

Abstract
The rapid development of programmable site-specific endonucleases has led to a dramatic increase in genome engineering activities for research and therapeutic purposes. Specific loci of interest in the genomes of a wide range of organisms including mammals can now be modified using zinc-finger nucleases (ZFNs), transcription activator-like endonucleases (TALENs), and CRISPR-associated Cas9 endonucleases in a site-specific manner, in some cases requiring relatively modest effort for endonuclease design, construction and application. While these technologies have made genome engineering widely accessible, the ability of programmable nucleases to cleave off-target sequences can limit their applicability and raise concerns about therapeutic safety. In this article we review methods to evaluate and improve the DNA cleavage activity of programmable site-specific endonucleases and describe a procedure for a comprehensive off-target profiling method based on the in vitro selection of very large (~1012-membered) libraries of potential nuclease substrates.
1.1. Introduction to programmable nucleases for genome editing
Programmable site-specific nucleases such as zinc-finger nucleases (ZFNs), transcription activator-like endonucleases (TALENs), and CRISPR-associated Cas9 nucleases can be designed to target any gene of interest and therefore are powerful research tools with significant therapeutic implications. In cells, a targeted double-strand break can lead to gene modification or insertion through homology-directed repair (HDR) with exogenous DNA or to gene knockout via non-homologous end-joining (NHEJ). In the HDR pathway, the creation of a double-strand break at a chromosomal DNA locus by a sequence-specific endonuclease can increase the efficiency of insertion of an exogenous donor DNA template by several orders of magnitude (Choulika, Perrin et al. 1995). If no donor template is provided, endogenous NHEJ pathways that repair the break will often introduce missense mutations that abrogate production of functional protein product (Lukacsovich, Yang et al. 1994; Rouet, Smih et al. 1994). Programmable nucleases have been used to modify the genomes of a variety of organisms and human cell lines, as has been reviewed extensively (Carroll 2011; Joung and Sander 2013; Sander and Joung 2014). In addition to engineering the genomes of cells or organisms for direct biological interrogation, genetic screens have recently been performed with these enzymes in human tissue culture to uncover genetic factors underlying specific cellular processes in an unbiased manner (Koike-Yusa, Li et al. 2014; Shalem, Sanjana et al. 2014; Wang, Wei et al. 2014; Zhou, Zhu et al. 2014).
These nucleases also serve as the promising basis of a new generation of human therapeutics. Clinical trials of two site-specific nucleases are currently underway as potential treatments for HIV and glioblastoma. Researchers at Sangamo BioSciences are conducting two phase 1 and one phase 1/2 clinical trials using a zinc finger nuclease (ZFN) that targets a sequence in the CCR5 gene (Tebas, Stein et al. 2014). CCR5 is a co-receptor used by HIV in early stage infection (Scarlatti, Tresoldi et al. 1997), and mutation of CCR5 (CCR5Δ32) is known to confer resistance to HIV infection (Huang, Paxton et al. 1996; Liu, Paxton et al. 1996; Samson, Libert et al. 1996).
The second ZFN in clinical trials, also led by Sangamo BioSciences, disrupts the gene for the glucocorticoid receptor (Reik, Zhou et al. 2008) as part of a potential treatment for glioblastoma. The target cells for the ZFN are T cells modified by other methods to express a cell-surface receptor that specifically recognizes malignant glioblastoma cells (Kahlon, Brown et al. 2004). The therapeutic cells, however, are rendered inactive by glucocorticoids, which are often also a component of therapy. ZFN-mediated modification of the glucocorticoid receptor in the therapeutic cells confers resistance to glucocorticoid treatment, while maintaining anti-glioblastoma activity, allowing the cells to recognize their malignant targets. These and other examples demonstrate that, in addition to serving as powerful research tools, programmable nucleases are promising platforms for clinically relevant genetic manipulation.
1.2. Overview of methods to study specificity of genome editing agents
Specificity is a crucial feature of programmable endonucleases, and a high (though currently undefined) level of specificity is desired for the vast majority of therapeutic applications. Until recently, however, few methods existed to study the DNA cleavage specificity of active, site-specific nucleases. An ideal study of off-target activities of site-specific endonucleases would measure nuclease activity against each of the >109 potential off-target sites for every target site in the human genome. While whole exome sequencing has been used in studies of site-specific endonuclease specificity (Li, Huang et al. 2011; Ding, Lee et al. 2013; Cho, Kim et al. 2014), sequencing offers limited sensitivity in detecting rare off-target events and exomes represent only a small fraction of genomic DNA containing potential off-target sites. Therefore, the general study of off-target activities of site-specific endonucleases has relied on the experimental identification of likely off-target sites. Off-target studies have taken one of three general forms: discrete off-target site testing, genome-wide selections, and minimally biased in vitro selections (Figure 1).
Figure 1. Overview of methods to study the specificity of nucleases.
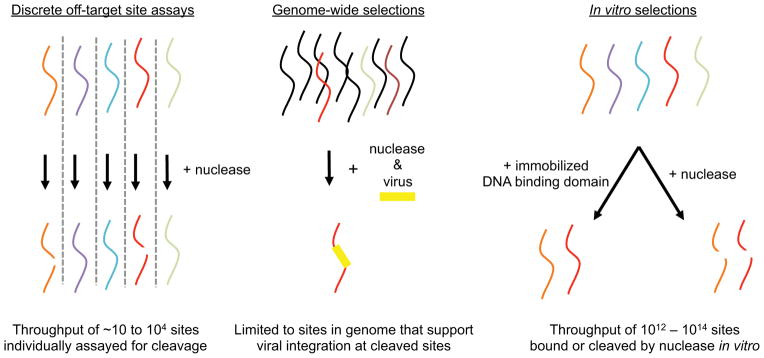
Potential substrate sequences of interest (colored strands) are subjected to nuclease cleavage to identify cleaved sequences (broken red and orange strands). In discrete off-target site assays, sequences are individually subjected to nuclease cleavage in a low- or high-throughput manner. In genome-wide selections, a few potential off-target sites are cleaved within predominantly uncleaved genomic DNA (black strands) and detected by viral integration. Using in vitro selection, many potential off-target sites in a vast DNA library are selected for binding or for their ability to be bound or cleaved site-specific nucleases in vitro.
1.2.1. Discrete off-target site testing
Perhaps the most obvious approach to evaluating the sequence specificity of nucleases is by assaying discrete potential off-target substrates, either in a low- or high-throughput format. While the methods summarized below are not a comprehensive list of such efforts, they are representative examples of this strategy.
Homing endonucleases such as I-SceI were the subjects of some of the earliest studies of the specificity of nucleases that recognize sites sufficiently long to be unique in the human genome, even though the presence of integrated binding and cleavage domains complicates engineering homing endonucleases with tailor-made specificities (Gimble, Moure et al. 2003; Chen and Zhao 2005; Doyon, Pattanayak et al. 2006; Chen, Wen et al. 2009). In early studies of I-SceI homing endonuclease specificity, Dujon and coworkers interrogated a subset of the 54 potential single-mutant individual off-target sequences of the 18 base pair target site (Colleaux, D'Auriol et al. 1988).
