

Abstract
How do viruses spread from cell to cell? Enveloped viruses acquire their surrounding membranes by budding. If a newly enveloped virus has budded through the plasma membrane, it finds itself outside the cell immediately. If it has budded through the bounding membrane of an internal compartment such as the ER, the virus finds itself in the lumen, from which it can exit the cell via the conventional secretion pathway. Thus, although some enveloped viruses destroy the cells they infect, there is no topological need to do so.
On the other hand, naked viruses such as poliovirus lack an external membrane. They are protein-nucleic acid complexes within the cytoplasm or nucleus of the infected cell, like a ribosome, a spliceosome or an aggregate of Huntingtin protein. The simplest way for such a particle to pass through the single lipid bilayer that separates it from the outside of the cell would be to violate the integrity of that bilayer. Thus, it is not surprising that the primary mode of exit for non-enveloped viruses is cell lysis. However, more complex exit strategies are possible, such as the creation of new compartments whose complex topologies allow the exit of cytoplasm and its contents without violating the integrity of the cell. Here we will discuss the non-lytic spread of poliovirus and recent observations of such compartments during viral infection with several different picornaviruses.
Keywords: Unconventional secretion, multi-vesicular bodies, autophagy, viral spread, picornavirus
Life cycles of poliovirus and other non-enveloped viruses inevitably show virus particles bursting from the cell, releasing hundreds if not thousands of virions that spread to neighboring cells (Fig 1A). Appropriately enough, experiments that provide the basis for these drawings were first published in the pages of Virology. In the first issue, published in 1955, Lwoff and colleagues described the kinetics of poliovirus release from single infected monkey kidney cells (1). From each cell, the bulk of the virus was released in one large burst within a time period of less than one hour (Fig. 1B). That virus was released from individual cells in a bolus was consistent with the hypothesis that the infected cells lysed, releasing the virus all at once.
Figure 1. Cell degeneration correlates with extracellular release of poliovirus.
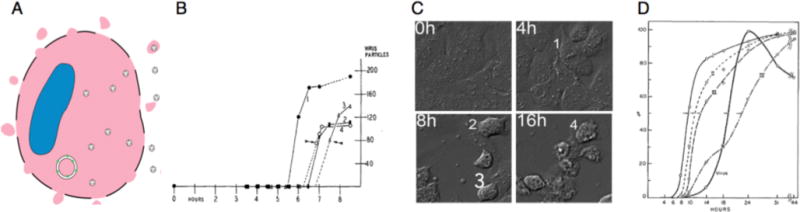
A) The conceptually simplest way for a molecular aggregate to escape the cell cytoplasm is to violate the plasma membrane. B) The appearance of infectious poliovirus in the medium of isolated cells was found to occur rapidly, in a bolus (1). C) To exemplify the stages of CPE described by Dunnebakke (2), Huh-7 cells were infected with a slowly growing poliovirus variant and monitored by differential interference contrast time-lapse photography (12). Still images from four time points are shown, with cells typical of stages 1–4 labeled. D) Analysis of cell populations (Dunnebakke, 1956) over time showed the sequential development of the stages of CPE (I–IV) and the presence of extracellular infectious virus that correlated with the onset of stage IV.
In a Virology paper in 1956, Thelma Dunnebacke dissected poliovirus-induced CPE into four discrete stages (2). These stages are labeled in Figure 1C on a contemporary image of infection.
Cell nuclear membranes wrinkle but the cells still lie flat against the culture dish.
The nucleus shrinks, the cytoplasm begins to draw in, and small vacuoles appear in the cytoplasm.
The cell rounds up and the nuclear material condenses.
The cell begins to detach from the dish, thus coming out of focus.
Using these descriptors, Dunnebacke followed virus-infected cells over time and categorized them into groups, while also keeping track of the total amount of virus in the supernatant. When 50% of the cells were in stage 3 (III), only 5% of the virus could be detected in the supernatant. Strikingly, by the time 50% of the cells had passed through stage 4 (IV), 95% of the virus had been released. Such large and rapid increases in viral titer, correlating with the final stages of CPE, could only be explained by cellular rupture and release of hundreds of infectious virus particles from individual infected cells.
