

Abstract
Objective: To investigate the antitumor activity of metformin combined with valproic acid (VPA) on renal cell carcinoma (RCC) cell lines. Methods: The effects of metformin combined with VPA on the viability of 786-O and caki-2 cell lines were evaluated by MTT assay. The inhibitory effect of combination of the two drugs was analyzed by the Chou and Talalay method. Flow cytometry was employed to analyze cell cycle and cell apoptosis. Results: MTT assay showed that both metformin and VPA decreased 786-O and Caki-2 cells viability in a dose-dependent and time-dependent manner. In 786-O cells, metformin combined with VPA had a synergistic inhibitory effect (CI < 1) when the inhibition effect was ≥ 0.3. In Caki-2 cells, metformin combined with VPA had a synergistic inhibitory effect (CI < 1) when the inhibition effect was ≥ 0.4. Metformin and VPA combination elicited significant apoptosis compared to drug used alone (P < 0.05). Furthermore, metformin and VPA acted synergistically to arrest 786-O and Caki-2 cells in G0/G1 phase. Conclusion: We highlighted for the first time that metformin combined with VPA could significantly increase anti-ccRCC effect through synergetic effect; its possible mechanisms were inducing apoptosis and adjusting cell cycle.
Keywords: Combination, metformin, valproic acid, renal cell carcinoma
Introduction
Renal cell carcinoma(RCC), accounts for about 90% of adult kidney cancers and 3% of adult malignancies [1]. The incidence of RCC has been increasing worldwide over the past three decades [2]. RCC is heterogeneous and comprises several histological types with different genetic and clinicopathologic features which determine clinical course and outcome. Clear cell renal cell carcinoma (ccRCC), also called conventional RCC, is the most common histological type of RCC, which represents approximately 80% of RCC [3]. Despite the development of imaging techniques and surgical innovations, ccRCC carries an extremely high risk of invasiveness and metastasis, with 50% of patients eventually developing metastatic disease. Further, ccRCC exhibits resistance to chemotherapy and radiation, and < 10% of patients suffering from metastatic disease survive 5 years after diagnosis [4,5]. So, new therapeutic regimens are desperately needed.
Metformin is a widely used antidiabetic drug prescribed for more than 30 years for the treatment of type II diabetes by modulating glucose and fatty acid metabolism. Metformin can reduce hepatic glucose production, increases insulin sensitivity and glucose utilization by muscles and adipocytes, resulting in decreased insulinemia and amelioration of insulin sensitivity [6]. Recently, observational studies and meta-analysis have demonstrated that metformin therapy could reduce the risk of cancer in type II diabetes patients [7]. More importantly, experimental studies showed that metformin was able to inhibit growth of several cancers, including prostate cancer, breast cancer, liver cancer, colon cancer, as well as renal cell carcinoma [8-12].
Acetylation and deacetylation of nucleosomal core histones play important roles in modulation of chromatin structure and regulation of gene expression, which is regulated by histone acetyltransferases (HATs) and histone deacetylases (HDACs). Therefore, disruption of balance between HATs and HDACs is known to be involved in cancer genesis and progression [13]. Over-expression of HDACs will lead to loss of differentiation, uncontrolled cell proliferation, tumorigenesis and progression [14-16]. Based on these findings, HDAC-inhibitors (HDAC-Is) may be a promising approach for cancer prevention. Valproic acid (VPA), a branched-chain fatty acid, is one of the HDAC-Is, which has been found to efficiently induce proliferation arrest, differentiation, and apoptosis of several cancer cells including breast, skin, prostate, colon, liver, cervix, lung, and ovarian cancer [17-19].
There is a growing body of evidence suggesting that combination of low dose of cancer chemopreventive and therapeutic agents with different modes of action may produce synergistic effects on efficacy and minimize possible side effects associated with high dose administration. In the present study, we used two human kidney cancer cell line 786-O and Caki-2 to demonstrate the synergistic interaction between metformin and VPA, and elucidate the mechanisms underlying the synergy.