The throughput of this approach was increased in the multitarget ELISA method developed by Barbas and coworkers (Segal, Dreier et al. 1999), in which 96 biotinylated oligonucleotides or oligonucleotide pools are plated individually in the streptavidin-coated wells of a 96-well plate. Fusions to maltose-binding protein of a DNA-binding domain of interest are incubated with the oligonucleotides in the wells. After a wash step to remove unbound protein, the wells are incubated with a primary antibody that recognizes maltose-binding protein, followed by a secondary antibody that allows visualization of wells containing bound protein.
Church and coworkers (Bulyk, Huang et al. 2001) have used a microarray approach to study zinc finger DNA-binding specificity. They prepared DNA microarrays containing all 64 possible three-base pair subsequences within a longer target site. The microarrays were incubated with M13 phage displaying the DNA-binding domain of interest, washed, and visualized with primary and secondary antibody staining to reveal DNA-binding specificities. A variant of this method, developed by Bulyk and coworkers (Philippakis, Qureshi et al. 2008), has been extended to profiling ten-base pair subsequences of transcription factor binding sites. Another microarray-based method (Carlson, Warren et al. 2010) has also been used to profile the DNA-binding specificity of engineered zinc fingers.
More recently, discrete testing of potential single- and double-mutant off-target sequences has been used in human cells to study the sequence preferences of Cas9. In these methods, a single target site in human cells is assayed for its ability to be modified through non-homologous end-joining by a set of endonucleases that are targeted to cleave either the target site or discrete single- or multiple-mutant variants of the target site. At least two separate studies have used this strategy. In one study by Joung and coworkers, an eGFP reporter is the target of a collection of Cas9:guide RNA complexes containing mutant (mismatched) guide RNAs (Fu, Foden et al. 2013). In this approach, off-target endonuclease activity leads to the loss of cellular GFP expression. A second study, developed by Zhang and coworkers (Hsu, Scott et al. 2013), assayed the ability of a set of Cas9:guide RNA complexes to cleave the EMX1 gene. Cleavage activity was detected as NHEJ events at the EMX1 locus using high-throughput sequencing. Although if Cas9 cleaved with perfect specificity the site would not be modified by Cas9:guide RNA complexes containing mutated guide RNAs, many of the mutated guide RNAs resulted in NHEJ, thereby demonstrating off-target activity. In both methods, other potential genomic off-target sites are extrapolated from the small set of off-target sites directly screened. The results of this approach applied to Cas9 are summarized in section 1.5 below and further demonstrate the utility of simple, discrete-off-target site testing to identify genomic off-target sites.
1.2.2. Genome-wide selections
In contrast to discrete screening assays of potential off-target sequences to be cleaved by a nuclease of interest, genome-wide selections have also been used to identify those sequences in a population of human cells that can bind to or are cleaved by a nuclease of interest. In assessments of genome-wide binding of Cas9, Adli, Sharp, Zhang, and their respective colleagues (Kuscu, Arslan et al. 2014; Wu, Scott et al. 2014) used chromatin immunoprecipitation followed by sequencing (ChIP-seq) to study the ability of inactive Cas9 to bind off-target sequences in the genome. In this method, hemagglutinin-tagged, catalytically inactive Cas9 is expressed in human cells. A crosslinking step covalently attaches the tagged Cas9 to any DNA target sites it is bound to in the cell. The bound DNA is then fractionated, the crosslinks are reversed, and high-throughput sequencing of the resulting DNA reveals the genomic sequences bound by Cas9. While these studies show that Cas9 is capable of extensively binding off-target sites, they also suggest that most of the off-target sites bound are not modified.
Genome-wide selections for DNA cleavage, rather than binding alone, have been achieved by exploiting the tendency of certain viruses to preferentially integrate at sites of double-strand breaks. The endonuclease of interest is expressed in cultured human cells, creating double-strand breaks at cleaved genomic sites. Cells are then exposed to a virus that preferentially integrates at double-strand breaks. Genomic DNA sequences containing integrated virus are then identified through selection or direct DNA sequencing. In a selection method developed by Miller and coworkers, adeno-associated virus packaged with antibiotic resistance markers and an E. coli plasmid origin is used as an integration marker (Petek, Russell et al. 2010). Any on-target and off-target substrates in the genome containing the integration marker would therefore contain a plasmid origin and antibiotic resistance markers. Genomic DNA is then isolated from infected cells, fragmented with a cocktail of restriction enzymes, circularized, and transformed into E. coli. Only fragments containing integrated adeno-associated virus have an E. coli origin of replication and the appropriate antibiotic resistant markers, and therefore only fragments containing integrated virus survive. Sequencing of the plasmid reveals the viral-chromosomal junctions, which contain the off-target sites of the endonuclease. Subsequent studies by von Kalle, Tolar, and their respective coworkers to study the specificities of ZFNs and TALENs have extended this approach, using instead integrase-deficient lentiviral vectors (IDLVs) and read-out of integration sites using high-throughput sequencing (Gabriel, Lombardo et al. 2011; Osborn, Starker et al. 2013).
Advantages of viral integration methods include their abilities to study specificity directly in the context of the target genome and the unbiased nature of the selection, allowing for identification of off-target sites that are not highly similar in sequence to the on-target site. Results from these methods should be interpreted carefully, however, as integration can occur at double-strand breaks that arise naturally, independent of nuclease activity. As with whole genome sequencing, viral integration methods may not be sufficiently sensitive to detect low-level off-target modification. In addition, abstraction of general properties of endonuclease specificity could be complicated by cellular factors such as DNA accessibility, which varies from site to site and between cell types (Maeder, Thibodeau-Beganny et al. 2008; Wu, Scott et al. 2014).
1.2.3. Minimally biased selections in vitro and in cells
The most general method to determine site-specific endonuclease specificity would test the activity of a given endonuclease against each potential off-target sequence. Since therapeutic endonucleases target long sequences (= ~20 base pairs) to ensure uniqueness in the genome, a truly comprehensive specificity study would require an assay with at least 420 (~ 1 x 1012) different substrates. Since libraries of this size are challenging to generate and process even using in vitro methods, selections to determine site-specific endonuclease specificity either rely on the use of “minimally biased” libraries or focus on smaller subsets of the DNA substrate to be studied. Minimally biased libraries are randomized across the nucleotide positions being studied, but the composition of nucleotides at each position is biased towards the target sequence rather than fully randomized. For example, if a particular target site of a three-base pair specific endonuclease is ATG, a fully randomized library would contain equal proportions of all sequences (NNN). A minimally biased library contains higher proportions of sequences that are similar to the target site. In this example, the most common sequence in the library would be ATG, followed by the single-mutant sequences (cTG, gTG, tTG, AaG, AcG, AgG, ATa, ATc, ATt), the double-mutant sequences, and then triple-mutant sequences, which are the rarest in the library. Biasing is accomplished through the incorporation of mixtures of phosphoramidites at each position during DNA synthesis, such that the on-target base is incorporated at a higher frequency than the other off-target bases (Figure 2a). In other variants of this approach, portions of the sequence are fixed, while subsets are fully randomized (for example, nTG, AnG, or ATn).