When lytic viruses don’t behave like lytic viruses
Although picornaviruses such as poliovirus and coxsackievirus were thus considered to be classic lytic viruses, experimental observations began to accumulate that were inconsistent with the necessity of cell destruction for virus spread.
Early evidence for nonlytic spread of poliovirus came from studies of persistently infected cell lines in the laboratory of Isabel Pelletier. When Sabin strains of poliovirus were used to infect two different human neuroblastoma cell lines, viral antigen was released into the medium continuously for nine months without death of the cells (3). Similarly, the laboratory of Richard Lloyd showed that poliovirus could persistently infect human K562 cells, an erythroblastoid lineage (4). During a three-month period of a persistent infection, the cell viability remained between 67 and 92%. If the infected cells were not dying, how was the virus escaping?
In 1993, Richard Compans and colleagues studied the release of poliovirus from polarized epithelial cells (5). Since poliovirus is an enteric microbe that traverses the gut, this cell type represented a physiologically relevant cell type. When polarized Caco-2 cells were infected with poliovirus, the virus was released almost exclusively from the apical surface. The authors hypothesized that this apical release must be mediated by a ‘vectorial transport mechanism’. One potential mechanism for this release was that aggregated viral particles trapped within vesicles traveled to the plasma membrane, fusing and releasing the aggregates. Images obtained by EM, which showed electron-dense material trapped within intracellular vesicles, were consistent with this hypothesis.
The most enigmatic picornavirus has been, and remains, hepatitis A virus. Unlike its more rapidly growing cousins poliovirus, rhinovirus and coxsackievirus, naturally occurring isolates of hepatitis A virus have not been observed to lyse infected cells in tissue culture or in infected humans [reviewed in (6)]. The liver of a person infected with hepatitis A virus can often be heavily infected, so the virus must spread from cell to cell. During acute human infection, virus appears in the stool before any evidence of any immune-mediated hepatocyte damage occurs. Therefore, hepatitis A virus would seem to spread exclusively non-lytically. Furthermore, such examples are not limited to picornaviruses; for example, simian virus 40 (SV40), a DNA virus, was shown to release from monkey kidney cells at very high titers in the apparent absence of lysis measured on a population scale (7).
There are two caveats to all such experiments. The first caveat also extends to reports of potentially nonlytic spread of aggregated proteins (8,9) and of other unconventional secretion pathways (10). Most such experiments are performed with cell populations, and it is not feasible to exclude the possibility that the lysis of only a few cells gave rise to the extracellular virus or other cargo. A caveat of ultrastructural analysis that is especially troublesome in the case of viruses is that the particle-to-PFU ratio is usually in the hundreds. Therefore, observing a virus particle in an intracellular compartment or in an extracellular vesicle does not mean that it is on an infectious pathway; in fact, any individual particle observed has a less than 1% chance of being infectious. For these reasons, we have chosen to perform experiments that monitor individual cells and to try to monitor infectious, rather than physical, viral populations (11).
