

Abstract
Objective: Dysferlin is a sarcolemmal protein that plays an important role in membrane repair by regulating vesicle fusion with the sarcolemma. Mutations in the dysferlin gene (DYSF) lead to multiple clinical phenotypes, including Miyoshi myopathy (MM), limb girdle muscular dystrophy type 2B (LGMD 2B), and distal myopathy with anterior tibial onset (DMAT). Patients with dysferlinopathy also show muscle inflammation, which often leads to a misdiagnosis as inflammatory myopathy. In this study, we examined and analyzed the dyferlinopathy-associated immunological features. Methods: Comparative immunohistochemical analysis of inflammatory cell infiltration, and muscle expression of MHC-I and C5b-9 was performed using muscle biopsy samples from 14 patients with dysferlinopathy, 7 patients with polymyositis, and 8 patients with either Duchenne muscular dystrophy or Becker muscular dystrophy (DMD/BMD). Results: Immunohistochemical analysis revealed positive staining for immune response-related CD4+ cells, macrophages, MHC-I and C5b-9 in dysferlinopathy, which is in a different mode of polymyositis and DMD/BMD. Conclusion: These results demonstrated the involvement of immune factors in the pathogenesis of dysferlinopathy.
Keywords: Becker muscular dystrophy, Duchenne muscular dystrophy, dysferlinopathy, immunohistochemical analysis, muscle pathology, polymyositis
Introduction
Dysferlinopathy is an autosomal recessive neuromuscular disease caused by mutations in dysferlin (DYSF) gene, which is critical to membrane repair and vesicle trafficking. This disease comprises of Miyoshi myopathy (MM), limb girdle muscular dystrophy type 2B (LGMD 2B), and distal myopathy with anterior tibial onset (DMAT) [1]. Although rare, dysferlinopathy accounts for up to 30% of progressive recessive muscular dystrophies in certain geographical areas such as the Middle East and the Indian subcontinent [2].
It is possible that dysferlin interacts with significant immunological pathways [3] as the clinical manifestations of dysferlinopathy resemble the idiopathic inflammatory myopathy. In addition, inflammatory cell infiltrates are commonly observed in dysferlinopathy [4,5], which often lead to a misdiagnosis of dysferlinopathy as inflammatory myopathy. In this study, we analyzed the clinical manifestation of immune factors in patients with dysferlinopathy, so that we may understand the role of immune factors in dysferlinopathy pathogenesis, and provide a differential diagnosis of dysferlinopathy.
Materials and methods
Patient data collection
This study consisted of 29 patients who underwent muscle biopsy from 2012 to 2013, and none of the patients received treatment during this period. Of these, 14 patients were diagnosed with dysferlinopathy based on their clinical manifestations and immunohistochemical analysis. Eight patients were diagnosed with Duchenne muscular dystrophy (DMD) or Becker muscular dystrophy (BMD) by histological and immunohistochemical analysis. Finally, seven patients with polymyositis (PM) were diagnosed according to clinical manifestations and histological analysis, which fulfilled the Bohan and Peter’s criteria. Complete clinical and laboratory data were collected, including detailed personal history, clinical features, dysmorphic features, and muscle power test. The DMD/BMD and PM patient samples were used as a control. The IRB of Chinese PLA General Hospital approved this study.
Muscle biopsy and specimen processing
The patients underwent open biopsy in quadriceps femoris, gastrocnemius, or biceps brachii under local anesthesia after obtaining a written informed consent. The muscle specimens were immediately frozen by immersing in isopentane prechilled in liquid nitrogen, and stored at -80°C until processed. Transverse serial 8-μm frozen muscle sections were routinely stained for haematoxylin and eosin (H&E) to determine muscle morphology, and additional serial sections (5-μm thick) were prepared separately for immunohistochemical staining.
