

Abstract
Morbidity and mortality from heart failure (HF) are high, and current risk stratification approaches for predicting HF progression are imperfect. Adenosine triphosphate (ATP) is required for normal cardiac contraction, and abnormalities in creatine kinase (CK) energy metabolism, the primary myocardial energy reserve reaction, have been observed in experimental and clinical HF. However, the prognostic value of abnormalities in ATP production rates through CK in human HF has not been investigated. Fifty-eight HF patients with nonischemic cardiomyopathy underwent 31P magnetic resonance spectroscopy (MRS) to quantify cardiac high-energy phosphates and the rate of ATP synthesis through CK (CK flux) and were prospectively followed for a median of 4.7 years. Multiple-event analysis (MEA) was performed for HF-related events including all-cause and cardiac death, HF hospitalization, cardiac transplantation, and ventricular-assist device placement. Among baseline demographic, clinical, and metabolic parameters, MEA identified four independent predictors of HF events: New York Heart Association (NYHA) class, left ventricular ejection fraction (LVEF), African-American race, and CK flux. Reduced myocardial CK flux was a significant predictor of HF outcomes, even after correction for NYHA class, LVEF, and race. For each increase in CK flux of 1 μmol g−1 s−1, risk of HF-related composite outcomes decreased by 32 to 39%. These findings suggest that reduced CK flux may be a potential HF treatment target. Newer imaging strategies, including noninvasive 31P MRS that detect altered ATP kinetics, could thus complement risk stratification in HF and add value in conditions involving other tissues with high energy demands, including skeletal muscle and brain.
Introduction
Heart failure (HF) affects about 5 million individuals in the United States and is associated with direct and indirect costs estimated at $32 billion per year (1). Risk stratification in HF is important for prognostic reasons and because of the challenges in timing aggressive, higher-risk therapeutic interventions that include ventricular-assist device (VAD) implantation and cardiac transplantation. Conventional clinical predictors of HF severity and progression, such as New York Heart Association (NYHA) class and left ventricular ejection fraction (LVEF), are readily acquired but imperfect. Hence, we sought to evaluate myocardial energy metabolism as a predictor of clinical HF events.
Myocardial energy metabolism is required for normal cardiac contractile function. It is impaired in experimental and clinical HF, and postulated to contribute to HF development and progression (2, 3). The creatine kinase (CK) reaction is the prime energy reserve of the heart, providing a rapid source of adenosine triphosphate (ATP) and enhancing its delivery from mitochondrial sites of production to sites of use, including the myofibrils (4, 5). In animal models, reduced CK ATP delivery impairs contractile function and contributes to arrhythmic susceptibility (6, 7). Because contractile dysfunction is associated with HF morbidity and mortality and has been linked to reduced CK ATP metabolism (8–10), we posited that reduced ATP delivery via CK is an independent contributor to HF progression and predicts future clinical events.
Phosphorus (31P) magnetic resonance spectroscopy (MRS) is the only noninvasive means to quantify the high-energy phosphates, ATP and creatine phosphate (PCr), in the human heart. Early 31P MRS studies identified a significant reduction in the ratio of PCr to ATP (PCr/ATP) in HF patients (11–13), and a subsequent report suggested that the myocardial PCr/ATP ratio may be a predictor of mortality in HF patients (14). However, the cardiac PCr/ATP ratio cannot detect ATP depletion or reductions in the rate of ATP delivery, which are arguably more sensitive and important indices of cardiac energy metabolism.
Indeed, 31P MRS studies of the absolute metabolite concentrations, [ATP] and [PCr], in patients with nonischemic dilated cardiomyopathy (DCM) suggest that decreases in cardiac PCr/ATP may underestimate the reduction in high-energy phosphate occurring in human HF (15). 31P MRS using saturation transfer (MRST) methods also permits quantification of the pseudo–first-order forward reaction rate constant, kf, for the CK reaction in the human heart (9, 16). Patient studies combining both MRST and concentration measurements have demonstrated not only that [PCr] is reduced, but also that the reduction in the rate of ATP synthesis through CK (CK flux), given by the product kf[PCr] (∼35 to 70%), is disproportionately greater than the reduction in the PCr/ATP ratio (∼10 to 20%) in patients with nonischemic DCM (9). In addition, in patients with pressure-overload left ventricular hypertrophy (LVH), the myocardial PCr/ATP ratio did not distinguish those with HF from those without HF, whereas kf was halved in those with HF, resulting in a markedly lower mean CK flux of only about 35% of normal (10).
The mechanistic importance of CK flux for contractile dysfunction in HF was recently highlighted in a study demonstrating that over-expression of the myofibrillar isoform of CK (CKM) (i) increased CK flux, (ii) significantly enhanced contractile function at rest and during adrenergic stress, and (iii) increased survival in failing mouse hearts (8). Given such evidence that reduced CK flux contributes to contractile dysfunction and mortality in experimental HF (8), as well as previous clinical observations linking reduced myocardial PCr/ATP ratio to mortality in HF patients (14), the more recent data reporting much larger reductions in CK flux than PCr/ATP in human HF (9, 10) suggest that reduced CK flux may be strongly related to outcomes and mortality in human HF.
We therefore prospectively quantified myocardial ATP and PCr concentrations, PCr/ATP ratios, the CK reaction rate constant kf, as well as the rate of ATP synthesis through CK (CK flux) in patients with nonischemic cardiomyopathy (NICM) and HF, and followed the patients for clinical HF events and mortality. In doing so, we tested the hypothesis that a reduced rate of ATP transfer through CK predicts HF-related events and mortality, with the translational relevance that myocardial metabolism quantified noninvasively with 31P MRS on clinical magnetic resonance (MR) scanners may be useful for better managing HF patients.
Results
Baseline demographic and metabolic parameters
Patient demographics and interquartile ranges (IQRs) are summarized in Table 1. NICM patients had a median age of 47.5 years, 32.8% (n = 19) were women, and 56.9% (n = 33) were African American. Healthy subjects (n = 17) had a median age of 38 years, and 29% (n = 5) were women. For NICM patients, the median LVEF was 20% and NYHA class II (IQR: II, III). Medication use, both at the time of the MRS and at the time of first event or last follow-up (in those with no events), is shown in table S1. Most patients were on β-blockers (81%), angiotensin-converting enzyme (ACE) inhibitors (88%), and diuretics (80%) at the time of study entry.
