

Abstract
UVB radiation is the major carcinogen responsible for skin carcinogenesis, thus elucidation of the molecular pathways altered in skin in response to UVB would reveal novel targets for therapeutic intervention. It is well established that UVB leads to upregulation of cyclooxygenase 2 (COX-2) in the skin which contributes to skin carcinogenesis. Overexpression of COX-2 has been shown to promote colon cancer cell growth through β-catenin signaling, however, little is known about the connection between UVB, COX-2 and β-catenin in the skin. In the present study, we have identified a novel pathway in which UVB induces β-catenin signaling in keratinocytes, which is modulated by COX-2 expression. Exposure of the mouse 308 keratinocyte cell line (308 cells) and primary normal human epidermal keratinocytes (NHEKs) to UVB resulted in increased protein levels of both N-terminally unphosphorylated and total β-catenin. In addition, we found that UVB enhanced β-catenin-dependent TOPflash reporter activity and expression of a downstream β-catenin target gene. We demonstrated that UVB-induced β-catenin signaling is modulated by COX-2, as treatment of keratinocytes with the specific COX-2 inhibitor NS398 blocked UVB induction of β-catenin. Additionally, β-catenin target gene expression was reduced in UVB-treated COX-2 knockout (KO) MEFs compared to wild-type (WT) MEFs. Furthermore, epidermis from UVB-exposed SKH-1 mice exhibited increased N-terminally unphosphorylated and total β-catenin protein levels and increased staining for total β-catenin, and both responses were reduced in COX-2 heterozygous mice. Taken together, these results suggest a novel pathway in which UVB induces β-catenin signaling in keratinocytes which is enhanced by COX-2 expression.
Keywords: UVB, β-catenin, COX-2, PGE2
Introduction
More than 2 million people in the United States alone are diagnosed with basal and squamous cell skin cancers annually [1] and extensive epidemiological, clinical and biological studies have identified UVB radiation as the major carcinogen responsible for the development of these forms of skin cancer [2-5]. The cellular response to UVB results in induction of a number of signaling pathways which play critical roles in keratinocyte proliferation, modulation of cell cycle, apoptosis and other downstream events important for skin carcinogenesis [6-9]. Several studies have demonstrated the induction of COX-2 expression in response to UVB radiation [9,10]. COX-2 is highly expressed in squamous cell carcinomas [9] and its expression is required for UVB-induced skin carcinogenesis in mice [11]. COX-2 catalyzes the rate-limiting first step in the conversion of arachidonic acid into prostaglandins, and prostaglandin E2 (PGE2) is the primary product of COX-2 in the skin. Studies in colon cancer, in which COX-2 is overexpressed, have demonstrated a link between COX-2/PGE2 and β-catenin signaling which contributes to cancer cell growth [12].
β-catenin is a 90kD cytosolic protein that was originally identified through its association with the cadherin class of proteins at the cell surface [13]. β-catenin also acts as a crucial component of the Wnt pathway. In the absence of Wnt ligands, β-catenin is recruited to the phosphorylation/destruction complex, which contains the tumor suppressor, adenomatous polyposis coli (APC), and Axin. The phosphorylation/destruction complex facilitates the phosphorylation of β-catenin by both casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β) [14], leading to the ubiquitination and subsequent proteasomal degradation of β-catenin. When extracellular Wnt ligands bind to their receptor complex which is composed of Frizzled and LDL-receptor-related proteins 5 and 6 (LRP 5/6), the phosphorylation/destruction complex is disrupted and β-catenin is stabilized. This stabilized form of β-catenin is specifically unphosphorylated at Ser37 and Thr41, which are two of the GSK3β phosphorylation sites, and is considered to be the transcriptionally active form of β-catenin (N-terminally unphosphorylated β-catenin) [15]. As N-terminally unphosphorylated β-catenin accumulates, it enters the nucleus where it interacts with transcription factors, such as T-cell factor (TCF), to activate transcription of target genes which are involved cell proliferation and survival, as well as cell fate [14].
One of the earliest events in sporadic colon cancer is the loss of a functional APC gene, which leads to stabilized β-catenin [16]. β-catenin has been implicated in the promotion of colon cancer by PGE2, in the absence of a functional APC [12]. Overexpression of COX-2 in colon cancer cells leads to increased PGE2 production. The binding of PGE2 to its receptor EP2 results in the activation of the G protein which is coupled to EP2. The Gαs subunit binds to the regulator of G protein signaling (RGS) domain of Axin, which promotes the release of GSK3β from the phosphorylation/destruction complex [12]. At the same time, the release of the Gβγ subunits stimulate the PI3K/Akt pathway leading to the phosphorylation and inactivation of GSK3β [12]. Together these events lead to the stabilization and nuclear translocation of β-catenin.
While COX-2 and β-catenin have been linked in colon cancer, little is known about the connection between COX-2 and β-catenin in the skin or how UVB could promote this signaling pathway. In the present study, we show that exposure of primary normal human epithelial keratinocytes (NHEKs) and a mouse keratinocyte cell line (308 cells) to UVB radiation results in increased β-catenin signaling and that this effect is mediated by COX-2 expression. We also demonstrate that mice exposed to UVB have increased levels of β-catenin in the skin. Our findings are the first to demonstrate a novel pathway connecting acute UVB exposure and β-catenin signaling in both human and mouse keratinocytes, which harbor no known mutations in the β-catenin pathway. Elucidation of this pathway provides further knowledge of the response of skin to UVB radiation, which is important to understanding the mechanism behind UVB-induced skin cancer.
Materials and Methods
Cell Culture
The mouse 308 keratinocyte cell line [17] was maintained in suspension minimum essential media (United States Biological, Swampscott, MA) supplemented with 8% chelexed (Bio-Rad Laboratories, Hercules, CA) fetal bovine serum, 0.05 mM Ca2+, 0.1 mM non-essential amino acids, 2 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (Invitrogen, Carlsbad, CA). NHEKs were isolated from normal human neonatal foreskin as previously described [18] and were maintained in M154CF keratinocyte medium supplemented with 10 μg/ml gentamicin/0.25 μg/ml amphotericin B and human keratinocyte growth supplement (Invitrogen). COX-2 WT and KO mouse embryonic fibroblasts (MEFs) were isolated from pregnant SKH-1 COX-2+/- females on embryonic day 13 according to standard protocols [19]. Experiments using NHEKs and MEFs were performed on cells at passage ≤4. For studies with specific inhibitors, cells were pre-treated for 1 hr with either NS398, AH6809 (Cayman Chemical, Ann Arbor MI) or melittin (Sigma, St. Louis, MO) prior to UVB irradiation. For PGE2 studies, cells were treated with prostaglandin E2 (Cayman Chemical) dissolved in DMSO and harvested after 15 mins.