Immunohistochemical analysis
To characterize inflammatory infiltrates, serial frozen muscle sections from all patients were stained with rabbit monoclonal antibodies against CD4+ T lymphocytes, CD8+ T lymphocytes, CD68+ macrophages, CD20+ B cells, major histocompatibility complex class I (MHC-I), and membrane attack complex (C5b-9; all from Abcam, Cambridge, UK). Approximately 5-μm thick consecutive transverse muscle sections from each of the 14 dysferlinopathy, 7 classical PM, and 8 DMD/BMD patients were fixed in ice-cold acetone prior to the immunohistochemical analysis. Serial sections were air-dried in room temperature, blocked with 3% H2O2, washed with 1% phosphate buffered saline (PBS), and blocked with 5% goat serum for 15 min each. After 1% PBS wash, the samples were incubated with mentioned primary antibodies (1:200) for 1 h at 37°C. Then, samples were treated with secondary antibody (polymerized HRP mouse anti-rabbit IgG) for 30 min at 37°C, and 3, 3’-diaminobenzidine (DAB) staining for 20 min. Finally, the slides were mounted with neutral balsam (all from Golden Bridge, Beijing) and observed under light microscope (BX51, Olympus, Japan). All incubation steps were performed in humidified chambers.
Evaluation of immunohistochemical staining
Because of high sample heterogeneity and low frequency of infiltrating inflammatory cells, quantification was performed with the hot spot method used for myositis and estimating new blood vessel formation in breast tumor [6]. This method allowed the quantification of CD4+, CD8+ and, B cells in hot spots defined as muscle areas containing the highest density of infiltrating cells. Accordingly, two hot spots per muscle section were selected randomly. In each spot, positive cells were counted in 10 consecutive fields (100×). The average of the number of positive cells per two hot spots was taken and expressed as the number of positive cells per field (100×).
For MHC-I and C5b-9 evaluation, we applied a semiquantitative grading system mentioned by M. Fanin [7] as follows: negative staining (-), slightly positive staining (+), moderately positive staining (++), and strongly positive staining (+++). The immunohistochemistry data was observed and interpreted independently by two persons.
Statistical analysis
The independent t-test was used to compare the means of two samples for continuous variables. Quantitative results are expressed as the mean ± SD. Levels of MHC-1 and C5b-9 expression were compared using the non-parametric Kruskal-Wallis H test.
Statistically significant values had a P-value of < 0.05. Statistical analysis was performed with SPSS (version 17.0, Chicago, IL, USA).
Results
Demographic and clinical features
In this study, of the 29 patients, 14, 8, and 7 patients were diagnosed with dysferlinopathy, DMD/BMD, and polymyositis respectively. The onset of dysferlinopathy in the 10 men and 4 women was from the age of 3-43 years (mean: 19.9 years), with disease duration of 1-20 years (mean: 9.4 years). The DMD/BMD (7 men and 1 woman) and polymyositis (2 men and 5 women) patients had disease onset between 2-35 years (mean: 8.8 years), and 8-73 years (mean: 40.0 years) respectively. The disease duration ranged from 1 to 15 years (mean: 8.8 years) and 0.2 to 2 years (mean: 0.8 years) for DMD/BMD and polymyositis, respectively. The demographic and clinical data of the patients are summarized in Table 1.
Table 1.
