

Abstract
Core elements of cell regulation are made up of protein-protein interaction (PPI) networks. However, many parts of the cell regulatory systems include unknown PPIs. To approach this problem, we have developed a computational method of high-throughput PPI network prediction based on all-to-all rigid-body docking of protein tertiary structures. The prediction system accepts a set of data comprising protein tertiary structures as input and generates a list of possible interacting pairs from all the combinations as output. A crucial advantage of this docking based method is in providing predictions of protein pairs that increases our understanding of biological pathways by analyzing the structures of candidate complex structures, which gives insight into novel interaction mechanisms. Although such exhaustive docking calculation requires massive computational resources, recent advancements in the computational sciences have made such large-scale calculations feasible. different rigid-body docking tools with different scoring models. We found that the predicted interactions were different between the results from the two tools. When the positive predictions from both of the docking tools were combined, all the core signaling interactions were correctly predicted with the exception of interactions activated by protein phosphorylation. Large-scale PPI prediction using tertiary structures is an effective approach that has a wide range of potential applications. This method is especially useful for identifying novel PPIs of new pathways that control cellular behavior.
Keywords: Interactome, Bacterial chemotaxis signaling pathway, Exhaustive docking, Pathway prediction, Protein-protein interaction network, Parallel computation, Rigid-body docking.
1. INTRODUCTION
Identifying protein-protein interaction (PPI) networks is useful in understanding molecular biological phenomena such as signal transduction and the regulation of gene expression [1]. The most powerful methods used for PPI network prediction includes sequence based searching [2-4] and evolutional information based methods [5]. In addition, there are approaches that also employ protein tertiary structure to investigate PPIs. Ogmen et al. developed a PPI prediction method based on the structural alignments of query proteins to known interface surfaces in the database [6]. Acuner Ozbabacan et al. applied this method to predict PPI in the human apoptosis pathway and identified several candidates that were not listed in the corresponding database [7]. These methods work well for most of the cases in which the interaction databases have similar sequences or interaction surface structures to those of the query proteins. However, there are examples, such as the epidermal growth factor receptor (EGFR) signaling pathway, where a complete set of interactions is unknown. In such cases it is difficult to predict novel PPIs using existing methods because they depend on known interaction regions. In order to discover novel interactions, the candidate interaction sites are searched throughout the entire surface of each protein structure.
Identifying large-scale PPI networks based on the full tertiary structure is computationally expensive. However, recent progress in computer science has facilitated the development of supercomputers that are able to perform such large-scale analyses. In the PPI research field, some pioneering studies have been done by taking advantage of this huge computational power. Mosca et al. presented a docking prediction database of the yeast interactome by using existing and modeled structures [8]. This idea was also later employed in the field of structural systems biology [9].
Recent studies have demonstrated novel methods of PPI network prediction by employing exhaustive rigid-body protein-protein docking and post-docking analysis [10-12]. Protein-protein docking is usually used to predict possible binding conformation between two proteins that are already known to undergo direct interaction. However, these docking calculations have now been extended to include all combinations of proteins, including protein pairs that were not previously known to directly interact with one another. In these works, the affinities of given protein pairs are evaluated using groups of models with high interaction energy scores. In our previous study, we examined up to ten thousand possible structure pairs and showed prediction performance that was better than random [12]. The same procedure was subsequently applied to RNA molecules [13]. A similar procedure has been used by Wass et al. with a larger dataset (about 50,000 pairs), using a different rigid-body docking tool Hex with shape complementarity score function and post-docking analysis [14]. Rigid-body docking software, where the relative coordinates of atoms comprising the query protein structures remain unchanged, is commonly used in this kind of approach.
The rigid-body docking method is the most feasible procedure for analyzing the interactome based on protein structures using current computational technology [14]. However, flexibility and dynamics are known to play an important role in PPIs such as in the induced fit. Soft docking software (e.g., RosettaDock [15] and FiberDock [16]), which consider backbone or side-chain flexibility, are advantageous in terms of obtaining structures of protein complexes with high resolution accuracy. One possible strategy for analyzing large-numbers of PPIs, such as the interactome, is to initially perform rigid-body docking based PPI predictions and then use particular protein pairs of interest for flexible docking or structural refinements.
To explore how the current rigid-body docking based method performs on real biological data, we applied a method used in our previous studies [12][13] to reconstruct the well-known bacterial chemotaxis signaling pathway (Fig. 1). The bacterial chemotaxis pathway has been studied for several decades and most of the functional relationships among the proteins involved in this signal process have been identified, especially those involving the core part of the signaling system. However there are still uncertainties concerning how flagellar motor proteins are assembled and operate (reviewed in [17]). Also in the existing databases there are some interactions not listed in conventional network descriptions (Fig. 2).
Fig. (1).
