

Abstract
Acute endothelial cell apoptosis and microvascular compromise couple GI tract irradiation to reproductive death of intestinal crypt stem cell clonogens (SCCs) following high-dose radiation. Genetic or pharmacologic inhibition of endothelial apoptosis prevents intestinal damage, but as the radiation dose is escalated, SCCs become directly susceptible to an alternate cell death mechanism, mediated via ceramide synthase (CS)-stimulated de novo synthesis of the pro-apoptotic sphingolipid ceramide, and p53-independent apoptosis of crypt SCCs. We previously reported that ATM deficiency resets the primary radiation lethal pathway, allowing CS-mediated apoptosis at the low-dose range of radiation. The mechanism for this event, termed target reordering, remains unknown. Here we show that inactivation of DNA damage repair pathways signal CS-mediated apoptosis in crypt SCCs, presumably via persistent unrepaired DNA double strand breaks (DSBs). Genetic loss-of-function of sensors and transducers of DNA DSB repair confers the CS-mediated lethal pathway in intestines of sv129/B6Mre11ATLD1/ATLD1 and C57BL/6Prkdc/SCID (SCID) mice exposed to low-dose radiation. In contrast, CS-mediated SCC lethality was mitigated in irradiated gain-of-function Rad50S/S mice, and epistasis studies order Rad50 upstream of Mre11. These studies suggest unrepaired DNA DSBs as causative in target re-ordering in intestinal SCCs. As such, we provide an in vivo model of DNA damage repair that is standardized, can be exploited to understand allele-specific regulation in intact tissue, and is pharmacologically tractable.
Keywords: Gastrointestinal tract, Crypt, Radiation, Gastrointestinal syndrome, Bone marrow syndrome
INTRODUCTION
Permanent damage to intestinal mucosa requires depletion of the crypt stem cells and early undifferentiated progenitors (termed here stem cell clonogens, or SCCs), located at positions 6–9 from the bottom of the crypt of Lieberkühn(1, 2). When exposed to 8–16Gy whole body radiation (WBR), intestinal SCC lethality occurs via reproductive cell death(2), linked to and regulated by an early wave of acid sphingomyelinae (ASMase)-dependent endothelial apoptosis and ensuing dysfunction of the crypt microvascular network(3, 4). Reproductive cell death is generic to irradiated mammalian cells, resulting from unrepaired or misrepaired DNA double strand breaks (DSBs), which confer genomic instability and post-replication generation of mutations and chromosomal aberrations, eventually leading to death of injured progeny(5).
DNA damage initiates a coordinated signaling network that senses exposed DSBs, arrests cell cycle progression, and either activates DSB repair via non homologous end joining (NHEJ) or homologous recombination (HR)(6), or signals cell death(7). Regulation of pathway choice is unknown, nor is there evidence that the DNA damage signaling network distinguishes between reparable and irreparable DSBs. Evidence suggests that in the early phase repair is preferentially expressed(7), while DSB persistence, when cellular repair capacity is exceeded or dysregulated, eventually confers cell death. Consistent with this latter notion, although HR is significantly enhanced when NHEJ is suppressed pharmacologically or genetically(8), NHEJ-deficient cells are nonetheless radiation hypersensitive(9, 10), resulting from residual unrepaired DSBs, as HR incompletely complements NHEJ deficiency.
While DSB-mediated reproductive cell death is primary in radiation lethality in crypt SCCs, some tissues employ apoptosis in pathogenesis of tissue damage. The most heralded apoptotic death pathway signaled by IR-induced DSBs is mediated via p53, which in C. elegans germ cells involves the sphingolipid ceramide during the mitochondrial phase of the apoptotic cascade(11). Ceramide is metabolically produced either via sphingomyelinase-mediated hydrolysis of sphingomyelin, or via ceramide synthase (CS)-mediated de novo synthesis(12, 13). In irradiated C. elegans germ cells, radiation activates simultaneously the ceramide synthetic pathway via CS, and independently the CEP-1/p53–EGL-1 (BH3-only protein) pathway via DNA damage-activated cell cycle checkpoint genes(11, 14). These pathways converge on mitochondrial membranes, where a rise in ambient ceramide regulates EGL-1-mediated displacement of CED-4 (homolog of Apaf-1) from the CED-4:CED-9 (Bcl-2 homolog) complex, effecting cytosolic CED-3 caspase activation and apoptosis(11). The mitochondrial response in C. elegans germ cells is equivalent to induction of mitochondrial outer membrane permeabilization (MOMP) and cytochrome c release in mammalian systems, which irreversibly commits cells to the final effector phase of apoptosis.
CS engagement in mammalian apoptosis is cell-type specific in response to ionizing radiation (IR), drugs, and cytokines(15–18). An extensive literature has accumulated regarding signaling of apoptotic cell death through CS. As opposed to ceramide generation at the plasma membrane via acid sphingomyelinase (ASMase), signaling through CS is delayed in almost every system defined to date. For example, ceramide generation and CS activity in bovine aortic endothelial cells (BAEC) exposed to 5 Gy were observed beginning at 8 hrs and peaking at 16 hrs post exposure(17), while daunorubicin-induced ceramide generation and CS activity were observed beginning at 6 hrs and peaking at 8 hrs post exposure in P388 murine leukemia cells(15). Studies utilizing metabolic incorporation of 125I-labeled 5-iodo-2′-deoxyuridine ([125I]dURd) into DNA of endothelial cells provided evidence that damage confined to DNA may signal post-transcriptional CS activation. 125I decays by electron capture, emitting cascades of low-energy Auger electrons, which when incorporated into DNA deposit their total radiation energy within less than 40 Å, producing DSBs within six base pairs of the site of incorporation(19–21). In bovine aortic endothelial cells (BAEC), [125I]dURd-mediated apoptosis induced post-transcriptional activation of CS, de novo ceramide synthesis and apoptosis, inhibitable by a natural specific competitive CS inhibitor, Fumonisin B1 (FB1)(17). The biologic significance of CS as transducer of apoptosis has been confirmed in vivo, as CS transactivates disease pathogenesis in gastric ulcer, ischemia-reperfusion and emphysema rodent models of human disease(1, 22–26). In these reports, epithelial apoptosis occurred 18–24 hrs post radiation exposure, or about 6–10 hrs after CS activation, and was inhibited by FB1.