Mice
SKH-1 WT and SKH-1 COX-2 +/- mice were housed in climate-controlled quarters (22 ± 1°C at 50% humidity) with 12/12 hr light/dark cycle in yellow fluorescent lights. The homozygous SKH-1 COX-2 -/- mice do not survive beyond a few weeks of age, thus, heterozygous COX-2 deficient animals (6-8 week old females) were used for these studies [11]. All housing and procedures were carried out in an animal facility at Science Park-Research Division, University of Texas MD Anderson Cancer Center, Smithville, TX. This facility is accredited by the American Association for the Assessment and Accreditation of Laboratory Animal Care, in accordance with Institutional Animal Care and Use Committee guidelines. Mice were maintained on chow ad libitum.
UVB irradiation
Prior to UVB exposure, the cell culture media was removed and saved, cells were washed with PBS and irradiated, and the medium was replaced. UVB radiation was provided by FS40T12-UVB lamps (National Biological Corporation, Twinsburg, OH) with peak emission at 313 nm, and a Kodacel K6808 filter (Eastman Kodak, Rochester, NY) was used to filter out any UVC wavelengths (below 295 nm). For mice, the UV apparatus consisted of eight Westinghouse FS20 sunlamps, an IL-1400 radiometer, and an attached UVB photometer. The spectral irradiance for the UVB lamps was 280-400 nm and the peak intensity was 297 nm. The mice were housed in individual compartments in a plastic holder on a rotating base to nullify any differences across the UVB bulbs. The mice were exposed to 2200 J/m2 UVB and were sacrificed 12, 24, or 36 hrs after UVB exposure. The dorsal skin was removed and the epidermis was either fixed in 10% neutral buffered formalin for histology or scraped into lysis buffer and frozen. Lysates were sonicated, cleared by centrifugation and used for Western blotting as described below.
Western blot and immunoprecipitation analysis
Cells were harvested at the indicated time points in lysis buffer (20 mM/liter Tris [pH 7.5], 1.5 mM NaCl, 2 mM EDTA, 10% glycerol, 1% Triton X-100, 1 mM PMSF, 0.5 mM DTT, protease inhibitor cocktail (Sigma), and phosphatase inhibitor cocktail (Sigma)). The bicinchroninic acid protein assay reagent (Pierce, Rockford, IL) was used to determine protein concentration and equal amounts of protein were resolved on sodium dodecyl sulfate (SDS)-polyacrylamide gels followed by electrophoretic transfer onto nitrocellulose membranes. Membranes were then blocked for 1 hr using 5% nonfat dry milk in Tris-buffered saline-0.05% Tween 20. The membranes were probed with primary antibodies overnight at 4°C, and then incubated with the appropriate HRP-conjugated secondary antibodies. The signal was detected using an ECL system (GE Healthcare, Piscataway, NJ). When detecting a phospho-protein, membranes were stripped using Restore Western Blot Stripping Buffer (Pierce, Rockford, IL) and re-probed using an antibody against the total protein. The band intensity was measured by densitometry and the fold change was calculated by normalizing proteins to GAPDH or actin, and representative blots are shown. The mean fold change of at least three independent experiments is reported in Figure 1 C and F and statistical significance was determined by a one-sample t test. For phospho-proteins, fold change was calculated by normalizing the phospho-protein to total protein, which was normalized to GAPDH. For immunoprecipitation, whole cell extract was incubated overnight at 4°C with the appropriate primary antibody, followed by 30 minute incubation at 4°C with Protein A/G-PLUS Agarose beads (Santa Cruz Biotechnology, Santa Cruz, CA). The beads were washed with lysis buffer and boiled for 5 minutes prior to being used for Western blotting as described above.
Figure 1. UVB exposure induces accumulation of N-terminally unphosphorylated β-catenin, total β-catenin and COX-2 in keratinocytes in vitro.
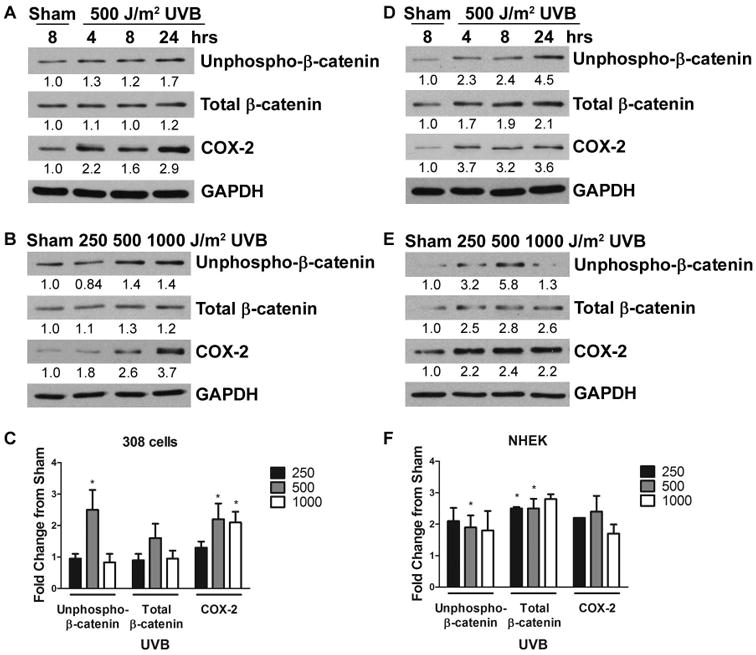
Keratinocytes were exposed to increasing doses of UVB and incubated for the indicated time. Cell lysates were harvested and proteins were detected by Western blot with the indicated antibodies. The band intensities were determined by densitometry and the fold change of proteins normalized to GAPDH is indicated. Panels A, B, D and E are representative blots from at least 3 independent experiments. (A) 308 cells and (D) NHEKs were sham irradiated or exposed to 500 J/m2 UVB and incubated for 4, 8, or 24 hrs. (B) 308 cells and (E) NHEKs were sham irradiated or exposed to 250, 500, or 1000 J/m2 UVB and incubated for 8 hrs. The mean fold change from sham of N-terminally unphosphorylated β-catenin (unphospho-β-catenin), total β-catenin and COX-2 for (C) 308 cells and (F) NHEKs was determined. *, P < 0.05.
Antibodies
The following antibodies were used for immunoblot, immunoprecipitation and/or immunohistochemistry: N-terminally unphosphorylated-β-catenin (unphospho-β-catenin, clone 8E7), total β-catenin (clone 2H4A7) (Millipore, Billerica, MA); Axin (H-98), COX-2 (C-20) (Santa Cruz Biotechnology); COX-2 (Cat# 160126, Cayman Chemical); phospho-Akt (Ser473, Cat# 9271), Akt (Cat# 9272), phospho-GSK3β (Ser9, Cat# 9336), GSK3β (27C10), Erk (Cat# 9102), mouse anti-rabbit IgG light chain specific (L57A3) (Cell Signaling Technology, Beverly, MA); GAPDH (MAB374) (Chemicon, Temecula, CA); goat anti-mouse and goat anti-rabbit IgG-HRP secondary antibodies (Bio-Rad Laboratories, Hercules, CA); donkey anti-goat and bovine anti-goat IgG-HRP secondary antibodies (Santa Cruz Biotechnology).