Figure 2. In vitro selection scheme for profiling the specificity of site-specific nucleases.

(a) Example sequences biased towards a target sequence for both the left- and right-half sites of TALEN targeting the human CCR5 gene. The on-target sequences are in bold and below are examples of variant sequences from minimally biased libraries. (b) A single-stranded library of DNA oligonucleotides containing partially randomized target sites (grey box) and constant region (thick black line) is circularized, then transformed into concatemeric repeats by rolling circle amplification. The concatemeric repeats of double stranded DNA (double arrows) target site variants are incubated in vitro with a site-specific nuclease of interest. The resulting and blunted ends are ligated to adapter #1. The ligation products are amplified by PCR using one primer consisting of adapter #1 and the other primer consisting of adapter #2–constant sequence, which anneals to the constant regions of the library. From the resulting ladder of amplicons containing 0.5, 1.5, 2.5, … repeats of a target site, amplicons corresponding to 1.5 target-sites in length are isolated by gel purification and subjected to high-throughput DNA sequencing and computational analysis.
Using minimally biased libraries, several methods have studied the binding specificities of monomeric zinc-finger domains, in the absence of cleavage domains and dimeric binding partners. In the bacterial one-hybrid system developed by Wolfe and coworkers (Meng, Brodsky et al. 2005), a DNA target site library is placed upstream of a selectable marker on a plasmid. The DNA-binding domain of interest is expressed in E. coli as a fusion to the α-subunit of RNA polymerase. In each individual bacterium, which each contains only one member of the target site library, RNA polymerase is recruited to the promoter of the selectable marker if the DNA-binding domain is able to bind to the library DNA sequence present. Only cells that have target sites that can be bound by the DNA-binding domain will express the selectable marker and survive. Wolfe and coworkers have used this approach to assay libraries of up to 108 molecules for DNA binding (Meng, Thibodeau-Beganny et al. 2007). A computational structure-based approach developed by Bradley and colleagues (Yanover and Bradley 2011) using the Rosetta algorithm has also been used to study monomeric zinc-finger domain specificity and has accurately predicted DNA-binding profiles that were obtained by the bacterial one-hybrid system.
Church and coworkers recently used a variant of the bacterial one-hybrid approach to study the specificity of Cas9 in human cells (Mali, Aach et al. 2013). In this method, a library of target sites was placed upstream of a reporter gene in a plasmid. Instead of using active Cas9 as an endonuclease in the selection, an inactive variant was expressed as a DNA-binding domain alone, fused to the VP64 activation domain. Therefore, any inactive Cas9 that could bind to a library member caused expression of the reporter gene. The results of this study are summarized in Section 1.5.
Larger libraries, covering more potential off-target sites, have been used to evaluate DNA-binding domain specificity in vitro. Applying an in vitro SELEX approach (Oliphant, Brandl et al. 1989) to large (~1014-membered) randomized DNA target site libraries (Miller, Tan et al. 2011), Struhl and coworkers, and later several other groups, enriched DNA sequences that can bind a given DNA-binding domain of interest (Thiesen and Bach 1990; Zykovich, Korf et al. 2009). In this approach, the DNA-binding domain is immobilized and incubated with a randomized target site library. After washing steps to remove unbound DNA, the bound DNA is eluted, amplified, and cycled through the procedure several times before being sequenced.
The bacterial one-hybrid and SELEX methods described above study DNA-binding domains alone, outside of the context of catalysis. Since site-specific endonucleases involve DNA cleavage in addition to DNA binding, and since DNA-binding specificities may not exactly predict DNA-cleavage specificities, methods to study the specificity of DNA cleavage reactions are desirable. Monnat and coworkers developed a gel electrophoresis-based method (Argast, Stephens et al. 1998) in which an active endonuclease is incubated in vitro with a target site library that was cloned into a circular plasmid. Cleavage of library members results in linearization of the plasmid, and the pool of cleavable, linearized DNA sequences is separated from uncleaved, circular DNA through agarose gel electrophoresis and gel purification. The linear DNAs containing bona fide substrate sequences are ligated back into circles and amplified in E. coli. After several rounds of enrichment of a pre-selection library with a theoretical complexity of 108 to 109 members (constrained by the need to introduce library members into E. coli), the post-selection library is sequenced and analyzed.
To combine the benefits of both large library sizes and the context of cleavage selection, Liu and coworkers developed a fully in vitro selection strategy to profile the DNA cleavage specificity of ZFNs, TALENs, and Cas9 using libraries of 1011–1012 potential off-target sites (Pattanayak, Ramirez et al. 2011; Pattanayak, Lin et al. 2013; Guilinger, Pattanayak et al. 2014). In this strategy, library construction is performed entirely in vitro and therefore is not bottlenecked by cell transformation efficiency. This method, which is described in detail in Section 2 below, uses the generation of 5’ phosphates upon DNA cleavage to selectively tag and amplify library members that are cleaved by nucleases. These cleaved library members are then revealed by high-throughput DNA sequencing (Figure 2b).
When applied to ZFNs, this in vitro DNA cleavage specificity profiling strategy demonstrated that a SELEX study on the specificity of individual DNA-binding domains, in the absence of dimerization and cleavage, did not detect some genomic off-target sites. Analysis of hundreds of thousands of off-target sites cleaved in vitro suggested that interactions between ZFN monomers affect DNA cleavage specificity and explain differences with the SELEX study (Pattanayak, Ramirez et al. 2011). For the CCR5-targeting ZFN described above, the in vitro cleavage selection also identified more genomic off-target sites than a genome-wide selection method on the same ZFN using IDLVs reported by von Kalle and coworkers (Gabriel, Lombardo et al. 2011). However, each method identified off-target sites that were missed by the other method. As was also demonstrated with SELEX(Perez, Wang et al. 2008), the computational analysis of in vitro selection results improves the sensitivity of the in vitro cleavage selection method to determine off-target sites. Joung, Liu, and coworkers (Sander, Ramirez et al. 2013) applied a machine-learning classifier algorithm to in vitro cleavage selection results for the CCR5-targeting ZFN, and identified 26 more off-target sites than had previously been identified, including all of the previously determined off-target sites. These studies collectively demonstrate how in vitro selection methods and genome-wide selection methods can serve as complementary tools in the determination of gene-editing nuclease specificities.
1.3. Insights and improvements from ZFN specificity studies
ZFNs(Kim, Cha et al. 1996) are dimeric fusions of the non-specific FokI restriction endonuclease cleavage domain (Hirsch, Wah et al. 1997) with zinc finger DNA-binding domains (Figure 3a). The FokI cleavage domain must dimerize to be active, therefore ZFNs can cleave DNA only after dimerizing and bridging two half-sites (Vanamee, Santagata et al. 2001) that are separated by an unspecified DNA spacer sequence. Target site specificity is therefore determined by two zinc-finger DNA-binding domains, each of which consist of three or more tandem repeats of individual zinc fingers. Each individual zinc finger recognizes three base pairs (Beerli, Segal et al. 1998), and a zinc finger DNA-binding domain in total recognizes at least nine base pairs. Therefore, in total, zinc-finger nucleases recognize sites that are at least 18 bp long (not including the spacer).
