

Abstract
The generation of reactive oxygen species (ROS) triggered by bacterial endotoxin lipopolysaccharide (LPS) plays a key role during the pathogenesis of sepsis. Given the key role that the interleukin-1 receptor associated kinase-1 (IRAK-1) plays in LPS-mediated Toll-like-receptor 4 (TLR4) pathway, we herein tested whether deletion of IRAK-1 gene in mice may render protection from LPS-induced oxidative tissue damage. In this report, we studied the levels of oxidative stress in vital organs including liver, kidney, and brain from wild type (WT) and IRAK-1 deficient mice injected with a lethal dose of LPS (25 mg/kg), a TLR4-specific agonist. We demonstrated that LPS challenge induced marked elevation of lipid peroxidation and nitrite levels in the plasma and tissues of WT mice, as well as elevated pro-inflammatory mediators. In contrast, IRAK-1 deficient mice had significantly lower lipid peroxidation and nitrite levels, as well as lower levels of pro-inflammatory mediators. Mechanistically, LPS triggered higher levels of iNOS activity and elevated membrane translocation of p47phox, a key component of NADPH oxidase in immune cell derived from WT mice compared to IRAK-1 deficient mice. Additionally, tissues harvested from WT mice injected with LPS exhibited reduced activities of anti-oxidant enzymes including glutathione peroxidase (GPx), catalase, and superoxide dismutase (SOD). In contrast, LPS challenge failed to reduce the activities of GPx and SOD in IRAK-1 deficient tissues. As a consequence, LPS caused significantly pronounced damage to liver and kidney tissues in WT mice as compared to IRAK-1 deficient mice.
Keywords: IRAK-1, Lipopolysaccharide, Sepsis, Free radicals, Oxidative stress, Antioxidant enzymes
1. Introduction
A highly lethal systemic multi-organ dysfunction syndrome (MODS) can occur following a diverse array of clinical insults, including endotoxemia due to disseminated infection, trauma, and injury (Glaros et al., 2009; Ghosh et al., 1993; Carden and Granger, 2000). The systemic inflammation that results from these challenges can spread throughout the intravascular space of humans and animals, and ultimately lead to the failure of multiple organ-systems (Glaros et al., 2009; Wenzel et al., 1996). MODS due to endotoxemia and sepsis has a mortality rate ranging from 40% to 80% and is the major cause of death in critical care units (CCU) and intensive care units (ICU) throughout the world. Antibiotics have been proven ineffective in controlling disseminated endotoxemia, since the bacterial endotoxin lipopolysaccharide (LPS), rather than the live bacteria, plays a major role in inducing MODS (Bone, 1996). LPS alone can trigger multi-organ failure in both humans and laboratory animals (Wenzel et al., 1996; Karima et al., 1999). Unfortunately, little improvement has been made in the treatment of human MODS has since the syndrome was first defined (O’Brien et al., 2000). A lack of systematic understanding of the inflammatory complications during the acute yet complex pathogenesis of MODS has contributed to the failure to successfully treat this devastating syndrome (O’Brien et al., 2000).
A plethora of inflammatory mediators are rapidly induced in the blood circulation of both human endotoxemic patients and experimentally-induced endotoxemia in animals (Andreasen et al., 2008; Wong et al., 2000). Such mediators include inflammatory cytokines and chemokines (e.g. IL-6, TNF-α, MIP-1a, GRO-a), acute phase proteins (e.g. lipocalin 2, endothelin-1, PAI-1), reactive oxygen species (ROS) and reactive nitrogen species (RNS). Elevated chemokines contribute to the inflammation by recruiting toxic neutrophils and activated monocytes to vital organs, while ROS production and release causes damaging modifications to host proteins and other macromolecules (Crimi et al., 2006). Collectively, high levels of these inflammatory mediators can contribute to multi-organ injury and death (Maier, 2000). Mechanistically, the levels of ROS produced and released within tissues and circulation are controlled by two families of enzymes with opposing functions. Oxidases such as NADPH oxidases are induced and/or activated by LPS, leading to the generation of ROS (Bedard and Krause, 2007; Rada and Leto, 2008) anti-oxidases such as glutathione peroxidase (GPx), superoxide dismutase (SOD), and catalase are suppressed by LPS, compromising the clearance of the generated ROS (Valenca et al., 2008; Ueda et al., 2008). In addition, LPS induces elevated activities of the inducible nitric oxide synthase (iNOS), which subsequently contribute to the formation of RNS. Collectively, systemically elevated levels of ROS and RNS can cause extensive oxidative tissue damage and contribute to the pathogenesis of multi-organ failures during septic shock.
The TLR4 pathway is the primary innate immune pathway responsible for the host response to LPS (Akira, 2001). Upon activation by LPS, components downstream of TLR4 can activate multiple pro-inflammatory transcription factors including NFkB, which eventually lead to the expression of oxidative enzymes such as NOX1 and iNOS. In the meantime, we have demonstrated that LPS can also suppress the expression of anti-oxidative enzymes such as GPx and catalase in cultured macrophages (Maitra et al., 2009a). Mechanistically, the key TLR4 intracellular kinase IRAK-1 has been shown to be critical for mediating the diverse cellular effects of LPS (a,Maitra et al., 2009b; Ringwood and Li, 2008).