Material and methods
Cell culture and drug treatment
The human renal cancer cell line 786-O and Caki-2 were purchased from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). It was routine maintained in RPMI1640 containing 100 ml/L fetal bovine serum (FBS), 100 U/mL penicillin, 100 U/mL streptomycin at 37°C in a humidified atmosphere containing 50 mL/L CO2. VPA was obtained from Sigma and dissolved in phosphate-buffered saline (PBS) at concentration of 1, 4, 8, 16, 24, 32 and 40 mM, PBS was added to culture media a negative control. Metformin was purchased from Sigma and diluted in purified water to make a 1 M stock solution and stored at 20°C. It was used at concentrations of 0.1, 0.2, 0.5, 1, 5, 10, 20, and 50 mM diluted in culture media.
Cell viability assay
The viability of 786-O and Caki-2 cells after metformin, VPA and their combination treatment were determined by MTT assay. Briefly, cells were seeded into 96-well plates at a density of 2 × 103 cells per well (200 µL), and incubated for 24 h till all cells sufficient attached to the plate. Before the drug treatment, the culture medium changed as RPMI 1640 medium contained 2% FBS. After 24 h, 48 h, and 72 h treatment with varying doses of single drug or their combination. After treatments, medium were discarded, washed with PBS and incubated with MTT (5 mg/mL in phosphate buffered saline) in 100 l L fresh medium, incubated at 37°C for another 4 h. Discard the medium, 200 µL DMSO was added to dissolve MTT formazan crystals and absorbance at 570 nm was measured using a multiwell plate reader (BioTek, Winooski, VT, USA). Wells containing only RPMI1640 and MTT were used as negative control (NC). Cell viability was calculated as percentage of viable cells in total population. Each experiment was performed three replicates.
Detection of apoptosis
Apoptotic cells were quantified by Annexin V/PI double staining assay. Annexin V/PI staining was done using apoptotic detection kit (Zymed, South San Francisco, CA) following manufacture’s instruction. Cells were gently detached by brief trypsinization (any floating cells were also collected), and then washed with ice cold PBS. Cells (7 × 104) were then suspended in 200 µL binding buffer containing Annexin V, and incubated for 10 min at room temperature. After centrifugation (1,500 g, 1 min), cell pellet was resuspended in 200 µL binding buffer containing 5 µg/mL propidium iodide. Early apoptotic cells were detected as Annexin V positive/PI negative using a Beckman Coulter flow cytometer (FC500), and data were processed using AXP acquisition and analysis software.
Cell cycle distribution analysis
Flow cytometric analysis was performed to determine the effects of single drug or their combination on cell cycle distribution. Briefly, 786-O and Caki-2 cells, grown in 6-well plates (2 × 105 cells/well), were synchronized at the G1/S boundary after starvation with basal medium for 24 hours, followed by incubation in single drug or their combination with 10% FBS for 48 hours. At the indicated intervals, cells were harvested by trypsinization and fixed with 70% ethanol, and measured following the manufacturer’s protocol (KEY GEN, Nanjing, China). Cell cycle distribution was analyzed by flow cytometry (FACSCalibur, BD Biosciences, Bedford, MA). DNA histograms were measured using FCS Express software and the percentage of G0/G1, S, and G2/M cells were calculated.
Analyses of synergy
For the study of synergism between metformin and VPA on cell growth inhibition of 786-O and Caki-2 cells, a combination index (CI) was performed using the data obtained from MTT assay. Drug combination studies were based on concentration effect curves generated as a plot of the fraction of unaffected cells versus drug concentration, in accordance to the Chou and Talalay method [20], using the following CI equation: CI = (D)1/(Dx)1 + (D)2/(Dx)2 + (D)1(D)2/(Dx)1(Dx)2, where (D)1 and (D)2 are the concentrations of metformin and VPA that exhibit a determined effect when applied simultaneously to the cells and (Dx)1 and (Dx)2 are the concentrations of the same drugs that exhibit the same determined effect when used in isolation. The CI values indicate a synergistic effect when < 1, an antagonistic effect when > 1, and an additive effect when equal to 1.
Statistical analysis
Differences between groups were assessed for statistical significance using the Mann-Whitney test or paired Student’s t-test, depending on the distribution of the data. Values of P < 0.05 were considered as statistically significant. Statistical analysis was carried out using the SPSS 18.0 statistical software (SPSS Inc., USA).