PGE2 measurement
After treatment with NS398 and/or UVB, the cell-free culture media from 308 cells was collected and PGE2 levels were determined by competitive enzyme-linked immunosorbent assay (ELISA) as directed by the manufacturer (Cayman Chemical).
Luciferase assay
Mouse 308 keratinocytes were transfected with TOPflash, which contains four TCF-consensus binding sites, or FOPflash, which contains four mutated TCF-consensus binding sites, luciferase reporter plasmids (Millipore) and Renilla (pRL-TK; Promega) using Lipofectamine and Plus Reagent (Invitrogen). Luciferase activity was determined using the Dual-Luciferase reporter assay system (Promega) according to manufacturer's protocol. Briefly, cells were rinsed with PBS and removed by scraping in 100 μl of passive lysis buffer. The lysate was then subjected to a freeze-thaw cycle and transferred to a tube. Assays were performed by using a Monolight 3010 luminometer (Analytical Luminescence Laboratory, Ann Arbor, MI). The results of three independent experiments are presented as a ratio of TOP to FOP and were normalized to Renilla luciferase activity. Statistical significance was determined by t test.
Real Time RT-PCR assays
Total RNA was isolated from cells using TRIzol reagent (Invitrogen) according to manufacturer's instructions. Reverse transcription of equal amounts of total RNA was performed using the SuperScript III first-strand synthesis system with random hexamer primers (Invitrogen). Real-time PCR was carried out in triplicate after reverse transcription using the TaqMan Gene Expression assay specific for AXIN2 (assay ID AXIN2 - Mm00443610_m1, Hs01063168_m1, Applied Biosystems, Foster City, CA). Fluorescence was detected using an ABI Prism 7900HT real-time PCR system and normalized using a TaqMan primer for eukaryotic 18S rRNA endogenous control (Applied Biosystems). The relative change in mRNA expression was calculated using the ΔΔCt method. The results are reported as fold change of at least three independent experiments. Statistical significance was determined by t test for 308 cells and NHEKs, and by two-way ANOVA for COX-2 WT and KO MEFs.
Immunohistochemistry
Skin tissues were fixed in 10% neutral-buffered formalin, processed for histology and embedded lengthwise in paraffin. Sections (5 μm) were stained for β-catenin (clone 2H4A7) or COX-2 (Cayman Chemical) overnight. The bound antibody was visualized using the DAKO EnVison + System-HRP (Dako, Carpinteria, CA) for use with mouse or rabbit primary antibodies.
Results
UVB induces β-catenin signaling in keratinocytes in vitro
To investigate the effect of UVB radiation on β-catenin signaling, we exposed mouse 308 keratinocytes (308 cells), a cell line derived from dimethylbenz[α]anthracene-initiated BALB/c mouse skin [17], or primary normal human epidermal keratinocytes (NHEKs), isolated from neonatal foreskin [18], to UVB radiation. We examined the expression of both N-terminally unphosphorylated and total β-catenin by Western blot analysis. To examine the expression of the transcriptionally active form of β-catenin (N-terminally unphosphorylated β-catenin), we used an antibody which specifically recognizes Ser37 and Thr41 of β-catenin in the unphosphorylated state [15]. UVB radiation of 308 cells resulted in increased N-terminally unphosphorylated β-catenin levels 8 hours after exposure to 500 J/m2 UVB, as well as a slight increase in total β-catenin levels (Figure 1A and B). NHEKs demonstrated increased N-terminally unphosphorylated β-catenin levels 8 hours after exposure to 250, 500, and 1000 J/m2 UVB (Figure 1D and E). Total β-catenin levels were more robustly increased in NHEKs than in 308 cells following UVB exposure (Figure 1D and E). Therefore, exposure to UVB radiation resulted in increased protein levels of both N-terminally unphosphorylated and total β-catenin in both 308 cells and in NHEKs (Figure 1). In order to account for variance between experiments, we calculated the mean fold change and determined that the increase in N-terminally unphosphorylated β-catenin was statistically significant 8 hours after 500 J/m2 UVB in both 308 cells and NHEKs (Figure 1C and F). Additionally, the mean fold change in total β-catenin levels in NHEKS was statistically significant at 8 hours after 250 and 500 J/m2 UVB (Figure 1F). Based on these results, we chose to use a dose of 500 J/m2 UVB for conducting all subsequent experiments.
β-catenin is a dual-function protein which can interact with cadherin-type adhesion receptors at the cell surface, while the cadherin-independent pool of β-catenin is involved in transcription of target genes that control cell proliferation [14]. In order to determine if the UVB-induced accumulation of β-catenin could enhance TCF-mediated transcriptional activity, we used the β-catenin/TCF-dependent gene reporter system, TOPflash. 308 cells exposed to UVB exhibited a nearly 2-fold increase in TOPflash luciferase activity compared to sham-irradiated cells, demonstrating that UVB irradiation induces β-catenin signaling (Figure 2A). To further confirm that UVB irradiation activates the expression of β-catenin-responsive downstream genes, we next examined the expression of AXIN2, a β-catenin target gene, in response to UVB radiation. AXIN2 is an endogenous β-catenin target gene and negative regulator of the β-catenin signaling pathway [20], which has been used as a read out of Wnt/β-catenin signaling. Cultures of 308 cells and NHEKs were irradiated with 500 J/m2 UVB and harvested after 8 hours. Real time RT-PCR analysis demonstrated that exposure to UVB significantly upregulated AXIN2 mRNA expression in both 308 cells and NHEKs (Figure 2B). Taken together, our data confirm that UVB radiation induces accumulation of β-catenin and enhances β-catenin signaling and downstream target gene expression.
Figure 2. UVB induces β-catenin/TCF-dependent transcriptional activity in keratinocytes.