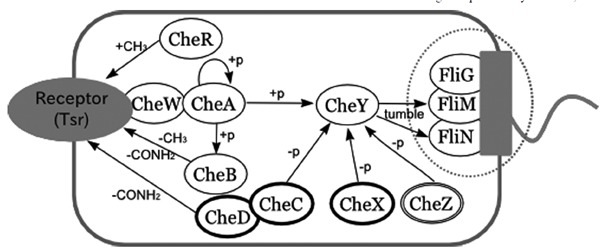
A pathway diagram of bacterial chemotaxis. Proteins in this map are taken from three species: E. coli, S. typhimurium, and T. maritima. The dotted circle highlights proteins that make up the flagellar motor. CheC, D and X (circled in bold) are only found in T. maritima, while CheZ (circled by a double line) is only found in E. coli and S. typhimurium. When bacteria sense external signals, such as the presence of chemicals or changes in temperature, chemotactic receptors in the form of a complex with histidine kinase CheA linked to CheW transmit a signal to the cytoplasm by changing the autophosphorylation rate of CheA.In this study, a serine receptor Tsr structure was used. In the unstimulated state, the phosphorylated CheA transfers its phosphate group to CheB and CheY. The phosphorylated form of CheY displays increased affinity for the flagellar motor switch protein FliM. This enhances the clockwise rotation of the flagellar motor, which causes the cell body to change direction in a random fashion (i.e., tumbling motion). When the external signal is of a favorable type, such as the presence of nutrients, it works to reduce the CheA phosphorylation rate. The net effect of this is to lower the CheY phosphorylation level, thus reducing the frequency of changing swimming direction. Consequently, the cell quickly enters a suitable environment for growth. The CheY phosphorylation level is also regulated by phosphatases (CheC, D, X, Z). CheR and CheB play a role in adaptation. Specifically, CheR methylates the activated receptor and CheB demethylates the receptor when phosphorylated. Methylated receptors have a reduced ability to suppress the rate of autophosphorylation of CheA.
Fig. (2).
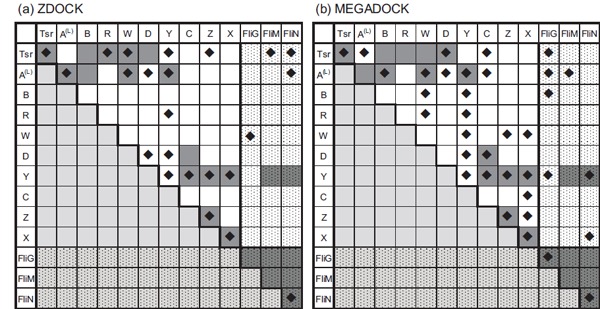
Predicted interactions among chemotaxis proteins. Predicted interactions among chemotaxis proteins by using (a) ZDOCK and (b) MEGADOCK as docking engines. The dark grey coloured cells indicate known interacting pairs based on conventional studies. Cells with diamond marks indicate predicted interactions. Cells filled with small dots show flagella protein related combinations. Proteins related to the flagellar motor are listed on the right/bottom side. The short form of CheA is known to interact with CheZ [34] but it was not included because the structure was unavailable. A total of seven interactions that are not coloured dark grey were found in the STRING database [35] by (i) searching interactions associated with experimental reports or (ii) those annotated in databases (KEGG, BioCyc). The interactions are: CheY-FliG, CheY-CheW, CheB-CheW, Tsr-CheZ, Tsr-CheA, CheR-FliN, CheR-CheZ. These interactions were not considered as “correct” in this study because they have not been characterized.
Here, we conducted PPI network prediction by exhaustive docking using two different docking engines: ZDOCK 3.0.1 and MEGADOCK 2.5. ZDOCK, which is currently one of the most popular rigid-body docking engines, uses a scoring function that includes shape complementarity, electrostatics and a heuristic potential called atomic contact energy [18]. MEGADOCK is a similar system to ZDOCK that searches probable docking structures in a grid-based 3D space using fast Fourier transform (FFT). MEGADOCK employs a much simpler score function in which only shape complementarity and electrostatics is considered and thus makes the calculations 8.8 times faster than ZDOCK. The method is set up to perform massive numbers of calculations that are run on parallel computing systems. There are also other types of useful rigid-body docking tools without a 3D grid-base searching system, such as Hex [19, 20], LZerD [21] and PatchDock [22]. Here we used FFT-based docking software because it thoroughly searches whole 3D space whilst at the same time being computationally efficient, which makes it suitable for large-scale exhaustive docking on parallel computing environments. We also considered using Hex for these experiments because it is one of the fastest docking engines for shape complementarity score function. Nonetheless, we used MEGADOCK as our docking engine with a simple score function that was mainly based on shape complementarity. Specifically, MEGADOCK is designed to be highly efficient in the exhaustive docking calculations and is implemented using parallelizing techniques. MEGADOCK computes each of the docking jobs in parallel using threads by OpenMP. The docking jobs are distributed to thousands of nodes by MPI. In this way, MEGADOCK usually completes the exhaustive docking calculations in just a few minutes, depending on the number of dockings needed and the number of available nodes. MEGADOCK has a further advantage in that the calculation time is almost unaffected by adding an electrostatic term to the score function [23].