Figure 3. Architecture of ZFN, TALEN, and Cas9 programmable nucleases.

(a) A ZFN monomer is a fusion of a FokI nuclease cleavage domain (purple) to (typically) four adjoined zinc-fingers each targeting three base pairs for a total of 12 base pairs recognized. Two different ZFNs bind their corresponding half-sites, allowing FokI dimerization and DNA cleavage between the half-sites. (b) A TALEN monomer contains an N-terminal domain followed by an array of TALE repeats (filled boxes), a C-terminal domain, and a FokI nuclease cleavage domain (purple). The 12th and 13th amino acids (the RVD, red) of each TALE repeat recognize a specific DNA base pair. Two different TALENs bind their corresponding half-sites, allowing FokI dimerization and DNA cleavage between the half-sites. (c) Cas9 protein (yellow) binds to target DNA in complex with a single guide RNA (sgRNA, green). The S. pyogenes Cas9 protein and sgRNA complex recognizes the PAM sequence NGG (blue). Black triangles indicate the cleavage points in the target DNA three bases from the PAM on both DNA strands.
The DNA-binding specificity of zinc finger nucleases is programmed by the composite individual zinc fingers. Each individual zinc finger consists of a compact ββα fold with a hydrophobic core stabilized by a zinc ion coordinated by two cysteines and two histidines. While a great deal of progress has been reported in the design of zinc fingers that can target any DNA triplet, primarily by Barbas, Joung, Klug, Pabo and their respective coworkers (Choo, Sanchez-Garcia et al. 1994; Rebar and Pabo 1994; Wu, Yang et al. 1995; Beerli, Segal et al. 1998; Dreier, Segal et al. 2000; Dreier, Beerli et al. 2001; Dreier, Fuller et al. 2005; Maeder, Thibodeau-Beganny et al. 2008; Sander, Dahlborg et al. 2011), designing the multi-finger domains of a zinc finger nuclease often requires selection (Greisman and Pabo 1997; Isalan, Klug et al. 2001; Maeder, Thibodeau-Beganny et al. 2008) or computational approaches (Sander, Dahlborg et al. 2011) such as those described by Joung and coworkers.
Initial studies of ZFN specificity using SELEX on zinc finger binding domains alone (Perez, Wang et al. 2008) suggested that ZFNs are highly specific, especially when heterodimeric versions, first developed by Rebar and Cathomen, are used (Miller, Holmes et al. 2007; Szczepek, Brondani et al. 2007). The heterodimeric ZFNs have mutations in the FokI cleavage domain that only allow dimerization between different ZFN monomers (Miller, Holmes et al. 2007; Szczepek, Brondani et al. 2007; Doyon, Vo et al. 2011). While many CCR5 ZFN off-target sites have been identified (Gabriel, Lombardo et al. 2011; Pattanayak, Ramirez et al. 2011; Sander, Ramirez et al. 2013), to date, no toxicity has been reported in clinical trials (Tebas, Stein et al. 2014).
In addition to identifying genomic off-target sites, in vitro selections on two different ZFNs by Liu and coworkers also illuminated several general properties of ZFN specificity (Pattanayak, Ramirez et al. 2011). Like other enzymes, ZFNs exhibit concentration-dependent specificity, such that a larger set of off-target sites can be cut when the ZFN is at higher concentration. In general, ZFN off-target sites with a small number of mutations (for example, for the CCR5 ZFN, three or fewer mutations out of 24 target base pairs) can be recognized and cleaved. Although no sequence preference in the spacer region between half-sites was observed, sites with disfavored four- and seven-base pair spacers were generally recognized with greater specificity than sites with the more favored five- and six-base pair spacers. Finally, off-target sites with several mutations in one half-site likely contain few to no mutations in the other half-site. All of these observations are consistent with a model in which ZFN:DNA binding energy must meet a minimum threshold for cleavage to take place, and that off-target cleavage activity arises from excess binding energy between a ZFN and DNA that can tolerate the energetic penalty incurred by protein-DNA mismatches.
1.4. Insights and improvements from TALEN specificity studies
Like ZFNs, TALENs are engineered fusions of DNA-binding domains with FokI nuclease domains (Figure 3b). In the case of TALENs, the DNA-binding domains consist of TALE repeat arrays (Christian, Cermak et al. 2010; Li, Huang et al. 2011; Miller, Tan et al. 2011). TALE repeats are naturally found in the plant pathogen Xanthomonas and are part of transcriptional activator proteins that lead to gene expression upon binding to specific promoter elements in the plant host cell (Gu, Yang et al. 2005; Yang, Sugio et al. 2006; Kay, Hahn et al. 2007). Canonical TALE repeats are 34-amino acid sequence that each recognize one base pair of DNA. The DNA-binding specificity of each repeat is determined by two amino acids referred to as the repeat-variable di-residue (RVD) (Boch, Scholze et al. 2009; Moscou and Bogdanove 2009). Examples of RVDs that recognize each of the four DNA base pairs are known. The only known sequence constraint on TALE repeat domains is a requirement for the 5’ end of the target site to contain T. Beyond this requirement, TALEs can be designed to target virtually any DNA sequence, and have been successfully used to manipulate genomes in a variety of organisms (Cermak, Doyle et al. 2011; Tesson, Usal et al. 2011; Wood, Lo et al. 2011; Moore, Reyon et al. 2012) and cell lines (Hockemeyer, Wang et al. 2011; Mussolino, Morbitzer et al. 2011; Reyon, Tsai et al. 2012).
Multiple studies, using genome-wide studies and minimally biased selections, have demonstrated that TALEN-mediated genome modification can be accompanied by very rare off-target effects. Whole genome sequencing of TALEN-treated yeast strains (Li, Huang et al. 2011) and whole exome sequencing of human cell lines derived from TALEN-treated cells (Ding, Lee et al. 2013) revealed no evidence of TALE-induced genomic off-target mutations. However, whole genome sequencing may not be sensitive to detecting rare mutations in the absence of sequencing the genomic DNA from an impractically large number of treated cells.
Discrete DNA cleavage studies using homology to on-target sequences to predict potential off-target sites found no TALEN-induced modification of potential off-target sites in Xenopus (Lei, Guo et al. 2012) and human cell lines (Kim, Kweon et al. 2013). Several groups have studied the specificity of the TALE repeat DNA-binding domains in isolation, in the absence of cleavage domains. Initial minimally biased selection experiments using SELEX and TALE activator binding (Hockemeyer, Wang et al. 2011; Miller, Tan et al. 2011; Tesson, Usal et al. 2011; Mali, Aach et al. 2013) on monomeric TALE repeat array domains demonstrated strong preferences for the intended target base pair at each position in the binding site, and a study by Duchateau and coworkers using cellular GFP reporter assay found that relatively few mismatches can be accommodated(Juillerat, Dubois et al. 2014).