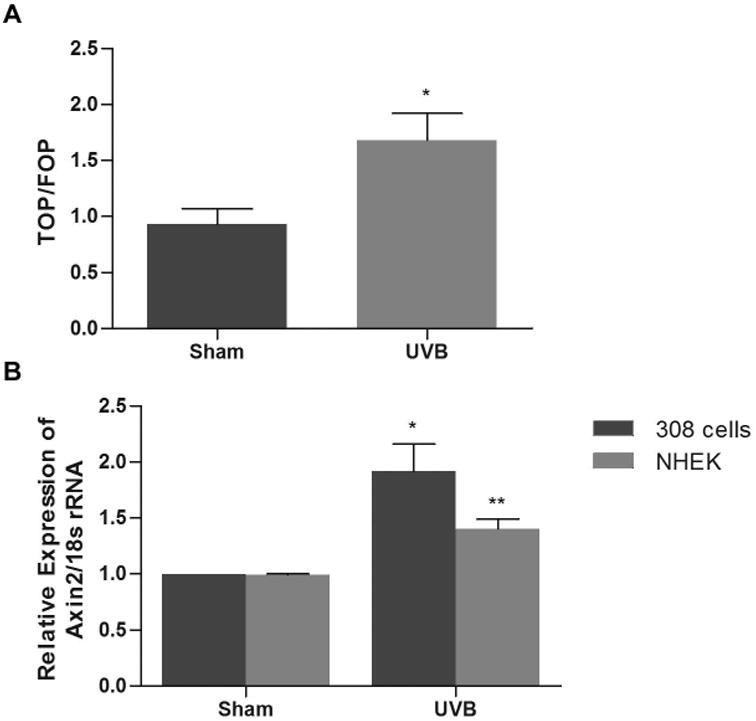
(A) The dual luciferase assay was performed on 308 cells which were transiently transfected with TOP- or FOPflash reporter constructs and Renilla and 24 hrs later sham irradiated or exposed to 500 J/m2 UVB (8 hrs). Results are mean ratio of TOPflash luciferase activity to FOPflash luciferase activity (TOP/FOP) from three independent experiments ± SEM. (B) 308 cells and NHEKs were exposed to sham or 500 J/m2 UVB and incubated for 8 hrs. Total RNA was harvested and real time RT-PCR was performed. Results are the mean of at least three independent experiments ± SEM. *, P < 0.02; **, P < 0.01.
UVB-induced β-catenin signaling in keratinocytes is mediated by COX-2 expression
A link between COX-2 and β-catenin has been established in colon cancer in which the product of COX-2 expression, PGE2, stimulates cancer cell growth through β-catenin signaling [12]. We show in Figure 1, consistent with previous studies by our lab and others [10,21,22], that UVB exposure stimulates COX-2 protein expression in both 308 cells and NHEKs. We next determined if COX-2 expression induced by UVB radiation could enhance UVB-induced β-catenin signaling in keratinocytes. We pre-treated cultures of 308 cells with multiple different doses of NS398, a COX-2 specific inhibitor, and exposed them to UVB. To ensure that NS398 inhibited the activity of COX-2, we examined the production of PGE2 by 308 cells treated with NS398. The UVB-induced COX-2-dependent increase in PGE2 production was inhibited by treatment with NS398 (Figure 3A). NS398 also inhibited the UVB-induced increase in both N-terminally unphosphorylated and total β-catenin protein levels in 308 cells (Figure 3B). To determine if COX-2 mediates UVB-induced β-catenin signaling, we investigated the ability of UVB to induce expression of the β-catenin target gene AXIN2 in COX-2 KO MEFs. Exposure to UVB resulted in increased AXIN2 expression in both COX-2 WT and COX-2 KO MEFs (Figure 3C). Importantly, UVB treated COX-2 KO MEFs had significantly less AXIN2 expression than UVB treated COX-2 WT MEFs, confirming that COX-2 plays a significant role in UVB-induced β-catenin target gene expression (Figure 3C). These data demonstrate that inhibition of COX-2, either by a chemical inhibitor or by genetic manipulation, resulted in substantial reduction in β-catenin signaling following UVB irradiation. These findings provide strong evidence that COX-2 modulates UVB-induced β-catenin accumulation and β-catenin signaling.
Figure 3. COX-2 modulates UVB-induced β-catenin signaling.
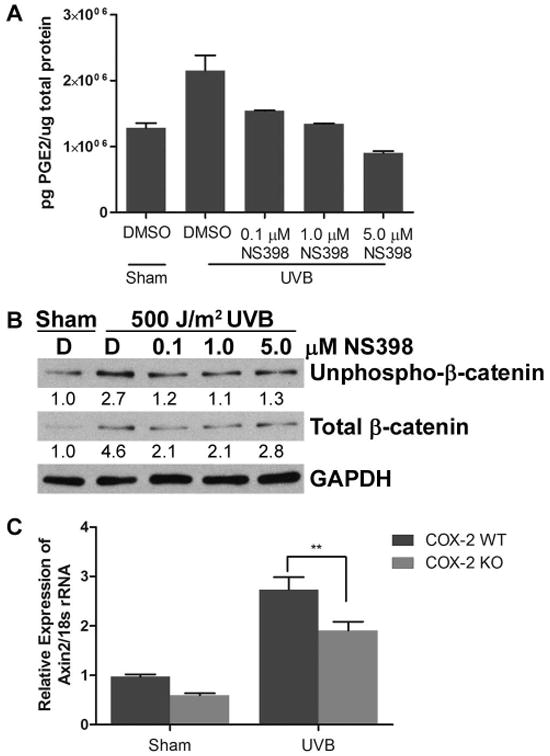
(A, B) 308 cells were pretreated with DMSO vehicle control [D] or the indicated doses of NS398 for 1 hr. Cells were then sham irradiated or exposed to 500 J/m2 UVB and incubated for 8 hrs. (A) Cell culture media was collected and PGE2 levels were measured by competitive ELISA. (B) Protein lysates were probed for unphospho-β-catenin, total β-catenin and GAPDH. Representative blots are shown and numbers indicate fold change of proteins normalized to GAPDH. (C) COX-2 WT and KO MEFs were sham irradiated or exposed to 500 J/m2 UVB and incubated for 8 hrs. Real time RT-PCR was performed to determine expression levels of AXIN2 mRNA. **, P < 0.01.
The PGE2 receptor, EP2, mediates UVB-induced β-catenin signaling
PGE2 is the major COX-2 derived prostaglandin implicated in cancer [23], and COX-2 is the rate limiting enzyme in the production of PGE2. PGE2 has been shown to promote colon cancer cell growth through β-catenin signaling, in the absence of a functional APC [12]. We show in Figure 4 that treatment of NHEKs with exogenous PGE2 resulted in increased accumulation of both N-terminally unphosphorylated and total β-catenin (Figure 4A). PGE2 acts by binding to one of four E prostanoid receptors (EP1-4), and EP2 has been shown to be important in both UVB-induced and chemically-induced skin carcinogenesis [24,25]. We next investigated the involvement of the EP2 receptor in UVB-induced β-catenin signaling in keratinocytes by pre-treating keratinocytes with AH6809, an EP2 antagonist. In both 308 cells and NHEKs, pre-treatment with AH6809 prevented UVB-induced accumulation of N-terminally unphosphorylated and total β-catenin (Figure 4B and C). Together, these results provide evidence that the EP2 receptor is involved in UVB-induced β-catenin signaling.