2. MATERIALS AND METHODS
2.1. Protein Structure Data
We selected the well characterized species E. coli, S. typhimurium and T. maritima as the targets for this study of bacterial chemotaxis. The following procedures were performed to retrieve the data. Firstly, we retrieved the PDB ID list that corresponded to the proteins in the bacterial chemotaxis pathway. Pathway data were obtained from KEGG [24] (KEGG pathway ID: eco02030, stm020230, tma02030 for each species). PDB IDs were obtained through LinkDB [25]. The collected protein structure files were prescreened according to the following criteria, as in the recently published protein-protein docking benchmark version 3.0 [26]: (i) experimental method: X-ray diffraction, resolution better than 3.25 Å; (ii) polypeptides consisting of more than 30 residues. In principle, mutant and synthetic data were excluded with the one exception of CheZ, for which only mutant data were available. Structural data for ligand binding domains of membrane proteins, which are located in the periplasm, were also excluded. Each PDB file was divided into data for one polypeptide chain. Using this procedure we obtained 101 protein structures, which corresponded to 13 protein species (CheA, CheB, CheC, CheD, CheR, CheW, CheX, CheY, CheZ, Tsr, FliM, FliG, FliN). Where possible we used multiple structural data of each protein species. The protein structure data used in this study are shown in Table 1.
Table 1.
List of protein structures used in this study.
| Protein name | Structures | ||
|---|---|---|---|
| CheA (21) | 1A0O_B,D,F,H | 1FFW_B,D | 1U0S_A |
| 1EAY_C,D | 1I5N_A,B,C,D | 2CH4_A,B | |
| 1FFG_B,D | 1I5D_A | ||
| 1FFS_B,D | 1TQG_A | ||
| CheB (3) | 1A2O_A,B | 1CHD_A | |
| CheC (3) | 1XKR_A | 2F9Z_A,B | |
| CheD (2) | 2F9Z_C,D | ||
| CheR (2) | 1AF7_A | 1BC5_A | |
| CheW (2) | 2CH4_W,Y | ||
| CheX (4) | 1SQU_A,B | 1XKO_A,B | |
| CheY (51) | 1A0O_A,C,E,G | 1KMI_Y | 2FMF_A |
| 1BDJ_A | 1TMY_A | 2FMH_A | |
| 1CHN_A | 1U0S_Y | 2FMI_A | |
| 1EAY_A,B | 1ZDM_A,B | 2FMK_A | |
| 1F4V_A,B,C | 2B1J_A,B | 2PL9_A,B,C | |
| 1FFG_A,C | 2CHE_A | 2PMC_A,B,C,D | |
| 1FFS_A,C | 2CHF_A | 2TMY_A | |
| 1FFW_A,C | 2CHY_A | 3CHY_A | |
| 1FQW_A,B | 2FKA_A | 3TMY_A,B | |
| 1HEY_A | 2FLK_A | 4TMY_A,B | |
| 1JBE_A | 2FLW_A | ||
| CheZ (1) | 1KMI_Z | ||
| FliG (3) | 1LKV_X | 1QC7_A,B | |
| FliM (1) | 2HP7_A | ||
| FliN (4) | 1O6A_A,B | 1YAB A,B | |
| Tsr (4) | 2CH7_A,B | 1QU7_A,B |
The first four letters of each protein structure indicate PDB ID followed by chain IDs.
2.2. Protein-protein Interaction Prediction
The PPI prediction scheme employed in this study consists of two stages. First, we conducted rigid-body docking simulations on all the possible binary combinations of target proteins. Using this process we obtained a group of high scoring docking complexes for each pair of proteins. Next, we analyzed each group of interactions by clustering based on the distance matrix calculated from each central coordinate of the complex. The sufficiently populated clusters were then taken and the docking energy score of the highest ranked complex involved in the selected clusters were used for PPI prediction. The deviation of the selected docking energy score from the score distribution of a high scoring group as Z-score was used to assess possible interactions. This prediction protocol has been described in detail elsewhere [12, 13]. Potential complexes that have no other high scoring interactions nearby were rejected. Thus, we consider likely binding pairs have at least one populated area of high scoring structures, one of which may be the true binding site.
2.2.1. Exhaustive Protein-protein Docking
We applied two FFT-based rigid-body protein-protein docking engines, ZDOCK 3.0.1 [27] and MEGADOCK 2.5 [13,23]. Both docking engines exhaustively search 3-D grid space around one protein (receptor) for probable docking interactions by rotating and translating another protein (ligand). The 3D-grid was constructed by a pitch of 1.2 Å. The rotation angle of ligand protein was set as every 15 degrees. The highest docking scores were recorded for each rotation angle throughout all the translation patterns. Using this procedure we obtained 3600 high scoring interactions. In this study we conducted dockings of 101 as receptors × 101 as ligands = 10201 structure data combinations.
2.2.2. Clustering of High-scoring Docking Conformations
We used the top 1000 ranked conformations for the post-docking analysis. Before reducing the number of potential hits, results of MEGADOCK were re-ranked by using ZRANK [28] to eliminate the energetically unlikely interactions.