In previous studies, we showed that although acute (ASMase)-dependent endothelial apoptosis and ensuing microvascular dysfunction couple GI tract irradiation to reproductive death of crypt SCC following 8–16 Gy, dose escalation to ≥17 Gy results in switching the target for radiation GI lethality to CS-mediated apoptosis directly within crypt SCCs (1). Unlike the C. elegans germ cell response, this CS-mediated apoptosis is p53-independent(1). Notably, ATM deficiency resets lethality, allowing CS-mediated apoptosis to initiate crypt SCC lethality at the low-dose radiation range. Accordingly, ATM deficiency hypersensitizes mice to the radiation GI syndrome, shifting the ED50 from 16.5Gy to 4.5Gy (3.7-fold; p<0.05), pre-empting microvascular-mediated damage(1). The mechanism for this change in the crypt SCC lethal pathway in ATM-deficient mice, termed target re-ordering(1), remains unknown.
The present studies address the mechanism of target re-ordering from the coupled endothelial apoptosis/reproductive death pathway to CS-mediated lethality of irradiated crypt SCCs. Whereas ATM is a core kinase transducer of signals generated by Mre11 complex sensing of DSBs in the HR pathway(27), we tested the hypothesis that signals, generated by genetic manipulation of various sensors/transducers of DSB repair, effect DSB-induced CS activation. We used the irradiated GI model in mice carrying loss- or gain-of-function mutations (lf and gf, respectively) in the Mre11 complex, which consists of Mre11, Rad50 and NBS1(27). The lf Mre11ATLD1/ATLD1 mice harbor a Mre11 C-terminally truncated mutation and exhibit reduced DNA DSB repair and IR hypersensitivity(27, 28), while gf Rad50S/S mice encode a hypermorphic variant of Rad50 and express constitutive ATM kinase activity(29, 30). We also studied C57Bl/6prkdc/SCID severe combined immunodeficiency (SCID) mice, which carry a germ-line lf mutation in DNA-dependent protein kinase (DNA-PK), a transducing kinase of the NHEJ DNA repair pathway(9, 31, 32). We find a generic relationship between inactivation of DNA damage repair genes and CS-mediated apoptotic death of crypt SCCs. Furthermore, CS-mediated SCC lethality was mitigated in irradiated gf Rad50S/S mice, and epistasis studies ordered Rad50 upstream of Mre11. These studies thus suggest that unrepaired DNA DSBs are causative in target re-ordering in intestinal SCCs.
MATERIALS and METHODS
Mice
C57BL/6, ATM knockout (129s6/SvEvTac-Atm tm1awb/j) and SCID (in C57BL/6 background) male mice, 8–12 weeks old, were purchased from Jackson Laboratories. Mre11ATLD1/ATLD1, Chk2−/− and Rad50s/s hypermorphic mice (in sv129 C57BL/6 background) were propagated using heterozygous breeding pairs provided by the laboratory of John Petrini and genotyped as described (29, 33, 34). Mice were housed at the animal core facility of Memorial Sloan-Kettering Cancer Center. This facility is approved by the American Association for Accreditation of Laboratory Animal Care and is maintained in accordance with the regulations and standards of the United States Department of Agriculture and the Department of Health and Human Services, National Institutes of Health. Protocols for conducting animal experiments were approved by the Memorial Sloan-Kettering Cancer Center Research Animal Resource Center.
Irradiation, delivery of bFGF and FB1, BM transplantation and tissue preparation
Whole body irradiation (WBR) was delivered with a Shepherd Mark-I unit (Model 68, SN643) operating 137Cs sources. The dose rate was 2.35Gy/min. Human recombinant bFGF from SCIOS was delivered intravenously by retro-orbital injection as described(35). FB1 from BIOMOL was solubilized in normal saline and injected intra-peritoneally (25mg/gram mouse body weight) 10 min before WBR. To obtain tissue specimens, mice were sacrificed by hypercapnia asphyxiation and 2.5 cm segments of proximal jejunum (2 cm distal to the ligament of Trietz), as well as the head and proximal femur, were obtained. Intestinal specimens were fixed by overnight incubation in 4% neutral buffered formaldehyde, while bone specimens were fixed in Formical-4 (Chemical Decal Corp.). Fixed tissues were embedded in paraffin blocks and sections of full organ circumference (5 μm thick) were obtained by microtomy, adhered to polylysine-treated slides and deparaffinized by heating at 90°C for 10 min and at 60°C for 5 min, washed twice in xylene for 5 min each, and stained with hematoxylin and eosin. Bone marrow cells were prepared and BMT carried out according to a standard protocol, as described(4).
Survival of mice after WBR and autopsy evaluation of animal death
Actuarial survival was calculated by the product limit Kaplan-Meier method(36) and statistical significance of differences in survival were calculated by the Mantel log-rank test(37). Causes of death were evaluated in autopsy specimens and classified as described(4).
Measurement of tissue levels of ceramide and CS activity
Tissue CS activity was measured in microsomal membranes prepared from scraped mucosa specimens. Scraped mucosa was washed with cold PBS, pelleted, and resuspended in 4 ml homogenization buffer (25mM HEPES, pH7.4, 5mM EGTA, 50mM NaF, and 10μg/ml each of leupeptin and soybean trypsin inhibitor). Cells were then disrupted, microsomal membrane obtained and ceramide levels and CS activity were measured in freshly-prepared microsomal membranes as described(1).
Quantitation of crypt clonogen apoptosis
Intestinal tissue specimens were obtained 4–24 h after WBR, fixed in 4% fresh formaldehyde, embedded in paraffin, and 5μm sections were evaluated for apoptosis by terminal deoxytransferase-mediated deoxyuridine triphosphate nick end labeling (TUNEL), as described(4). The apoptotic index was scored as published(4) in full longitudinal sections of crypts containing at least 17 cells, including four Paneth cells. Frequency of apoptosis for each cell position from the crypt bottom was scored in 200 half crypt sections and reported as mean±SEM.
Quantitation of intestinal microvascular endothelial apoptosis
Intestinal tissue specimens were obtained at 4 h after WBR, fixed in 4% fresh formaldehyde, embedded in paraffin, and 5μm sections evaluated for endothelial apoptosis by TUNEL-CD34 double staining as described(4).
Crypt microcolony assays
Crypts were identified histologically according to established criteria(4). Surviving crypts were defined as containing 10 or more adjacent chromophilic non-Paneth cells, at least one Paneth cell and a lumen. The number of surviving crypts was counted in the whole circumference of the transverse cross-section of the intestine. We scored nine circumferences per mouse and used 4–5 mice to generate each data point. Data are reported as mean±SEM.