Table 1. Baseline characteristics of patients (n = 58) enrolled in the study.
IQRs are (25%, 75%).
| Median age (IQR), years | 47.5 (42.8, 55.8) |
| Gender, n (%) | |
| Male | 39 (67.2) |
| Female | 19 (32.8) |
| Median LVEF (IQR) | 20 (15, 30) |
| NYHA class, n (%) | |
| I | 12 (20.7) |
| II | 25 (43.1) |
| III | 20 (34.5) |
| IV | 1 (1.7) |
| Race, n (%) | |
| African American | 33 (56.9) |
| White | 24 (41.4) |
| Other | 1 (1.7) |
Figure 1 shows representative cardiac 31P MRST spectra (i to viii) from two HF patients, as well as their corresponding anatomical locations in the heart, as seen on cardiac 1H magnetic resonance images (MRIs). Red arrows depict the location of spectral saturation at control locations [Fig. 1, A (spectra i and v) and B (spectra iii and vii)], and with γ-phosphate of ATP (γ-ATP) resonance saturated [Fig. 1, A (spectra ii and vi) and B (spectra iv and viii)]. The drop in PCr with γ-ATP saturation (green lines) is directly proportional to CK flux (the higher the slope, the greater the flux). The CK flux for the patient in Fig. 1A who had no events was 2.8 μmol g−1 s−1, whereas the flux for the patient in Fig. 1B who subsequently died was only 0.5 μmol (g wet weight)−1 s−1. For all subjects, the median myocardial PCr/ATP ratio was 1.56, and the [PCr] and [ATP] pools were 7.4 and 4.56 μmol (g wet weight)−1, respectively (Table 2). The median cardiac CK kf (0.24 s−1) and CK flux [1.59 μmol (g wet weight)−1 s−1] were significantly lower in HF patients than in healthy controls analyzed in the same way [kf = 0.38 s−1; CK flux = 3.50 μmol (g wet weight)−1 s−1], consistent with previous reports (9, 10).
Fig. 1. Cardiac 31P MRST and transaxial scout MRI.
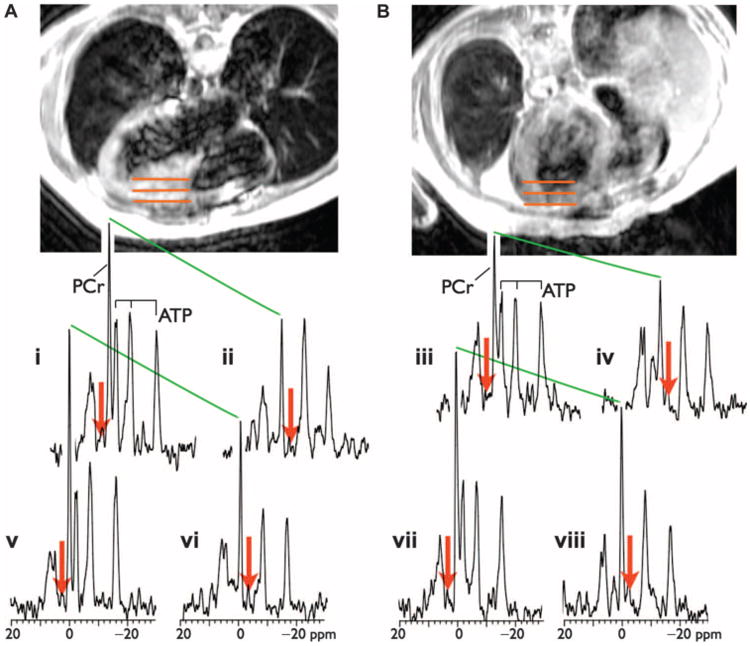
(A) An image from a 30-year-old man who had no HF events during the follow-up period. (B) A 61-year-old man who experienced cardiovascular death. The images are annotated (orange bars, 5 cm long) to show the locations of two (upper and lower) anterior myocardial 1-cm-thick volumes sampled by 31P MRS, with corresponding upper and lower spectra plotted below the respective image. In (i and v) and (iii and vii), spectra were acquired with control saturation (red arrows). In (ii and vi) and (iv and viii), spectra were acquired with the γ-ATP saturated. Each pair of spectra in (i/v), (ii/vi), (iii/vii), and (iv/viii) is from a 32-spectrum set (flip angle, α = 60°). The change in the height of the PCr peak (green lines) between control saturation and ATP saturation is due to the forward flux through CK and is proportional to the rate constant, k. The cardiac PCr/ATP ratios were similar for these patients, but the CK flux was higher in the patient without an HF event.
Table 2. Anterior myocardial high-energy phosphate concentrations and CK flux in healthy subjects and HF patients measured by 31P MRS.
Data are medians and IQRs (25%, 75%) for healthy subjects (n = 17) and HF patients (n = 58).
| Parameter | Healthy subjects | HF |
|---|---|---|
| [PCr] [μmol (g wet weight)−1] | 9.81 (8.90, 10.69) | 7.4 (5.47, 9.94)** |
| [ATP] [μmol (g wet weight)−1] | 5.60 (4.51, 6.52) | 4.56 (3.91, 5.52)* |
| kf (s−1) | 0.38 (0.32, 0.43) | 0.24 (0.16,0.33)*** |
| PCr/ATP | 1.77 (1.37, 2.04) | 1.56 (1.23, 1.95) |
| CK flux [μmol (g wet weight)−1 s−1] | 3.50 (3.01, 4.15) | 1.59 (1.06, 2.41)*** |
P < 0.02,
P < 0.001,
P < 0.0001 versus healthy subjects, two-tailed Mann-Whitney-Wilcoxon test.
HF-related events and mortality
About 95% of enrolled subjects were followed over a median period of 4.7 years (range up to 8.2 years): three HF subjects could not be located for follow-up. The all-cause and cardiovascular mortalities in HF patients were 33% (19 deaths) and 24% (14 deaths), respectively, during the follow-up period. Figure 2 shows all-cause and cardiovascular mortality for these subjects after the time of the MRS examination. Thirty-eight percent (n = 22) of patients had one or more HF-related hospitalization, 3% (n = 2) underwent VAD implantation, and 10% (n = 6) underwent cardiac transplantation.