Results
Isolated effects of metformin and VPA on786-O and Caki-2 cells viability
786-O and Caki-2 cell lines in the exponential growth phases were exposed to different concentrations of metformin and VPA, and the effect on cell viability was examined after 24, 48, and 72 hours of culture. MTT assay showed that metformin decreased cell viability in a dose-dependent and time-dependent manner (shown in Tables 1, 2). VPA also showed an inhibition on the cell viability in a dose-dependent and time-dependent manner (shown in Tables 3, 4).
Table 1.
Inhibition effect of metformin on 786-O cell lines examined after 24, 48, and 72 hours of culture
| Metformin (mM) | Inhibition rate (%) | ||
|---|---|---|---|
|
|
|||
| 24 h | 48 h | 72 h | |
| 0 | 0 | 0 | 0 |
| 1 | 14.27 | 18.43 | 21.33 |
| 5 | 28.87 | 35.13 | 38.93 |
| 10 | 42.77 | 48.57 | 51.70 |
| 20 | 59.00 | 67.00 | 70.33 |
| 50 | 75.33 | 79.33 | 91.00 |
Table 2.
Inhibition effect of metformin on Caki-2 cell lines examined after 24, 48, and 72 hours of culture
| Metformin (mM) | Inhibition rate (%) | ||
|---|---|---|---|
|
|
|||
| 24 h | 48 h | 72 h | |
| 0 | 0 | 0 | 0 |
| 1 | 15.33 | 17.33 | 20.67 |
| 5 | 31.67 | 33.67 | 37.33 |
| 10 | 44.67 | 47.33 | 51.67 |
| 20 | 61.00 | 62.67 | 68.67 |
| 50 | 78.33 | 83.00 | 87.3 |
Table 3.
Inhibition effect of VPA on 786-O cell lines examined after 24, 48, and 72 hours of culture
| VPA (mM) | Inhibition rate (%) | ||
|---|---|---|---|
|
|
|||
| 24 h | 48 h | 72 h | |
| 0 | 0 | 0 | 0 |
| 1 | 9.33 | 14.00 | 14.33 |
| 4 | 18.67 | 27.33 | 30.67 |
| 8 | 32.33 | 37.67 | 43.33 |
| 16 | 41.67 | 49.00 | 54.00 |
| 24 | 49.00 | 57.00 | 61.00 |
| 32 | 61.00 | 66.33 | 75.33 |
| 40 | 70.00 | 76.00 | 85.67 |
Table 4.
Inhibition effect of VPA on Caki-2 cell lines examined after 24, 48, and 72 hours of culture
| VPA (mM) | Inhibition rate (%) | ||
|---|---|---|---|
|
|
|||
| 24 h | 48 h | 72 h | |
| 0 | 0 | 0 | 0 |
| 1 | 12.00 | 17.33 | 20.33 |
| 4 | 21.00 | 30.67 | 38.67 |
| 8 | 30.67 | 38.67 | 48.33 |
| 16 | 40.33 | 45.67 | 52.00 |
| 24 | 49.33 | 57.67 | 63.33 |
| 32 | 59.67 | 72.67 | 81.33 |
| 40 | 67.33 | 78.67 | 92.67 |
Analysis of synergistic effects between metformin and VPA on786-O and Caki-2 cells
In 786-O cells, metformin combined with VPA had a synergistic inhibitory effect (CI < 1) when the inhibition effect was ≥ 0.3 (shown in Table 5). In Caki-2 cells, metformin combined with VPA had a synergistic inhibitory effect (CI < 1) when the inhibition effect was ≥ 0.4 (shown in Table 5).
Table 5.
Combination index of metformin and VPA on786-O and Caki-2 cells at different inhibition effects
| Inhibition effect | 0.9 | 0.8 | 0.7 | 0.6 | 0.5 | 0.4 | 0.3 | 0.2 | 0.1 |
|---|---|---|---|---|---|---|---|---|---|
| CI (786-O) | 0.21 | 0.33 | 0.41 | 0.42 | 0.51 | 0.66 | 0.73 | 1.11 | 1.13 |
| CI (caki-2) | 0.34 | 0.41 | 0.49 | 0.54 | 0.67 | 0.79 | 1.03 | 1.32 | 1.48 |
Combination of metformin and VPA induce 786-O and Caki-2 cell lines apoptosis
To confirm the induction of apoptosis by combination of metformin and VPA, 786-O and Caki-2 cells were treated with the drugs alone or in combination and examined by apoptosis analysis. At the concentrations tested, metformin and VPA combination elicited significant apoptosis compared to drug used alone (P < 0.05, shown in Figures 1, 2).