RESULTS
Mre11-deficient and SCID mice exposed to WBR are hypersensitive to lethality from the GI syndrome
We first defined baseline radiosensitivity parameters of mice harboring lf mutations of DNA repair enzymes. Table 1 shows that Mre11ATLD1/ATLD1 mice displayed severe hypersensitivity to WBR, associated with shortened mean survival (4.8 and 4.0 days after 10 and 12 Gy, respectively) compared to wild-type littermates (10.5 and 10.2 days after 10 and 12 Gy, respectively; p<0.01 for each). In contrast to wild type mice, which die from marrow aplasia at 10 and 12 Gy, autopsies of Mre11ATLD1/ATLD1 mice succumbing to these doses revealed complete destruction of the crypt-villus system with denudation of intestinal mucosa (Fig. 1A, middle left panel). There was also progressive, though incomplete, hemorrhagic marrow destruction as the dose escalated from 8 Gy to 12 Gy, although hematopoietic elements were still identifiable at time of death (Fig. 1A, middle right panel). Furthermore, autologous BM transplantation (BMT) failed to rescue these mice (Supplemental Fig. 1, left panel), confirming that intestinal destruction was the predominant cause of death. SCID mice were previously reported to exhibit 1.8-fold increase in intestinal radiosensitivity(9). Here we confirmed a hypersensitive intestinal radiation phenotype in our strain of C57BL/6Prkdc/SCID mice that displayed mean survivals of 4.7 and 4.4 d after 8 and 10 Gy WBR, respectively (Table 1). Autopsies revealed extensive and lethal GI damage (Fig. 1A, lower left panel, Table 1), similar to that described above in Mre11ATLD1/ATLD1 mice, and autologous BMT failed to rescue these mice (Supplemental Fig. 1, right panel). These data indicate that DNA repair deficiency results in radiation hypersensitive intestinal phenotypes that constitute the prime cause of animal lethality, as the WBR-treated mice could not be rescued by syngeneic marrow transplantation.
Table 1.
Survival and autopsy results of mice exposed to WBR.
| Survival (%) | Mean Survival | GI | BM | |
|---|---|---|---|---|
|
| ||||
| sv129/B6 8 Gy (5) | 100 | - | - | - |
| sv129/B6 10 Gy (5) | 0 | 10.5 | - | +++ |
| sv129/B6 12 Gy (5) | 0 | 10.2 | +, R | +++ |
| Mre11ATLD1/ATLD1 8 Gy (5) | 0 | 12.2 | - | +++ |
| Mre11ATLD1/ATLD1 10 Gy (10) | 0 | 4.8* | +++ | ++ |
| Mre11ATLD1/ATLD1 12 Gy (6) | 0 | 4.0* | +++ | ++ |
| C57BL/6 8 Gy (10) | 100 | - | - | - |
| C57BL/6 10 Gy (10) | 0 | 12.4 | R | +++ |
| Prkdc/scid 8 Gy (10) | 0 | 4.7 | +++ | ++ |
| Prkdc/scid 10 Gy (10) | 0 | 4.4 | +++ | +++ |
| Rad50+/+ 18 Gy (5) | 0 | 4.8 | +++ | ++ |
| Rad50+/+ + bFGF 18 Gy (5) | 0 | 4.0 | +++ | ++ |
| Rad50s/s 18 Gy (10) | 0 | 4.5 | ++ | ++ |
| Rad50s/s + bFGF 18 Gy (10) | 0 | 7.7* | +, R | +++ |
Survival is expressed as mean duratio n after WBR. Statistical significance of differences in survival was derived fr om product-limit Kaplan-Meier survival curves (Fig. 1A and not shown) and P values calculated by the Mantel log-rank test. Statistical significance of DNA damage mutants compared to wild type controls (p<0.01) is indicated by *. Autopsy findings were classified as in Methods, wit h + to +++ designations representing increasing severity and the R designation representing GI regeneration. GI, gastrointestinal tract; BM, bone marrow. The numbers in parentheses indicate animals treated per group. Note that 10 Gy and 8 Gy represent minimal effective doses for 100% GI lethality for Mre11ATLD1/ATLD1 and Prkdc/scid strains, respectively.
Figure 1. DNA damage repair mutant mice are sensitized to the radiation-induced GI syndrome and radiation-induced apoptosis of the crypt SCC compartment, but not the microvascular endothelium.


(A) Hematoxylin and eosin-stained intestinal and BM sections of a representative wild-type animal succumbing to BM aplasia 12 days post 10 Gy and an Mre11ATLD1/ATLD1 and Prkdc/scid animal dying from the GI syndrome 5 days following 10 and 8 Gy, respectively. (B) Frequency histograms of the microvascular endothelial apoptotic response in the lamina propria of sv129/B6, C57BL/6 and mutant mice 4 hrs following 8–15 Gy WBR. Apoptotic endothelial cells were identified as containing a TUNEL-positive nuclear signal coupled with blue immunohistochemical staining for CD31. Apoptosis was scored in 300 villi per point. Data represent collated scores from two experiments. (C) Representative 5-μm histologic section of proximal jejunal crypts 18 hrs following 8 Gy WBR. Apoptotic cells are identified by brown TUNEL staining of condensed and fragmented nuclei. Original magnification x400. (D) Quantitative comparison of the crypt epithelial apoptotic response relative to the most distal paneth cell at the base of the half crypt at 18 hrs after 10 Gy WBR in wild-type SV129/B6 and SV129B6Mre11ATLD1/ATLD1 mice and after 8 Gy in wild-type C57BL/6 and C57BL/6Prkdc/SCID mice. Apoptotic response was scored as the mean percent apoptotic cells at each cell position derived from 200 half crypts sections, pooled from 4 mice.
Mre11-hypomorphs and SCID mice exposed to lethal doses of WBR exhibit crypt SCC but not intestinal endothelial apoptosis
To assess the mechanism of GI lethality in the lf mutant mice studied here, we selected 10 Gy for Mre11ATLD1/ATLD1 mice and 8 Gy for C57BL/6Prkdc/SCID mice, the minimum radiation doses that induce 100% lethal GI damage in each strain, respectively. We first evaluated whether endothelial apoptosis is triggered by radiation dose levels that induce the GI syndrome, using double immunostaining with anti-CD-34 and TUNEL. Intestinal mucosa specimens from Mre11ATLD1/ATLD1 and C57Bl/6prkdc/scid mice exposed to 8–10 Gy showed no evidence of significant GI endothelial apoptosis at 4 h after WBR, while concomitant wild-type control C57BL/6 mice exposed to their GI lethal dose of 15 Gy (positive controls) showed extensive endothelial apoptosis (≥10 apoptotic lesions/villus lamina propria) as published(3, 4). Similar to Mre11ATLD1/ATLD1 and C57Bl/6prkdc/scid mice, C57BL/6ATM−/− mice exposed to 8Gy [negative controls(1)], did not display an apoptotic endothelial response (Fig. 1B).