Targeting the 1000 top ranked interactions, we conducted clustering based on the positional differences between the various models. The group average method was used for clustering. Through this method we created at least 20 clusters per docking. The parameters used for the clustering as well as the clustering method itself were carefully chosen in order to obtain the best possible prediction performance by employing a general benchmark dataset as described in our previous study [12]. The distance matrix of interactions was calculated as described previously [12]. Specifically, we take the sum of the two Euclid distances as follows: (i) 3-D coordinates of the ligand proteins when receptor protein positions are superimposed; (ii) 3-D coordinates of the receptor proteins when ligand protein positions are superimposed.
2.2.3. Prediction of Binary Interactions
After clustering analysis we calculated a predicted affinity index E for each protein pair as follows. First we defined sufficiently populated clusters C’ from {C | Ci to Cnumber of clusters} with the cutoff value of m*, where for the cluster Ci, the mi was calculated as the Z-score of the population of Ci throughout all the clusters. Second, the interaction with the highest docking score among all the data included in C’ was selected as the representative interaction. Finally we calculated the deviation of the docking score of the representative interaction, “E”, as the Z-score using scores of 1000 models as the population.
Using the value E we made predictions as to whether a pair of proteins interacts by means of the cutoff value E*. The value of E* was determined independently for each docking engine as the value that yielded the best F-measure when applied to a general benchmark dataset as described previously [12].
In many cases, there were multiple X-ray protein structures available for the docking procedure. For example when we investigate CheA (21 structures) and CheY (51), our docking results provided 21×51=1071 satisfactory results. We then calculate the affinity score (E) for each of these docking results. If the E values are larger than the cutoff (E*) among any of the 1071 docking results, we consider that CheA and CheY interact.
3. RESULTS
3.1. Predicted PPIs
Figure 2 shows the predicted PPIs by ZDOCK and MEGADOCK. The best F-measure value was 0.52 (ZDOCK, TP=12, TN=57, FP=11, FN=11, recall=0.52, precision=0.52, when E*=7.9 and m*=2.0) and 0.48 (MEGADOCK, TP=14, TN=47, FP=21, FN=9, recall=0.61, precision=0.40, when E*=5.5 and m*=0.0). For both the ZDOCK and MEGADOCK predictions, parameter values E* and m* were set as the same values that yielded the best F-measure value when applied to general benchmark data used in a previous study [12].
Previously known PPIs are colored gray in (Fig. 2). Relevant PPIs are defined based on published data [29-33]. The interactions of short form CheA [34] were not considered because its structure was unavailable. In addition, interactions based on genetic observations alone were excluded. FliG, FliM and FliN were considered as binding to the protein species because they make solid flagellar motor machinery. For in vitro studies, large numbers of interactions are listed in public databases such as the STRING database [35]. However, these data sets were not included in this study because the physical interactions for those PPIs are not characterized.
3.2. Considering Protein Localization
In the real cell, FliG, FliM and FliN proteins are closely associated with the membrane and only CheY is considered capable of interacting with these proteins. When we take into account protein localization, resulting in removing flagellar proteins (Fig. 1, proteins circled by the dotted line) in the dataset, the best F-measure value was 0.69 (ZDOCK, TP=11, TN=34, FP=6, FN=4, recall=0.73, precision=0.65) and 0.54 (MEGADOCK, TP=11, TN=25, FP=15, FN=4, recall=0.73, precision=0.42).
By restricting target proteins using localization information, both ZDOCK and MEGADOCK yielded better F-measure values, with both precision and recall values higher than that of the whole dataset. These results show that more accurate PPI predictions are made if protein localization is taken into consideration.
3.3. Comparison of the Prediction by using ZDOCK and MEGADOCK
Figure 3 shows a comparison of the results obtained by using ZDOCK and MEGADOCK. In total, 17 out of 23 relevant PPIs were detected when at least one of the docking software programs are used. Among 17 true positives, 9 were predicted by both of the software packages. Among the 28 false positives, 4 were common for both software packages and 7 were specific to ZDOCK, while 17 were specific to MEGADOCK. Thus, a lower precision value was obtained using MEGADOCK when compared to ZDOCK.
Fig. (3).
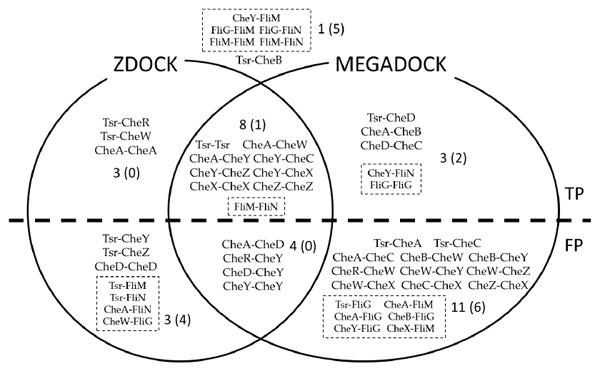
Predicted protein-protein interactions. Interactions listed inside the circles and above the dotted line show ‘True Positive’ pairs, those below the dotted line are ‘False Positive’ pairs. Pairs that are listed outside both circles are ‘False Negative’ pairs. Dotted boxes show flagella protein related interactions.