Fig. 2. Kaplan-Meier survival estimates.
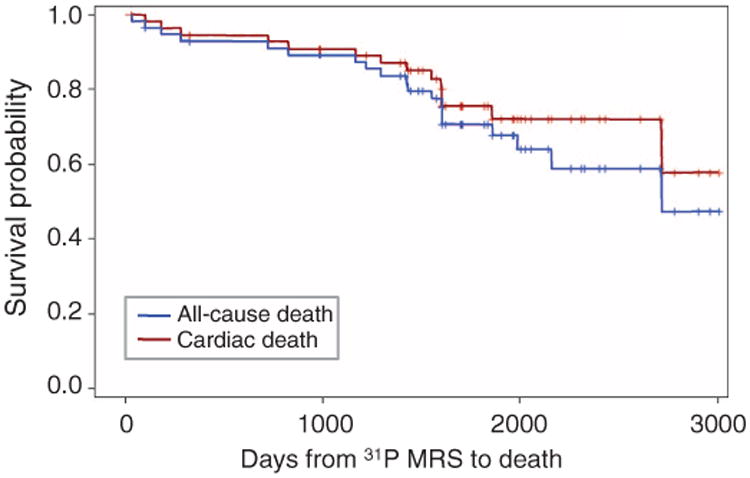
Survival curves for all-cause (n = 19) and cardiac (n = 14) mortality in the overall population.
Relationship between baseline parameters and HF events
Multiple-event analysis (MEA) was performed for the composite of all-cause death and HF-related events (HF hospitalization, VAD implantation, and cardiac transplantation). An initial model was constructed that included clinical predictors of HF from the literature and cardiac energetic variables measured here (Table 3, “Initial model”). Among baseline demographic, clinical, and metabolic parameters, MEA identified four independent predictors of HF events and all-cause mortality: African-American race, NYHA class ≥3, LVEF, and myocardial CK flux (Table 3, “Final model”). Relative (PCr/ATP) and absolute myocardial metabolite pool sizes ([PCr] and [ATP]) were not significant predictors for the composite HF end points. When entered into a multivariate model, myocardial CK flux was found to be an independent predictor of all-cause mortality and HF-related composite outcomes (hazard ratio, 0.61; P = 0.001, by MEA) after adjustment for baseline variables including age, LVEF, race, and NYHA class (Table 3, “Final model”). To put this in perspective, an increase of 1 μmol (g wet weight)−1 s−1 in ATP flux through CK was associated with ∼39% decrease in risk of HF-related composite outcomes and all-cause mortality.
Table 3. Clinical variables as predictors of all-cause mortality and HF-related events by MEA.
HF-related events were HF hospitalization, VAD implantation, and cardiac transplantation. All variables included in the initial and final models are shown at left and right, respectively. Hazard ratios and P values were determined by MEA, whereby males were compared with females, and African Americans with non–African Americans. NYHA class II and class III–IV were compared with NYHA class I subjects. All other variables were treated as continuous. CI, confidence interval.
| Initial model | Final model | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Variable | Hazard ratio (95% CI) | P | Variable | Hazard ratio (95% CI) | P |
| Female | 1.0 | — | |||
| Male | 0.97 (0.47–1.98) | 0.93 | |||
| Non–African American | 1.0 | — | Non–African American | 1.0 | — |
| African American | 5.98 (2.91–12.3) | <0.0001 | African American | 4.51 (2.28–8.92) | <0.0001 |
| Age | 1.02 (0.98–1.05) | 0.34 | |||
| LVEF | 0.966 (0.94–0.99) | 0.004 | LVEF | 0.97 (0.95–0.98) | 0.0002 |
| NYHA class I | 1.0 | — | NYHA class I | 1.0 | — |
| NYHA class II | 1.41 (0.59–3.39) | 0.44 | NYHA class II | 1.10 (0.57–2.14) | 0.77 |
| NYHA class III–IV | 4.10 (1.63–10.3) | 0.003 | NYHA class III–IV | 2.87 (1.38–5.94) | 0.005 |
| [PCr] [μmol (g wet weight)−1] | 0.65 (0.27–1.54) | 0.32 | |||
| [ATP] [μmol (g wet weight)−1] | 2.26 (0.58–8.81) | 0.24 | |||
| kf (s−1) | 54 (0.02–186,702) | 0.34 | |||
| PCr/ATP | 13.6 (0.34–540) | 0.16 | |||
| CK flux [μmol (g wet weight)−1 s−1] | 0.34 (0.11–1.04) | 0.059 | CK flux | 0.61 (0.45–0.82) | 0.001 |
Myocardial CK flux was also an independent predictor of the composite end point that included cardiovascular death instead of all-cause death (Table 4). Specifically, the risk for cardiovascular mortality and the HF-related end points (HF hospitalization, cardiac transplantation, and VAD implantation) were independently predicted by African-American race, NYHA class >3, LVEF, and CK flux (Table 4, “Final model”). CK flux predicted HF outcomes (P = 0.002, by MEA) after adjustment for race, NYHA class, and LVEF. An increase in ATP flux through CK of 1 μmol (g wet weight)−1 s−1 was associated with ∼36% decrease in cardiovascular mortality and HF-related composite outcomes.
Table 4. Clinical variables as predictors of cardiac mortality and HF-related events by MEA.