Single-cell analysis can identify individual donor and recipient cells
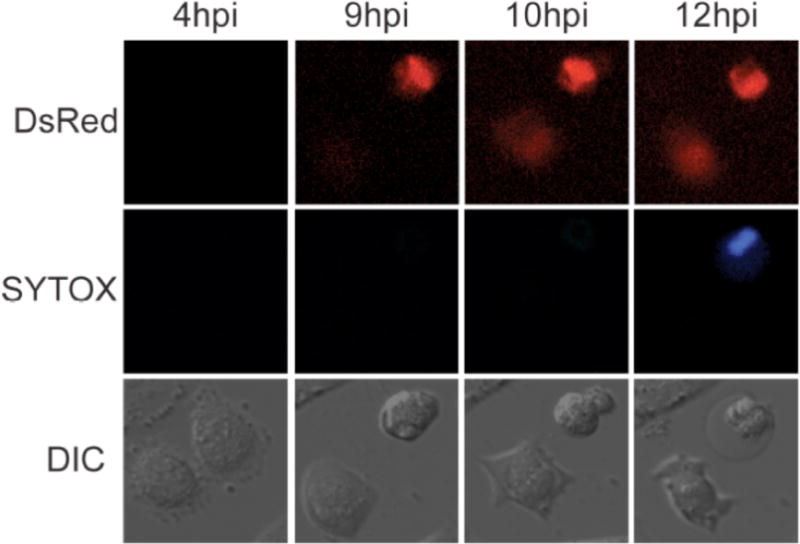
To observe viral spread directly, we performed live monitoring of poliovirus infections on a single-cell level for 48 hours (12). For live imaging we employed a DsRed-expressing poliovirus variant (PV-DsRed) from the laboratory of Ellie Ehrenfeld (13). Following infection of cells with PV-DsRed, viral protein translation results in hundreds of copies of 2A-DsRed which renders infectious cells detectable by fluorescence. Although virions are not labeled, infected cells can be visualized as early as 3.5 hours post infection, with peak fluorescence around 8 hours post infection. When Huh7 cells were infected sparsely with PV-DsRed and viewed under agar, many different modes of viral transfer between neighboring cells could be visualized (12). In the case shown in Figure 2, a single cell that is red at all time points, which we will call the ‘donor’ cell, can be seen. This cell passed through stages 1, 2, 3 and 4 of cytopathic effect in the time points presented. Strikingly, a neighbor cell became infected when the donor cell was only in stage 2, and clearly continued living thereafter. Figure 2 thus reveals a clear example of the transfer of poliovirus infection between living cells. Events such as that shown in Figure 2 were rare in untreated cells, but increased greatly in frequency when cells were treated with either loperimide or nicardipine to induce cellular autophagy (12), which will be discussed below.
Figure 2. Visualization of non-lytic spread of poliovirus.
Huh7.A.1/GFP-LC3 cells were infected with PV-DsRed (13) at an MOI of 0.1 PFU/cell and imaged every 15 minutes for 48 hours. Single cells images show an infected cell with an intact membrane at 8hpi. By 10hpi, infection has spread to a neighboring cell. By 12hpi, membrane is ruffled in the originally infected cell and the membrane has ruptured by 14hpi, after nonlytic spread occurred to a neighboring cell.
Potential role for autophagy pathway and constituents in non-lytic viral spread
Macroautophagy, referred throughout as autophagy, is a highly conserved recycling pathway characterized by the formation of double-membraned vesicles and the subsequent degradation and recycling of their cytoplasmic contents (Fig. 3). The topology of a double membrane allows the inner compartment to be cytoplasmic, which the compartment between the bilayers being luminal. Therefore, the damaged and long-lived proteins and organelles enveloped in these vesicles are found within the innermost compartment.
Figure 3. Overview of signaling, induction and execution of cellular autophagy.
The highly regulated pathway of autophagy results in the formation of a double-membraned phagophore that sequesters long-lived proteins or components destined for degradation. The fully formed autophagosome fuses with a lysosome, an acidic compartment that degrades the components of the autophagsome. Small molecules that activate autophagy are shown in green; those that inhibit are shown in red. Originally discovered as the Target Of Rapamycin, mTORC1 is inhibited by rapamycin or by nutrient starvation, thus alleviating its normal repression of cellular autophagy. Alternative autophagy activation pathways focus on beclin-1, which is downstream of the mTORC1 complex but can be activated independently. Beclin1 is present in several intracellular complexes, including the beclin1/Vps34/Atg14 complex responsible for initiating autophagosome formation. The formation of this complex is normally inhibited because of the preferential association of beclin1 and Bcl2, which is reported to be stabilized by eugenol. The function of the beclin1/Vps34/Atg14 complex can be disrupted by 3-methyladenine, which blocks the PI3 kinase activity of Vps34, and by spautin-1, which inhibits the deubiquitylases that stabilize beclin1. Subsequent recruitment of the lipidated form of LC3 (LC3-II) and the ATG5/12 complex allow the growth, elongation and curvature of the isolation membrane. This process requires high intracellular calcium concentrations, which can be depleted by calpain. Calpain can be inhibited by nicardipine and loperimide, which thus have the effect of stimulating autophagy. The double-membraned autophagosome matures to become a degradative organelle via fusion with lysosomes. The inner membrane is destroyed by lysosomal lipases and proteases to generate the single-membraned autolysosome. It is not yet clear whether the double-membraned form of the autophagosome-like membranes or the partially degraded single-membraned autolysosome-like vesicles, or both, fuse with the plasma membrane during unconventional secretion (see text for references).