Clinical features of patients with dysferlinopathy, DMD/BMD, and polymyositis
| NO. | Sex/age | Age of onset (yrs) | Disease duration (yrs) | Symptoms at onset | Disease progression | CK value (2-200 u/l) |
|---|---|---|---|---|---|---|
| Dysferlinopathy | ||||||
| 1 | Male/17 | 15 | 2 | Lower limbs weakness | slow | 8489.6 |
| 2 | Male/22 | 20 | 2 | Lower limbs weakness | slow | 19619 |
| 3 | Female/49 | 19 | 30 | Lower limbs weakness | slow | 1807.3 |
| 4 | Male/8 | 3 | 5 | Unsymptomic hyperckemia | unprogressive | 4822 |
| 5 | Female/57 | 27 | 30 | Upper and lower limbs weakness | slow | 1542.6 |
| 6 | Male/17 | 13 | 4 | Lower limbs weakness | slow | 13297 |
| 7 | Male/33 | 30 | 3 | Lower limbs weakness | slow | 4791.5 |
| 8 | Male/15 | 3 | 12 | Delayed motor milestone | unprogressive | |
| 9 | Male/29 | 28 | 1 | Upper and lower limbs weakness | unprogressive | 1432.7 |
| 10 | Female/16 | 10 | 6 | Upper and lower limbs weakness; spine deformity | intermediate | 96.3 |
| 11 | Male/20 | 25 | 5 | Lower limbs weakness | slow | 1528 |
| 12 | Male/26 | 16 | 10 | Upper and lower limbs weakness | slow | 4747 |
| 13 | Female/45 | 43 | 2 | Upper and lower limbs weakness | slow | 7081 |
| 14 | Male/46 | 26 | 20 | Lower limb weakness | intermediate | 77.4 |
| polymyositis | ||||||
| 1 | Female/60 | 59 | 0.7 | upper and lower limbs weakness; skin rash | intermediate | 22.3 |
| 2 | Female/48 | 46 | 2 | Muscle weakness; dysarthria | intermediate | 451.5 |
| 3 | Male/11 | 10 | 1 | Muscle weakness; dysphagia | intermediate | 461.5 |
| 4 | Female/20 | 19 | 0.25 | upper and lower limbs weakness | intermediate | 4218.1 |
| 5 | Male/8 | 8 | 0.2 | upper and lower limbs weakness; myalgia; skin rash | rapid | 4429 |
| 6 | Female/73 | 73 | 0.7 | upper and lower limbs weakness; skin rash | intermediate | 492.4 |
| 7 | Female/65 | 65 | 0.8 | upper and lower limbs weakness; myalgia | intermediate | 233.5 |
| DMD/BMD | ||||||
| 1 | Male/20 | 5 | 15 | Lower limb weakness; delayed motor milestone | slow | 2631.1 |
| 2 | Male/12 | 2 | 10 | Lower limb weakness; delayed motor milestone | slow | 6734.3 |
| 3 | Male/16 | 2 | 14 | Muscle fatigue; hyperckemia | slow | 1849.3 |
| 4 | Female/12 | 2 | 10 | delayed motor milestone | slow | 3827 |
| 5 | Male/10 | 7 | 3 | Lower limbs weakness | intermediate | 5811 |
| 6 | Male/27 | 15 | 12 | Lower limbs weakness | slow | 4709.4 |
| 7 | Male/7 | 2 | 5 | Lower limb weakness; delayed motor milestone | slow | 4763.1 |
| 8 | Male/36 | 35 | 1 | upper and lower limbs weakness | intermediate | 827 |
The most common disease onset symptom for all the three groups was weakness in the limbs or motor delay. The dysferlinopathy patients showed particular weakness in the lower limbs, with additional asymptomatic hyperCKemia. The clinical course was relatively benign as many patients were able to walk even years after the onset of the disease. The polymyositis patients exhibited skin rash or myalgia in addition to the weakened limbs with the clinical progression of the onset symptoms. Furthermore, elevated levels of creatine kinase (CK) were found in all the three groups. The CK values were the highest in the dysferlinopathy patients, who showed up to 10-100 times the normal range. The DMD/BMD patients showed slow disease progression and had 10-30 times elevated CK values. However, not all the polymyositis patients showed increased levels of CK; those patients with elevated CK had 20 times CK than the normal. Patients from all the groups were confined to wheelchairs during muscle biopsy.