HF-related events were HF hospitalization, VAD implantation, and cardiac transplantation. All variables included in the initial and final models are shown at left and right, respectively. Hazard ratios and P values were determined by MEA, whereby males were compared with females, and African Americans with non-African Americans. NYHA class II and class III–IV were compared with NYHA class I subjects. All other variables were treated as continuous.
| Initial model | Final model | ||||
|---|---|---|---|---|---|
|
|
|
||||
| Variable | Hazard ratio (95% CI) | P | Variable | Hazard ratio (95% CI) | P |
| Female | 1.0 | — | |||
| Male | 1.01 (0.50–2.04) | 0.98 | |||
| Non–African American | 1.0 | — | Non–African American | 1.0 | — |
| African American | 5.19 (2.51–10.74) | <0.0001 | African American | 4.72 (2.44–9.08) | <0.0001 |
| Age | 1.02 (0.99–1.04) | 0.24 | |||
| LVEF | 0.944 (0.92–0.97) | <0.0001 | LVEF | 0.95 (0.93–0.97) | <0.0001 |
| NYHA class I | 1.0 | — | NYHA class I | 1.0 | — |
| NYHA class II | 1.06 (0.46–2.40) | 0.90 | NYHA class II | 1.09 (0.63–1.89) | 0.76 |
| NYHA class III–IV | 3.53 (1.58–7.93) | 0.002 | NYHA class III–IV | 2.78 (1.52–5.08) | 0.001 |
| [PCr] [μmol (g wet weight)−1] | 1.08 (0.47–2.50) | 0.85 | |||
| [ATP] [μmol (g wet weight)−1] | 1.17 (0.31–4.49) | 0.81 | |||
| kf (s−1) | 4,441 (1.59–12,440,228) | 0.038 | |||
| PCr/ATP | 3.42 (0.09–137) | 0.51 | |||
| CK flux [μmol (g wet weight)−1 s−1] | 0.18 (0.06–0.52) | 0.002 | CK flux | 0.64 (0.48–0.85) | 0.002 |
MEA was also performed by a statistician not involved with this study, using a different statistical software platform, but a comparable approach to that described above—except that LVEF was treated as a dichotomous variable and NYHA class III–V was compared to class I—II. The results of this independent analysis are shown in tables S2 and S3. This second analysis confirmed that myocardial CK flux is an independent predictor of all-cause mortality and HF end points and of cardiovascular mortality and HF end points, whereby an increase in CK flux of 1 μmol g−l min−1 resulted in a decrease in risk of ∼32 to 36%.
Discussion
ATP is required for normal myocardial contraction. This study shows that a decreased rate of myofibrillar ATP delivery by CK is an independent multivariate predictor of events in HF patients. These observations suggest that the rate of ATP synthesis through CK can be used, along with established predictors like ejection fraction and NYHA class, to predict future HF events and, by extension, may help guide the timing of intensive intervention in nonischemic patients with HF. Moreover, the findings in this cohort suggest that impaired cardiac energetics and ATP delivery are fundamentally linked to the progression of clinical HF and its attendant morbidity and mortality.
Although HF is the final consequence of a constellation of different etiologies, impaired myocardial energy metabolism has been proposed as one common underlying process. ATP is required for both cell viability and myocardial pump function. Chemical energy released from cleavage of the terminal phosphate is converted into the work of contraction and relaxation. The amount of ATP in myocytes is small relative to the rates of myofibrillar ATP utilization and therefore must be continually generated and transported to the sites of utilization to maintain a constant concentration of ATP and normal function. In the myocardium, ATP is primarily synthesized in the mitochondria through oxidative phosphorylation and moves to the contractile apparatus, where it is consumed by myosin adenosine triphosphatase (ATPase). Although ATP levels may be reduced in the later stages of advanced HF (15, 17), the reductions are still rather modest and not more than ∼30 to 40% below normal, and not to the level of the Km (Michaelis constant) for most ATP-consuming reactions (2, 3). However, impaired ATP transfer and utilization via CK may limit contractile function and reserve at rest and during increased stress (8). Perhaps not surprisingly, inotropes, which increase myocardial energetic demand, are related to worse HF outcomes (18, 19). Conversely, energy-sparing therapies, such as ACE inhibitors and β-blockers, reduce morbidity and mortality in HF patients (20, 21).
The CK reaction also plays an important role in myocardial energy metabolism by maintaining high ADP (adenosine 5′-diphosphate) levels at the mitochondria, where ATP is generated, and low ADP levels at the myofibrils, where ATP is used. Thus, a reduction in ATP transfer capacity through CK (CK flux) in HF could impair contractile function and reserve. Previous studies showed a 50 to 60% reduction in cardiac CK flux in patients with DCM or LVH who had only mild to moderate HF even before a reduction in ATP could be detected (9, 10). This reduction in CK flux in mild to moderate HF, coupled with a lack of increase during stress in the normal heart (9), suggests that CK energy reserve is compromised in human HF. The potential importance of CK metabolism in HF is also suggested by the finding that the hearts of CK-depleted mice are more prone to fail in response to ischemia or injury and to exhibit electrical vulnerability (22, 23).
Causal evidence that the failing heart is energy-starved, as it relates to CK metabolism, comes from very recent studies demonstrating that contractile function, both at baseline and in response to adrenergic stimulation, is improved in failing mouse hearts when the CKM is overexpressed and the rate of ATP synthesis through CK is increased (8). In addition, survival in murine HF is significantly increased with cardiac CKM over-expression and augmented CK flux (8). Another strategy to augment CK flux in the failing heart is xanthine oxidase inhibition (24), which was recently shown to nearly normalize the rate of ATP synthesis through cardiac CK in patients with NICM (25). Together, ATP synthesis through CK is markedly reduced in experimental and human HF: augmenting ATP flux through CK improves energy availability and, potentially, outcomes.
Our current findings differ somewhat from those in a previous report suggesting that a low cardiac PCr/ATP ratio predicts mortality in patients with DCM (14). The mean PCr/ATP ratios were comparable in the two studies, with patients in the current report receiving contemporary HF therapies (∼81% of patients taking β-blockers and 81 to 88% taking ACE inhibitors) (table S1). The total and cardiovascular mortality rates (33 and 24%, respectively, over ∼4.6 years) as well as medication usage in our study are comparable to those of more recent large clinical HF trials (26, 27). Note also that the present sample size is greater and the patient-years of follow-up were about two- to threefold longer than in the previous study (14). Furthermore, we did not find a significant association between either lower [PCr] or [ATP] and the risk for HF-related events or mortality. Although ATP depletion might occur in advanced or end-stage disease (17), we did not study such critically ill patients. Thus, although [PCr] and kf both factor directly into the CK energy supply, it appears that it is the rate of delivery of ATP—the CK flux—that is the best, of all of the CK indices studied, in predicting future HF events. It will be crucial in future studies to determine the factors that impair CK energy metabolism in failing hearts and the mechanisms by which altered CK metabolism affects HF prognosis.