Despite convincing in vitro data, relatively little information is available regarding the in vivo relevance of IRAK-1 during LPS-induced tissue oxidative damage. In this report, we have performed systematic evaluations of in vivo oxidative damage in vital organs of both WT and IRAK-1 deficient mice. Our present study reveals that IRAK-1 serves as a critical mediator for LPS-induced oxidative tissue damage. These data indicate that IRAK-1 may serve as a novel therapeutic target for future intervention strategies in limiting the occurrence of multi-organ failure during septic events.
2. Material and methods
2.1. Ethics statement
Wild type C57BL/6 mice and IRAK-1 deficient mice with C57BL/6 background were bred in a pathogen-free environment in accordance with Virginia Tech guidelines. IRAK-1 deficient mice had been back-crossed with C57BL/6 mice for 14 generations. All animal experiments were performed according to the Institutional Animal Care and Use Committee (IACUC) guidelines from Virginia Tech and were approved by Virginia Tech IACUC under the protocol number BIO 09-203.
2.2. Reagents
LPS (E. coli O111:B4) was obtained from Sigma. 2-(4-Iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazo-lium (WST-1) was purchased from Dojindo Molecular Technologies Inc., Maryland, USA. The polyclonal anti-P47phox antibody was purchased from Millipore, MA, USA.
2.3. Mice
Wild-type C57BL/6 mice were obtained from the Charles River Laboratory. IRAK1−/− mice with C57BL/6 background were kindly provided by Dr. James Thomas from the University of Texas Southwestern Medical School. All mice were housed and bred at Derring Hall Animal Facility in compliance with approved Animal Care and Use Committee protocols at Virginia Polytechnic Institute and State University. Bone marrow-derived macrophages (BMDM), murine embryonic fibroblasts (MEF) and microglia were harvested and cultured as previously described (Wang et al., 2008). Wild type and IRAK-1−/− mice of matched gender and age were injected with LPS (E. coli O111:B4, Sigma) (25 mg/kg body weight) or PBS intraperitoneally. Total blood samples were drawn 10 h after the injection. Plasma samples were collected for subsequent analyses. Various tissues were harvested and used for described assays.
2.4. Lipid peroxidation assay
Tissue samples were thawed on ice and homogenized in ice-cold 50 mM PBS buffer containing 10 μl of 0.5 M butylated hydroxy toulene (BHT). The crude homogenate were centrifuged at 3000 × g for 10 min at 4 °C. The clear supernatant was aliquoted and stored on ice until further analysis. Malondialdehyde and 4-hydroxynonenal (MDA-HNE), by-products of membrane bound polyunsaturated fatty acid peroxidation were measured as indicators of lipid peroxidation. Lipid peroxidation in tissue samples was measured as previously described (Carrillo-Vico et al., 2005).
2.5. Measurement of tissue antioxidant and anti-oxidative enzymes
Liver, kidney, and brain samples were removed and washed in ice-cold PBS, snap frozen in liquid nitrogen and stored at −80 °C until further analysis. The crude homogenates were centrifuged at 10,000 × g for 15 min at 4 °C. The clear supernatant was aliquoted and stored on ice until further analysis. Samples were used to measure the activities of antioxidant enzymes. GPx activity was determined using commercially available BIOXYTECH GPx-340™ kit (Oxis International Inc., Portland, OR, USA). SOD activity was determined as described previously by Basini et al. (2008). Catalase activity was measured by the Oxis Bioxytech Catalase-520 assay kit (Oxis International Inc., Portland, OR, USA). The levels of reduced glutathione were determined as previously reported (Stojiljkovic et al., 2007). The protein concentration of the samples was measured as described (Hill and Straka, 1988) and enzymatic activities were expressed as per mg protein.
2.6. Superoxide assay
The amount of extracellular superoxide produced was determined by measuring superoxide-mediated reduction of WST-1 dye (Peskin and Winterbourn, 2000; Walrand et al., 2003) as previously reported (Liu et al., 2001).
2.7. Real time RT-PCR
Total RNA was extracted from tissues or cultured cells using the TRIzol reagent (Invitrogen) according to the manufacturer’s protocol. Reverse transcription was carried out using the high capacity cDNA reverse transcription kit (Applied Biosystems), and subsequent real time RT-PCR analyses were performed using the SYBR green supermix on an IQ5 thermocycler (Bio-Rad). The relative levels of transcripts were calculated using the ΔDela;Ct method after normalizing with GAPDH as the internal control. The primer sets were obtained from IDT. The primer sequences are as follows- GPX3 (+): 5′ CCAGCTACTGAGGTCTGACAGA 3′; GPX3 (−): 5′ CAAATGGCCCAAGTTCTTCTTG 3′; catalase (+): 5′ TTCAGAA-GAAAGCGGTCAAGAAT 3′; catalase (−): 5′ GATGCGGGCCCCATAGTC 3′; GAPDH (+): 5′ AACTTTGGCATTGTGGAAGGGCTC 3′; GAPDH (−):5′ TGGAAGAGTGGGAGTTGCTGTTGA 3′.