Figure 4. Accumulation of β-catenin induced by UVB is mediated by the EP2 receptor.

(A) NHEKs were treated with DMSO vehicle control or the indicated doses of PGE2 for 15 mins and cell lysates were immunoblotted with antibodies for the indicated proteins. (B) 308 cells and (C) NHEKs were pre-treated with DMSO vehicle control [D] or the indicated doses of AH6809. Cells were then sham irradiated or exposed to 500 J/m2 UVB and incubated for 8 hrs. Cell lysates were analyzed by immunoblotting for unphospho-β-catenin, total β-catenin and GAPDH. Representative blots are shown and numbers indicate fold change of proteins normalized to GAPDH.
UVB radiation disrupts the β-catenin phosphorylation/destruction complex
In the absence of a Wnt signal, β-catenin is recruited to a phosphorylation/destruction complex that consists of APC, Axin and GSK3β, which facilitates the phosphorylation of β-catenin by GSK3β leading to its ubiquitinization and proteasomal degradation [14]. The EP2 receptor is coupled to G proteins of the Gαs family and previous studies have shown that after activation by PGE2, Gαs directly binds Axin resulting in the release of GSK3β [12]. To determine if Gαs was involved in UVB-induced β-catenin signaling, we treated 308 cells with melittin, a Gαs specific inhibitor [26]. Inhibition of Gαs by melittin prevented UVB-induced accumulation of N-terminally unphosphorylated and total β-catenin in 308 cells, demonstrating that the EP2 receptor is involved in UVB-induced activation of β-catenin signaling (Figure 5A).
Figure 5. UVB disrupts the β-catenin phosphorylation/destruction complex.

(A) 308 cells were pre-treated with 0.25 μM melittin for 1 hr and then sham irradiated or exposed to 500 J/m2 UVB for 8 hrs. (B) 308 cells were sham irradiated or exposed to 250, 500, or 1000 J/m2 UVB and incubated for 8 hrs. Cell lysates were subjected to immunoblot analysis with antibodies specific for the indicated proteins. Representative blots are shown and numbers indicate fold change of proteins normalized to GAPDH. (C) 308 cells were sham irradiated or exposed to 500 J/m2 UVB for 8 hrs. Cell lysates were immunoprecipitated with an antibody against Axin and probed for GSK3β by Western blot. WCL, whole cell lysate.
GSK3β is a target of Akt, and UVB has been shown to activate the PI3K/Akt pathway [22]. Additionally, after activation of EP2 by PGE2, the Gβγ subunits activate the PI3K/Akt pathway [12]. Exposure of 308 cells to UVB resulted in increased phospho-Akt, as well as increased phosphorylation of GSK3β at Ser9 (Figure 5B). Phosphorylation of GSK3β at Ser9 inhibits its kinase activity, thereby preventing GSK3β from being able to phosphorylate β-catenin in the phosphorylation/destruction complex, thus preventing β-catenin degradation. To determine the status of the phosphorylation/destruction complex after UVB exposure, we performed co-immunoprecipitation studies in 308 cells with an anti-Axin antibody after exposure to UVB. UVB radiation strongly inhibited the co-immunoprecipitation of GSK3β with Axin illustrating that UVB exposure promotes the release of GSK3β from the phosphorylation/destruction complex (Figure 5C). Collectively, our observations suggest that UVB induces phosphorylation of Akt and GSK3β via activation of the G protein coupled to the EP2 receptor, which leads to stabilization of β-catenin and disruption of the phosphorylation/destruction complex.
UVB induces β-catenin accumulation in vivo
To determine if UVB induces β-catenin signaling in vivo, we examined β-catenin accumulation in SKH-1 mice exposed to UVB. SKH-1 WT mice showed increased accumulation of both N-terminally unphosphorylated and total β-catenin at 12 hours post UVB, confirming our in vitro data (Figure 6A). Similarly, immunohistochemical analysis demonstrated increased total β-catenin staining after UVB exposure at 12 hours (Figure 6B). To examine whether UVB-induced β-catenin accumulation in vivo is associated with COX-2 expression, we exposed SKH-1 COX-2 WT and COX-2 +/- heterozygote mice to UVB. COX-2 -/- KO mice could not be used for these experiments because they only survive for a short time after birth [11]. Western blot analysis of lysates from mouse epidermis demonstrate that the UVB-induced accumulation of N-terminally unphosphorylated and total β-catenin was diminished in UVB irradiated COX-2 +/- heterozygous mice as compared to UVB irradiated WT mice (Figure 6A). In agreement with these findings, staining for total β-catenin was also reduced in COX-2 +/- heterozygous mice compared with COX-2 WT mice (Figure 6B). We also examined COX-2 expression in both SKH-1 WT and COX-2 +/- heterozygous mice. COX-2 expression is increased in both SKH-1 WT and COX-2 +/- heterozygous mice after UVB exposure (Figure 6C). Previous studies have demonstrated increased PGE2 production in COX-2 +/- heterozygous mice following UVB exposure; however, the level of UVB-induced PGE2 production in COX-2 +/- heterozygous mice is significantly less than the level of UVB-induced PGE2 production in SKH-1 WT mice [11]. These data confirm our in vitro results, and we conclude that COX-2 expression contributes to the UVB-induced accumulation of N-terminally unphosphorylated and total β-catenin in vivo.
Figure 6. Expression of β-catenin in COX-2 WT and COX-2 +/- mice.
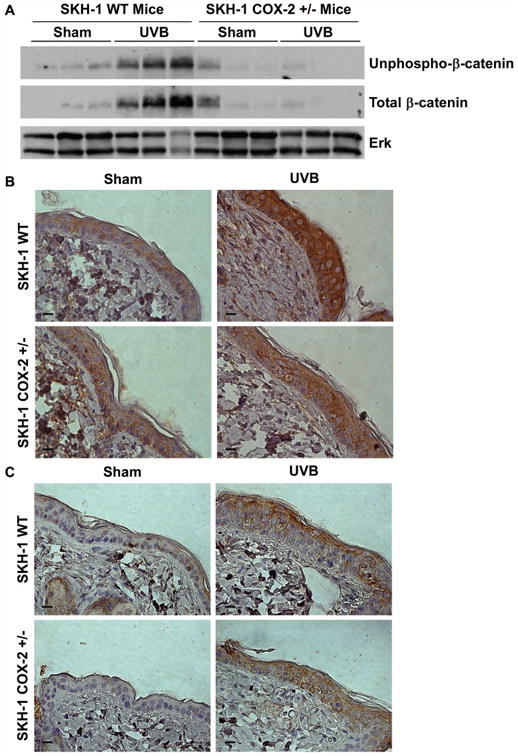
UVB-induced β-catenin accumulation is inhibited in COX-2 +/- mice. SKH-1 WT mice and SKH-1 COX-2 +/- mice were sham irradiated or exposed to 2200 J/m2 UVB and sacrificed 12 hrs post-UVB. (A) Cell lysates from the epidermis were immunoblotted for unphospho-β-catenin, total β-catenin, and Erk as a loading control. Each lane represents a single animal. Dorsal epidermis was stained for (B) total β-catenin or (C) COX-2. Scale bar = 10 μm.