We used MEA to study the impact of myocardial metabolism on HF-related outcomes. Previous studies have used this analysis strategy in prospective randomized controlled trials to investigate the impact of anti-platelet and lipid-lowering therapy on cardiovascular events (28, 29). The utility of MEA for assessing relevant HF-related cardiovascular events is of increasing importance because repeat HF hospitalizations are closely related to poor short- and long-term outcomes and high health care costs. Although MEA may raise concerns about overemphasizing the effects of repeat events (30), this method provides an understanding of the overall morbidity and impact of interrelated events in diseases such as HF, which would be ignored in traditional time to first event survival analyses. Because repeat hospitalizations are closely related to increasing risk of “hard” events including death (31), transplantation, and VAD implantation, including them in a MEA offers a more comprehensive understanding of the impact of HF on mortality, health care utilization, and quality of life.
The present work should be interpreted as exploratory owing to the relatively limited sample size and number of events. A validation study in a larger cohort is needed to address the ability of CK flux to predict each separate component of the combined end point, which is not possible here. Nevertheless, the study was large enough with a sufficiently long follow-up to demonstrate that ATP flux through CK predicts HF events, even after adjustment for NYHA class and LVEF. This study does not address changes over time, if any, in myocardial CK flux and other high-energy phosphate parameters. Left ventricular function was not measured systematically by MRI at the time of the MRS examination, and thus, the indices were derived from contemporary clinical examinations using different imaging modalities. Finally, as noted, MEA may overemphasize risk. However, we believe that the recurrent events included in this analysis are closely interrelated, and related as well to the total morbidity and mortality burden associated with HF.
In conclusion, myocardial high-energy phosphate metabolism is altered in NICM. Unlike the relative or absolute sizes of the cardiac PCr and ATP pools, the rate of ATP synthesis through CK, the heart's primary energy reserve, is shown here to be a predictor of HF outcomes in NICM patients over about 5 years of follow-up, even after correction for NYHA class and LVEF. Thus, 31P MRS offers a noninvasive window into energy metabolism and the rates of myofibrillar ATP delivery via CK in the failing human heart, and merits further investigation as a tool to complement existing risk assessment metrics for HF patients. These observations strongly support the development and investigation of strategies to augment CK ATP metabolism in the failing human heart. The recent study demonstrating augmentation of cardiac CK flux in HF patients by 40% (from 1.9 to 2.7 μmol g−1 s−1) after acute administration of allopurinol (25), for example, shows that this may be feasible and, in the context of the present work, suggests that the potential impact on HF-related composite outcomes could be significant. Finally, the demonstration that a metabolic imaging tool like 31P MRS has the potential to predict risk of future clinical events merits investigation in neurodegenerative, muscular, and other diseases in tissues with high metabolic energy demands.
Materials and Methods
Study design
This was a prospective, nonrandomized study to test the hypothesis that cardiac energetic abnormalities in patients with HF, as detected by 31P MRS, predict future events, including death (all-cause and cardiovascular), HF hospitalization, cardiac transplantation, and left VAD placement (clinicaltrials.gov: NCT00181259). The 31P MRS data were analyzed by an investigator (P.A.B.) blinded to clinical outcomes. The clinical events were adjudicated by investigators (G.S.P. and A.S.) blinded to the 31P MRS results. All human studies were approved by the Johns Hopkins Institutional Review Board (IRB) for human investigation. All subjects gave informed consent after explanation of the study and protocol.
Patient population
Fifty-eight patients with a clinical diagnosis of chronic HF (NYHA class I–IV) and 17 healthy subjects were studied, including some participants reported previously (9, 10). HF patients were referred for study by their cardiologists in the inpatient or outpatient settings at Johns Hopkins, and healthy subjects were recruited with IRB-approved posters. None of the HF patients had coronary disease as defined by >50% luminal stenosis assessed by cardiac catheterization, computed tomography angiography, or positive stress nuclear or echocardiogram test. Those patients who had a recent HF decompensation were only enrolled after at least 2 weeks of clinical stabilization. Patients with any contraindication to MR scanning, inability to lie flat or complete the MR study, pregnancy, or those receiving investigational drugs were excluded from this study. Healthy subjects, who had no history of heart disease, hypertension, or diabetes mellitus, were studied by MRS contemporaneously with the patients and served as controls for the CK flux and metabolite measurements.
31P MRS methods
Patients were studied at rest in a General Electric 1.5-T MRI/MRS system (GE Healthcare Technologies). MRS studies were performed with subjects prone on a 6.5-cm 31P receive/25-cm 31P transmit surface coil set. Because the duration of conventional unlocalized MRST studies is impractical for localized studies of heart patients, we used the four-angle saturation transfer method (FAST) to measure the kf of the CK reaction (9, 32, 33). FAST measures kf in four short-repetition time (TR) acquisitions (with TR < T1), wherein PCr is excited with two different (adiabatic) pulse flip angles (α), both with and without saturation of γ-ATP.
The complete patient cardiac MRS protocol thus comprised (i) conventional scout proton (1H) MRI to position the anterior myocardium over the coil and shim the magnet over the heart; (ii) acquisition of the four 31P FAST data sets localized by the one-dimensional (1D) chemical shift imaging (CSI) method [32 transaxial 1-cm-thick slices; TR = 1 s; number of excitations per phase encodes, NEX = 12, α = 60°; and NEX = 24, α = 15° with chemical-selective saturation at ±2.7 parts per million (ppm)] (33); (iii) acquisition of a fifth 31P 1D CSI set with saturation turned off (α = 60°; NEX = 12; cardiac-gated with TR ≈1 s) for phosphate metabolite quantification (9, 10, 25) and to correct for spillover irradiation (33, 34); and (iv) acquisition of a sixth 1H 1D CSI data set with the 31P coil(α = 60°; NEX = 4; cardiac-gated with TR ≈2s) to provide a water concentration reference, also for metabolite quantification (9). The total MRI/MRS examination time was about 70 min, including ∼36, ∼6, and ∼4 min for MRS acquisitions in steps (ii) to (iv), respectively. After the subject MRS examination, the 31P and 1H 1D CSI acquisitions were repeated under fully relaxed conditions in steps (v) and (vi) (TR = 4 s for 1H; TR = 8 s for 31P; saturation < 3% versus TR < 24 s) on a homogeneous phantom of inorganic phosphate to provide reference signals for determining the cardiac metabolite concentrations (9, 10, 32).