2.8. Membrane fractionation and protein analyses
Membrane fractions were extracted as described (Dusi et al., 1995). Briefly, pelleted BMDMs were resuspended in relaxation buffer (100 mM KCl, 10 mM Hepes, 3.5 mM MgCl2, 3 mM NaCl, 1.2 mM ethyleneglycotetraacetic acid [EGTA], 25 mM NaF, 5 mM Na3VO4, 1 mM p-nitrophenyl phosphate, 2 mM PMSF and complete protease inhibitory cocktail,). The cells were briefly sonicated with a daigenode Bioruptor (UCD-200TM-Ex, TOSHO Dengi co Ltd). Unbroken cells and nuclei, were pelleted by gentle centrifugation at 1300 rpm for 10 min at 4 °C. The supernatant was centrifuged at 100,000 × g for 15 min at 4 °C. The pellet was resuspended in 0.5 ml relaxation buffer and recentrifuged at 100,000 × g for another 15 min at 4 °C. The final pellet representing the membrane fraction was solubilized in 0.5 ml solubilization buffer (25 mM Tris, pH 7.5, 150 mM NaCl, 1% (v/v) Triton X-100, 1% (w/v) DOC, 0.1% (w/v) SDS), and quantified using the BIO-RAD Bradford reagent. Equal amount of proteins were subsequently resolved on SDS-PAGE and blotted with antibody specific for p47phox. Immunoblots were developed by using Amersham Biosciences ECL Plus chemiluminescent detection system (GE Healthcare). The intensities of the bands were quantified using the Fujifilm MultiGuage software.
2.9. Measurement of plasma levels of nitrite/nitrate, endothelin-1, and cytokines
The nitrite/nitrate levels from plasma and cell culture supernatants were analyzed using commercially available Griess reagents as previously described (Datta and Lianos, 1999). The plasma levels of inflammatory cytokines (IL-6, TNF-α, MCP-1) were measured using BIO-RAD multiplex reagents as per manufacturer’s protocol (Bio-rad Endogen, Rockford, IL, USA). The plasma levels of endothelin-1 (ET-1) were measured by ELISA using the kit purchased from Assay Designs (Ann Arbor, MI).
2.10. Histological evaluation
Liver and kidney tissue samples were fixed in 10% neutral buffered formalin, dehydrated and embedded in paraffin. 5 μm sections were cut and stained with hematoxylin and eosin. The evaluation of tissue samples slides was carried out by an un-biased pathologist. Histological changes due to acute tubular damage were quantified by counting the percentage of tubules that displayed tubular vacuolization and dilatation are as follows: 0 = none, 1 = <10%, 2 = 11–25%, 3 = 26–45%, 4 = 46–75%, and 5 = >76%. At least 20 fields (200×) were reviewed for each slide.
2.11. Statistical analysis
Results were expressed as mean ± standard deviation (SD). The statistical analyses were performed using Sigmaplot software version 11 (Systat Software, San Jose, CA, USA). P values were determined using ANOVA followed by Tukey test. P < 0.05 was considered statistically significant.
3. Results
3.1. LPS induces lipid peroxidation in plasma and tissues from WT, but not IRAK-1 deficient mice
We have previously documented that IRAK-1 deficient mice are partially protected from LPS-induced mortality (Maitra et al., 2009a). To determine whether LPS may differentially contribute to tissue oxidative damage in WT and IRAK-1 deficient mice, we measured the lipid peroxidation in plasma collected from mice, 10 h after LPS injection. As shown in Fig. 1A, the levels of plasma lipid peroxidation rose significantly in WT, but not IRAK-1 deficient mice. Similarly, we examined the levels of lipid peroxidation in vital organs including liver, brain, and kidney. We observed consistently and significantly elevated lipid peroxidation levels in vital organs from WT, but not IRAK-1 deficient mice (Fig. 1B–D).
Fig. 1.

The effect of IRAK-1 deletion on lipid peroxidation levels in vital organs. Organs were harvested 10 h after either PBS or LPS (25 mg/kg) injection. Lipid peroxidation levels in (A) plasma, (B) liver, (C) kidney and (D) brain were measured and expressed as mean ± SD. (N = 4) from three independent experiments. *P < 0.05.
3.2. LPS suppresses anti-oxidative enzymes in tissues from WT, but not IRAK-1deficient mice
Since tissue oxidation is regulated by opposing forces of oxidative and anti-oxidative activities, we measured the levels of anti-oxidative activities in vital organs harvested from mice injected with LPS. First, we measured the levels of reduced glutathione, a key reducing agent in vital organs. As shown in Fig. 2, LPS injection significantly reduced the levels of glutathione in liver (~24% reduction), kidney (~13% reduction) and brain from WT mice. In contrast, LPS injection had no significant effect on the levels of glutathione from tissues harvested from IRAK-1 deficient mice.