Several studies, in human cell lines (Mussolino, Morbitzer et al. 2011), zebrafish (Dahlem, Hoshijima et al. 2012), and rats (Tesson, Usal et al. 2011) have demonstrated TALEN-mediated off-target modification of multiple genomic sites that differ from the on-target site at two to six base pairs. The detection of these sites is not thought to be a general problem of TALEN specificity, since for many applications a TALEN on-target site (up to 36 bp long) can be chosen to be at least seven mutations from any other site in the human genome. However, at least three studies have uncovered off-target sites modified in cells with more than seven mutations from the target site. In one study, Jaenisch and coworkers used DNA-binding SELEX results on TALE repeat domains in isolation to computationally predict potential genomic off-target sites of a fully active heterodimeric TALEN. Of the 19 predicted sites assayed, two off-target sites containing nine or 10 mutations relative to the on-target site, were modified in cultured human cells (Hockemeyer, Wang et al. 2011). Tolar and coworkers used genome-wide selection with IDLVs (Gabriel, Lombardo et al. 2011; Osborn, Starker et al. 2013) to capture off-target double-strand break sites in cells, resulting in the identification of three off-target sites in the genome with up to 12 mutations from the target sequence.
Finally, Liu and colleagues applied the in vitro cleavage selection method described above to reveal 16 sites confirmed to be off-target sites in human cells with modification efficiencies ranging from 0.03% to 2.3% (Guilinger, Pattanayak et al. 2014). The 16 off-target sites contained eight to 12 mutations compared to the on-target site, demonstrating that TALENs can have appreciable off-target activities in human cells even at loci that are quite distant from the on-target sequence. Similar to the model developed to describe ZFN specificity, the in vitro cleavage results of Liu and coworkers suggested that reducing the cationic charge of the canonical 63-aa TALE C-terminal domain or the canonical N-terminal TALE domain could improve specificity by reducing non-specific DNA-binding energy. Consistent with this hypothesis, the ability of off-target sites to survive the in vitro selection decreased as these cationic residues were mutated to neutral amino acids. Many of these charge-engineered TALENs demonstrated improved specificity across all positions in the target site. Specificity profiles generated using the in vitro selection method applied to charge-engineered TALENs indeed showed ~10 to ~100-fold improved specificity from assays of on-target and off-target activity both in vitro and in cells.
1.5. Insights and improvements from Cas9 specificity studies
In contrast to ZFNs and TALENs, RNA-guided Cas9 nucleases (referred to below as Cas9) do not require the design of separate DNA-binding domains for each new target site (Figure 3c). Cas9 is a member of the CRISPR/Cas family of proteins that naturally defend bacterial genomes through endonuclease activity against foreign DNA sequences. In contrast to ZFNs and TALENs, the target DNA specificity of Cas9 is programmed by hybridization of the target DNA to a Cas9-bound guide RNA sequence (sgRNA) (Jinek, Chylinski et al. 2012). Similar to TALENs, Cas9 target sequences are constrained at one end. All Cas9-targeted sequences require a sequence motif called a protospacer adjacent motif (PAM), the identity of which depends on the species of the Cas9 protein. For example, the most commonly used Cas9, from S. pyogenes, cleaves most efficiently target sequences containing an NGG PAM. Unlike ZFNs and TALENs, Cas9 target sites described to date consist of at most 20 base pairs, not including the PAM sequence.
Early studies of Cas9 specificity in nature by Siksyns, Severinov, Maraffini, and their respective coworkers (Sapranauskas, Gasiunas et al. 2011; Semenova, Jore et al. 2011; Jinek, Chylinski et al. 2012; Cong, Ran et al. 2013; Jiang, Bikard et al. 2013) suggested that specific recognition of target DNA by Cas9 was limited to a 7–12 base pair subsequence adjacent to the PAM end of the target site. Further in vitro study using discrete off-target site testing by Doudna, Charpentier, and colleagues (Jinek, Chylinski et al. 2012) also supported the model. In this model of Cas9 specificity, mismatches were thought to be tolerated at the non-PAM end of the molecule. This model would suggest that Cas9 could not be used for specific genome-modification, since a 12-base pair sequence plus two base pair PAM is not long enough to specify a unique sequence in the human genome. Several studies had shown, however, that Cas9 could be used for genome modification in several organisms without adverse effects; for example, Joung and coworkers reported that Cas9-mediated gene modification in zebrafish embryos exhibited a similar rate of off-target toxicity as ZFNs and TALENs (Hwang, Fu et al. 2013).
Four subsequent studies, two using discrete off-target testing in human cell culture by Joung (Fu, Foden et al. 2013), Zhang(Hsu, Scott et al. 2013), and their respective coworkers, one using a minimally biased selection in cells by Church and coworkers (Mali, Aach et al. 2013), and one using a minimally biased selection in vitro by Liu and coworkers (Pattanayak, Lin et al. 2013), investigated Cas9 specificity and showed that while Cas9 specificity is sufficient for at least some genome editing applications, several off-target cleavage sites could be detected for most Cas9 target sites tested. While the magnitude of off-target activity varied in the four studies, Joung and coworkers observed that some off-target sites could be modified at similar frequencies to the on-target site.
All four studies showed that Cas9 specificity extended past the 7–12 base pair subsequence near the PAM, and that specificity is decreased at the end of the target site farthest from the PAM. The subsequence near the PAM, while highly specified, tolerates certain single-base pair mismatches in an unpredictable fashion depending on the target site. These functional observations of cleavage specificity have been supported both by a molecular dynamics study of Cas9 by Doudna and colleagues (Sternberg, Redding et al. 2014), as well as crystallographic models of Cas9 elucidated by Doudna, Nureki, and their respective colleagues (Jinek, Jiang et al. 2014; Nishimasu, Ran et al. 2014). Genomic binding site profiling of inactive Cas9 by Adli, Sharp, Zhang, and their respective colleagues, in addition to confirming that the entire Cas9 target site is necessary for cleavage, suggests that Cas9 can bind many more sites in the genome than it actually cleaves (Kuscu, Arslan et al. 2014; Wu, Scott et al. 2014).
Within the PAM itself, certain mismatches can also be tolerated. While the S. pyogenes Cas9 specifies an NGG PAM, observations by Church, Liu, and their respective coworkers showed that an NAG PAM can also be recognized, and in vitro, an NNG or an NGN PAM can be recognized with weak activity when the rest of the target sequence is fully complementary to the guide RNA sequence (Pattanayak, Lin et al. 2013). A more recent study on Cas9 specificity by Bao and colleagues (Lin, Cradick et al. 2014) also suggests that Cas9 can tolerate single-base pair insertions or deletions in the target sequence relative to the guide RNA sequence, though with reduced activity. In addition, several studies have established that specificity is dependent on Cas9 concentration (Fu, Foden et al. 2013; Hsu, Scott et al. 2013; Pattanayak, Lin et al. 2013) and guide RNA architecture (Hsu, Scott et al. 2013; Pattanayak, Lin et al. 2013; Fu, Sander et al. 2014).
Given the significant off-target activity of Cas9 endonucleases, numerous groups have engineered Cas9 or guide RNA variants with enhanced specificity. Joung and co-workers improved the specificity of the Cas9:sgRNA complex by truncating the sgRNA to target less than the canonical 20-bp target sites (Figure 4a) (Fu, Sander et al. 2014). By analogy to a study by Kim and colleagues on dimeric zinc finger nickases (Kim, Kim et al. 2012), Church, Zhang, and their respective coworkers demonstrated that mutant Cas9 proteins that cleave only a single strand of dsDNA can be used to nick opposite strands of two nearby target sites, generating what is effectively a double strand break with reduced off-target activity (Figure 4b) (Beurdeley, Bietz et al. 2013; Mali, Aach et al. 2013; Cho, Kim et al. 2014).