Discussion
UVB radiation has been identified as the major carcinogen responsible for skin cancer [2,3]. Therefore, understanding the signaling pathways activated by UVB in keratinocytes is essential for identification of targets for better therapies and/or prevention of skin cancer. Upregulation of COX-2 by UVB, and the subsequent elevation in PGE2, have both been shown to be critical for murine skin carcinogenesis [11]. Previous work by Castellone and coworkers identified a connection between COX-2 and β-catenin by demonstrating that PGE2 can promote the growth of colon cancer cells by activating β-catenin signaling [12]. Little is known about the connection between COX-2 and β-catenin in the skin. In colon cancer cells, COX-2 is constitutively expressed and the tumor suppressor APC is non-functional [12]. In this report, we provide evidence that acute UVB exposure induces β-catenin signaling through upregulation of COX-2 in both human and mouse keratinocytes in vitro and in mouse skin in vivo in the presence of an intact β-catenin signaling pathway. A schematic of the proposed relationship between UVB, COX-2 and β-catenin is shown in Figure 7. The data presented herein provide evidence that COX-2 expression modulates UVB-induced β-catenin signaling in multiple in vitro cell lines and in a relevant in vivo model (Figure 6). Our work presented here is the first to demonstrate the activation of β-catenin signaling in response to UVB in keratinocytes.
Figure 7. UVB induces β-catenin signaling in keratinocytes.
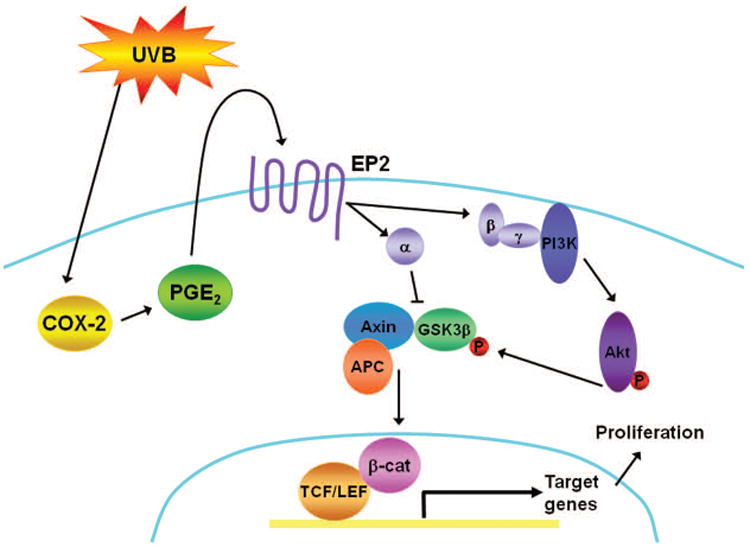
Our proposed pathway: UVB exposure leads to increased COX-2 expression which results in increased PGE2 production. PGE2 binds and activates its G-protein coupled receptor, EP2. Upon the binding of PGE2, Gαs binds to Axin resulting in the release of GSK3β from the phosphorylation/destruction complex. Concurrently, the Gβγ subunits stimulate Akt, through PI3K, leading to the phosphorylation and inactivation of GSK3β. This allows for stabilization and accumulation of β-catenin which translocates to the nucleus and activates transcription of target genes, such as AXIN2. Activation of β-catenin signaling could contribute to the proliferative response of keratinocytes to UVB radiation.
Hildesheim and coworkers previously reported that Gadd45α suppresses β-catenin signaling by maintaining p38 MAPK activation after UV radiation by promoting activation of GSK3β following UVB exposure leading to degradation of β-catenin [27]. Our results, however, differ with this study. We show inactivation of GSK3β by increased phosphorylation at Ser9 (Figure 5B), disruption of the GSK3β/Axin interaction (Figure 5B), increased β-catenin signaling by a reporter assay (Figure 2A), as well as increased expression of a β-catenin target gene following UVB exposure (Figure 2B). In accordance with our data, Bikkavillli et. al. show that activation of p38 MAPK activates Wnt-β-catenin signaling by inactivating GSK3β [28]. Additionally, previous work on keratinocytes and UVA exposure, which is considerably less energetic than UVB, has shown that UVA can cause the disassociation of β-catenin from E-cadherin which enhances keratinocyte invasiveness.
UVB exposure causes DNA damage and apoptosis in the skin [29]; however, there is also a proliferative response. UVB induces survival mechanisms and proliferation by activating various growth factors [30,31]. UVB-induced activation of the epidermal growth factor receptor (EGFR) is mediated by PGE2 and has been shown to be critical for keratinocyte survival and proliferation [31,32]. EGFR activation has been shown to stimulate β-catenin signaling by promoting disassociation of β-catenin from α-catenin and enhancing β-catenin transactivation [33]. Additionally, phosphorylation of β-catenin at Ser552 by Akt enables some β-catenin to disassociate from cell-cell contacts and accumulate in the nucleus [34]. In view of the fact that UVB radiation leads to activation of EGFR and Akt, both of which have been shown to activate β-catenin signaling leading to proliferation, these could be additional pathways by which UVB induces β-catenin signaling. Activation of these other signaling pathways could explain why inhibition of COX-2 only partially inhibited UVB-induced β-catenin signaling (Figure 3).
In addition to survival and proliferation in vitro, UVB induces epidermal hyperplasia in mice and COX-2 expression has been shown to contribute to this proliferative response [11]. PGE2 has been identified as the link between overexpression of COX-2 and cell proliferation in the mouse skin [35]. COX-2 has been shown to inhibit UVB-induced apoptosis in mouse skin via the PGE2 receptor EP2 [36]. A recent study by Chun and co-workers has shown that EP2 regulates the expression of survivin in UVB-exposed mouse skin via an EGFR pathway [37]. Survivin, a β-catenin target gene, is a member of the inhibitor of apoptosis (IAP) family and could be a mechanism by which COX-2 and EP2 protect against UVB-induced apoptosis [37]. The data presented here provide evidence that β-catenin signaling could be the link between UVB and COX-2/PGE2 and cell proliferation. Given that many signaling pathways activated in response to UVB can lead to increased β-catenin signaling and that activation of β-catenin signaling leads to cell survival and proliferation, it is likely that β-catenin signaling contributes to the proliferative response of keratinocytes to UVB.