Detection of CD4+ cells, CD8+ cells, B cells, and macrophages
The detection of inflammatory cells in muscle biopsy samples from the 29 patients are shown in Table 2 and Figure 1. Patients with polymyositis had the most severe inflammatory cell infiltration, specifically CD4+ cells and macrophages. The frequency of CD4+ cells infiltration was similar in dysferlinopathy and DMD/BMD patients, and both were fewer than that in the polymyositis patients (5.7 ± 4.4 vs. 12.3 ± 6.4, P = 0.009; 4.9 ± 5.7 vs. 12.3 ± 6.4, P = 0.009). Consistently, more macrophage infiltration was observed in the polymyositis patients than the DMD/BMD patients (10.8 ± 6.5 vs. 3.7 ± 3.1, P = 0.006) and dysferlinopathy patients. Unlike the CD4+ cell infiltration, the number of macrophages infiltrating dysferlinopathy muscle sample was greater than that of DMD/BMD patients (7.8 ± 4.3 vs. 3.7 ± 3.1, P = 0.047). Thus, DMD/BMD patients showed the least amount of macrophage infiltration. The dysferlinopathy patients showed lesser CD8+ cell infiltration than polymyositis patients did (1.3 ± 1.1 vs. 3.3 ± 1.8, P = 0.005). There were no other statistically significant differences in CD8+ cell or B cell infiltration in the three groups.
Table 2.
Comparison of inflammatory cells in muscle biopsy samples of dysferlinopathy, DMD/BMD, and polymyositis patients
| CD4+ cells (mean ± SD) | CD8+ cells (mean ± SD) | B cells (mean ± SD) | Macrophages (mean ± SD) | |
|---|---|---|---|---|
| Dysferlinopathy | 5.7 ± 4.4a | 1.3 ± 1.1c | 2.3 ± 2.2 | 7.8 ± 4.3d |
| Polymyositits | 12.3 ± 6.4 | 3.3 ± 1.8 | 2.6 ± 1.9 | 10.8 ± 6.5 |
| DMD/BMD | 4.9 ± 5.7b | 2.0 ± 1.6 | 2.5 ± 3.4 | 3.7 ± 3.1e |
Dysferlinopathy versus polymyositis; P = 0.009;
DMD/BMD versus polymyositis; P = 0.009;
dysferlinopathy versus polymyositis; P = 0.005;
dysferlinopathy versus DMD/BMD; P = 0.047;
DMD/BMD versus polymyositis; P = 0.006.
No other statistically significant differences were found among the different subgroups.
Figure 1.
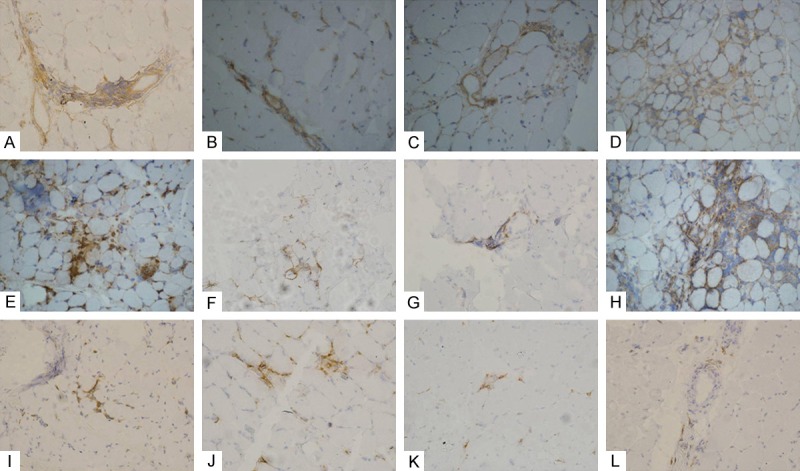
Detection of inflammatory infiltrating cells. Immunohistochemical staining for inflammatory cells in dysferlinopathy (A-D), polymyositis (E-H), and DMD/BMD (I-L) patients. Immunohistochemical staining for CD4+ cells (A, E, I), CD8+ cells (B, F, J), B cells (C, G, K), and macrophages (D, H, L).