Regulation and upstream signaling of autophagy are highly complex. It is worthwhile for the virologist to understand this pathway, however, because of the numerous opportunities for viral exploitation or inhibition, and the many potential therapeutic opportunities. The induction of autophagy by starvation, for example, is mediated by mTOR, or mammalian target of rapamycin. mTOR present in one of its complexes, mTORC1, requires nutrient signals such as amino acids for activation. The repression of autophagy by mTORC1 is mediated by its direct phosophorylation and inhibition of serine/threonine kinase ULK1. It is the unfettered action of the ULK1 complex that is required for the recruitment and stabilization of beclin 1 in its autophagy-specific complex with VPS1 and ATG14. Competing complexes exist, such as the complex between beclin 1 and Bcl-2, whose stabilization can inhibit autophagy.
The executation phase of autophagy begins when the ‘isolation membrane’, bounded by a single bilayer begins to recruit the ATG5-ATG12/ATG16 complex, thus mediating the lipidation of a small microtubule-associated protein, LC3. Lipidated LC3, termed LC3-II, is required for the growth and curvature of the autophagosome and its eventual double-membraned structure. Opportunities for the regulation of this step can be found in a requirement for calpains, whose promotion of calcium ion flux inhibits the formation of the ATG5-ATG12/ATG16 complex. Thus, the inhibition of calpains by loperamide and nicardipine, for example, activates autophagosome formation at a downstream step that bypasses much of the upstream signaling.
The autophagosome becomes degradative by tracking along microtubules to fuse with lysosomes, which deliver lipases and proteases into the luminal compartment between the two bilayers. The inner bilayer must be destroyed, forming the autolysosome, for the degradation of the cytosolic contents to occur. The transition from autophagosome to autolysosome therefore has the unusual effect of pooling luminal and cytosolic compartments and changing the topology of the cytoplasmic contents relative to the plasma membrane.
Given that the cellular autophagy pathway is a cell cleanup and clearance mechanism, it comes as no surprise that it is an important component of the innate immune response to many intracellular microbes. Naturally, given the arms race between microbes and their hosts, several viruses, bacteria and eukaryotic parasites have evolved to avoid destruction by cellular autophagy, to subvert its complex features, or both. This topic and the autophagy pathway itself have been extensively reviewed (14–17). Here, we will focus on the potential role of autophagy and related pathways on the non-lytic transfer of viruses and other cytoplasmic components.
It was first noted 50 years ago (18) and re-discovered 19 years ago (19) that many of the vesicles that accumulate in poliovirus-infected cells are double membraned, with cytosolic contents that can contain viral particles. However, when the abundance of crucial autophagy proteins ATG5 and LC3 were reduced by siRNA treatment of the host cells, only a modest decrease in the amount of intracellular virus was observed (20). However, the amount of extracellular virus being released from the cells, especially at very early time points before lysis should canonically occur, was observed. This led to the interpretation that a mechanism by which poliovirus can exit the cell non-lytically could result from exploitation of the autophagy pathway and its constituents, and the pathway was termed AWOL (autophagosome-mediated exit without lysis; 21). Interestingly, reduction of the abundance of ATG12 and LC3, but not inhibition of beclin 1, reduced the amount of extracellular poliovirus early in infection. It is likely that the subversion of the autophagy pathway or components by poliovirus occurs downstream of the signaling steps that require beclin 1. Such a bypass of the beclin 1 step could be mediated by the direct interaction of LC3 with poliovirus proteins (22), but that remains conjecture at this point.