In contrast, significant apoptosis was detected at 18 h after 8–10 Gy in the crypt SCC compartment of Mre11ATLD1/ATLD1 and SCID mice (Figs. 1C, 1D), doses significantly lower than 17 Gy, the threshold for activating SCC apoptosis in wild-type littermates(1). Apoptotic SCCs were identified in the epithelial lining by brown TUNEL staining of condensed and fragmented nuclei at positions 6–9 above Paneth cells (Fig 1C). In the wild-type specimen at 8–10 Gy, crypt epithelial apoptosis was restricted to positions 4–5 (Fig. 1D), as reported(2, 3). This apoptotic response has been defined as p53-dependent, induced by as little as 1 Gy IR, and not involved in the GI syndrome(38). Similar apoptotic changes were observed at positions 4–5 of SCID and Mre11ATLD1/ATLD1 crypts exposed to 8 and 10 Gy, respectively. However, in addition, there was an increase in the apoptotic index at positions 6–9 at 18 h, from a mean of 13±3.8% in sv129 wild-type mice (Mre11+/+) after 10 Gy to 31.7±9.6% Mre11ATLD1/ATLD1 mice (p<0.05; Fig. 1D), and from 6.3±3.8% in C57Bl/6 controls after 8 Gy to 21.3±5.5% in SCID mice (p<0.05).
CS activation mediates crypt SCC apoptosis in irradiated Mre11-deficient and SCID mice
Biochemical evidence for CS activation was confirmed by tissue measurements of CS enzymatic activity and ceramide levels in fresh extracts of intestinal mucosa before and at various times after WBR. In Mre11ATLD1/ATLD1 intestinal mucosa, the Vmax of CS activity increased at 12 h after 10 Gy 1.43-fold over unirradiated controls (from 172±4.9 to 246±23.6 pmol/min/mg protein, p<0.01; Fig. 2A), while there was no significant increase following 10 Gy in wild-type littermates (not shown). An increase in intestinal ceramide content was also observed, which at 15 h after 10 Gy reached a level of 145.0±7.5% of unirradiated controls (p<0.05; Fig. 2B), absent in sv129/B6 wild-type animals. Similarly, C57Bl/6prkdc/scid exhibited 1.54-fold-increased CS activity at 9 h following 10 Gy WBR (p<0.05; Fig. 2C), unchanged in wild-type C57BL/6 littermates. Ceramide content also significantly increased within this same time frame in C57Bl/6prkdc/scid mice (p<0.05; Fig. 2D). These CS hypersensitive phenotypes of irradiated Mre11ATLD1/ATLD1 and SCID SCCs are similar to that previously reported in atm−/− mice(1).
Figure 2. DNA damage repair mutants are sensitized to ceramide synthase (CS)-mediated ceramide generation.
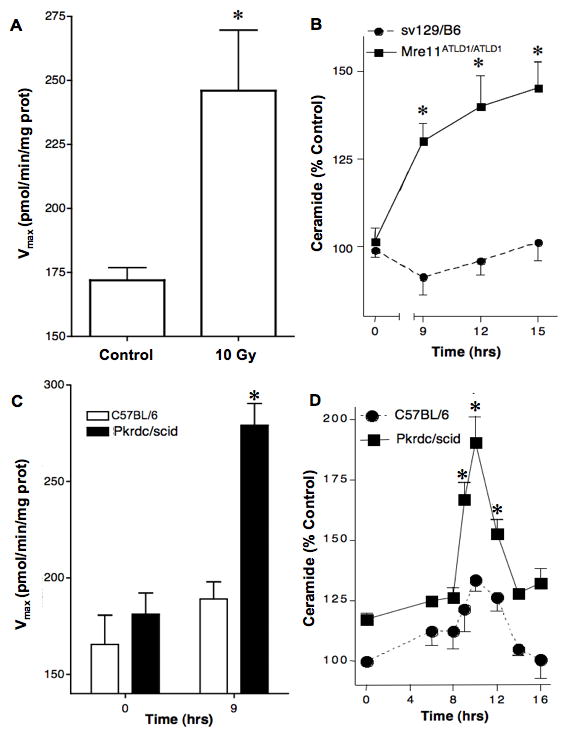
(A) CS is activated and (B) ceramide generated in the GI mucosa of sv129/B6Mre11ATLD1/ATLD1 mice exposed to 10 Gy WBR. CS activity was measured in microsomes under Michaelis-Menten kinetics. Ceramide levels were quantified by diacyglycerol kinase assay. Data (mean±SEM) are pooled from triplicate determination from two experiments. (C) CS activity and (D) ceramide levels determined in the GI mucosa of Prkdc/scid mice and wild-type C57Bl/6 mice exposed to 8 Gy WBR, performed as in Fig. 2A and 2B, respectively.
Whereas a recent study reported that transient transfection with DNA-PKcs small interfering RNA (siRNA) sequences reduced ATM mRNA and protein levels in normal human fibroblasts(39), we questioned whether the hypersensitive CS phenotype of irradiated SCID or Mre11ATLD1/ATLD1 intestines might be secondary to ATM deficiency. However, Western immunoblots of GI mucosal extracts from SCID or Mre11ATLD1/ATLD1 mice showed no reduction in ATM protein levels when compared to wild-type littermates (not shown), indicating that crypt apoptosis in these mice was not associated with ATM deficiency.