Figure 1.
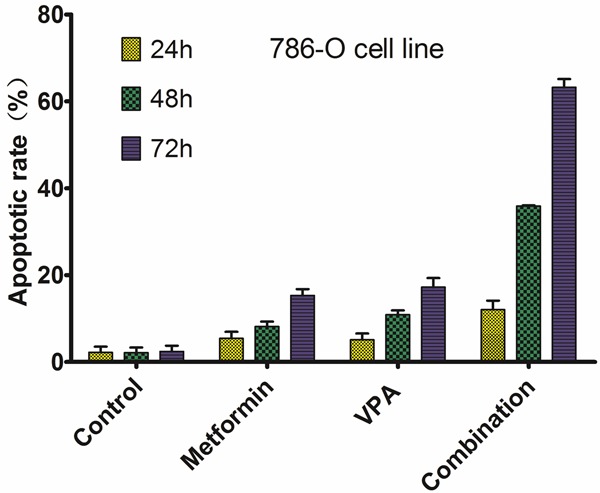
Apoptotic rate of 786-O cells on different treatment.
Figure 2.
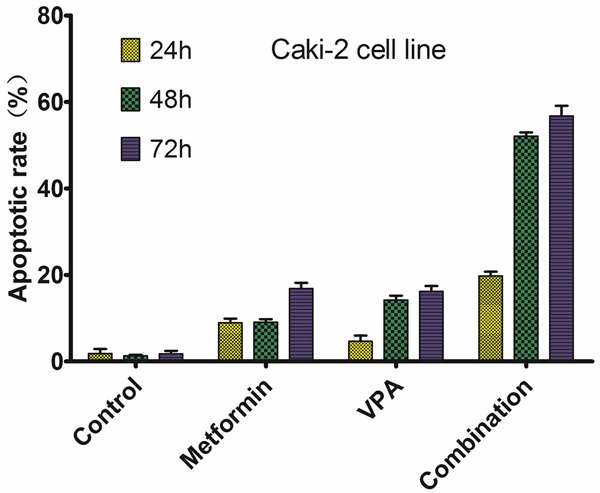
Apoptotic rate of Caki-2 cells on different treatment.
Combined treatment potentiates cell cycle arrest in G0/G1 phase caused by metformin or VPA
We studied mechanisms underlying the synergy between metformin and VPA in growth inhibition in 786-O and Caki-2 cells. Cell cycle analyses by flow cytometry were performed on both 786-O and Caki-2 cells after 48h of drug exposure. As shown in Figures 3, 4, the combined treatment caused a much more extensive cell cycle arrest in G0/G1 phase than either agent alone at the same concentrations. These results demonstrated that metformin and VPA acted synergistically to arrest 786-O and Caki-2 cells in G0/G1 phase.
Figure 3.
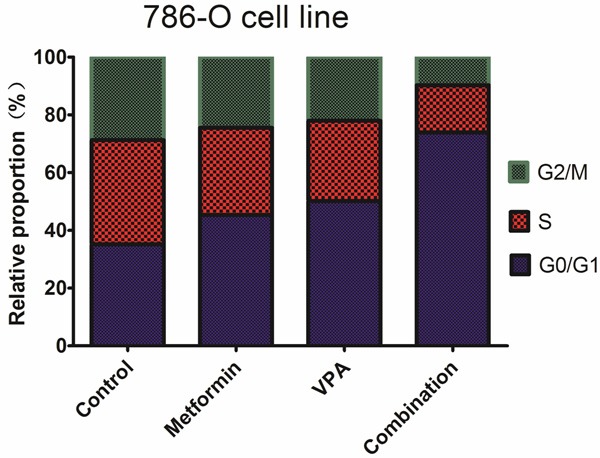
Effects of metformin, VPA, and their combinations on cell cycle of 786-O cell line.