Fig. 2.

IRAK-1 deletion prevents LPS-induced GSH reduction. The levels of reduced GSH following either PBS or LPS injection were measured in liver (A), kidney (B) and brain (C) of WT and IRAK-1 deficient mice. Data are expressed as mean ± SD (N = 4) from three independent experiments. *P < 0.05.
Secondly, we measured the enzymatic activities of anti-oxidative enzymes including glutathione peroxidase (GPx), catalase, and superoxide dismutase in vital organs. As shown in Fig. 3, LPS injection caused a significant reduction of GPx activities in kidney (35.5%), liver tissues (16.5%), and brain tissues(8.5%), as well as a reduction of catalase activities (Fig. 4) in kidney and liver tissues. Additionally, we observed a reduction in superoxide dismutase activities (kidney, 25%; brain, 10.5%)in WT mice (Fig. 5). In contrast, LPS failed to reduce these enzymatic activities in all of the tissues from IRAK-1 deficient mice. Mechanistically, we observed that LPS injection led to reduced expression of GPx and catalase in WT, but not IRAK-1 deficient tissues (Fig. 6).
Fig. 3.

IRAK-1 is required for LPS-mediated suppression of GPx. The activities of GPx following either LPS or PBS injection were measured in liver (A), kidney (B) and brain (C) tissues of WT and IRAK-1 deficient mice. Data is expressed as mean ± SD (N = 4) of at least three independent experiments. *P < 0.05.
Fig. 4.
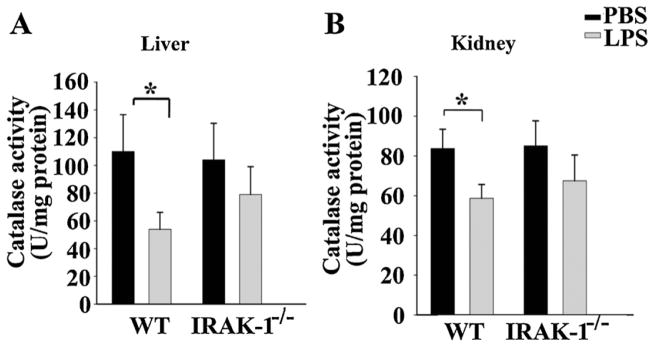
IRAK-1 is involved in LPS-mediated suppression of tissue catalase activity. Mice were injected i.p. with either LPS or PBS. Organs were harvested after 10 h. The catalase activities in liver (A) and kidney (B) tissues were measured. Each data point represents the mean ± SD (N = 4) of at least three independent experiments. *P < 0.05.
Fig. 5.
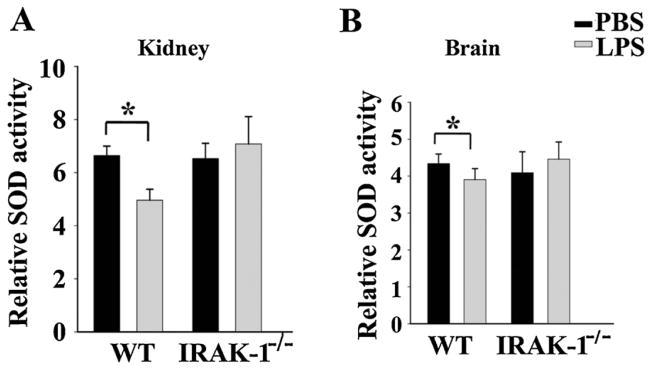
IRAK-1 is involved in LPS-mediated suppression of SOD. The levels of SOD activity in the kidney (A) and brain (B) tissues from WT and IRAK-1 deficient mice injected with either PBS or LPS were measured. Data is expressed as mean ± SD (N = 4) from at least three independent experiments. *P < 0.05.
Fig. 6.

IRAK-1 is required for LPS-mediated suppression of GPX3 and catalase. The message levels of GPX3 and catalase from liver and kidney tissues of WT and IRAK-1 deficient mice injected with either PBS or LPS were measured by real-time RTPCR. Each data point represents the mean ± SD (N = 4) of at least three independent experiments. *P < 0.05.
3.3. IRAK-1 is involved in ROS production following LPS challenge
To determine whether IRAK-1 is involved in LPS-induced production of ROS, we measured the amount of superoxide produced in response to LPS in bone-marrow-derived macrophages (BMDMs) harvested from WT and IRAK-1 deficient mice. As shown in Fig. 7, LPS challenge (100 ng/ml) resulted in a significant increase in superoxide production in WT BMDM (4.5 fold increase), as compared to a minimal increase (1.8 fold) in IRAK-1−/− BMDM.
Fig. 7.