CS-mediated lethality of crypt SCCs confers the intestinal radiation phenotype of Mre11-deficient and SCID mice
While these data demonstrated correlation between CS-mediated apoptosis in the SCC compartment and the GI syndrome in Mre11ATLD1/ATLD1 and C57Bl/6prkdc/scid mice, they are not evidence for a causative relationship. Hence, we explored the effect of the specific CS competitive inhibitor FB1 on number of surviving crypts assessed by the microcolony assay. The 3.5 day microcolony assay (also termed crypt survival assay) is commonly used as a surrogate for quantitative evaluation of dose-dependent lethality of crypt SCCs post-radiation(2, 40). Previous studies showed that surviving fractions of ~35% (40 regenerating crypts in total) correlates with 50% of animals succumbing to the GI syndrome (2, 40). The dose resulting in 10% microcolony crypt survival (D10) has frequently been used for inter-strain comparisons of mouse sensitivity to the GI syndrome (4, 35). Fig. 3 shows 3.5 day microcolony dose-response curves for SCID and Mre11ATLD1/ATLD1 mice, compared with atm−/− mice used as positive controls. Curves for atm−/− mice (Fig. 3A) show a left shift, with D10 value decreasing from 13.0±0.5 Gy in wild-type controls to 4.7±0.5 Gy, yielding a dose-modifying factor (DMF) of 2.8±0.1 (p<0.005; Table 2). Similar sensitization was observed in Mre11ATLD1/ATLD1 mice (Fig. 3B), which showed a D10 of 6.9±1.2 Gy, reduced from 14.8±1.3 Gy in wild-type SV129/BL6 mice (p<0.005; DMF of 2.0±0.2; Table 2), and in C57BL/6Prkdc/SCID mice (Fig. 3C), exhibiting a D10 of 5.2±0.4 Gy (p<0.005; DMF of 2.5±0.2; Table 2). Pretreatment with 25 mg/kg FB1, the maximal non-toxic dose in these mice, delivered 30 min prior to WBR increased the Mre11ATLD1/ATLD1 crypt surviving fraction after 10 Gy by 3.4-fold, from 1.9±0.4% observed in control FB1-untreated mice to 6.7±0.6% (p<0.001; Fig. 3D). Similarly, FB1 pretreatment of C57BL/6Prkdc/SCID mice prior to 8 Gy WBR increased crypt survival 4.0-fold, from 2.8±0.4% in FB1-untreated controls to 11.0±0.8% (p<0.001; Fig. 3D). Similar effects of FB1 on crypt survival were reported in atm−/− mice(1), permitting mucosal regeneration that was not observed in control FB1-untreated atm−/− mice. These data support the notion that CS-mediated crypt epithelial apoptosis contributes to Mre11ATLD1/ATLD1 and SCID SCC lethality post-radiation. It should, however, be noted that in both the present and prior atm−/− studies, FB1 provided only partial rescue of SCCs from radiation lethality.
Figure 3. DNA damage repair mutants are sensitized to CS-mediated crypt SCC lethality.

Dose response parameters of crypt survival from full transverse sections of proximal jejunum obtained 3.5 days following WBR treatment of (A) C57BL/6ATM−/−, (B) SV129B/6Mre11ATLD1/ATLD1, and (C) C57BL/6Prkdc/SCID mice, assessed by the crypt microcolony assay, as described (4). (D) Crypt survival in SV129B/6Mre11ATLD1/ATLD1 (12 Gy) and C57BL/6Prkdc/SCID (10 Gy) mice with and without administration of 25 mg/kg of the CS inhibitor Fumonisin B1 (FB1) 30 min prior to irradiation. Data for computation of the surviving fraction at each dose level were compiled from 2–4 animals irradiated concomitantly, with 9–20 circumferences scored per mouse. Surviving fraction per dose was calculated with the FIT software program (44). Data (mean±SEM) were obtained from two independent experiments.
Table 2.
Jejunal crypt survival curve parameters D0 and D10.
| Strain | D0 | D10 (Gy) | DMF | Lethal GI Dose (Gy) |
|---|---|---|---|---|
|
| ||||
| C57BL/6 | 1.5±0.1 | 13.0±0.5 | 15 | |
| C57BL/6Prkdc/scid | 0.7±0.1 | 5.2±0.4 | 2.5±0.2 (−) | 8 |
| sv129/BL6 | 2.2±0.5 | 14.8±1.3 | 15 | |
| sv129/BL6atm−/− | 1.1±0.2 | 4.7±0.5 | 2.8±0.1 (−) | 6 |
| sv129/BL6Mre11+/+ | 2.1±0.7 | 14.0±1.3 | 15 | |
| sv129/BL6MRE11ALTD1/ALTD1 | 1.6±0.1 | 6.9±1.2 | 2.0±0.2 (−) | 10 |
| sv129/BL6Rad50+/+ | 1.9±0.2 | 15.4±1.9 | 15 | |
| sv129/BL6Rad50s/s | 3.0±0.3 | 20.6±2.0 | 1.3±0.1 (+) | >18 |
Samples of proximal jejunum were obtained before or 3.5 days following WBR, fixed in paraformaldehyde, and embedded in paraffin blocks. Transverse tissue sections of the full jejunal circumference (5 μM thick) were stained with hematoxylin and eosin and scored for the number of regenerative crypts, as described in Methods. D0 and D10 values are derived from the FIT software program. (−) indicates a DMF for radiosensitization, (+) indicates a DMF for radioprotection.
Whereas these data indicated a causative engagement of CS activation in Mre11ATLD1/ATLD1 apoptotic crypt lethality, we explored whether Chk2 activation, a downstream element in Mre11/ATM signaling pathway of p53-mediated apoptosis(33) is involved in CS-mediated crypt SCC apoptosis. However, Chk2−/− mice exhibited the same SCC radiosensitivity as wild-type sv129/B6 mice (35.3±1.9% vs. 38.6±1.6% microcolony crypt surviving fractions after 8 Gy and 1.6±0.2% vs. 1.5±0.2% surviving fraction after 18 Gy, respectively), consistent with DSB-induced activation of the Chk2/p53 pathway as not involved in regulating CS-mediated apoptosis in crypt SCC.
