angiotensin II production and protects against chlorhexide digluconate-induced liver peritoneal fibrosis in rats. Cytokine. 2014;65:105–118. doi: 10.1016/j.cyto.2013.10.003. [DOI] [PubMed] [Google Scholar]
- 16.Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGF-β-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19–27. doi: 10.1016/j.cytogfr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 18.Wang A, Ziyadeh FN, Lee EY, Pyagay PE, Sung SH, Sheardown SA, Laping NJ, Chen S. Interference with TGF-β signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. Am J Physiol Renal Physiol. 2007;293:F1657–F1665. doi: 10.1152/ajprenal.00274.2007. [DOI] [PubMed] [Google Scholar]
- 19.Harrison IF, Dexter DT. Epigenetic targeting of histone deacetylase: therapeutic potential in Parkinson’s disease? Pharmacol Ther. 2013;140:34–52. doi: 10.1016/j.pharmthera.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard F, Chipoy C. Histone deacetylase inhibitors: new drugs for the treatment of inflammatory diseases? Drug Discov Today. 2005;10:197–204. doi: 10.1016/S1359-6446(04)03309-4. [DOI] [PubMed] [Google Scholar]
- 21.Adcock I. HDAC inhibitors as anti-inflammatory agents. Br J Pharmacol. 2007;150:829–831. doi: 10.1038/sj.bjp.0707166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roche J, Nasarre P, Gemmill R, Baldys A, Pontis J, Korch C, Guilhot J, Ait-Si-Ali S, Drabkin H. Global decrease of histone H3K27 acetylation in ZEB1-induced epithelial to mesenchymal transition in lung cancer cells. Cancers. 2013;5:334–356. doi: 10.3390/cancers5020334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen X, Xiao W, Chen W, Luo L, Ye S, Liu Y. The epigenetic modifier trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and epithelial-mesenchymal transition of lens epithelial cells. Cell Death Dis. 2013;4:e884. doi: 10.1038/cddis.2013.416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shestakova EA, Mansuroglu Z, Mokrani H, Ghinea N, Bonnefoy E. Transcription factor YY1 associates with pericentromeric gamma-satellite DNA in cycling but not in quiescent (G0) cells. Nucleic Acids Res. 2004;32:4390–9. doi: 10.1093/nar/gkh737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Blick T, Widodo E, Hugo H, Waltham M, Lenburg M, Neve R, Thompson E. Epithelial mesenchymal transition traits in human breast cancer cell lines. Clin Exp Metastasis. 2008;25:629–642. doi: 10.1007/s10585-008-9170-6. [DOI] [PubMed] [Google Scholar]
- 26.Liu J, Zeng L, Zhao Y, Zhu B, Ren W, Wu C. Selenium suppresses lipopolysaccharide-induced fibrosis in peritoneal mesothelial cells through inhibition of epithelial-to-mesenchymal transition. Biol Trace Elem Res. 2014;161:202–209. doi: 10.1007/s12011-014-0091-8. [DOI] [PubMed] [Google Scholar]
- 27.Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY, Kao WW, Roberts AB. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164:651–663. doi: 10.1016/S0002-9440(10)63153-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486–1494. doi: 10.1172/JCI19270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flanders KC, Sullivan CD, Fujii M, Sowers A, Anzano MA, Arabshahi A, Major C, Deng C, Russo A, Mitchell JB, Roberts AB. Mice lacking Smad3 are protected against cutaneous injury induced by ionizing radiation. Am J Pathol. 2002;160:1057–1068. doi: 10.1016/S0002-9440(10)64926-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bonniaud P, Kolb M, Galt T, Robertson J, Robbins C, Stampfli M, Lavery C, Margetts PJ, Roberts AB, Gauldie J. Smad3 null mice develop airspace enlargement and are resistant to TGF-β-mediated pulmonary fibrosis. J Immunol. 2004;173:2099–2108. doi: 10.4049/jimmunol.173.3.2099. [DOI] [PubMed] [Google Scholar]
- 31.Lin RJ, Nagy L, Inoue S, Shao W, Miller WH, Evans RM. Role of the histone deacetylase complex in acute promyelocytic leukaemia. Nature. 1998;391:811–814. doi: 10.1038/35895. [DOI] [PubMed] [Google Scholar]
- 32.Bieliauskas AV, Pflum MKH. Isoform-selective histone deacetylase inhibitors. Chem Soc Rev. 2008;37:1402–1413. doi: 10.1039/b703830p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 34.Johnstone RW. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov. 2002;1:287–299. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 35.Marks PA, Rifkind RA, Richon VM, Breslow R, Miller T, Kelly WK. Histone deacetylases and cancer: causes and therapies. Nat Rev Cancer. 2002;1:194–202. doi: 10.1038/35106079. [DOI] [PubMed] [Google Scholar]
- 36.Vigushin DM. FR-901228 Fujisawa/National Cancer Institute. Curr Opin Invest Drugs. 2002;3:1396–402. [PubMed] [Google Scholar]
- 37.Prakash S, Foster BJ, Meyer M, Wozniak A, Heilbrun LK, Flaherty L, Zalupski M, Radulovic L, Valdivieso M, LoRusso PM. Chronic oral administration of CI-994: a phase 1 study. Invest New Drugs. 2001;19:1–11. doi: 10.1023/a:1006489328324. [DOI] [PubMed] [Google Scholar]
- 38.Zerr P, Palumbo-Zerr K, Huang J, Tomcik M, Sumova B, Distler O, Schett G, Distler JH. Sirt1 regulates canonical TGF-β signalling to control fibroblast activation and tissue fibrosis. Annal Rheum Dis. 2014 doi: 10.1136/annrheumdis-2014-205740. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 39.Pang M, Zhuang S. Histone deacetylase: a potential therapeutic target for fibrotic disorders. J Pharmacol Exp Ther. 2010;335:266–272. doi: 10.1124/jpet.110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nural-Guvener HF, Zakharova L, Nimlos J, Popovic S, Mastroeni D, Gaballa MA. HDAC class I inhibitor, Mocetinostat, reverses cardiac fibrosis in heart failure and diminishes CD90+ cardiac myofibroblast activation. Fibrogenesis Tissue Repair. 2014;7:10. doi: 10.1186/1755-1536-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Advani A, Huang Q, Thai K, Advani SL, White KE, Kelly DJ, Yuen DA, Connelly KA, Marsden PA, Gilbert RE. Long-term administration of the histone deacetylase inhibitor vorinostat attenuates renal injury in experimental diabetes through an endothelial nitric oxide synthase-dependent mechanism. Am J Pathol. 2011;178:2205–14. doi: 10.1016/j.ajpath.2011.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Io K, Nishino T, Obata Y, Kitamura M, Koji T, Kohno S. SAHA SuppreSSeS peritoneAl fibroSiS in mice. Perit Dial Int. 2014 doi: 10.3747/pdi.2013.00089. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]