Data analysis
Details of the MRS analysis have been described previously (9, 10, 32–34). The CK rate constants, kf (s−1), were calculated in accordance with the latest numerical analysis of errors caused by spillover irradiation (25, 34) using Eqs. 1 and 2 in (25, 34), as applied to the T1-corrected PCr signal amplitudes recorded in the presence and absence of γ-ATP saturation from MRS protocol steps (ii) and (iii). For quality control purposes, only spectra with PCr signal-to-noise ratios >5 were quantified.
The metabolite concentrations, [PCr] and [ATP] in μmol/g wet weight of tissue, were noninvasively measured in anterior myocardium identified by scout MRI in two ways (9, 10, 32). First, concentrations were calculated from the ratios of metabolite peak areas acquired in step (iii) to the water signal from step (iv), multiplied by the 1H/31P calibration factor determined from steps (v) and (vi), and by the cardiac tissue water proton concentration. The latter was taken as 86 mol/kg tissue wet weight (9, 10, 32). Second, [PCr] and [ATP] were determined from the ratio of the peak areas from step (iii), to the signal from the phosphate reference phantom in step (v) (9, 10, 25). Metabolite signals were T1-corrected assuming literature-average relaxation times, and [ATP] was corrected for blood contamination (12, 35).
The forward CK flux was determined from the product, kf[PCr], in mmol/g wet weight per second at each depth. Then, for each subject, the CK flux, kf, and all of the [ATP] and [PCr] measurements were averaged from the same two to four adjacent MRS slices in the anterior myocardium for use in the statistical analyses.
Subject follow-up for mortality and clinical HF events
HF clinical events were tracked by direct phone contact with the patients and/or their primary care physician or cardiologist. Hospitalizations, cardiac transplantation, and VAD placement were documented with clinical records. A team composed of a research nurse and a cardiologist blinded to the MRS results reviewed all clinical records and reports pertaining to hospitalizations and death, and adjudicated the causes of hospitalization or death.
Hospitalization for HF required two or more symptoms (shortness of breath, chest tightness, fatigue, leg swelling, or edema), clinical findings (elevated jugular venous pressure, rales, ascites, lower extremity edema, and persistent hepatojugular reflux), and radiographic (pulmonary edema) or biochemical findings (worsening renal function and elevated brain natriuretic peptide) documented in clinical records. In addition, hospitalizations in which patients required intravenous diuretics or inotropes were considered HF-related. Death was confirmed by reports from the medical examiner's office and cross-checked against the National Death Index and Social Security database. Death was considered to be cardiovascular-related if there was deterioration of congestive HF preceding the terminal event or clinical documentation of a cardiovascular cause in the hospital records. Death was also adjudicated as cardiovascular if the patient went to hospice for end-stage HF and there was no competing etiology such as advanced malignancy or metastatic disease at the time of death. When there was a competing diagnosis, the decision was made on the basis of active symptoms or conditions being treated at that time. If no clear etiology was found, the cause of death was not considered to be cardiovascular.
Statistical analysis
Results are presented as median with IQR (25%, 75%). The Mann-Whitney-Wilcoxon test was used with two-tailed testing and exact P test to compare baseline metabolic variables between healthy subjects and HF patients. Categorical variables were compared using χ2 testing.
The marginal approach of Wei, Lin, and Weissfeld (WLW) was implemented in SAS software and used (by S.L.) to perform MEA of time to a composite event, defined as recurrent HF hospitalizations, VAD implantation, cardiac transplantation, and cardiac or all-cause death (36, 37). The WLW approach fits a Cox proportional hazards model to each component time to event and makes statistical inference of the regression parameters based on a robust “information sandwich”–type estimate of the SE to accommodate within-subject correlations between events (38). The importance of each variable included in this model was evaluated with the following: (i) an examination of its Wald statistic and (ii) a comparison of its regression coefficient, as determined from the multivariate model, with that from the corresponding univariate model. Variables that ceased to make significant contributions to the model according to these two criteria were deleted in a stagewise manner, and a new model was refitted. This process of eliminating, refitting, and verifying continued until only the statistically significant variables remained, yielding a final model (39). The initial model included literature-identified HF risk factors (age, gender, LVEF, race, and NYHA class) and metabolic predictors tested here (PCr/ATP, [PCr], [ATP], kf, and CK flux) as co-variates. Age, LVEF, PCr/ATP, [PCr], [ATP], and myocardial CK flux were treated as continuous variables; gender and race (African American or others) were treated as dichotomous variables; and NYHA class was categorized (class I, class II, and combined class III + IV, because there was only one class IV subject).
MEA was also performed independently by a statistician not involved with this study. It was implemented on a different software platform (STATA) with a comparable approach to that described above, except that LVEF was treated as a dichotomous variable (<30% versus ≥30%) and NYHA class III–V was compared to class I–II. The results of this independent analysis are shown in tables S2 and S3.
Supplementary Material
Table S1. Medication profile at study entry and at first event or last follow-up.
Table S2. All-cause mortality and the composite of HF events by MEA run on STATA software from final model.
Table S3. Cardiovascular mortality and the composite of HF events by MEA run on STATA software from final model.
Acknowledgments
We thank C. Thompson of the Johns Hopkins Biostatistics Center for the statistical review.
Funding: This work was supported by NIH grants RO1-HL61912 and R01-HL056882, DW Reynolds Cardiovascular Clinical Research Center at Johns Hopkins University, and the Russell H. Morgan (P.A.B.) and Clarence Doodeman (R.G.W.) Endowments.
Footnotes
Author contributions: P.A.B., G.G., and R.G.W. designed the study; P.A.B. and R.G.W. acquired the 31P MRS data; P.A.B. analyzed the 31P MRS data; G.S.P. and A.S. collected and tabulated the clinical data; S.L. performed the statistical analysis; S.S.N., G.A.H., and K.W. contributed to the recruitment of patients and conduct of the study; G.S.P. wrote the manuscript; P.A.B., G.G., and R.G.W. critically revised the manuscript; and all authors contributed to the final version of the revised manuscript.
Competing interests: The authors declare that they have no competing financial interests.