The pro-apoptotic Rad50s allele protects against CS-mediated apoptosis and crypt lethality
Whereas genetic lf in Mre11 complex function generates a hypersensitive CS phenotype, we hypothesized that Mre11 complex gf might exhibit an opposite phenotype. The gf Rad50S allele confers constitutive ATM kinase activity, and Rad50S/S mice exhibit a phenotypic stress response, consisting of Chk2/p53-mediated apoptosis of hematopoietic cells, semeniferous tubules and intestinal crypt epithelium(41). The hematopoietic phenotype is severe, as apoptosis leads to hematopoietic stem cell attrition and animal demise from anemia at 2–3 months of age, although at time of death there is no evidence of testicular or intestinal crypt attrition(29). Fig. 4A quantifies crypt apoptosis in unirradiated Rad50S/S mice, exhibiting increased constitutive apoptosis at all crypt positions from 3 to 9, reaching a cumulative apoptotic index of 11.0±2.0% compared to 2.6±0.7% in wild-type controls (p<0.05). However, when exposed to 18 Gy WBR, Rad50S/S mice exhibited significant abrogation of SCC apoptosis at positions 6–9 at 18 h post-radiation. Specifically, wild-type mice exhibited a cumulative apoptotic index at positions 6–9 of 28.5±3.2%, while Rad50s/s mutants exhibited an index of 13.0±4.0% (p<0.001). The anti-apoptotic effects of the Rad50S allele in 18 Gy-irradiated crypts was matched by significant protection against crypt lethality at 3.5 days after WBR (Fig. 4B), increasing the Do and D10 values (p<0.005; Table 2), translating into a protective DMF value of 1.3±0.1. Although animal survival after 18 Gy WBR was not prolonged in Rad50S/S mice (Fig. 4C, Table 1), autopsies revealed extensive and significant increase of crypt regeneration and mucosal repair in Rad50s/s as compared to wild-type mice (not shown and Table 1), with mice succumbing to severe marrow aplasia and incomplete repair of intestinal damage. Pretreatment with bFGF, which attenuates the endothelial component of radiation-induced GI damage(3), significantly prolonged survival of Rad50s/s mutant, but not wild-type mice, to 7.7 days (p<0.005; Fig. 4C), with mice succumbing to BM aplasia (Table1) while exhibiting near complete intestinal mucosa recovery (not shown and Table 1). Taken together these data indicate that the Rad50S allele abrogates crypt SCC apoptosis and crypt lethality, normally observed in wild-type littermates.
Figure 4. Rad50s/s hypermorphic mice are resistant to ionizing radiation-induced epithelial apoptosis and crypt lethality.

(A) Quantification of baseline crypt epithelium apoptosis and the apoptotic response 18 hrs following 18 Gy WBR in Rad50s/s mice compared to wild-type littermates. Crypt apoptosis was quantified as in Fig. 1D. (B) Dose response parameters of crypt survival in Rad50s/s mice compared to wild-type littermates, as in Fig. 3. (C) Actuarial survival of wild-type and Rad50s/s mice treated with or without bFGF, performed as described in Methods. (D) Mre11 signals upstream of Rad50 in radiation-induced crypt epithelial apoptosis. Wild-type sv129B/6, Mre11ATLD1/ATLD1, Rad50s/s, or Mre11ATLD1/ATLD1/Rad50s/s double mutants were irradiated with 8 Gy WBR. Crypt survival was quantified from 2–4 animals irradiated concomitantly, with 9–20 circumferences scored per mouse as in Fig. 3.
Since genetic mutations in two elements of the Mre11 complex, Mre11ATLD1 and Rad50s alleles, conferred opposing intestinal crypt apoptotic phenotypes upon irradiation, we carried out epistasis studies using the 3.5 day microcolony assay as a readout. Figure 4D shows that the anti-apoptotic protective effect of the gf Rad50S allele on irradiated crypt SCCs was abrogated in double mutant Rad50s/s/Mre11ATLD1/ATLD1 mice, which manifest a crypt surviving fraction of 0.1±0.1 at 3.5 days after 8 Gy WBR, not significantly different from the surviving fraction in Mre11ATLD1/ATLD1 littermates, but significantly reduced from the 0.6±0.2 surviving fraction in Rad50s/s mice (Rad50s/s vs. sv129/B6 p<0.001, Rad50s/s vs. Rad50s/s/Mre11ATLD1/ATLD1 p<0.001). Similar results were found following 18 Gy WBR (not shown). These data indicate that Rad50 orders upstream of Mre11 in effecting the radiation-induced CS response.
DISCUSSION
The major finding of the present studies is that genetic manipulation of sensors and transducers of DNA damage repair affect GI tract sensitivity via DSB-induced CS-mediated apoptosis of crypt SCCs. At doses lethal to the GI tract in Mre11ATLD1/ATLD1 and SCID mice, up to half of the log cell kill is CS-dependent, based on the level of FB1 protection of crypts in the microcolony assay. It should be noted that we found different lethal doses ranges for these strains. As the Mre11ATLD1 allele is hypomorphic while the Prkdc/scid and atm−/− mice harbor null alleles, these differences cannot be construed to indicate differential contribution of these genes in conferring radiation sensitivity. This CS-mediated apoptotic response is Chk2/p53-independent. Other modes of the SCC death response are apparently non-apoptotic and may be, at least in part, regulated by Chk2/p53 signaling, consistent with previous reports that p53 inactivation is radiosensitizing to the GI in the absence of enhanced apoptosis(42), apparently involving classic reproductive/clonogenic death as defined in the radiobiologic literature. While we have no data indicating that the non-apoptotic component relies on p53 inactivation, the current studies do indicate that ceramide-dependent apoptotic and ceramide-independent non-apoptotic damage to SCCs in NHEJ or HR deficiency are subject to target switching.
Target switching is a recently proposed concept, which indicates that tissues operate a set of primary and secondary targets for stress-induced damage, such as occurs with IR, and that access to secondary targets is engaged when primary targets are repressed genetically or pharmacologically(1). This concept differs from traditional concepts regarding tissue damage, which suggest that tissues operate one mechanism for regulating damage responsiveness/repair that is either on or off. In the case of the GI tract, the primary response mechanism in a wild-type mouse engages a coordinated endothelial cell apoptosis and resultant microvascular dysfunction coupled to direct DNA damage to the SCC compartment(3, 4). This mechanism is operative up to 16 Gy in C57/Bl6 mice. Upon inactivating this mechanism by protecting against endothelial apoptosis, the GI tract is protected against radiation lethality(3, 4). However, further dose escalation restores GI lethality as radiation switches to CS-mediated apoptosis and non-apoptotic damage to the SCC compartment to signal lethal damage(1). This set of events, appears regulated by repair/sensor enzymes and likely by persistent unrepaired DSBs, and is subject to target reordering as inactivation of ATM, MRE11 or DNA-PKcs reset CS activation as the primary response mechanism, yet do not impact endothelial cell apoptosis (see Supplemental Fig. 2).