IRAK-1 deletion inhibits the production of ROS in vitro in cultured cells following LPS challenge. WT and IRAK-1 deficient BMDMs were seeded at a density of 1.5 × 105/well in 96 well plate. Cells were treated with or without LPS (100 ng/ml) in the presence of WST-1 (250 μM). Change in absorbance over time was calculated and results are expressed as folds of relative induction. *P < 0.05.
3.4. LPS-mediated p47phox membrane translocation is compromised in IRAK-1 deficient cells
Mechanistically, the generation of reactive oxygen species can be partly attributed to the activation of NADPH oxidase. In order to determine whether LPS may differentially activate NADPH oxidase in tissues from WT and IRAK-1 deficient mice, we examined the membrane translocation of p47phox following LPS challenge in WT and IRAK-1 deficient murine BMDMs. As shown in Fig. 8, LPS treatment led to significant membrane accumulation of p47phox. In contrast, there was much less p47 membrane translocation in IRAK-1 deficient cells.
Fig. 8.

IRAK-1 is involved in the translocation of p47phox to the cellular membrane. WT and IRAK-1 deficient BMDM cells (10 × 106 cells) were treated with or without LPS (100 ng/ml) for 30 min at 37 °C. Equal amount of cell membrane fraction were resolved on SDS-PAGE and probed with an antibody against p47phox. The intensities of the p47phox bands were quantified by densitometry. These experiments were performed in duplicate.
3.5. NO production induced by LPS was significantly reduced in IRAK-1 deficient cells and plasma
Since both ROS and RNS contribute to oxidative tissue damage, we examined plasma nitrite levels as an indication of RNS. As shown in Fig. 9A, LPS injection led to a significant elevation of plasma nitrite levels in WT mice. Meanwhile, the induced nitrite levels were dramatically lower in IRAK-1 deficient mice (~50% reduction as compared to WT). LPS contributes to the accumulation of RNS through inducing the expression of inducible nitric oxide synthase (iNOS). Thus, we subsequently measured the levels of iNOS in WT and IRAK-1 deficient cells treated with LPS. As shown in Fig. 9B, LPS induced iNOS levels were dramatically reduced in IRAK-1 deficient murine embryonic fibroblasts (MEF) cells (~70% reduction as compared to WT cells). Similar reduction in nitrite levels was observed in IRAK-1−/− BMDM and microglia cells (Fig. 9C and D).
Fig. 9.

The induction of NO by LPS was significantly reduced in IRAK-1 deficient cells and plasma. (A) The plasma nitrite levels in WT and IRAK-1 deficient mice (N = 4) were analyzed 10 h after LPS challenge (25 mg/kg). The experiments were repeated 3 times. Data is represented as mean ± SD. *P < 0.05. (B–D) In vitro production of NO in cultured primary cells is IRAK-1 dependent. Cultured MEF (B), BMDM (C) and microglia (D) from WT and IRAK-1 deficient mice were treated with or without LPS for 16 h. The levels of nitrite were measured using Griess reagent. Experiments were performed in triplicate at least 4 times. Data is expressed as mean ± SD. *P < 0.05.
3.6. Reduced production of pro-inflammatory mediators in IRAK-1 deficient mice
Oxidative damage and tissue inflammation often accompanies the expression of pro-inflammatory mediators. Thus, we examined the levels of representative cytokines in plasma samples from WT and IRAK-1 deficient mice treated with LPS. As shown in Fig. 10, LPS injection led to a significant induction of IL-6, MCP-1, and TNF in WT mice. In contrast, the magnitude of induction by LPS was dramatically reduced in IRAK-1 deficient mice TNF: ~33% reduction, MCP-1: ~50% reduction and IL-6: ~40% reduction. In addition, we examined the plasma levels of endothelin-1 (ET-1), a key pro-inflammatory mediator involved in tissue damage during septic shock (Haynes and Webb, 1998). As shown in Fig. 10D, LPS treatment significantly induced plasma levels of ET-1 in WT, but not in IRAK-1 deficient mice (60% reduction).
Fig. 10.

Reduced production of pro-inflammatory mediators in IRAK-1 deficient mice. The plasma levels of TNF-α (A), MCP-1(B), IL-6(C) and ET-1 (D) from WT and IRAK-1 deficient mice injected with either PBS or LPS were measured (N = 4). The data is represented as a dot plot, indicating data for each individual sample. *P < 0.05.
3.7. Reduced tissue damage in IRAK-1 deficient mice
Elevated tissue oxidative damages can eventually lead to multi-organ failure. Thus, we examined the morphological changes of vital organs from mice injected with LPS. As shown in Fig. 11, LPS injection caused significant neutrophil infiltration (10 h) and necrotic foci (16 h) in hepatic parenchyma whereas extensive tubular vacuolization and dilatation with few neutorphilic cells were evident in liver and kidney, respectively, in WT mice. In contrast, there was significantly less neutrophil infiltration and tubular changes were observed in liver and kidney tissues from IRAK-1 deficient mice.
Fig. 11.