Figure 4. Engineered Cas9 components with improved DNA cleavage specificity.

(a) A truncated guide RNA (tru-gRNA, right), contains 17–18 base pairs of complementarity to its DNA target site, rather than 20 base pairs in a canonical sgRNA (left). The base pairs in the sgRNA that are not present in the tru-gRNA are colored black. (b) Mutant Cas9 proteins that cleave only a single strand of dsDNA (nCas9) can be targeted to opposite strands of adjacent sites as pairs to cause double strand breaks. (c) Monomers of FokI nuclease (red) fused to catalytically inactive dCas9 bind to separate sites within a target locus. Only adjacently bound FokI-dCas9 monomers can assemble a catalytically active FokI nuclease dimer, triggering dsDNA cleavage.
Nickases even when bound to off-target loci as monomers retain their ability to nick DNA, which can result in a low level of undesired genome modification (Ran, Hsu et al. 2013; Cho, Kim et al. 2014; Fu, Sander et al. 2014), as has previously been described for single zinc finger nickases (Ramirez, Certo et al. 2012; Wang, Friedman et al. 2012). Therefore, Liu, Joung and their respective coworkers developed engineered Cas9 variants that are only able to cleave DNA when two monomers are adjacently bound to a target locus by fusing a FokI restriction endonuclease cleavage domain to a catalytically inactive Cas9 (dCas9) (Figure 4c) (Guilinger, Thompson et al. 2014; Tsai, Wyvekens et al. 2014), analogous to dimeric zinc-finger nucleases (ZFNs) and TALENs. In discrete off-target studies, the FokI-dCas9 fusions maintain substantial on-target DNA modification with a large reduction in off-target modification at known Cas9 off-target sites.
Collectively, studies that reveal in detail the DNA cleavage specificity of Cas9, together with the engineering of improved Cas9 variants, demonstrate the potential of Cas9 as an accessible and specific genome engineering tool.
2.1. Overview of in vitro selection-based nuclease specificity profiling
The in vitro selection method developed by our group to profile the DNA cleavage specificity of a nuclease comprises three major steps: pre-selection library construction, in vitro selection, and high-throughput sequencing and analysis. Briefly, synthetic 5’-phosphorylated oligonucleotides are converted into concatemeric repeats of a library of potential off-target sites through intramolecular circularization followed by rolling-circle amplification. The resulting pre-selection libraries are then incubated in vitro with the appropriate nuclease, either in purified form or used directly from in vitro translation systems. Cleaved library members, which contain free 5’ phosphates, are captured by adapter ligation enabling their separation from uncleaved pre-selection library members, which do not contain 5’ phosphates. Cleaved post-selection library members are then amplified by PCR prior to high-throughput DNA sequencing.
2.2. Pre-selection library design
While it would be ideal to use a pre-selection library that consists of all possible off-target sequences of a given length (for example, an N22 library for Cas9, including PAM), the in vitro selection method has an upper limit of approximately 1012 sequences in the pre-selection library. Since an N22 library would contain 422 (~1013 sequences), a library biased in favor of sequences resembling the on-target recognition site is used instead. Library biasing is accomplished through the use of randomized nucleotide mixtures at all target-site base pairs during library construction. We and others (Argast, Stephens et al. 1998; Doyon, Pattanayak et al. 2006; Pattanayak, Ramirez et al. 2011; Mali, Aach et al. 2013; Pattanayak, Lin et al. 2013; Guilinger, Pattanayak et al. 2014) have had success using mixtures that contain 79% on-target base pair at each targeted position, with the remaining 21% of the mixture comprising of the three off-target base pairs. For Cas9, this approach results in a pre-selection library that in theory contains at least ten copies of each potential off-target sequence containing eight or fewer mutations relative to the on-target site. For a 36-base pair TALEN on-target site, the preselection library provides at least ten-fold coverage of all sequences with six or fewer mutations. The partially randomized on-target site is also flanked by fully randomized base pairs on each side to test for patterns of specificity beyond the canonical target site.
The concentration of nuclease used to digest the pre-selection library should be enough to produce sufficient cleaved sequences for robust detection but not enough to completely digest highly cleaved sequences. The use of high nuclease concentrations will augment the detection of rare off-target cleavage events and could result in lower apparent nuclease specificity. Therefore, careful consideration of the nuclease concentrations used, or at least the percentage of on-target sequences cleaved under the assay conditions, is required when studying and describing specificity.
2.3 In vitro selection protocol
Before day 1: Design and synthesize pre-selection library oligonucleotides
For a selection using a guide RNA (CLTA4) targeted to the human clathrin gene (CLTA) (Pattanayak, Lin et al. 2013), the library oligonucleotide was ordered from Integrated DNA Technologies (IDT) and was of the form: 5Phos/TTG TGT NNN NC*C* NT*G* T*G*G* A*A*A* C*A*C* T*A*C* A*T*C* T*G*C* NNN NAC CTG CCG AGT TGT GT ‘/5Phos/’ refers to a 5’ phosphate modification. The underlined sequence refers to the target site library, where each asterisk denotes a position that was ordered as a mixture of bases with 79% of the mixture corresponding to the base preceding the asterisk and 7% each corresponding to the other three bases. For Cas9, we found that this target site orientation (with the reverse complement of the PAM at the 5’ end of the oligonucleotide) yielded higher quality data than the reverse complement of this orientation. The italicized sequences denotes a repeated six-base pair barcode that can be used to identify the target site used in the selection, if multiple selections are assayed at once. Other barcodes that we have used include AAC ACA, TCT TCT, and AGA GAA. Any barcodes can be used, with a minimum of two base pairs differing between each barcode. The sequence in bold denotes a constant region that remains the same for all selections. The constant sequence includes a BspMI restriction site that is used for pre-selection library preparation for high-throughput sequencing.
Day 1: Circularize library oligonucleotides
Dilute library oligonucleotides to 10 3M in 1 mM Tris, pH 8.0. In PCR tubes, add 1 3L of library oligo (10 pmol), 2 3L 10x CircLigase II 10x Reaction Buffer, 1 3L 50 mM MnCl2, 15 3L water, and 1 3L CircLigase II ssDNA Ligase (100 U) (Epicentre #CL9021K). Incubate 16 hours at 60 °C, followed by a 10 min inactivation step at 85 °C.
Day 2: Confirm circularization of library oligonucleotides and perform rolling-circle amplification
On a 15% TBE-Urea polyacrylimide gel, load 2.5 pmol of uncircularized library oligo and 2.5 3L (1.25 pmol) of the CircLigase mixture without purification. Run for 75 min at 200 V. Stain the gel in 100 mL 0.5x TBE containing 10 3L SYBR Gold (Invitrogen #11494) for 2 hours. Rinse with water before imaging the gel. Under these conditions, the circularized oligonucleotides should migrate more slowly than the linear oligonucleotide control.