At present there is no information regarding the trans-regulatory pathways initiated by DNA DSBs that lead to post-translational activation of CS. Such a pathway is, however, assumed, as we have recently found that following IR exposure of HeLa cells, CS is activated in mitochondrial-associated membrane (MAM), an ER-like structure that co-purifies with mitochondria, and serves as a site for lipid synthesis and as a conduit for lipid transfer to the outer mitochondrial membrane. Consistent with MAM CS as the source of radiation-induced ceramide, our preliminary data indicate that MAM CS-generated neo-synthesized ceramide is in fact transferred to outer mitochondrial membranes where it regulates BAX oligomerization (Lee, Kolesnick and Fuks, unpublished). MAM CS appears distinct from ER CS, which constitutively provides ceramide for trafficking to golgi, where headgroups are attached to generate the plethora of sphingolipids involved in membrane biogenesis. Rather MAM CS appears to be in the “off” state prior to DSB-induced signaling, similar to the activation sequence during heat shock in Saccharomyces cerevisiae, where yeast CS activation is abrupt and requisite for completion of the heat shock adaptive response(43). Furthermore, while currently it is technically infeasible to isolate MAM from C. elegans, the genetic and biochemical localization of CS-generated ceramide post-radiation to mitochondria(11) suggest that MAM CS activation may serve as a generic source for registering stress at mitochondrial membranes.
The current data are consistent with two different models by which unrepaired or misrepaired DSBs might activate CS. In the most parsimonious, HR and NHEJ pathways normally mediate DSB repair reducing the DSB load to below a threshold for CS signaling. Repair system insufficiency would yield persistent DSB levels sufficient for CS activation. Alternatively, a dynamic balance may exist between CS activating and CS-attenuating signaling pathways, regulated by activation status of the DNA damage sensing and repair pathways. In wild-type intestines exposed to doses ≤16Gy, the attenuating function would prevail, preventing crypt SCC apoptosis while DSBs are being repaired, whereas at higher doses the balance would shift to favor CS activation, enabling apoptosis. Irrespective of the mechanism used by DNA sensing and repair enzymes to regulate CS, which will require extensive future investigation to sort out, the present studies define robust and standardized in vivo readout systems for examination of allele specific regulation of the DNA damage response. Evaluation of radioprotection or radiation sensitization of the intestines should be multiparametric, involving at a minimum the 3.5 day microcolony assay, Kaplan-Meier dose survival analysis with autopsy evaluation of every experimental animal at the time of death, coupled to assessment of endothelial and SCC apoptosis and their respective ceramide generating mechanisms.
Supplementary Material
Acknowledgments
We would like to acknowledge John Petrini for critical comments and valuable discussion during the writing of this manuscript. These studies were supported by US National Institutes of Health grants CA105125 (AF) and CA85704 (RK) and a gift from the Virginia and D.K. Ludwig Fund for Cancer Research (ZF).
Footnotes
Conflicts of Interest Notification: There are no conflicts of interest
References
- 1.Ch’ang HJ, Maj JG, Paris F, et al. ATM regulates target switching to escalating doses of radiation in the intestines. Nature medicine. 2005;11(5):484–90. doi: 10.1038/nm1237. [DOI] [PubMed] [Google Scholar]
- 2.Potten CS. A comprehensive study of the radiobiological response of the murine (BDF1) small intestine. Int J Radiat Biol. 1990;58(6):925–73. doi: 10.1080/09553009014552281. [DOI] [PubMed] [Google Scholar]
- 3.Paris F, Fuks Z, Kang A, et al. Endothelial apoptosis as the primary lesion initiating intestinal radiation damage in mice. Science (New York, NY. 2001;293(5528):293–7. doi: 10.1126/science.1060191. [DOI] [PubMed] [Google Scholar]
- 4.Rotolo JA, Maj JG, Feldman R, et al. Bax and Bak do not exhibit functional redundancy in mediating radiation-induced endothelial apoptosis in the intestinal mucosa. International journal of radiation oncology, biology, physics. 2008;70(3):804–15. doi: 10.1016/j.ijrobp.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 5.Bedford JS, Dewey WC Radiation Research Society. 1952–2002. Historical and current highlights in radiation biology: has anything important been learned by irradiating cells? Radiat Res. 2002;158(3):251–91. doi: 10.1667/0033-7587(2002)158[0251:hachir]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 6.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 7.Borges HL, Linden R, Wang JY. DNA damage-induced cell death: lessons from the central nervous system. Cell research. 2008;18(1):17–26. doi: 10.1038/cr.2007.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delacote F, Han M, Stamato TD, Jasin M, Lopez BS. An xrcc4 defect or Wortmannin stimulates homologous recombination specifically induced by double-strand breaks in mammalian cells. Nucleic Acids Res. 2002;30(15):3454–63. doi: 10.1093/nar/gkf452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Biedermann KA, Sun JR, Giaccia AJ, Tosto LM, Brown JM. scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(4):1394–7. doi: 10.1073/pnas.88.4.1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nussenzweig A, Sokol K, Burgman P, Li L, Li GC. Hypersensitivity of Ku80-deficient cell lines and mice to DNA damage: the effects of ionizing radiation on growth, survival, and development. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(25):13588–93. doi: 10.1073/pnas.94.25.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng X, Yin X, Allan R, et al. Ceramide biogenesis is required for radiation-induced apoptosis in the germ line of C. elegans. Science (New York, NY) 2008;322(5898):110–5. doi: 10.1126/science.1158111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hannun YA, Luberto C, Argraves KM. Enzymes of sphingolipid metabolism: from modular to integrative signaling. Biochemistry. 2001;40(16):4893–903. doi: 10.1021/bi002836k. [DOI] [PubMed] [Google Scholar]
- 13.Mandon EC, Ehses I, Rother J, van Echten G, Sandhoff K. Subcellular localization and membrane topology of serine palmitoyltransferase, 3-dehydrosphinganine reductase, and sphinganine N-acyltransferase in mouse liver. The Journal of biological chemistry. 1992;267(16):11144–8. [PubMed] [Google Scholar]
- 14.Deng X, Hofmann ER, Villanueva A, et al. Caenorhabditis elegans ABL-1 antagonizes p53-mediated germline apoptosis after ionizing irradiation. Nat Genet. 2004;36(8):906–12. doi: 10.1038/ng1396. [DOI] [PubMed] [Google Scholar]