4. DISCUSSION
4.1. Performance of PPI Prediction
On the prediction of binary interactions, both ZDOCK and MEGADOCK yielded F-measure values of more than 0.4. When localizations of proteins were considered, ZDOCK performed better than MEGADOCK. It should be noted that MEGADOCK employs shape complementarity and an electrostatic score function whereas ZDOCK also takes into account heuristic score function based on atomic contact energy (IFACE) [18].
As shown in (Fig. 3), eight interactions were detected only by one of the docking software programs. Tsr-CheR, Tsr-CheW and CheA-CheA interactions were detected only by ZDOCK. Tsr-CheD, CheA-CheB, CheD-CheC, CheY-FliN and FliG-FliG interactions were detected only by MEGADOCK. Two out of three of the interactions detected by ZDOCK, Tsr-CheW and CheA-CheA, are tight binding interactions that constitute the receptor complex. In the case of MEGADOCK, with the exception of FliG-FliG, all the five detected interactions are transient. These results suggest there are differences in the type of interactions detected when different score functions are applied. It is very interesting to see the difference of the predicted PPIs by using different score functions.
Applying other score functions for docking or conducting re-ranking calculations with more sophisticated score functions to the generated decoys would be useful for analyzing the effects of score function on PPI prediction. To investigate this further we require a more thorough dataset such as that used by Kastritis and Bonvin [36] to evaluate any correlation between score function types and known protein-protein binding affinities.
One such example of applying different PPI prediction procedures is given in (Fig. 4), which shows PPI prediction results for a chemotaxis dataset using the PRISM protocol [37]. PRISM uses a template dataset of known protein-protein binding interfaces extracted from PDB. The surface of the target monomer protein, for which we want to identify binding partners, is analyzed against all the interface templates by structural alignment. Target protein pairs whose surface structures are aligned to any known interface pairs in the template dataset are then refined and scored by FiberDock [16].
Fig. (4).

Predicted interactions among chemotaxis proteins identified by using PRISM. The cells with a diamond mark indicate the predicted interacting pairs. The prediction was performed by defining an interacting pair of proteins according to the following criteria: (i) if the two potential binding partners have an interaction surface that is aligned to a template dataset constructed from known crystal structures, (ii) the predicted binding event yields less than zero energy by FiberDock calculations. The dark grey coloured cells indicate known interacting pairs based on conventional studies.
PRISM identified four candidates that potentially interact with most of the proteins in the dataset. Specifically, of the 13 proteins involved in the chemotaxis dataset, CheA and CheZ were predicted to interact with 11 while Tsr and CheY were predicted to interact with 10. Interestingly, the docking-based procedure applied to CheZ gave fewer predicted binding partners i.e., ZDOCK (three partners) and MEGADOCK (four partners). In this chemotaxis dataset only two interactions are confirmed for CheZ i.e., oligomerization of CheZ and interaction with CheY-p. However, CheZ is also known to interact with the short form of CheA [34] and localize in the cell pole area where receptor complexes are located [38]. CheY, the main target for CheZ, moves between the receptor area and flagellar motor area. The template-based PPI prediction suggests that CheZ may undergo non-specific interactions. Thus, it would be of interest to further analyze the role of CheZ in the receptor complex area.
4.2. Protein Localization
Both docking tools yielded better performances when flagellar motor related proteins are excluded from the target, while random prediction with a recall value of 0.5 yielded similar F-measure values (0.34 for the whole dataset and 0.35 for the restricted targets). It should be noted that direct binary interactions among flagellar motor proteins are still unclear and the true interacting pairs might be different from the “correct” interactions used here. Combining protein localization prediction methods such as PSORT [39] and SOSUI [40], especially for forecasting whether a given protein is membrane associated or soluble, to our PPI prediction would be useful when applying our method to large numbers of target proteins.
4.3. False Negative Interactions
When using both ZDOCK and MEGADOCK predictions, 7 interactions were not detected; FliG-FliG, FliM-FliM, FliG-FliM, FliG-FliN, FliM-FliN, Tsr-CheB and CheY-FliM (Fig. 3).
When we removed interactions among flagellar motors to consider protein localization, only the interaction between Tsr-CheB and CheY-FliM were not detected by both ZDOCK and MEGADOCK. It is known that for these interactions to occur CheB and CheY both need to be phosphorylated [41, 42]. In our dataset there were no protein structures of the phosphorylated forms. However, CheY structures used here include the mimicked activated state by using BeFe3-, such as PDB ID: 1ZDM [43]. This activated structure showed only modest differences with the native structure in terms of backbone geometry [43]. Based on these results, we cannot assess whether a rigid-body docking method is capable of distinguishing the phosphorylated from the non-phosphorylated state. Nonetheless, our findings are understandable because we did not use a flexible docking tool that considers phosphorylation mediated conformational changes of CheY and CheB.
One possible mean of obtaining increased sensitivity in our PPI prediction model is to construct likely structural variations of target proteins and then use the ensemble set as a docking target.
4.4. False Positive Interactions
There are four common false positive PPIs (CheD-CheY, CheA-CheD, CheR-CheY, CheY-CheY) predicted both by ZDOCK and MEGADOCK, three of which include CheY. In total, there are 51 structures of CheY in the dataset and 7 interactions out of 13 target proteins were predicted for CheY by both docking tools. Positive predictions were obtained using various structures of single protein species. This, however, does not mean that only specific protein structure pairs generate positive interactions. CheA has 21 structures in the dataset. Both of the docking tools predicted 5 interactions out of 13. The availability of more structural data for a given protein enriches structural variation and serves to increase sensitivity. In such cases we can consider using higher E* value to get better precision.