We use the illustra TempliPhi Amplification Kit (GE Healthcare #25–6400-10) for the rolling-circle amplification reaction. Combine 5 3L (2.5 pmol) of each unpurified CircLigase reaction and 50 3L of TempliPhi sample buffer. Incubate 3 min at 95 °C. Cool at 0.5 °C/second to 4 °C. Add 50 3L TempliPhi reaction buffer and 1 3L TempliPhi enzyme mix. Incubate 16 hours at 30 °C. Heat-inactivate for 10 min at 65 °C. The rolling-circle amplification step can be halved or doubled in scale, with the final pre-selection library size determined by the amount of CircLigase reaction that is used.
Day 3: Quantify and digest pre-selection library
Since the pre-selection library is used in the selection without purification, a double-stranded DNA quantification reagent (Quant-it PicoGreen dsDNA Assay Kit, Invitrogen) is used to quantify the amplified double-stranded DNA. To quantify, add 1 3L rolling-circle amplified DNA or 10–200 ng of lambda DNA standard to 200 3L 1 mM Tris, pH 8.0. Incubate 10 min at room temperature in the dark, before reading fluorescence in a plate reader (excitation wavelength ~ 480 nM, emission wavelength ~ 520 nM). Create a standard curve relating DNA concentration to fluorescence using, for example, pre-quantitated phage lambda DNA and calculate the concentration of the rolling-circle amplified preselection library.
To perform the in vitro selection, digest the pre-selection library with purified or in vitro translated site-specific endonuclease. For Cas9, 200 nM amplified pre-selection library was incubated with 100 nM Cas9 and 100 nM sgRNA or 1,000 nM Cas9 and 1,000 nM sgRNA in Cas9 cleavage buffer (20 mM HEPES, pH 7.5, 150 mM potassium chloride, 10 mM magnesium chloride, 0.1 mM EDTA, 0.5 mM dithiothreitol) for 10 min at 37 °C. Separately, the pre-selection library is also incubated with 2 U of BspMI restriction endonuclease (NEB) in NEBuffer 3 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 1 mM dithiothreitol, pH 7.9) for 1 hours at 37 °C. Both nuclease-digested and restriction-digested libraries are purified with the QIAQuick PCR Purification Kit (Qiagen).
For site-specific nucleases that leave overhangs, such as ZFNs and TALENs, an additional step to convert the cut overhangs into blunt ends is performed before adapter ligation. In this step, 50 3L of purified, digested DNA is incubated with 3 3L of 10 mM dNTP mix (10 mM dATP, 10 mM dCTP, 10 mM dGTP, 10 mM dTTP) (NEB), 6 3L of 10x NEBuffer 2, and 1 3L of 5 U/3L Klenow Fragment DNA Polymerase (NEB) for 30 min at room temperature. The blunt-ended mixture is purified with the QIAQuick PCR Purification Kit (Qiagen).
Once the cut ends have been made blunt, either by Cas9, or for TALENs by Klenow polymerase, sequencing adapters are ligated. For post-selection blunt libraries, adapter1 (5’ AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC TAA CA) and adapter2 (5’ TGT TAG ATC GGA AGA GCG TCG TGT AGG GAA AGA GTG TAG ATC TCG GTG G) are used to incorporate sequences for Illumina sequencing. The reverse complementary sequences in italics can be varied to barcode multiple reaction conditions. The rest of the adapter sequences can also be varied depending to be used with other high-throughput sequencing platforms. In the ligation step, 10 pmol each of adapter1 and adapter2 are incubated with the blunt-ended post-selection library and 1,000 U T4 DNA Ligase (NEB) in in NEB T4 DNA Ligase Reaction Buffer (50 mM Tris-HCl, pH 7.5, 10 mM magnesium chloride, 1 mM ATP, 10 mM dithiothreitol) overnight at room temperature. For the restriction-digested pre-selection library, the ligation protocol is the same, with the exception of the use of lib adapter1 (5’ GAC GGC ATA CGA GAT) and lib adapter2 (5’ TTG TAT CTC GTA TGC CGT CTT CTG CTT G). Of note, the first four bases of lib adapter2 (in italics) must match the first four bases of the library oligonucleotide barcode (see Before day 1, above), since BspMI digestion will leave an overhang that is specific to the barcode used. Therefore, if multiple target sites are tested in the same selection run, multiple lib adapter2’s must be used.
Day 4: PCR of pre- and post-selection libraries
The PCR amplification step prior to high-throughput sequencing must be well controlled to minimized the potential effects of PCR bias on the final sequencing results. Prior to PCR, the adapter ligation mixtures from Day 3 are purified with the QiaQuick PCR Purification Kit (Qiagen) and eluted with 50 3L 1 mM Tris, pH 8.0. We use Phusion Hot Start Flex DNA Polymerase (NEB) in Buffer HF with an annealing temperature of 60 °C and an extension termperature of 72 °C for 1 min per cycle. For the nuclease-digested post-selection PCR, primers sel PCR (5’ CAA GCA GAA GAC GGC ATA CGA GAT ACA CAA CTC GGC AGG T) and PE2 short (5’ AAT GAT ACG GCG ACC ACC GA) are used. For the restriction-digested pre-selection library PCR, use the same PCR cycling conditions with primers lib fwd PCR (5’ CAA GCA GAA GAC GGC ATA CGA GAT) and lib seq PCR (5’ AAT GAT ACG GCG ACC ACC GAG ATC TAC ACT CTT TCC CTA CAC GAC GCT CTT CCG ATC TNN NNA CCT ACC TGC CGA GTT GTG T). Four Ns are included in the lib seq PCR to provide a randomized initiation sequence to maintain compatibility with Illumina sequencing requirements. Of note, if multiple target sites are used in the same selection run, multiple sel PCR primers must be used, with the four base sequence in sel PCR and the six base sequence in lib seq PCR listed in italics should be modified to maintain complementarity to the original library oligonucleotide backbone (see Before day 1, above).
Before PCR of the full volume of post-selection library, we suggest performing a test PCR or test qPCR with 1 3L of purified post-selection library under the PCR conditions listed above to determine the number of cycles required to reach saturation. If doing a test PCR, remove aliquots every five cycles for 35 cycles and visualize the reaction products. Determine the point at which the PCR amplification saturates and subtract an appropriate number of cycles when the PCR is scaled up. For example, if scaling from 1 3L to 32 3L of purified post-selection library, subtract five cycles (25 = 32).
After PCR, there may be a ladder of products, corresponding to amplified post-selection library members that contain 0.5, 1.5, 2.5, etc. repeats of a given library member (Figure 5). The variation in PCR product size results from the concatemeric nature of the pre-selection library. During PCR, the sel PCR primer can also anneal to one of multiple repeats, also leading a distribution of PCR product sizes. To standardize the analysis, only those PCR products that contain 1.5 repeats are analyzed. Therefore, before high-throughput sequencing, a final gel purification step is used to enrich the amplified post-selection library members that contain exactly 1.5 repeats and to remove any remaining free adapters and primers.
Figure 5. Sample processing during in vitro selection-based nuclease specificity profiling.