Figure 4.
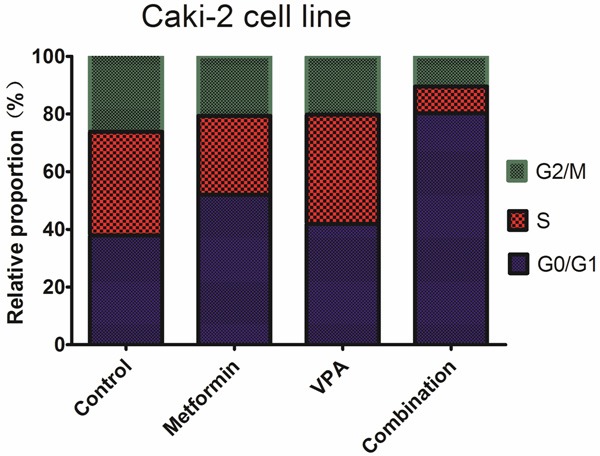
Effects of metformin, VPA, and their combinations on cell cycle of Caki-2 cell line.
Discussion
Surgical intervention is the primary treatment for RCC, which includes radical nephrectomy and nephron sparing surgery (NSS). However, these treatment options are not suitable for all patients since there are subsets of patients who develop metastases or recurrent disease. Furthermore, RCC is resistant to conventional treatment (chemotherapy, hormonal therapy, and radiotherapy). Thus, there is a tremendous need for the development of new treatment strategies. Currently, targeted therapies, which interfere with specific signal transduction pathways of tumor formation and progression, are an area of great clinical and research interest. For example, sorafenib and sunitinib, which target the vascular endothelial growth factor (VEGF) pathways, and temsirolimus, a mammalian target of rapamycin (mTOR)-inhibitor, have been approved for treatment of metastatic RCC (mRCC). Treatment using these targeted agents has shown impressive improvements in time to progression and survival. Unfortunately, these drugs are not effective for all mRCC patients [21]. They are expensive, and only a small population of patients will respond to such treatment. It is necessary to develop new targeted agents to manage RCC. There is a growing body of evidence suggesting that combination of low dose of cancer chemopreventive and therapeutic agents with different modes of action may produce synergistic effects on efficacy and minimize possible side effects associated with high dose administration.
In the present study, we used two human kidney cancer cell line 786-O and Caki-2 to demonstrate the synergistic interaction between metformin and VPA, and elucidate the mechanisms underlying the synergy. MTT assay showed that both metformin and VPA decreased 786-O and Caki-2 cells viability in a dose-dependent and time-dependent manner. In 786-O cells, metformin combined with VPA had a synergistic inhibitory effect (CI < 1) when the inhibition effect was ≥ 0.3. In Caki-2 cells, metformin combined with VPA had a synergistic inhibitory effect (CI < 1) when the inhibition effect was ≥ 0.4. These results suggested that metformin combined with VPA could significantly increase anti-ccRCC effect through synergetic effect. We then explored the possible mechanisms underlying the synergy. It was reported that metformin was able to down-regulated cyclin D1 expression and induced G0/G1 cell cycle arrest in RCC cells [12]. Our results showed that metformin and VPA acted synergistically to arrest 786-O and Caki-2 cells in G0/G1 phase. Furthermore, metformin and VPA combination elicited significant apoptosis compared to drug used alone.