This result is also understandable from the fact that CheY has multiple binding partners. Bacterial chemotaxis is a two-component signal transduction system consisting of a histidine kinase (CheA) and response regulators (CheB, CheY). CheA operates in the form of a complex with receptors and CheW. Phosphorylated CheB works as a modifier of receptor proteins, which accumulate at the cell pole [44]. While CheB operates in the local area around the receptor complex, CheY accepts signals from CheA and transmits them to the flagellar motors, which are evenly distributed around the entire surface of the cell. There are several processes that modify CheY activity during transmission of the signal; CheC, CheX (T. maritima) and CheZ (E. coli, S. typhimurium) have activity that dephosphorylates CheY [33,45,46]. Thus, CheY undergoes transient interactions with several different proteins during the signal transduction process. Indeed, our conclusion that CheY undergoes non-specific binding with many types of proteins is in agreement with our findings given in (Fig. 2). It should also be noted that both docking tools predict CheY undergoes dimerization. Moreover, sequence homology based interlog search using PiSite [2] also suggests that dimerization of CheY is likely.
5. CONCLUSION
We conducted a reconstruction of the protein-protein interaction network using two distinct physical docking tools. The predicted interactions generated from the two tools were slightly different. However, when the positive predictions from both tools were combined, the vast majority of relevant interactions were represented. Indeed, there were only two exceptions, both of which required phosphorylation to activate the corresponding interaction.
Large-scale PPI prediction using tertiary structures is an effective approach that has a wide range of applications. This approach is especially useful for identifying PPIs associated with novel pathways that control cellular behavior. We are currently attempting to use a PPI network prediction method to find novel interactions in the signaling pathway involving epidermal growth factor receptor (EGFR) with related proteins whose expression levels change in response to specific drugs. In such an application, it is realistic to perform the prediction with a higher threshold (E*) value where a high level of precision is expected. Using this approach, we can evaluate the likelihood of novel interactions that were previously unconfirmed. In such cases, it would be helpful to employ multiple docking tools to identify as many probable interactions as possible.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
Docking and post-docking analysis calculations were conducted on the TSUBAME supercomputer system at the Global Scientific Information and Computing Center, Tokyo Institute of Technology, RIKEN Integrated Cluster of Clusters (RICC) and K computer at RIKEN, Japan, through the HPCI System Research Project (Project ID:hp120131). This work was supported in part by a Grant-in-Aid for Research and Development of The Next-Generation Integrated Life Simulation Software, a Grant-in-Aid for JSPS Fellows 238750, all from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT).
ABBREVIATIONS
- PPI =
Protein-Protein Interaction
- FFT =
Fast Fourier Transform
- TP =
True Positive
- FP =
False Positive
- TN =
True Negative
- FN =
False Negative
- CheA =
Chemotaxis protein CheA
- CheB =
Chemotaxis protein CheB
- CheC =
Chemotaxis protein CheC
- CheD =
Chemotaxis protein CheD
- CheR =
Chemotaxis protein CheR
- CheW =
Chemotaxis protein CheW
- CheX =
Chemotaxis protein CheX
- CheY =
Chemotaxis protein CheY
- CheZ =
Chemotaxis protein CheZ
- Tsr =
Methyl-accepting chemotaxis receptor Tsr (Serine receptor)
- FliG =
Flagellar motor switch protein FliG
- FliM =
Flagellar motor switch protein FliM
- FliN =
Flagellar motor switch protein FliN
- EGFR =
Epidermal Growth Factor Receptor
- E. coli =
Escherichia coli
- S. typhimurium =
Salmonella enterica, Serovar Typhimurium
- T. maritima =
Thermotoga maritima
REFERENCES
- 1.Gomperts BD, Kramer IM, Tatham PER. 2nd ed. USA: Academic Press: MA; 2009. Signal Transduction. [Google Scholar]
- 2.Higurashi M, Ishida T, Kinoshita K. Identification of transient hub proteins and the possible structural basis for their multiple interactions. Protein Sci. 2008;17:72–78. doi: 10.1110/ps.073196308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kakuta M, Nakamura S, Shimizu K. Prediction of protein-protein interaction sites using only sequence information and using both sequence and structural information. IPSJ Trans. Bioinform. 2008;49:25–35. [Google Scholar]