Reduced tissue damage in IRAK-1 deficient mice following endotoxemia. (A) H&E staining of liver tissues from WT and IRAK-1 deficient mice injected with LPS (25 mg/kg) or PBS. Histopathological changes in the liver were evaluated 10 and 16 h following LPS challenge. Time-dependent hepatic injury was observed in the liver, characterized by marked neutrophilic infiltration and extensive necrotic foci throughout WT liver parenchyma. (B) H&E staining of kidney tissues from WT and IRAK-1 deficient mice injected with LPS (25 mg/kg) or PBS. Light microscopic evaluation of the H&E stained kidney in both control groups (WT and IRAK-1−/−) showed regular morphology of renal parenchyma with well-defined glomeruli and tubules. Degenerative changes in tubules along with neutrophilic infiltration were observed in LPS-injected WT kidney tissues.
4. Discussion
In this report, we have provided first-hand evidence demonstrating the in vivo beneficial effect of IRAK-1 deficiency in LPS-induced oxidative tissue damage and injury. Mechanistically, we observed that LPS dramatically reduces the levels and activities of anti-oxidative enzymes in vital organs in WT mice, but to a significantly lesser degree in IRAK-1 deficient mice. IRAK-1 deficiency also reduces LPS-triggered p47phox membrane translocation, a process necessary for the assembly of active NADPH oxidase. Furthermore, IRAK-1 is involved in LPS induced expression of iNOS and generation of reactive nitrogen species. Collectively, our findings demonstrate the reduced damage to vital organs following LPS challenge in IRAK-1 deficient mice.
Free radicals including ROS and RNS generated during endotoxemia play a vital role in the pathogenesis of sepsis (Crimi et al., 2006). Potential sources of ROS include the respiratory burst associated with activated NADPH oxidase in neutrophils, macrophages and other innate immune cells which generate reactive oxygen species (Parihar et al., 2008; Guo and Ward, 2007). In addition, excessive NO generated by elevated iNOS in the vicinity of ROS can lead to the formation of peroxynitrite, a key form of RNS (Fink, 2002). Both ROS and RNS can in turn react with cellular macromolecules and lead to oxidative damage. Although the local generation of free radicals is a function of the host defense system to kill invading pathogens, excessive production of free radicals during disseminated bacterial infection and sepsis can backfire and cause significant damage to host tissues. Strategies aimed at reducing the production of ROS and RNS during acute sepsis may hold promise in decreasing the high mortality rate. To this regard, our current findings reveal that IRAK-1 may be a viable therapeutic target for such circumstances, since deletion of IRAK-1 significantly reduced the generation of free radicals and tissue damages following a lethal LPS challenge. Our study extends previous in vitro studies performed by us and others demonstrating the involvement of IRAK-1 in LPS-mediated intracellular signaling events (Arcaroli et al., 2006; Glaros et al., 2009; Liu et al., 2008; Maitra et al., 2009b). However, the in vivo relevance of these in vitro findings in terms of oxidative tissue damage has never been addressed. This current study provides in vivo data demonstrating reduced levels of lipid peroxidation in circulating blood as well as in vital organs including brain, liver, and kidney of IRAK-1 deficient mice challenged with LPS. Furthermore, we demonstrated that the plasma nitrite levels were dramatically reduced in IRAK-1 deficient mice as compared to WT controls. Consequently, we observed significantly less tissue damage and higher survival rates in IRAK-1 deficient mice as compared to WT mice challenged with a lethal dose of LPS.
Our study also revealed the novel contribution of IRAK-1 to the regulation of antioxidative enzymes and scavenging activities for free radicals in vivo. We demonstrated that LPS significantly decreased the levels and activities of critical antioxidative enzymes including SOD, GPx and catalase in vital organs such as brain, kidney and liver. In contrast, LPS failed to suppress these enzyme activities in IRAK-1 deficient mice.
In addition to the generation of ROS and RNS, LPS challenge also leads to elevated expression of pro-inflammatory cytokines and other inflammatory mediators such as endothelin-1. ET-1 can further exacerbate tissue damage through the activation of neutrophils and other leukocytes, resulting in endothelial cell death and the induction of ROS (Wong et al., 2000). Here, we have demonstrated that IRAK-1 is critically involved in LPS-induced expression of pro-inflammatory cytokines, as well as ET-1. Our data indicate that IRAK-1 serves as a key control point during the LPS signaling processes that contribute to the elevated oxidative tissue damage during septic shock.
Collectively, our results demonstrate that IRAK-1 is an important regulator of the host inflammatory and oxidative stress response during LPS-mediated sepsis and septic shock. By inducing the expression of inflammatory cytokines, oxidant enzymes, and iNOS, as well suppressing the activities of anti-oxidative enzymes, IRAK-1 plays a crucial role in the pathogenesis of septic shock and multi-organ failure.
Acknowledgments
We thank Samantha Chang for technical assistance and critical reading. This work is partially supported by grants from NIH to LL.