As well as being a major player in the Wnt signaling pathway, β-catenin is also found at the cell membrane in the adherens junction where it interacts with E-cadherin. UVB has been shown to cleave E-cadherin ([38] and our unpublished observation) and loss of membranous E-cadherin has been shown in human squamous cell carcinoma (SCC) [39,40]. These events could contribute to the accumulation of β-catenin in the cytoplasm in response to UVB radiation. Loss of membranous β-catenin expression and nuclear/cytoplasmic localization of β-catenin have also been demonstrated in human SCC [39,41]. While it has been shown that β-catenin is not required for proliferation and differentiation of keratinocytes in normal skin homeostasis [42], there is evidence that β-catenin signaling contributes to chemically-induced skin cancer. In two-stage chemical skin carcinogenesis in mice, activation of β-catenin signaling as indicated by nuclear β-catenin staining was seen [43] and cutaneous cancer stem cell maintenance has been shown to be dependent on β-catenin signaling [44]. These findings, in combination with our data, support our hypothesis that UVB-induced β-catenin signaling contributes to keratinocyte proliferation and skin carcinogenesis.
Altogether, these data presented herein are the first to identify a novel pathway in which UVB induces β-catenin signaling in mouse and human keratinocytes in vitro and mouse skin in vivo. The activation of β-catenin signaling in keratinocytes could contribute to the proliferative response of cells to UVB exposure and to skin carcinogenesis. With this report, we provide further understanding of the complex response of skin to UVB radiation with the goal of providing new targets for the development of better treatment and prevention strategies for skin cancer.
Acknowledgments
Grant Support: This work was supported by NIH grant CA104768 and AR057579 to J.C.P., the Zell Foundation (J.C.P was a Zell Scholar), NIH T32 grant CA09560 to K.A.S., a Malkin Scholars Award to K.A.S, in part by the Robert H. Lurie Comprehensive Cancer Center and in part by resources provided by the Northwestern Skin Disease Research Center 5P30AR057216, Chicago, IL with support from the NIH/NIAMS.
Abbreviations
- APC
adenomatous polyposis coli
- COX-2
cyclooxygenase-2
- GSK3β
glycogen synthase kinase 3β
- MEFs
mouse embryo fibroblasts
- NHEKs
normal human epidermal keratinocytes
- PGE2
prostaglandin E2
References
- 1.Society AC. Cancer Facts & Figures 2010. Atlanta: American Cancer Society; 2010. [Google Scholar]
- 2.Miller DL, Weinstock MA. Nonmelanoma skin cancer in the United States: incidence. J Am Acad Dermatol. 1994;30(5 Pt 1):774–778. doi: 10.1016/s0190-9622(08)81509-5. [DOI] [PubMed] [Google Scholar]
- 3.Urbach F. Incidence of nonmelanoma skin cancer. Dermatol Clin. 1991;9(4):751–755. [PubMed] [Google Scholar]
- 4.Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88(22):10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gailani MR, Leffell DJ, Ziegler A, Gross EG, Brash DE, Bale AE. Relationship between sunlight exposure and a key genetic alteration in basal cell carcinoma. J Natl Cancer Inst. 1996;88(6):349–354. doi: 10.1093/jnci/88.6.349. [DOI] [PubMed] [Google Scholar]
- 6.Hussein MR. Ultraviolet radiation and skin cancer: molecular mechanisms. J Cutan Pathol. 2005;32(3):191–205. doi: 10.1111/j.0303-6987.2005.00281.x. [DOI] [PubMed] [Google Scholar]
- 7.de Gruijl FR. Skin cancer and solar UV radiation. Eur J Cancer. 1999;35(14):2003–2009. doi: 10.1016/s0959-8049(99)00283-x. [DOI] [PubMed] [Google Scholar]
- 8.Brash DE. Roles of the transcription factor p53 in keratinocyte carcinomas. Br J Dermatol. 2006;154(Suppl 1):8–10. doi: 10.1111/j.1365-2133.2006.07230.x. [DOI] [PubMed] [Google Scholar]
- 9.Bode AM, Dong Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci STKE. 2003;2003(167):RE2. doi: 10.1126/stke.2003.167.re2. [DOI] [PubMed] [Google Scholar]
- 10.Buckman S, Gresham A, Hale P, et al. COX-2 expression is induced by UVB exposure in human skin: implications for the development of skin cancer. Carcinogenesis. 1998;19(5):723–729. doi: 10.1093/carcin/19.5.723. [DOI] [PubMed] [Google Scholar]
- 11.Fischer SM, Pavone A, Mikulec C, Langenbach R, Rundhaug JE. Cyclooxygenase-2 expression is critical for chronic UV-induced murine skin carcinogenesis. Mol Carcinog. 2007;46(5):363–371. doi: 10.1002/mc.20284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310(5753):1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 13.Fuchs SY, Ougolkov AV, Spiegelman VS, Minamoto T. Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle. 2005;4(11):1522–1539. doi: 10.4161/cc.4.11.2129. [DOI] [PubMed] [Google Scholar]
- 14.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5(9):691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 15.Staal FJ, Noort Mv M, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep. 2002;3(1):63–68. doi: 10.1093/embo-reports/kvf002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 17.Strickland JE, Greenhalgh DA, Koceva-Chyla A, et al. Development of Murine Epidermal Cell Lines Which Contain an Activated rasHa Oncogene and Form Papillomas in Skin Grafts on Athymic Nude Mouse Hosts. Cancer Res. 1988;48(1):165–169. [PubMed] [Google Scholar]
- 18.Abu-Yousif AO, Smith KA, Getsios S, Green KJ, Van Dross RT, Pelling JC. Enhancement of UVB-induced apoptosis by apigenin in human keratinocytes and organotypic keratinocyte cultures. Cancer Res. 2008;68(8):3057–3065. doi: 10.1158/0008-5472.CAN-07-2763. [DOI] [PubMed] [Google Scholar]
- 19.Langenbach R, Morham SG, Tiano HF, et al. Prostaglandin synthase 1 gene disruption in mice reduces arachidonic acid-induced inflammation and indomethacin-induced gastric ulceration. Cell. 1995;83(3):483–492. doi: 10.1016/0092-8674(95)90126-4. [DOI] [PubMed] [Google Scholar]