- 4.Shen J, Zhang J, Luo X, Zhu W, Yu K, Chen K, Li Y, Jiang H. Predicting protein-protein interactions based only on sequences information. Proc. Natl. Acad. Sci. USA. 2007;104:4337–4341. doi: 10.1073/pnas.0607879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valencia A, Pazos F. Structural Bioinformatics. Second Edition. pp. New York: Wiley and sons; 2009. Prediction of protein-protein interactions from evolutionary information; pp. 617–634. [Google Scholar]
- 6.Ogmen U, Keskin O, Aytuna AS, Nussinov R, Gursoy A. PRISM: protein interactions by structural matching. Nucleic Acids Res. 2005;33:W331–336. doi: 10.1093/nar/gki585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Acuner Ozbabacan SE, Keskin O, Nussinov R, Gursoy A. Enriching the human apoptosis pathway by predicting the structures of protein-protein complexes. J. Struct. Biol. 2012;179((3)):338–46. doi: 10.1016/j.jsb.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mosca R, Pons C, Fernández-Recio J, Aloy P. Pushing Structural Information into the Yeast Interactome by High-Throughput Protein Docking Experiments. PLoS Comput. Biol. 2009;5:e1000490. doi: 10.1371/journal.pcbi.1000490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wass MN, David A, Sternberg MJE. Challenges for the prediction of macromolecular interactions. Curr. Opin. Struct. Biol. 2011;21:382–390. doi: 10.1016/j.sbi.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Tsukamoto K, Yoshikawa T, Hourai Y, Fukui K, Akiyama Y. Development of an affinity evaluation and prediction system by using the shape complementarity characteristic between proteins. J. Bioinform. Comput. Biol. 2008;6:1133–1156. doi: 10.1142/s0219720008003904. [DOI] [PubMed] [Google Scholar]
- 11.Yoshikawa T, Tsukamoto K, Hourai Y, Fukui K. Improving the accuracy of an affinity prediction method by using statistics on shape complementarity between proteins. J. Chem. Inform. Model. 2009;49:693–703. doi: 10.1021/ci800310f. [DOI] [PubMed] [Google Scholar]
- 12.Matsuzaki Y, Matsuzaki Y, Sato T, Akiyama Y. In silico screening of protein-protein interactions with all-to-all rigid docking and clustering: an application to pathway analysis. J. Bioinform. Comput. Biol. 2009;7:991–1012. doi: 10.1142/s0219720009004461. [DOI] [PubMed] [Google Scholar]
- 13.Ohue M, Matsuzaki Y, Akiyama Y. Docking-calculation-based method for predicting protein-RNA interactions. Genome Informatics. 2011;25:25–39. [PubMed] [Google Scholar]
- 14.Wass MN, Fuentes G, Pons C, Pazos F, Valencia A. Towards the prediction of protein interaction partners using physical docking. Mol. Syst. Biol. 2011;7:469–0. doi: 10.1038/msb.2011.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gray JJ, Moughon S, Schueler-Furman O, Kuhlman B, Rohl CA, Baker D. Protein-protein docking with simultaneous optimization of rigid-body displacement and side-chain conformations. J. Mol. Biol. 2003;331:281–299. doi: 10.1016/s0022-2836(03)00670-3. [DOI] [PubMed] [Google Scholar]
- 16.Mashiach E, Nussinov R, Wolfson HJ. FiberDock: Flexible induced-fit backbone refinement in molecular docking. Proteins. 2010;78:1503–1519. doi: 10.1002/prot.22668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Macnab R. The bacterial flagellum: reversible rotary propellor and type III export apparatus. J. Bacteriol. 1999;181:7149–7153. doi: 10.1128/jb.181.23.7149-7153.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mintseris J, Pierce B, Wiehe K, Anderson R, Chen R, Weng Z. Integrating statistical pair potentials into protein complex prediction. Proteins. 2007;69:511–520. doi: 10.1002/prot.21502. [DOI] [PubMed] [Google Scholar]
- 19.Ritchie DW, Kemp GJ. Protein docking using spherical polar Fourier correlations. Proteins. 2000;39:178–194. [PubMed] [Google Scholar]
- 20.Ritchie DW, Venkatraman V. Ultra-fast FFT protein docking on graphics processors. Bioinformatics. 2010;26:2398–2405. doi: 10.1093/bioinformatics/btq444. [DOI] [PubMed] [Google Scholar]
- 21.Venkatraman V, Yang YD, Sael L, Kihara D. Protein-protein docking using region-based 3D Zernike descriptors. BMC Bioinformatics. 2009;10:407. doi: 10.1186/1471-2105-10-407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–7. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohue M, Matsuzaki Y, Uchikoga N, Ishida T, Akiyama Y. MEGADOCK: An all-to-all protein-protein prediction system using tertiary structure data. Protein Pept. Lett. 2014;20((7)):766–78. doi: 10.2174/09298665113209990050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujibuchi W, Goto S, Migimatsu H, Uchiyama I, Ogiwara A, Akiyama Y, Kanehisa M. DBGET/LinkDB: An integrated database retrieval system. Pac. Symp. Biocomput. 1998:683–694. [PubMed] [Google Scholar]