In conclusion, we demonstrated that metformin and VPA produced a strong synergy in growth inhibition of human ccRCC cells, and this was associated with their synergistic actions on cell cycle arrest and apoptosis. It may, thus, have potential in treating RCC in patients, a finding that demands further clinical testing.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Chow WH, Dong LM, Devesa SS. Epidemiology and risk factors for kidney cancer. Nat Rev Urol. 2010;7:245–257. doi: 10.1038/nrurol.2010.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Decastro GJ, McKiernan JM. Epidemiology, clinical staging, and presentation of renal cell carcinoma. Urol Clin North Am. 2008;35:581–592. vi. doi: 10.1016/j.ucl.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 4.Escudier B, Eisen T, Porta C, Patard JJ, Khoo V, Algaba F, Mulders P, Kataja V ESMO Guidelines Working Group. Renal cell carcinoma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2012;23(Suppl 7):vii65–71. doi: 10.1093/annonc/mds227. [DOI] [PubMed] [Google Scholar]
- 5.Escudier B, Szczylik C, Porta C, Gore M. Treatment selection in metastatic renal cell carcinoma: expert consensus. Nat Rev Clin Oncol. 2012;9:327–337. doi: 10.1038/nrclinonc.2012.59. [DOI] [PubMed] [Google Scholar]
- 6.Bost F, Sahra IB, Le Marchand-Brustel Y, Tanti JF. Metformin and cancer therapy. Curr Opin Oncol. 2012;24:103–108. doi: 10.1097/CCO.0b013e32834d8155. [DOI] [PubMed] [Google Scholar]
- 7.Thakkar B, Aronis KN, Vamvini MT, Shields K, Mantzoros CS. Metformin and sulfonylureas in relation to cancer risk in type II diabetes patients: a meta-analysis using primary data of published studies. Metabolism. 2013;62:922–934. doi: 10.1016/j.metabol.2013.01.014. [DOI] [PubMed] [Google Scholar]
- 8.Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008;27:3576–3586. doi: 10.1038/sj.onc.1211024. [DOI] [PubMed] [Google Scholar]
- 9.Hirsch HA, Iliopoulos D, Tsichlis PN, Struhl K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009;69:7507–7511. doi: 10.1158/0008-5472.CAN-09-2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kisfalvi K, Eibl G, Sinnett-Smith J, Rozengurt E. Metformin disrupts crosstalk between G protein-coupled receptor and insulin receptor signaling systems and inhibits pancreatic cancer growth. Cancer Res. 2009;69:6539–6545. doi: 10.1158/0008-5472.CAN-09-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu B, Fan Z, Edgerton SM, Deng XS, Alimova IN, Lind SE, Thor AD. Metformin induces unique biological and molecular responses in triple negative breast cancer cells. Cell Cycle. 2009;8:2031–2040. doi: 10.4161/cc.8.13.8814. [DOI] [PubMed] [Google Scholar]
- 12.Liu J, Li M, Song B, Jia C, Zhang L, Bai X, Hu W. Metformin inhibits renal cell carcinoma in vitro and in vivo xenograft. Urol Oncol. 2013;31:264–270. doi: 10.1016/j.urolonc.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 13.Marchion D, Munster P. Development of histone deacetylase inhibitors for cancer treatment. Expert Rev Anticancer Ther. 2007;7:583–598. doi: 10.1586/14737140.7.4.583. [DOI] [PubMed] [Google Scholar]
- 14.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 15.Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, Perez-Cardenas E, de la Cruz-Hernandez E, Herrera LA. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev. 2008;34:206–222. doi: 10.1016/j.ctrv.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Witt O, Deubzer HE, Milde T, Oehme I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 17.Blaheta RA, Michaelis M, Driever PH, Cinatl J Jr. Evolving anticancer drug valproic acid: insights into the mechanism and clinical studies. Med Res Rev. 2005;25:383–397. doi: 10.1002/med.20027. [DOI] [PubMed] [Google Scholar]
- 18.Kwiecinska P, Tauboll E, Gregoraszczuk EL. Effects of valproic acid and levetiracetam on viability and cell cycle regulatory genes expression in the OVCAR-3 cell line. Pharmacol Rep. 2012;64:157–165. doi: 10.1016/s1734-1140(12)70742-9. [DOI] [PubMed] [Google Scholar]
- 19.Zhang L, Wang G, Wang L, Song C, Leng Y, Wang X, Kang J. VPA inhibits breast cancer cell migration by specifically targeting HDAC2 and down-regulating Survivin. Mol Cell Biochem. 2012;361:39–45. doi: 10.1007/s11010-011-1085-x. [DOI] [PubMed] [Google Scholar]
- 20.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 21.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, Negrier S, Chevreau C, Solska E, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, Freeman S, Schwartz B, Shan M, Simantov R, Bukowski RM TARGET Study Group. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125–134. doi: 10.1056/NEJMoa060655. [DOI] [PubMed] [Google Scholar]