Expression of MHC-I and C5b-9 in muscle biopsy
The staining data of MHC-I and C5b-9 expression in all the muscle biopsies is shown in Table 3 and Figure 2. MHC-1 expression was observed in 71.4% (10/14) of the dysferlinopathy patients and 100% of polymyositis patients. In contrast, only 25% (2/8) stained slightly positive for MHC-I in DMD/BMD patients. All the dysferlinopathy and polymyositis patient samples stained positive for muscle-specific C5b-9 expression, with 28% (4/14) of dysferlinopathy patients showing strongly positive fibers. The C5b-9 staining in the muscles of DMD/BMD patients was negative or slightly positive.
Table 3.
Immunohistochemical results of MHC-I and C5b-9
| - | + | ++ | +++ | |
|---|---|---|---|---|
| MHC-I* | ||||
| Dysferlinopathy | 4 | 7 | 3 | 0 |
| Polymyositis | 0 | 5 | 2 | 0 |
| DMD/BMD | 6 | 2 | 0 | 0 |
| C5b9# | ||||
| Dysferlinopathy | 0 | 6 | 4 | 4 |
| Polymyositis | 0 | 4 | 2 | 1 |
| DMD/BMD | 4 | 4 | 0 | 0 |
P = 0.011 for MHC-I and C5b-9 staining respectively within the three groups;
P = 0.003 for MHC-I and C5b-9 staining respectively within the three groups.
Figure 2.
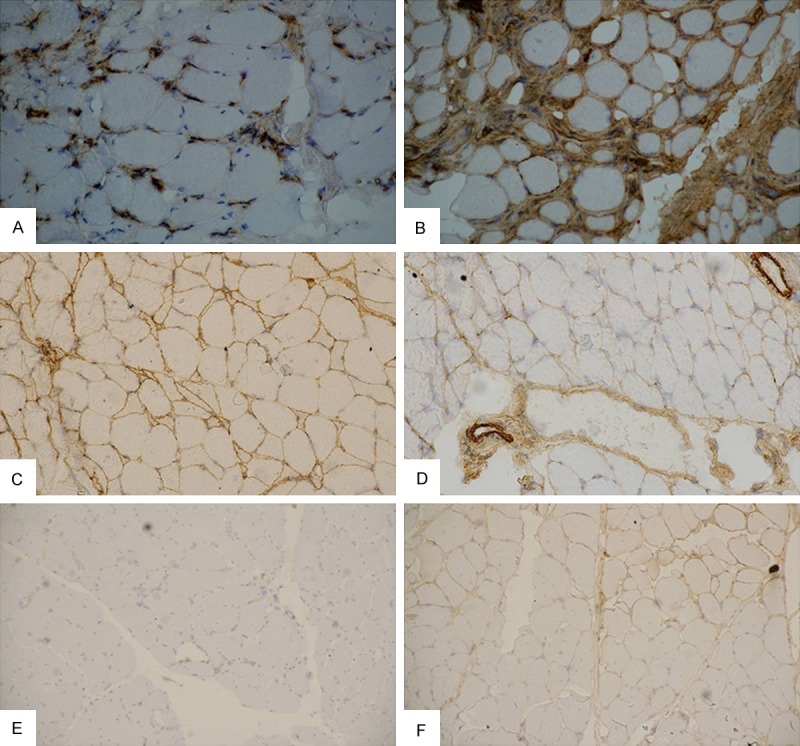
Expression of MHC-I and C5b-9. Immunohistochemical staining for MHC-I (A, C, E) and C5b-9 (B, D, F) in dysferlinopathy (A, B), polymyositis (C, D), DMD/BMD (E, F) patients. Magnification is 100×.