- 26.Hwang H, Pierce B, Mintseris J, Janin J, Weng Z. Protein-protein docking benchmark version 30. Proteins. 2008;73:705–709. doi: 10.1002/prot.22106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pierce BG, Hourai Y, Weng Z. Accelerating protein docking in ZDOCK using an advanced 3D convolution library. PLoS ONE. 2011;6:e24657–0. doi: 10.1371/journal.pone.0024657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierce B, Weng Z. ZRANK: reranking protein docking predictions with an optimized energy function. Proteins. 2007;67:1078–1086. doi: 10.1002/prot.21373. [DOI] [PubMed] [Google Scholar]
- 29.Bren A, Eisenbach M. How signals are heard during bacterial chemotaxis: protein-protein interactions in sensory signal propagation. J. Bacteriol. 2000;182:6865–6873. doi: 10.1128/jb.182.24.6865-6873.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sarkar MK, Paul K, Blair D. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl. Acad. Sci. USA. 2010;107:9370–9375. doi: 10.1073/pnas.1000935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blat Y, Eisenbach M. Oligomerization of the phosphatase CheZ upon interaction with the phosphorylated form of CheY The signal protein of bacterial chemotaxis. J. Biol. Chem. 1996;271:1226–1231. doi: 10.1074/jbc.271.2.1226. [DOI] [PubMed] [Google Scholar]
- 32.Zhao R, Collins EJ, Bourret RB, Silversmith RE. Structure and catalytic mechanism of the E coli chemotaxis phosphatase CheZ. Nat. Struct. Biol. 2002;9:570–575. doi: 10.1038/nsb816. [DOI] [PubMed] [Google Scholar]
- 33.Park S-Y, Chao X, Gonzalez-Bonet G, Beel BD, Bilwes AM, Crane BR. Structure and function of an unusual family of protein phosphatases: the bacterial chemotaxis proteins CheC and CheX. Mol. Cell. 2004;16:563–574. doi: 10.1016/j.molcel.2004.10.018. [DOI] [PubMed] [Google Scholar]
- 34.Wang H, Matsumura P. Characterization of the CheAS/CheZ complex: a specific interaction resulting in enhanced dephosphorylating activity on CheY-phosphate. Mol. Microbiol. 1996;19:695–703. doi: 10.1046/j.1365-2958.1996.393934.x. [DOI] [PubMed] [Google Scholar]
- 35.Szklarczyk D, Franceschini A, Kuhn M, Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork P, Jensen LJ. The STRING databese in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561–568. doi: 10.1093/nar/gkq973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kastritis PL, Bonvin AM. Are scoring functions in protein-protein docking ready to predict interactomes? Clues from a novel binding affinity benchmark. J. Proteome Res. 2010;9:2216–2225. doi: 10.1021/pr9009854. [DOI] [PubMed] [Google Scholar]
- 37.Tuncbag N, Gursoy A, Nussinov R, Keskin O. Predicting protein-protein interactions on a proteome scale by matching structural similarities at the interface using PRISM. Nat. Protoc. 2011;6:1341–1354. doi: 10.1038/nprot.2011.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sourjik V, Berg HC. Localization of components of the chemotaxis machinery of Escherichia coli using fluorescent protein fusions. Mol. Microbiol. 2000;37:740–751. doi: 10.1046/j.1365-2958.2000.02044.x. [DOI] [PubMed] [Google Scholar]
- 39.Nakai K, Horton P. PSORT: a program for detecting the sorting signals of proteins and predicting their subcellular localization. Trends Biochem. Sci. 1999;24:34–35. doi: 10.1016/s0968-0004(98)01336-x. [DOI] [PubMed] [Google Scholar]
- 40.Hirokawa T, Boon-Chieng S, Mitaku S. SOSUI: classification and secondary structure prediction system for membrane proteins. Bioinformatics. 1998;14:378–379. doi: 10.1093/bioinformatics/14.4.378. [DOI] [PubMed] [Google Scholar]
- 41.Parkinson JS. Bacterial chemotaxis: a new player in response regulator dephosphorylation. J. Bacteriol. 2003;185:1492–1494. doi: 10.1128/JB.185.5.1492-1494.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Welch M, Oosawa K, Aizawa S, Eisenbach M. Phosphorylation-dependent binding of a signal molecule to the flagellar switch of bacteria. Proc. Natl. Acad. Sci. USA. 1993;90:8787–8791. doi: 10.1073/pnas.90.19.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee SY, Cho HS, Pelton JG, Yan D, Berry EA, Wemmer DE. Crystal structure of activated CheY Comparison with other activated receiver domains. J. Biol. Chem. 2001;276:16425–16431. doi: 10.1074/jbc.M101002200. [DOI] [PubMed] [Google Scholar]
- 44.Sourjik V. Receptor clustering and signal processing in E. coli chemotaxis. Trends Microbiol. 2004;12:569–576. doi: 10.1016/j.tim.2004.10.003. [DOI] [PubMed] [Google Scholar]
- 45.Szurmant H, Muff TJ, Ordal GW. Bacillus subtilis CheC and FliY are members of a novel class of CheY-P-hydrolyzing proteins in the chemotactic signal transduction cascade. J. Biol. Chem. 2004;279:21787–21792. doi: 10.1074/jbc.M311497200. [DOI] [PubMed] [Google Scholar]
- 46.Hess JF, Oosawa K, Kaplan N, Simon MI. Phosphorylation of three proteins in the signaling pathway of bacterial chemotaxis. Cell. 1988;53:79–87. doi: 10.1016/0092-8674(88)90489-8. [DOI] [PubMed] [Google Scholar]