- 20.Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22(4):1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong X, Van Dross RT, Abu-Yousif A, Morrison AR, Pelling JC. Apigenin prevents UVB-induced cyclooxygenase 2 expression: coupled mRNA stabilization and translational inhibition. Mol Cell Biol. 2007;27(1):283–296. doi: 10.1128/MCB.01282-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tang Q, Gonzales M, Inoue H, Bowden GT. Roles of Akt and glycogen synthase kinase 3beta in the ultraviolet B induction of cyclooxygenase-2 transcription in human keratinocytes. Cancer Res. 2001;61(11):4329–4332. [PubMed] [Google Scholar]
- 23.Wang D, DuBois RN. Prostaglandins and Cancer. Gut. 2006;55(1):115–122. doi: 10.1136/gut.2004.047100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brouxhon S, Konger RL, VanBuskirk J, et al. Deletion of Prostaglandin E2 EP2 Receptor Protects against Ultraviolet-Induced Carcinogenesis, but Increases Tumor Aggressiveness. 2006;127(2):439–446. doi: 10.1038/sj.jid.5700547. [DOI] [PubMed] [Google Scholar]
- 25.Sung YM, He G, Hwang DH, Fischer SM. Overexpression of the prostaglandin E2 receptor EP2 results in enhanced skin tumor development. 2006;25(40):5507–5516. doi: 10.1038/sj.onc.1209538. [DOI] [PubMed] [Google Scholar]
- 26.Fukushima N, Kohno M, Kato T, et al. Melittin, a metabostatic peptide inhibiting Gs activity. Peptides. 1998;19(5):811–819. doi: 10.1016/s0196-9781(98)00027-8. [DOI] [PubMed] [Google Scholar]
- 27.Hildesheim J, Belova GI, Tyner SD, Zhou X, Vardanian L, Fornace AJ., Jr Gadd45a regulates matrix metalloproteinases by suppressing DeltaNp63alpha and beta-catenin via p38 MAP kinase and APC complex activation. Oncogene. 2004;23(10):1829–1837. doi: 10.1038/sj.onc.1207301. [DOI] [PubMed] [Google Scholar]
- 28.Bikkavilli RK, Feigin ME, Malbon CC. p38 mitogen-activated protein kinase regulates canonical Wnt-beta-catenin signaling by inactivation of GSK3beta. J Cell Sci. 2008;121(Pt 21):3598–3607. doi: 10.1242/jcs.032854. [DOI] [PubMed] [Google Scholar]
- 29.Matsumura Y, Ananthaswamy HN. Toxic effects of ultraviolet radiation on the skin. Toxicol Appl Pharmacol. 2004;195(3):298–308. doi: 10.1016/j.taap.2003.08.019. [DOI] [PubMed] [Google Scholar]
- 30.Lu YP, Lou YR, Yen P, Mitchell D, Huang MT, Conney AH. Time course for early adaptive responses to ultraviolet B light in the epidermis of SKH-1 mice. Cancer Res. 1999;59(18):4591–4602. [PubMed] [Google Scholar]
- 31.Peus D, Vasa RA, Meves A, Beyerle A, Pittelkow MR. UVB-induced epidermal growth factor receptor phosphorylation is critical for downstream signaling and keratinocyte survival. Photochem Photobiol. 2000;72(1):135–140. doi: 10.1562/0031-8655(2000)072<0135:uiegfr>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 32.El-Abaseri TB, Putta S, Hansen LA. Ultraviolet irradiation induces keratinocyte proliferation and epidermal hyperplasia through the activation of the epidermal growth factor receptor. Carcinogenesis. 2006;27(2):225–231. doi: 10.1093/carcin/bgi220. [DOI] [PubMed] [Google Scholar]
- 33.Ji H, Wang J, Nika H, et al. EGF-induced ERK activation promotes CK2-mediated disassociation of alpha-Catenin from beta-Catenin and transactivation of beta-Catenin. Mol Cell. 2009;36(4):547–559. doi: 10.1016/j.molcel.2009.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fang D, Hawke D, Zheng Y, et al. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282(15):11221–11229. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ansari KM, Rundhaug JE, Fischer SM. Multiple signaling pathways are responsible for prostaglandin E2-induced murine keratinocyte proliferation. Mol Cancer Res. 2008;6(6):1003–1016. doi: 10.1158/1541-7786.MCR-07-2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chun KS, Akunda JK, Langenbach R. Cyclooxygenase-2 inhibits UVB-induced apoptosis in mouse skin by activating the prostaglandin E2 receptors, EP2 and EP4. Cancer Res. 2007;67(5):2015–2021. doi: 10.1158/0008-5472.CAN-06-3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chun KS, Langenbach R. The prostaglandin E(2) receptor, EP2, regulates survivin expression via an EGFR/STAT3 pathway in UVB-exposed mouse skin. Mol Carcinog. 2011 doi: 10.1002/mc.20728. [DOI] [PubMed] [Google Scholar]
- 38.Hung CF, Chiang HS, Lo HM, Jian JS, Wu WB. E-cadherin and its downstream catenins are proteolytically cleaved in human HaCaT keratinocytes exposed to UVB. Exp Dermatol. 2006;15(4):315–321. doi: 10.1111/j.0906-6705.2006.00411.x. [DOI] [PubMed] [Google Scholar]
- 39.Papadavid E, Pignatelli M, Zakynthinos S, Krausz T, Chu AC. Abnormal immunoreactivity of the E-cadherin/catenin (alpha-, beta-, and gamma-) complex in premalignant and malignant non-melanocytic skin tumours. J Pathol. 2002;196(2):154–162. doi: 10.1002/path.1019. [DOI] [PubMed] [Google Scholar]
- 40.Lyakhovitsky A, Barzilai A, Fogel M, Trau H, Huszar M. Expression of e-cadherin and beta-catenin in cutaneous squamous cell carcinoma and its precursors. Am J Dermatopathol. 2004;26(5):372–378. doi: 10.1097/00000372-200410000-00005. [DOI] [PubMed] [Google Scholar]
- 41.Brasanac D, Boricic I, Todorovic V, Tomanovic N, Radojevic S. Cyclin A and beta-catenin expression in actinic keratosis, Bowen's disease and invasive squamous cell carcinoma of the skin. Br J Dermatol. 2005;153(6):1166–1175. doi: 10.1111/j.1365-2133.2005.06898.x. [DOI] [PubMed] [Google Scholar]
- 42.Posthaus H, Williamson L, Baumann D, et al. beta-Catenin is not required for proliferation and differentiation of epidermal mouse keratinocytes. J Cell Sci. 2002;115(Pt 23):4587–4595. doi: 10.1242/jcs.00141. [DOI] [PubMed] [Google Scholar]
- 43.Bhatia N, Spiegelman VS. Activation of Wnt/beta-catenin/Tcf signaling in mouse skin carcinogenesis. Mol Carcinog. 2005;42(4):213–221. doi: 10.1002/mc.20077. [DOI] [PubMed] [Google Scholar]
- 44.Malanchi I, Peinado H, Kassen D, et al. Cutaneous cancer stem cell maintenance is dependent on beta-catenin signalling. Nature. 2008;452(7187):650–653. doi: 10.1038/nature06835. [DOI] [PubMed] [Google Scholar]