Discussion
Dysferlinopathy is an autosomal, recessive muscular dystrophy caused by the mutations in the DYSF gene, which encodes the dysferlin protein. Mutations in the dysferlin gene cause a heterogeneous group of muscular dystrophies, including Miyoshi myopathy (MM), limb girdle muscular dystrophy Type 2B (LGMD2B), and distal myopathy with anterior tibial onset (DMAT). These diseases are collectively referred as dysferlinopathy [8-10], and MM and LGMD 2B are the most common types of dysferlinopathy. The patients share the same clinical manifestations such as adolescent onset, progressive muscle weakness, and elevated CK level. The MM patients show distal muscular weakness at onset, frequently involving the gastrocnemius muscles and LGMD 2B patients show shoulder and pelvic girdle weakness at onset [11]. Epigenetic interactions are suspected to underlie some of the clinical symptomatic variations; however, this remains to be confirmed.
In our study, the patients with dysferlinopathy, usually demonstrated disease onset at adult stage, relatively benign clinical course, and elevated CK values compared to the DMD/BMD or polymyositis patients. The deficient dysferlin protein results in defective membrane patching causing leakage of intracellular contents from the inside of the muscle fibers, leading to a significant increase in the CK level. We hypothesize that the damage caused by dysferlin is less progressive than dystrophin, which is supported by the relatively benign clinical course, the asymptomatic hyperCKemia, and the late symptom onset. According to the “danger” hypothesis proposed by Andrew et al [12], intracellular contents that leaked due to membrane disruption in dysferlinopathy can act as endogenous adjuvants that trigger an inflammatory response.
Muscle inflammation was observed in the dysferlinopathy patients, making the muscle pathophysiology indistinguishable from that of idiopathic inflammatory myopathy. Moreover, SLJ/J mice that are used as animal models for experimental autoimmune myopathy carry mutations in dysferlin [13]. Several studies have investigated the causes underlying inflammation in dysferlinopathy, and have reported the upregulation of inflammasome, and the involvement of macrophages [14,15]. In fact, dysferlin-deficient monocytes display increased phagocytic activity [16]. In this study, we demonstrated that a significantly higher number of macrophages infiltrate muscles in dysferlinopathy patients than the DMD/BMD patients, implying a pivotal role for macrophages in dysferlinopathy pathogenesis.
Han et al. demonstrated that the complement system is important in muscle pathology in dysferlinopathy, and the genetic disruption of the central component (C3) of the complement system ameliorated muscle pathology in dysferlin-deficient mice [17]. Moreover, the decay-accelerating factor (CD55), the complement inhibitor, was downregulated in the skeletal muscle of dysferlin-deficient SJL/J mice, indicating the involvement of complement system in the dysferlinopathy pathogenesis [18]. Supporting previous studies, C5b-9, a marker of membrane attack complex (MAC) displayed the strongest expression in the dysferlinopathy patients. In addition, overexpression of MHC-I was observed in the muscle tissue of dysferlinopathy patients, suggesting that both muscle inflammation and the complement system participate in the pathogenesis of dysferlinopathy.
SJL/J (SJL) mice and A/J mice are two naturally occurring dysferlinopathy animal models [19]. In SJL/J mice, a 171-bp in-frame deletion in the DYSF mRNA is predicted to remove 57 amino acids from the corresponding protein, while A/J mice carry a unique ETn retrotransposon insertion near the 5’ end (intron 4) of the DYSF gene [20]. Interestingly, the SJL/J (SJL) and A/J mice display divergent phenotypes. For example, A/J mice display a later onset, and a slower progression of the muscular disease compared to the SJL mice [21]; this difference may be due to varying genetic background. The SJL/J mouse model has been extensively used to study autoimmune diseases such as encephalomyelitis [22], myositis [23], and hypophysitis [24]. Hence, the immunological data obtained using these animal models can be used to understand the immune response pathways involved in dysferlinopathy.
In conclusion, this study demonstrated, at least in part, the immune factors expressed dysferlinopathy patients. Dysferlin protein plays a role in the membrane fusion and repair and the deficient function cause leakage of intracellular contents, which may be an attractant for immunological responses. Thus, it is important for future studies to focus on understanding the co-relation of the immune system and muscle dystrophy. The presence of MHC-I and C5b-9 might aid in the differential diagnosis of dysferlinopathy patients.