In 2012, strong evidence for the involvement of the autophagy pathway in unconventional secretion of host proteins was provided. In yeast and Dictyostelium, the secretion of critical sporulation hormone Acb1 was shown to bypass the ER and the Golgi, but to be dependent on components of both the autophagy and the multivesicular body pathways (23, 24). In fact, these pathways are probably not so distinct, given that they both give rise to intracellular vesicles that contain both luminal material and additional vesicles with cytosolic contents. In fact, extracellular vesicles are often termed ‘exosomes’ and assumed to be uniform, while it has been shown that they can have multiple origins (25). Thus, the discussion in the next section is relevant to an origin of intracellular compartments of complex topology that resemble either autophagosomes or multivesicular bodies.
What are the agents of non-lytic spread?
Any pathway of generating extracellular vesicles that contain cytoplasm will provide a way to ‘secrete’ cytoplasmic material. Such pathways include the autophagy pathway, the multivesicular body pathway, and direct blebbing from the plasma membrane. To understand the mechanism or mechanisms of nonlytic viral spread, it will be useful to define which component in the extracellular milieu is responsible for infecting subsequent cells.
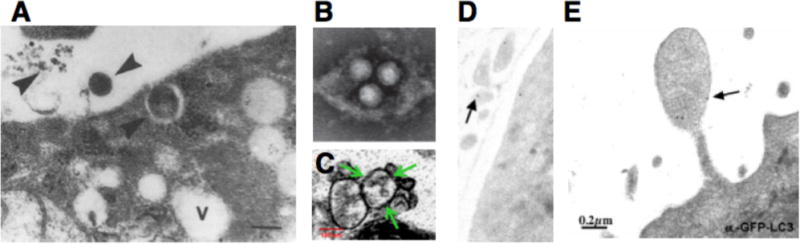
The first potential agent for nonlytic spread, structure #1 in Figure 4A, is an extracellular, single-membraned vesicle that contains virions. Several laboratories have directly observed such structures in medium of cells infected with poliovirus (3, 20), hepatitis A virus (26) and coxsackievirus B3 (27, 28). Figure 4 depicts a gallery of these observations. How were these structures generated? An image consistent with an autophagosome-like origin for an extracellular vesicle formed by a poliovirus-infected cell is shown in Figure 3E. The membranes surrounding coxsackievirus B3 particles similar to those shown in Figure 3D were shown in contain autophagy protein LC3 (28). Visual evidence consistent with the blebbing of LC3-containing membranes from a poliovirus-infected cell is shown in Figure 3F. On the other hand, the formation of infectious membranous fractions from hepatitis A-infected cells, which contain structures such as that shown in Figure 3B, is dependent on the presence of VPS4B and ALIX, components of the multivesicular body pathway (26). In fact, components of both the autophagy and multivesicular body pathways are used in unconventional secretion in fungi (23, 24), so perhaps these are not so separate after all.
Figure 4. Potential vehicles of non-lytic spread of positive-strand RNA viruses.