Abbreviations
- BHT
butylated hydroxy toulene
- CCU
critical care unit
- ET-1
endothelin-1
- GPx
glutathione peroxidase
- HNE
hydrononenal
- IL-6
interleukin-6
- iNOS
inducible nitric oxide synthase
- IRAK-1
interleukin-1 receptor associated kinases
- LPS
lipopolysaccharide
- MCP-1
monocyte chemotactic protein-1
- MDA
malondialdehdye
- MEF
murine embryonic fibroblast
- MODS
multiple organ dysfunction syndrome
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SOD
superoxide dismutase
- TLR-4
Toll-like receptor-4
- TNF-α
tumor necrosis factor-alpha
- WST-2
(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium
- WT
wild type
Footnotes
Author disclosure statement
No competing financial interests exist.
References
- Akira S. Toll-like receptors and innate immunity. Adv Immunol. 2001;78:1–56. doi: 10.1016/s0065-2776(01)78001-7. [DOI] [PubMed] [Google Scholar]
- Andreasen AS, Andreasen AS, Krabbe KS, Krogh-Madsen R, Taudorf S, Pedersen BK, Moller K. Human endotoxemia as a model of systemic inflammation. Curr Med Chem. 2008;15:1697–1705. doi: 10.2174/092986708784872393. [DOI] [PubMed] [Google Scholar]
- Arcaroli J, Silva E, Maloney JP, He Q, Svetkauskaite D, Murphy JR, Abraham E. Variant IRAK-1 haplotype is associated with increased nuclear factor-kappaB activation and worse outcomes in sepsis. Am J Respir Crit Care Med. 2006;173:1335–1341. doi: 10.1164/rccm.200603-341OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basini G, Basini G, Simona B, Santini SE, Grasselli F. Reactive oxygen species and anti-oxidant defences in swine follicular fluids. Reprod Fertil Dev. 2008;20:269. doi: 10.1071/rd07147. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases, physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bone RC. Why sepsis trials fail. JAMA. 1996;276:565–566. [PubMed] [Google Scholar]
- Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190:255–266. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Carrillo-Vico A, Lardone PJ, Naji L, Fernandez-Santos JM, Martin-Lacave I, Guerrero JM, Calvo JR. Beneficial pleiotropic actions of melatonin in an experimental model of septic shock in mice, regulation of pro-/anti-inflammatory cytokine network, protection against oxidative damage and anti-apoptotic effects. J Pineal Res. 2005;39:400–408. doi: 10.1111/j.1600-079X.2005.00265.x. [DOI] [PubMed] [Google Scholar]
- Crimi E, Crimi E, Sica V, Slutsky AS, Zhang H, Williams-Ignarro S, Ignarro LJ, Napoli C. Role of oxidative stress in experimental sepsis and multisystem organ dysfunction. Free Radic Res. 2006;40:665–672. doi: 10.1080/10715760600669612. [DOI] [PubMed] [Google Scholar]
- Datta PK, Lianos EA. Retinoic acids inhibit inducible nitric oxide synthase expression in mesangial cells. Kidney Int. 1999;56:486–493. doi: 10.1046/j.1523-1755.1999.00576.x. [DOI] [PubMed] [Google Scholar]
- Dusi S, Donini M, Rossi F. Mechanisms of NADPH oxidase activation in human neutrophils, p67phox is required for the translocation of rac 1 but not of rac 2 from cytosol to the membranes. Biochem J. 1995;308:991–994. doi: 10.1042/bj3080991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink MP. Reactive oxygen species as mediators of organ dysfunction caused by sepsis, acute respiratory distress syndrome, or hemorrhagic shock, potential benefits of resuscitation with Ringer’s ethyl pyruvate solution. Curr Opin Clin Nutr Metab. 2002;5:167–174. doi: 10.1097/00075197-200203000-00009. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Latimer RD, Gray BM, Harwood RJ, Oduro A. Endotoxin-induced organ injury. Crit Care Med. 1993;21:19–24. doi: 10.1097/00003246-199302001-00005. [DOI] [PubMed] [Google Scholar]
- Glaros T, Larsen M, Li L. Macrophages and fibroblasts during inflammation, tissue damage and organ injury. Front Biosci. 2009;14:3988–3993. doi: 10.2741/3506. [DOI] [PubMed] [Google Scholar]
- Guo RF, Ward PA. Role of oxidants in lung injury during sepsis. Antioxid Redox Signal. 2007;9:1991–2002. doi: 10.1089/ars.2007.1785. [DOI] [PubMed] [Google Scholar]
- Haynes WG, Webb DJ. Endothelin as a regulator of cardiovascular function in health and disease. J Hypertens. 1998;16:1081–1098. doi: 10.1097/00004872-199816080-00001. [DOI] [PubMed] [Google Scholar]
- Hill HD, Straka JG. Protein determination using bicinchoninic acid in the presence of sulfhydryl reagents. Anal Biochem. 1988;170:203–208. doi: 10.1016/0003-2697(88)90109-1. [DOI] [PubMed] [Google Scholar]