Acknowledgements
The National Natural Science Foundation of China under Grant No. 81271399 and the Natural Science Foundation of Beijing under Grant No. 7132216 supported this work.
Disclosure of conflict of interest
None.
References
- 1.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–13. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Urtizberea JA, Bassez G, Leturcq F, Nguyen K, Krahn M, Levy N. Dysferlinopathies. Neurol India. 2008;56:289–97. doi: 10.4103/0028-3886.43447. [DOI] [PubMed] [Google Scholar]
- 3.Glover L, Brown RH Jr. Dysferlin in membrane trafficking and patch repair. Traffic. 2007;8:785–94. doi: 10.1111/j.1600-0854.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 4.Confalonieri P, Oliva L, Andreetta F, Lorenzoni R, Dassi P, Mariani E, Morandi L, Mora M, Cornelio F, Mantegazza R. Muscle inflammation and MHC class I up-regulation in muscular dystrophy with lack of dysferlin: an immunopathological study. J Neuroimmunol. 2003;142:130–6. doi: 10.1016/s0165-5728(03)00255-8. [DOI] [PubMed] [Google Scholar]
- 5.Choi JH, Park YE, Kim SI, Kim JI, Lee CH, Park KH, Kim DS. Differential immunohistological features of inflammatory myopathies and dysferlinopathy. J Korean Med Sci. 2009;24:1015–23. doi: 10.3346/jkms.2009.24.6.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tournadre A, Porcherot M, Chérin P, Marie I, Hachulla E, Miossec P. Th1 and Th17 balance in inflammatory myopathies: interaction with dendritic cells and possible link with response to high-dose immunoglobulins. Cytokine. 2009;46:297–301. doi: 10.1016/j.cyto.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 7.Fanin M, Angelini C. Muscle pathology in dysferlin deficiency. Neuropathol Appl Neurobiol. 2002;28:461–70. doi: 10.1046/j.1365-2990.2002.00417.x. [DOI] [PubMed] [Google Scholar]
- 8.Han R. Muscle membrane repair and inflammatory attack in dysferlinopathy. Skelet Muscle. 2011;1:10. doi: 10.1186/2044-5040-1-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bushby KM. Making sense of the limb-girdle muscular dystrophies. Brain. 1999;122:1403–20. doi: 10.1093/brain/122.8.1403. [DOI] [PubMed] [Google Scholar]
- 10.Illa I, Serrano-Munuera C, Gallardo E, Lasa A, Rojas-García R, Palmer J, Gallano P, Baiget M, Matsuda C, Brown RH. Distal anterior compartment myopathy: a dysferlin mutation causing a new muscular dystrophy phenotype. Ann Neurol. 2001;49:130–34. [PubMed] [Google Scholar]
- 11.Liu J, Aoki M, Illa I, Wu C, Fardeau M, Angelini C, Serrano C, Urtizberea JA, Hentati F, Hamida MB, Bohlega S, Culper EJ, Amato AA, Bossie K, Oeltjen J, Bejaoui K, McKenna-Yasek D, Hosler BA, Schurr E, Arahata K, de Jong PJ, Brown RH Jr. Dysferlin, a novel skeletal muscle gene, is mutated in Miyoshi myopathy and limb girdle muscular dystrophy. Nat Genet. 1998;20:31–36. doi: 10.1038/1682. [DOI] [PubMed] [Google Scholar]
- 12.Andrews NW. Membrane repair and immunological danger. EMBO Reports. 2005;6:826–830. doi: 10.1038/sj.embor.7400505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vafiadaki E, Reis A, Keers S, Harrison R, Anderson LV, Raffelsberger T, Ivanova S, Hoger H, Bittner RE, Bushby K, Bashir R. Cloning of the mouse dysferlin gene and genomic characterization of the SJL-Dysf mutation. Neuroreport. 2001;12:625–629. doi: 10.1097/00001756-200103050-00039. [DOI] [PubMed] [Google Scholar]