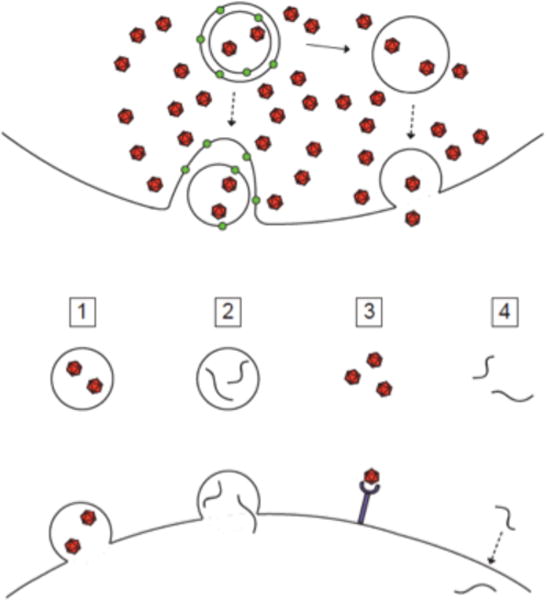
Possible mechanisms for nonlytic spread. A) Infectious material could exit a cell in least four different forms: 1, membrane-wrapped virions; 2, membrane-wrapped viral RNA; 3, naked virions; 4; free RNA, or some combination. Mechanisms 2 and 4 should be receptor-independent and non-neutralizable, mechanism 3 receptor-dependent and neutralizable, and mechanism 1 could be either, depending on the integrity of the ‘exosomal’ membrane and the stability of the virion particle within the newly infected cell. (B)–(F) Electron microscopic images consistent with the budding of picornaviruses; viral particles are all approximately 30–35 nm. B) Hepatitis A virions within extracellular vesicles (26). C) Coxsackie B3 virions within extracellular vesicles (28). D) Poliovirion protein VP1 detected by immunogold staining within extracellular vesicle; size bar as in panel F (20); E) Apparent release of cytoplasmic vesicle near putative autophagosome-like cytoplasmic structure in poliovirus-infected cell (5) size bar 200 μm; F) GFP-LC3-containing cytoplasmic protrusion from poliovirus-infected cell (20).
Possible route #2 is that the vesicles contain RNA in addition to virus particles, and that the RNA is the infectious component. The observation of virions in vesicles does not mean that they are infectious, especially given the high particle-to-PFU ratio on RNA viruses. Viral RNA molecules within a single-membraned vesicle in the extracellular milieu would not be visualized by electron microscopy, but the fusion of an RNA-containing vesicle would release infectious RNA into the cytoplasm of the recipient cell.
Another possibility for the infectious material responsible for non-lytic spread is that virions or RNA are present free outside the cell because the vesicles formed within autophagosomes, multivesicular bodies, or at the plasma membrane are unstable. In the case of hepatitis A and coxsackievirus, free virions (#3) are not likely to explain the infectivity of the secreted material because this infectivity is resistant to antibody neutralization. However, if free infectious RNA (#4) were present extracellularly, it could be the agent of viral spread and even be membrane-associated, due to the membranous nature of picornaviral RNA replication complexes.
What is the purpose of nonlytic spread?
For hepatitis A, non-lytic spread may be its only route of dissemination and thus the recent publications cited are likely to explain a long-debated phenomenon. For viruses capable of cell lysis, however, the potential purpose of an alternative exit route is a matter of conjecture. For example, although poliovirus infection is highly lytic in most cells in tissue culture, and paralytic poliomyelitis is caused by the destruction of neurons in the CNS, little is known about the mechanisms of poliovirus spread via the intestine, Peyer’s patches, bloodstream, muscle tissue, and peripheral neurons in a natural infection. Similar points can be made about the dissemination of coxsackieivirus. Therefore, potential roles for nonlytic spread in the propagation and transmission of lytic picornaviruses remain to be investigated.
During poliomyelitis, viral escape to the CNS can lead to the destruction of motor neurons coincident with the flaccid paralysis associated with the virus. However, nonlytic spread could play a role in viral transport through the CNS. A study in 1998 on the cell-to-cell spread of poliovirus in the spinal cord of Bonnet monkeys hints at a possible role for nonlytic spread in the CNS (29). The authors infected monkeys with poliovirus in the ulnar nerve and tracked viral spread at different times post-infection. Interestingly, neurons in the spinal cord were shown to recover at late times during infection, suggesting that the virus could spread without killing all the cells it infected. The authors went on to conclude: “From these studies it appears that poliovirus has a non-cytolytic cycle of infection in the cervical neurons.”
One role for nonlytic spread would be exemplified by the release of poliovirus exclusively from the apical side of polarized epithelial cells (3). As a fecal-oral pathogen, slow and directional release of viral particles into the intestinal lumen in the absence of cellular lysis would be a good way for the virus to be shed continuously by the host. This principle might hold true for other picornaviruses that infect polarized cells, allowing viral transmission to be skewed toward directions helpful in viral propagation and away from tissues that would harm the host.