- Karima R, Matsumoto S, Higashi H, Matsushima K. The molecular pathogenesis of endotoxic shock and organ failure. Mol Med Today. 1999;5:123–132. doi: 10.1016/s1357-4310(98)01430-0. [DOI] [PubMed] [Google Scholar]
- Liu B, Qin L, Yang SN, Wilson BC, Liu Y, Hong JS. Femtomolar concentrations of dynorphins protect rat mesencephalic dopaminergic neurons against inflammatory damage. J Pharmacol Exp Ther. 2001;298:1133–1141. [PubMed] [Google Scholar]
- Liu G, Park YJ, Abraham E. Interleukin-1 receptor-associated kinase (IRAK)-1-mediated NF-kappaB activation requires cytosolic and nuclear activity. FASEB J. 2008;22:2285–2296. doi: 10.1096/fj.07-101816. [DOI] [PubMed] [Google Scholar]
- Maier RV. Pathogenesis of multiple organ dysfunction syndrome – endotoxin, inflammatory cells, and their mediators, cytokines and reactive oxygen species. Surg Infect (Larchmt) 2000;1:197–204. doi: 10.1089/109629600750018123. (Discussion 204–205) [DOI] [PubMed] [Google Scholar]
- Maitra U, Chang S, Singh N, Li L. Molecular mechanism underlying the suppression of lipid oxidation during endotoxemia. Mol Immunol. 2009;47:420–425. doi: 10.1016/j.molimm.2009.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maitra U, Singh N, Gan L, Ringwood L, Li L. IRAK-1 contributes to LPS-induced ROS generation in macrophages by inducing NOX-1 transcription, Rac1 activation, and suppressing the expression of anti-oxidative enzymes. J Biol Chem. 2009b;284:35403–35411. doi: 10.1074/jbc.M109.059501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien JM, Jr, Ali NA, Abraham E. Year in review 2007, critical care -multiple organ failure and sepsis. Crit Care. 2000;12:228. doi: 10.1186/cc6950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar A, Parihar MS, Milner S, Bhat S. Oxidative stress and anti-oxidative mobilization in burn injury. Burns. 2008;34:6–17. doi: 10.1016/j.burns.2007.04.009. [DOI] [PubMed] [Google Scholar]
- Peskin AV, Winterbourn CC. A microtiter plate assay for superoxide dismutase using a water-soluble tetrazolium salt (WST-1) Clin Chim Acta. 2000;293:157–166. doi: 10.1016/s0009-8981(99)00246-6. [DOI] [PubMed] [Google Scholar]
- Rada B, Leto TL. Oxidative innate immune defenses by Nox/Duox family NADPH oxidases. Contrib Microbiol. 2008;15:164–187. doi: 10.1159/000136357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringwood L, Li L. The involvement of the interleukin-1 receptor-associated kinases (IRAKs) in cellular signaling networks controlling inflammation. Cytokine. 2008;42:1–7. doi: 10.1016/j.cyto.2007.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojiljkovic V, Todorovic A, Radlovic N, Pejic S, Mladenovic M, Kasapovic J, Pajovic SB. Antioxidant enzymes, glutathione and lipid peroxidation in peripheral blood of children affected by coeliac disease. Ann Clin Biochem. 2007;44:537–543. doi: 10.1258/000456307782268075. [DOI] [PubMed] [Google Scholar]
- Ueda J, Starr ME, Takahashi H, Du J, Chang LY, Crapo JD, Evers BM, Saito H. Decreased pulmonary extracellular superoxide dismutase during systemic inflammation. Free Radic Biol Med. 2008;45:897–904. doi: 10.1016/j.freeradbiomed.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenca SS, Silva Bezerra F, Lopes AA, Romana-Souza B, Marinho Cavalcante MC, Lima AB, Goncalves Koatz VL, Porto LC. Oxidative stress in mouse plasma and lungs induced by cigarette smoke and lipopolysaccharide. Environ Res. 2008;108:199–204. doi: 10.1016/j.envres.2008.07.001. [DOI] [PubMed] [Google Scholar]
- Walrand S, Valeir S, Rodriquez C, Ligot P, Chassagne J, Vasson M-P. Flow cytometry study of polymorphonuclear neutrophil oxidative burst: a comparison of three fluorescent probes. Clin Chim Acta. 2003;331(1–2):103–110. doi: 10.1016/s0009-8981(03)00086-x. [DOI] [PubMed] [Google Scholar]
- Wang D, Fasciano S, Li L. The interleukin-1 receptor associated kinase 1 contributes to the regulation of NFAT. Mol Immunol. 2008;45:3902–3908. doi: 10.1016/j.molimm.2008.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel RP, Pinsky MR, Ulevitch RJ, Young L. Current understanding of sepsis. Clin Infect Dis. 1996;22:407–412. doi: 10.1093/clinids/22.3.407. [DOI] [PubMed] [Google Scholar]
- Wong PM, Chugn SW, Sultzer BM. Genes, receptors, signals and responses to lipopolysaccharide endotoxin. Scand J Immunol. 2000;51:123–127. doi: 10.1046/j.1365-3083.2000.00689.x. [DOI] [PubMed] [Google Scholar]