References and Notes
- 1.Writing Group Members; Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, Mc Dermott MM, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics—2010 update: A report from the American Heart Association. Circulation. 2010;121:e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 2.Ingwall JS. Is cardiac failure a consequence of decreased energy reserve? Circulation. 1993;87:VII-58–VII-62. [Google Scholar]
- 3.Katz AM. Is the failing heart energy depleted? Cardiol Clin. 1998;16:633–644. doi: 10.1016/s0733-8651(05)70040-0. [DOI] [PubMed] [Google Scholar]
- 4.Ingwall JS, Kramer MF, Fifer MA, Lorell BH, Shemin R, Grossman W, Allen PD. The creatine kinase system in normal and diseased human myocardium. N Engl J Med. 1985;313:1050–1054. doi: 10.1056/NEJM198510243131704. [DOI] [PubMed] [Google Scholar]
- 5.Wallimann T, Dolder M, Schlattner U, Eder M, Hornemann T, O'Gorman EO, Rück AR, Brdiczka D. Some new aspects of creatine kinase (CK): Compartmentation, structure, function and regulation for cellular and mitochondrial bioenergetics and physiology. Biofactors. 1998;8:229–234. doi: 10.1002/biof.5520080310. [DOI] [PubMed] [Google Scholar]
- 6.Ye Y, Gong G, Ochiai K, Liu J, Zhang J. High-energy phosphate metabolism and creatine kinase in failing hearts: A new porcine model. Circulation. 2001;103:1570–1576. doi: 10.1161/01.cir.103.11.1570. [DOI] [PubMed] [Google Scholar]
- 7.Ye Y, Wang C, Zhang J, Cho YK, Gong G, Murakami Y, Bache RJ. Myocardial creatine kinase kinetics and isoform expression in hearts with severe LV hypertrophy. Am J Physiol Heart Circ Physiol. 2001;281:H376–H386. doi: 10.1152/ajpheart.2001.281.1.H376. [DOI] [PubMed] [Google Scholar]
- 8.Gupta A, Akki A, Wang Y, Leppo MK, Chacko VP, Foster DB, Caceres V, Shi S, Kirk JA, Su J, Lai S, Paolocci N, Steenbergen C, Gerstenblith G, Weiss RG. Creatine kinase-mediated improvement of function in failing mouse hearts provides causal evidence the failing heart is energy starved. J Clin Invest. 2012;122:291–302. doi: 10.1172/JCI57426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci USA. 2005;102:808–813. doi: 10.1073/pnas.0408962102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith CS, Bottomley PA, Schulman SP, Gerstenblith G, Weiss RG. Altered creatine kinase adenosine triphosphate kinetics in failing hypertrophied human myocardium. Circulation. 2006;114:1151–1158. doi: 10.1161/CIRCULATIONAHA.106.613646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Conway MA, Allis J, Ouwerkerk R, Niioka T, Rajagopalan B, Radda GK. Detection of low phosphocreatine to ATP ratio in failing hypertrophied human myocardium by 31P magnetic resonance spectroscopy. Lancet. 1991;338:973–976. doi: 10.1016/0140-6736(91)91838-l. [DOI] [PubMed] [Google Scholar]
- 12.Hardy CJ, Weiss RG, Bottomley PA, Gerstenblith G. Altered myocardial high-energy phosphate metabolites in patients with dilated cardiomyopathy. Am Heart J. 1991;122:795–801. doi: 10.1016/0002-8703(91)90527-o. [DOI] [PubMed] [Google Scholar]
- 13.Neubauer S, Krahe T, Schindler R, Horn M, Hillenbrand H, Entzeroth C, Mader H, Kromer EP, Riegger GA, Lackner K, Ertl G. 31P magnetic resonance spectroscopy in dilated cardiomyopathy and coronary artery disease. Altered cardiac high-energy phosphate metabolism in heart failure. Circulation. 1992;86:1810–1818. doi: 10.1161/01.cir.86.6.1810. [DOI] [PubMed] [Google Scholar]
- 14.Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, Pabst T, Ertl G, Hahn D, Ingwall JS, Kochsiek K. Myocardial phosphocreatine-to-ATP ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. 1997;96:2190–2196. doi: 10.1161/01.cir.96.7.2190. [DOI] [PubMed] [Google Scholar]
- 15.Beer M, Seyfarth T, Sandstede J, Landschütz W, Lipke C, Köstler H, von Kienlin M, Harre K, Hahn D, Neubauer S. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with 31P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40:1267–1274. doi: 10.1016/s0735-1097(02)02160-5. [DOI] [PubMed] [Google Scholar]
- 16.Schär M, El-Sharkawy AM, Weiss RG, Bottomley PA. Triple repetition time saturation transfer (TRiST) 31P spectroscopy for measuring human creatine kinase reaction kinetics. Magn Reson Med. 2010;63:1493–1501. doi: 10.1002/mrm.22347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Starling RC, Hammer DF, Altschuld RA. Human myocardial ATP content and in vivo contractile function. Mol Cell Biochem. 1998;180:171–177. [PubMed] [Google Scholar]
- 18.Packer M, Carver JR, Rodeheffer RJ, Ivanhoe RJ, DiBianco R, Zeldis SM, Hendrix GH, Bommer WJ, Elkayam U, Kukin ML, Mallis GI, Sollano JA, Shannon J, Tandon PK, DeMets DL. PROMISE Study Research Group, Effect of oral milrinone on mortality in severe chronic heart failure. N Engl J Med. 1991;325:1468–1475. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 19.Feldman AM, Bristow MR, Parmley WW, Carson PE, Pepine CJ, Gilbert EM, Strobeck JE, Hendrix GH, Powers ER, Bain RP, White BG Vesnarinone Srudy Group. Effects of vesnarinone on morbidity and mortality in patients with heart failure. N Engl J Med. 1993;329:149–155. doi: 10.1056/NEJM199307153290301. [DOI] [PubMed] [Google Scholar]
- 20.The SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 21.Packer M, Bristow MR, Cohn JN, Colucci WS, Fowler MB, Gilbert EM, Shusterman NH. U.S. Carvedilol Heart Failure Study Group, The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. N Engl J Med. 1996;334:1349–1355. doi: 10.1056/NEJM199605233342101. [DOI] [PubMed] [Google Scholar]