- 14.Rawat R, Cohen TV, Ampong B, Francia D, Henriques-Pons A, Hoffman EP, Nagaraju K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am J Pathol. 2010;176:2891–900. doi: 10.2353/ajpath.2010.090058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Luna N, Gallardo E, Sonnet C, Chazaud B, Dominguez-Perles R, Suarez-Calvet X, Gherardi RK, Illa I. Role of thrombospondin 1 in macrophage inflammation in dysferlin myopathy. J Neuropathol Exp Neurol. 2010;69:643–53. doi: 10.1097/NEN.0b013e3181e0d01c. [DOI] [PubMed] [Google Scholar]
- 16.Nagaraju K, Rawat R, Veszelovszky E, Thapliyal R, Kesari A, Sparks S, Raben N, Plotz P, Hoffman EP. Dysferlin Deficiency Enhances Monocyte Phagocytosis: A Model for the Inflammatory Onset of Limb-Girdle Muscular Dystrophy 2B. Am J Pathol. 2008;172:774–85. doi: 10.2353/ajpath.2008.070327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han R, Frett EM, Levy JR, Rader EP, Lueck JD, Bansal D, Moore SA, Ng R, Beltrán-Valero de Bernabé D, Faulkner JA, Campbell KP. Genetic ablation of complement C3 attenuates muscle pathology in dysferlin-deficient mice. J Clin Invest. 2010;120:4366–74. doi: 10.1172/JCI42390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wenzel K, Zabojszcza J, Carl M, Taubert S, Lass A, Harris CL, Ho M, Schulz H, Hummel O, Hubner N, Osterziel KJ, Spuler S. Increased susceptibility to complement attack due to down-regulation of decay-accelerating factor/CD55 in dysferlin-deficient muscular dystrophy. J Immunol. 2005;175:6219–25. doi: 10.4049/jimmunol.175.9.6219. [DOI] [PubMed] [Google Scholar]
- 19.Kobayashi K, Izawa T, Kuwamura M, Yamate J. Dysferlin and animal models for dysferlinopathy. J Toxicol Pathol. 2012;25:135–47. doi: 10.1293/tox.25.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho M, Post CM, Donahue LR, Lidov HG, Bronson RT, Goolsby H, Watkins SC, Cox GA, Brown RH Jr. Disruption of muscle membrane and phenotype divergence in two novel mouse models of dysferlin deficiency. Hum Mol Genet. 2004;13:1999–2010. doi: 10.1093/hmg/ddh212. [DOI] [PubMed] [Google Scholar]
- 21.Kobayashi K, Izawa T, Kuwamura M, Yamate J. The distribution and characterization of skeletal muscle lesions in dysferlin-deficient SJL and A/J mice. Exp Toxicol Pathol. 2010;62:509–517. doi: 10.1016/j.etp.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 22.Bernard CC, Carnegie PR. Experimental autoimmune encephalomyelitis in mice: Immunologic response to mouse spinal cord and myelin basic proteins. J Immunol. 1975;114:1537–1540. [PubMed] [Google Scholar]
- 23.Rosenberg NL, Ringel SP, Kotzin BL. Experimental autoimmune myositis in SJL/J mice. Clin Exp Immunol. 1987;68:117–129. [PMC free article] [PubMed] [Google Scholar]
- 24.Tzou SC, Lupi I, Landek M, Gutenberg A, Tzou YM, Kimura H, Pinna G, Rose NR, Caturegli P. Autoimmune hypophysitis of SJL mice: clinical insights from a new animal model. Endocrinology. 2008;149:3461–3469. doi: 10.1210/en.2007-1692. [DOI] [PMC free article] [PubMed] [Google Scholar]