- 15.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82(3):405–14. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 16.Kroesen BJ, Pettus B, Luberto C, et al. Induction of apoptosis through B-cell receptor cross-linking occurs via de novo generated C16-ceramide and involves mitochondria. The Journal of biological chemistry. 2001;276(17):13606–14. doi: 10.1074/jbc.M009517200. [DOI] [PubMed] [Google Scholar]
- 17.Liao WC, Haimovitz-Friedman A, Persaud RS, et al. Ataxia telangiectasia-mutated gene product inhibits DNA damage-induced apoptosis via ceramide synthase. The Journal of biological chemistry. 1999;274(25):17908–17. doi: 10.1074/jbc.274.25.17908. [DOI] [PubMed] [Google Scholar]
- 18.Truman JP, Gueven N, Lavin M, et al. Down-regulation of ATM protein sensitizes human prostate cancer cells to radiation-induced apoptosis. The Journal of biological chemistry. 2005;280(24):23262–72. doi: 10.1074/jbc.M503701200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.LeMotte PK, Little JB. DNA damage induced in human diploid cells by decay of incorporated radionuclides. Cancer research. 1984;44(4):1337–42. [PubMed] [Google Scholar]
- 20.Martin RF, Bradley TR, Hodgson GS. Cytotoxicity of an 125I-labeled DNA-binding compound that induces double-stranded DNA breaks. Cancer research. 1979;39(8):3244–7. [PubMed] [Google Scholar]
- 21.Radford IR, Hodgson GS. 125I-induced DNA double strand breaks: use in calibration of the neutral filter elution technique and comparison with X-ray induced breaks. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1985;48(4):555–66. doi: 10.1080/09553008514551611. [DOI] [PubMed] [Google Scholar]
- 22.Uehara K, Miura S, Takeuchi T, et al. Significant role of ceramide pathway in experimental gastric ulcer formation in rats. The Journal of pharmacology and experimental therapeutics. 2003;305(1):232–9. doi: 10.1124/jpet.102.045195. [DOI] [PubMed] [Google Scholar]
- 23.Hwang D, Popat R, Bragdon C, O’Donnell KE, Sonis ST. Effects of ceramide inhibition on experimental radiation-induced oral mucositis. Oral surgery, oral medicine, oral pathology, oral radiology, and endodontics. 2005;100(3):321–9. doi: 10.1016/j.tripleo.2004.09.018. [DOI] [PubMed] [Google Scholar]
- 24.Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nature medicine. 2005;11(5):491–8. doi: 10.1038/nm1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masini E, Giannini L, Nistri S, et al. Ceramide: a key signaling molecule in a Guinea pig model of allergic asthmatic response and airway inflammation. The Journal of pharmacology and experimental therapeutics. 2008;324(2):548–57. doi: 10.1124/jpet.107.131565. [DOI] [PubMed] [Google Scholar]
- 26.Cuzzocrea S, Di Paola R, Genovese T, et al. Anti-inflammatory and anti-apoptotic effects of fumonisin B1, an inhibitor of ceramide synthase, in a rodent model of splanchnic ischemia and reperfusion injury. The Journal of pharmacology and experimental therapeutics. 2008;327(1):45–57. doi: 10.1124/jpet.108.139808. [DOI] [PubMed] [Google Scholar]
- 27.Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA repair. 2004;3(8–9):845–54. doi: 10.1016/j.dnarep.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 28.Stewart GS, Maser RS, Stankovic T, et al. The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 1999;99(6):577–87. doi: 10.1016/s0092-8674(00)81547-0. [DOI] [PubMed] [Google Scholar]
- 29.Morales M, Theunissen JW, Kim CF, Kitagawa R, Kastan MB, Petrini JH. The Rad50S allele promotes ATM-dependent DNA damage responses and suppresses ATM deficiency: implications for the Mre11 complex as a DNA damage sensor. Genes Dev. 2005;19(24):3043–54. doi: 10.1101/gad.1373705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Usui T, Petrini JH, Morales M. Rad50S alleles of the Mre11 complex: questions answered and questions raised. Exp Cell Res. 2006;312(14):2694–9. doi: 10.1016/j.yexcr.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 31.Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell research. 2008;18(1):134–47. doi: 10.1038/cr.2007.111. [DOI] [PubMed] [Google Scholar]
- 32.Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell research. 2008;18(1):114–24. doi: 10.1038/cr.2008.3. [DOI] [PubMed] [Google Scholar]
- 33.Stracker TH, Couto SS, Cordon-Cardo C, Matos T, Petrini JH. Chk2 suppresses the oncogenic potential of DNA replication-associated DNA damage. Molecular cell. 2008;31(1):21–32. doi: 10.1016/j.molcel.2008.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stracker TH, Morales M, Couto SS, Hussein H, Petrini JH. The carboxy terminus of NBS1 is required for induction of apoptosis by the MRE11 complex. Nature. 2007;447(7141):218–21. doi: 10.1038/nature05740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maj JG, Paris F, Haimovitz-Friedman A, Venkatraman E, Kolesnick R, Fuks Z. Microvascular function regulates intestinal crypt response to radiation. Cancer Res. 2003;63(15):4338–41. [PubMed] [Google Scholar]
- 36.Kaplan EL, Meier p. Nonparametric estimation from incomplete observation. J Am Stat Assoc. 1958;53:457–81. [Google Scholar]
- 37.Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50(3):163–70. [PubMed] [Google Scholar]
- 38.Merritt AJ, Allen TD, Potten CS, Hickman JA. Apoptosis in small intestinal epithelial from p53-null mice: evidence for a delayed, p53-independent G2/M-associated cell death after gamma-irradiation. Oncogene. 1997;14(23):2759–66. doi: 10.1038/sj.onc.1201126. [DOI] [PubMed] [Google Scholar]
- 39.Peng Y, Woods RG, Beamish H, et al. Deficiency in the catalytic subunit of DNA-dependent protein kinase causes down-regulation of ATM. Cancer research. 2005;65(5):1670–7. doi: 10.1158/0008-5472.CAN-04-3451. [DOI] [PubMed] [Google Scholar]
- 40.Hendry JH, Potten CS, Roberts NP. The gastrointestinal syndrome and mucosal clonogenic cells: relationships between target cell sensitivities, LD50 and cell survival, and their modification by antibiotics. Radiat Res. 1983;96(1):100–12. [PubMed] [Google Scholar]
- 41.Bender CF, Sikes ML, Sullivan R, et al. Cancer predisposition and hematopoietic failure in Rad50(S/S) mice. Genes Dev. 2002;16(17):2237–51. doi: 10.1101/gad.1007902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Komarova EA, Kondratov RV, Wang K, et al. Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene. 2004;23(19):3265–71. doi: 10.1038/sj.onc.1207494. [DOI] [PubMed] [Google Scholar]
- 43.Jenkins GM. The emerging role for sphingolipids in the eukaryotic heat shock response. Cell Mol Life Sci. 2003;60(4):701–10. doi: 10.1007/s00018-003-2239-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Albright N. Computer programs for the analysis of cellular survival data. Radiat Res. 1987;112(2):331–40. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.