- 22.Abraham MR, Selivanov VA, Hodgson DM, Pucar D, Zingman LV, Wieringa B, Dzeja PP, Alekseev AE, Terzic A. Coupling of cell energetics with membrane metabolic sensing. Integrative signaling through creatine kinase phosphotransfer disrupted by M-CK gene knock-out. J Biol Chem. 2002;277:24427–24434. doi: 10.1074/jbc.M201777200. [DOI] [PubMed] [Google Scholar]
- 23.Saupe KW, Spindler M, Tian R, Ingwall JS. Impaired cardiac energetics in mice lacking muscle-specific isoenzymes of creatine kinase. Circ Res. 1998;82:898–907. doi: 10.1161/01.res.82.8.898. [DOI] [PubMed] [Google Scholar]
- 24.Cappola TP, Kass DA, Nelson GS, Berger RD, Rosas GO, Kobeissi ZA, Marbán E, Hare JM. Allopurinol improves myocardial efficiency in patients with idiopathic dilated cardiomyopathy. Circulation. 2001;104:2407–2411. doi: 10.1161/hc4501.098928. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch GA, Bottomley PA, Gerstenblith G, Weiss RG. Allopurinol acutely increases adenosine triphospate energy delivery in failing human hearts. J Am Coll Cardiol. 2012;59:802–808. doi: 10.1016/j.jacc.2011.10.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardy GH, Lee KL, Mark DB, Poole JE, Packer DL, Boineau R, Domanski M, Troutman C, Anderson J, Johnson G, McNulty SE, Clapp-Channing N, Davidson-Ray LD, Fraulo ES, Fishbein DP, Luceri RM, Ip JH Sudden Cardiac Death in Heart Failure Trial (SCD-HeFT) Investigators. Amiodarone or an implantable cardioverter–defibrillator for congestive heart failure. N Engl J Med. 2005;352:225–237. doi: 10.1056/NEJMoa043399. [DOI] [PubMed] [Google Scholar]
- 27.Zannad F, McMurray JJ, Krum H, van Veldhuisen DJ, Swedberg K, Shi H, Vincent J, Pocock SJ, Pitt B EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21. [Google Scholar]
- 28.Murphy SA, Cannon CP, Wiviott SD, McCabe CH, Braunwald E. Reduction in recurrent cardiovascular events with intensive lipid-lowering statin therapy compared with moderate lipid-lowering statin therapy after acute coronary syndromes from the PROVE IT–TIMI 22 (Pravastatin or Atorvastatin Evaluation and Infection Therapy–Thrombolysis In Myocardial Infarction 22) trial. J Am Coll Cardiol. 2009;54:2358–2362. doi: 10.1016/j.jacc.2009.10.005. [DOI] [PubMed] [Google Scholar]
- 29.Tikkanen MJ, Szarek M, Fayyad R, Holme I, Cater NB, Faergeman O, Kastelein JJ, Olsson AG, Larsen ML, Lindahl C, Pedersen TR IDEAL Investigators. Total cardiovascular disease burden: Comparing intensive with moderate statin therapy insights from the IDEAL (Incremental Decrease in End Points Through Aggressive Lipid Lowering) trial. J Am Coll Cardiol. 2009;54:2353–2357. doi: 10.1016/j.jacc.2009.08.035. [DOI] [PubMed] [Google Scholar]
- 30.Nissen SE. Cardiovascular outcomes in randomized trials: Should time to first event for “hard” end points remain the standard approach? J Am Coll Cardiol. 2009;54:2363–2365. doi: 10.1016/j.jacc.2009.09.021. [DOI] [PubMed] [Google Scholar]
- 31.O'Connor CM, Abraham WT, Albert NM, Clare R, Gattis Stough W, Gheorghiade M, Greenberg BH, Yancy CW, Young JB, Fonarow GC. Predictors of mortality after discharge in patients hospitalized with heart failure: An analysis from the Organized Program to Initiate Lifesaving Treatment in Hospitalized Patients with Heart Failure (OPTIMIZE-HF) Am Heart J. 2008;156:662–673. doi: 10.1016/j.ahj.2008.04.030. [DOI] [PubMed] [Google Scholar]
- 32.Bottomley PA, Wu KC, Gerstenblith G, Schulman SP, Steinberg A, Weiss RG. Reduced myocardial creatine kinase flux in human myocardial infarction: An in vivo phosphorus magnetic resonance spectroscopy study. Circulation. 2009;119:1918–1924. doi: 10.1161/CIRCULATIONAHA.108.823187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bottomley PA, Ouwerkerk R, Lee RF, Weiss RG. Four-angle saturation transfer (FAST) method for measuring creatine kinase reaction rates in vivo. Magn Reson Med. 2002;47:850–863. doi: 10.1002/mrm.10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabr RE, Weiss RG, Bottomley PA. Correcting reaction rates measured by saturation-transfer magnetic resonance spectroscopy. J Magn Reson. 2008;191:248–258. doi: 10.1016/j.jmr.2007.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bottomley PA, Hardy CJ, Weiss RG. Correcting human 31P heart spectra for partial saturation. Evidence that saturation factors for PCr/ATP are homogenous in normal and disease states. J Magn Reson. 1969;95:341–355. [Google Scholar]
- 36.Wei LJ, Lin DY, Weissfeld L. Regression analysis of multivariate incomplete failure time data by modeling marginal distributions. J Am Stat Assoc. 1989;84:1065–1073. [Google Scholar]
- 37.Lin DY. Cox regression analysis of multivariate failure time data: The marginal approach. Stat Med. 1994;13:2233–2247. doi: 10.1002/sim.4780132105. [DOI] [PubMed] [Google Scholar]
- 38.Kelly PJ, Lim LL. Survival analysis for recurrent event data: An application to childhood infectious diseases. Stat Med. 2000;19:13–33. doi: 10.1002/(sici)1097-0258(20000115)19:1<13::aid-sim279>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 39.Hosmer DW, Lemeshow S. Applied Survival Analysis: Regression Modeling of Time to Event Data. John Wiley & Sons; New York: 1999. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Medication profile at study entry and at first event or last follow-up.
Table S2. All-cause mortality and the composite of HF events by MEA run on STATA software from final model.
Table S3. Cardiovascular mortality and the composite of HF events by MEA run on STATA software from final model.