The study of viruses often reveals interesting cell biological phenomena. This is not only because viruses are adept at co-opting host cell processes, but also because there are very few measurement of cell constituents as sensitive as assays for viral infectivity. The study of viral exit from cells may continue to add to the list of mechanisms of unconventional secretion (30), which already includes tunneling nanotubules, dramatic conformational changes at the plasma membrane, and, now, the nonlytic release of infectious cytoplasm.
HIGHLIGHTS.
Many past findings led to a sharp distinction between lytic and nonlytic viruses
Defying this paradigm, some nonenveloped viruses seemed to spread without lysis
Measurements of cell populations cannot exclude lysis of a few cells
Single-cell microscopy has shown nonlytic cell-to-cell poliovirus spread
Poliovirus, hepatitis A and coxsackievirus B3 can be found in extracellular vesicles
Acknowledgments
We thank the BioX Interdisciplinary Initiatives Program Seed Grant, National Institutes of Health T32-GM007276 and NIH grant R56 AI-103500 for support of this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lwoff A, Dulbecco R, Vogt M, Llowff M. Kinetics of the release of poliomyelitis virus from single cells. Virology. 1955;1:128–139. doi: 10.1016/0042-6822(55)90010-6. [DOI] [PubMed] [Google Scholar]
- 2.Dunnebacke TH. Correlation of the stage of cytopathic change with the relase of poliomyelitis virus. Virology. 1956;2:399–410. doi: 10.1016/0042-6822(56)90034-4. [DOI] [PubMed] [Google Scholar]
- 3.Colbere-Garapin F, Christodoulou C, Crainic R, Pelletier I. Persistent poliovirus infection of human neuroblastoma cells. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:7590–7594. doi: 10.1073/pnas.86.19.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lloyd RE, Bovee M. Persistent infection of human erythroblastoid cells bypoliovirus. Virology. 1993;194:200–209. doi: 10.1006/viro.1993.1250. [DOI] [PubMed] [Google Scholar]
- 5.Tucker SP, Thornton CL, Wimmer E, Compans RW. Vectorial release of poliovirus from polarized human intestinal epithelial cells. J Virol. 1993;67:4274–4282. doi: 10.1128/jvi.67.7.4274-4282.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lemon SM. Type A viral hepatitis. New developments in an old disease. New Engl J Medi. 1985;313:1059–1067. doi: 10.1056/NEJM198510243131706. [DOI] [PubMed] [Google Scholar]
- 7.Clayson ET, Brando LV, Compans RW. Release of simian virus 40 virions from epithelial cells is polarized and occurs without cell lysis. J Virol. 1989;63:2278–2288. doi: 10.1128/jvi.63.5.2278-2288.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ren PH, Lauckner JE, Kachirskaia I, Heuser JE, Melki R, Kopito RR. Cytoplasmic penetration and persistent infection of mammalian cells by polyglutamine aggregates. Nat Cell Biol. 2009;11:219–225. doi: 10.1038/ncb1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions and prionoids. Neuron. 2009;64:783–790. doi: 10.1016/j.neuron.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 10.Zhang M, Schekman R. Unconventional secretion, unconventional solutions. Science. 2013;340:559–561. doi: 10.1126/science.1234740. [DOI] [PubMed] [Google Scholar]
- 11.Bird SW. PhD Thesis. Stanford University; 2015. Non-lytic spread of a naked virus. [Google Scholar]
- 12.Bird SW, Maynard ND, Covert MW, Kirkegaard K. Nonlytic viral spread enhanced by autophagy components. Proc Natl Acad Sci USA. 2014;111:13081–1308. doi: 10.1073/pnas.1401437111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teterina NL, Levenson EA, Ehrenfeld E. Viable polioviruses that encode 2A proteins with fluorescent protein tags. J Virol. 2010;84:1477–1488. doi: 10.1128/JVI.01578-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deretic V, Saitoh T, Akira S. Autophagy in infection, inflammation and immunity. Nat Rev Immunol. 2013;13:722–737. doi: 10.1038/nri3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richards AL, Jackson WT. How positive-strand RNA viruses benefit from autophagosome maturation. J Virol. 2013;87:9966–9972. doi: 10.1128/JVI.00460-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Choi AM, Ryter SW, Levine B. Autophagy in human health and disease. New Engl J Med. 2013;368:651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 17.Mizushima N, Yoshimori T, Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27:107–132. doi: 10.1146/annurev-cellbio-092910-154005. [DOI] [PubMed] [Google Scholar]
- 18.Dales S, Eggers HJ, Tamm I, Palade GE. Electron microscopic study of the formation of poliovirus. Virology. 1965;26:379–389. doi: 10.1016/0042-6822(65)90001-2. [DOI] [PubMed] [Google Scholar]
- 19.Schlegel A, Giddings TH, Ladinsky MS, Kirkegaard K. Cellular origin and ultrastructure of membranes induced during poliovirus infection. J Virol. 1996;70:6576–6588. doi: 10.1128/jvi.70.10.6576-6588.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jackson WT, Giddings TH, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, Kirkegaard K. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3:e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor MP, Burgon TB, Kirkegaard K, Jackson WT. Role of microtubules in extracellular relase of poliovirus. J Virol. 2009;83:6599–6609. doi: 10.1128/JVI.01819-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taylor MP, Kirkegaard K. Modification of cellular autophagy protein LC3 by poliovirus. J Virol. 2007;81:12543–12553. doi: 10.1128/JVI.00755-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duran JM, Anjard C, Stefan C, Loomis WF, Malhotra V. Unconventional secretion of Acb1 is mediated by autophagosomes. J Cell Biol. 2010;188:527–536. doi: 10.1083/jcb.200911154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manjithaya R, Anjard C, Loomis WF, Subramani S. Unconventional secretion of Pichia pastoris Acb1 is dependent of GRASP protein, peroxisomal functions and autophagosome formation. J Cell Biol. 2010;188:537–546. doi: 10.1083/jcb.200911149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pallet N, Sirois I, Bell C, Hanafi LA, Hamelin K, Dieudé M, Rondeau C, Thibault P, Desjardins M, Hebert MJ. A comprehensive characterization of membrane vesicles released by autophagic human endothelial cells. Proteomics. 2013;13:1108–1120. doi: 10.1002/pmic.201200531. [DOI] [PubMed] [Google Scholar]
- 26.Feng Z, Hensley L, McKnight KL, Hu F, Madden V, Ping L, Jeong SH, Walder C, Lanford RE, Lemon SM. A pathogenic picornavirus acquires an envelope by hijacking cellular membranes. Nature. 2013;496:367–371. doi: 10.1038/nature12029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alirezaei M, Flynn CT, Wood MR, Whitton JL. Pancreatic acinar cell-specific autophagy disruption reduces coxsackievirus replication and pathogenesis in vivo. Cell Host Microbe. 2012;11:298–305. doi: 10.1016/j.chom.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson SM, Tsueng G, Sin J, Mangale V, Rahawi S, McIntyre LL, Williams W, Kha N, Cruz C, Hancock BM, Nguyen DP, Sayen MR, Hilton BJ, Doran KS, Segall AM, Wolkowicz R, Cornell CT, Whitton JL, Gottlieb RA, Feuer R. Coxsackievirus B exits the host cell in shed microvesicles displaying autophagosomal markers. PLoS Path. 2014;10:e1004045. doi: 10.1371/journal.ppat.1004045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ponnuraj EM, John TJ, Levin MJ, Simoes EA. Cell-to-cell spread of poliovirus in the spinal cord of bonnet monkeys (Macada radiata) J Gen Virol. 1998;79:2393–2403. doi: 10.1099/0022-1317-79-10-2393. [DOI] [PubMed] [Google Scholar]
- 30.Rabouille C, Malhotra V, Nickel W. Diversity in unconventional protein secretion. J Cell Science. 2012;125:5251–5255. doi: 10.1242/jcs.103630. [DOI] [PubMed] [Google Scholar]