(a) Preselection DNA consisting of many repeats of a library member (gray boxes) becomes smaller in size due to nuclease digestion, depending on which target site along the pre-selection DNA is cleaved. (b) During post-selection library amplification, the PCR primer (blue arrows) can anneal to any one of the repeats, leading to a set of smaller PCR products. To simplify analysis, only PCR products with 1.5 repeats are purified and analyzed.
Day 5: High-throughput sequencing and analysis
The gel-purified post-selection and pre-selection libraries can be quantified using the KAPA Library Quantification Kit-Illumina (KAPA Biosystems) before subjecting to high-throughput sequencing. We used single-read sequencing on an Illumina HiSeq and Illumina MiSeq, though the selection should be compatible with any high-throughput sequencing platform as long as the adapter sequences and PCR primers are modified appropriately. For Cas9, a minimum of 66 bases must be sequenced to capture the entire library member. If using a selection condition barcode (for example, AACA below), we recommend spiking in a PhiX library control at 25% with the sequencing run to provide appropriate initial base-calling diversity if using Illumina sequencing.
The sequencing output can be binned using a simple scripting language, such as C++ or Python. The components of the sequencing read are illustrated below:
AACAcatgggtcgACACAAACACAACTCGGCAGGTACTTGCAGATGTAGTCTTTCCACATGGG TCGACACAAACACAACTCGGCAGGTATCTCGTATGCC
AACA is the four basepair barcode for selection conditions. catgggtcg is the cut “half” of the library target site. ACACAAACACAA is the Cas9 target barcode. CTCGGCAGGT is the constant sequence (the reverse complement of the bold sequence in the Library Design section). ACTTGCAGATGTAGTCTTTCCACATGGGTCG is the full sequence of the post-selection library member. This sequence can be recognized and cut in the selection. The non-underlined portion of the sequence consist of the eight random basepairs (four on each side) that flanked the target site library. Once sequences are binned, standard analyses can be performed on the set of target sites (bold and underlined). For example, specificity profiles can be represented as heat maps of specificity scores calculated as the enrichment level of each possible base pair at every position in the post-selection sequences relative to the pre-selection sequences, normalized to the maximum possible enrichment of that base pair (Figure 6).
Figure 6. An in vitro selection-derived specificity profile.

The heat map shows the specificity profile resulting from a selection performed on Cas9:sgRNA targeting the human CLTA gene. Specificity scores of 1.0 (dark blue) and -1.0 (dark red) corresponds to 100% enrichment for and against, respectively, a particular base pair at a particular position. Black boxes denote the intended target nucleotides.
2.4. Confirmation of in vitro-identified genomic off-target sites
To identify genomic off-target sites, the set of target sites identified in vitro by selection and high-throughput sequencing can be searched for sequences that appear in the human genome. In addition to this simple comparison, a machine-learning algorithm can be trained on the in vitro dataset to assist the identification of potential genomic off-target sequences (Sander, Ramirez et al. 2013). The tested site-specific nuclease is then expressed in cultured human cells, along with a parallel experiment with a control, inactive form of the same site-specific nuclease. Genomic DNA is isolated, followed by PCR with primers specific to each potential off-target site. A primer design tool, such as NCBI Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/), can be useful in the design of primers that lead to specific amplification of the target site of interest. Portions of the high-throughput adapter sequences can be incorporated into the primers (for example, for Illumina, 5’ ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TCT at the 5’ end of one primer and 5’ GTG ACT GGA GTT CAG ACG TGT GCT CTT CCG ATCT on the other) for the initial PCR. When assaying multiple off-target sites at once, PCRs can be pooled in equimolar ratios, purified, and then reamplified using primers PE1-barcode (5’ CAA GCA GAA GAC GGC ATA CGA GAT ATA TCA GTG TGA CTG GAG TTC AGA CGT GTG CT) and PE2-barcode (5’ AAT GAT ACG GCG ACC ACC GAG ATC TAC ACA TTA CTC GAC ACT CTT TCC CTA CAC GAC). The number of cycles of the re-amplification PCR should be minimized to avoid introducing significant PCR bias. The italicized bases in PE1-barcode and PE2-barcode correspond to barcodes that can be used in Illumina sequencing. Different barcodes should be used for PCR products derived from active-nuclease-treated DNA compared to inactive-nuclease-treated DNA.
Following high-throughput sequencing, nuclease-modified off-target sequences can be identified through sequence alignment or through computational methods. One algorithm for identifying modified-sequences involves searching for the 20 base pairs flanking each off-target site for each high-throughput sequencing read. For example: 5’CAATCTCCCGCATGCGCTCAGTCCTCATCTCCCTCAAGCAGGCCCCGCTGGTGCACTGA AGAGCCACCCTGTGAAACACTACATCTGCAATATCTTAATCCTACTCAGTGAAGCTCTT CACAGTCATTGGATTAATTATGTTGAGTTCTTTTGGACCAAACC The flanking sequences (underlined) can be used to identify the off-target site being assayed (in bold). In the reference genome sequence, the sequence between the underlined flanking regions is 5’ CCCTGTGGAAACACTACATCTGC. In this example, the sequence between the underlined flanking regions is 5’ CCCTGT-GAAACACTACATCTGC, where the dash indicates a one base pair deletion. For each potential off-target site tested, the fraction of sequences with insertions and deletions can be calculated and compared between active-nuclease and inactive-nuclease experiments. For target sites with high modification efficiencies, it may be necessary to use flanking sequences (the underlined sequences above) that are more distal to the target site, in case NHEJ leads to deletion of a region that is larger than the off-target site (the bold sequence).
3. Conclusion
Genome engineering in the last few years has become more facile through the use of programmable site-specific nucleases such as TALENs and Cas9, which can be designed target nearly any DNA sequence. As the use of ZFNs, TALENs, and Cas9 in research and clinical settings continues to grow, efforts to reveal in depth the DNA cleavage specificity of programmable nucleases will become increasingly important. Efforts to characterize programmable nuclease specificity have ranged from discrete target-site assays to in vitro selections to genome-wide selections, all of which have been applied recently to study TALEN and Cas9 specificity. The findings from these methods will continue to deepen our understanding of the basis of the DNA cleavage specificity of these important proteins, inform the development of programmable nucleases with improved specificity, and perhaps eventually enable the broad application of these or ther programmable nucleases to treat human genetic diseases.
Acknowledgments
V.P., J.P.G., and D.R.L. were supported by DARPA HR0011-11-2-0003, DARPA N66001-12-C-4207, NIH/NIGMS R01 GM095501 (DRL), and the Howard Hughes Medical Institute. D.R.L. was supported as a HHMI Investigator. V.P. was supported by NIGMS T32GM007753. We are grateful to J. Keith Joung for helpful comments.
References
- Argast GM, Stephens KM, et al. I-PpoI and I-CreI homing site sequence degeneracy determined by random mutagenesis and sequential in vitro enrichment. J Mol Biol. 1998;280(3):345–353. doi: 10.1006/jmbi.1998.1886. [DOI] [PubMed] [Google Scholar]
- Beerli RR, Segal DJ, et al. Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proc Natl Acad Sci U S A. 1998;95(25):14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurdeley M, Bietz F, et al. Compact designer TALENs for efficient genome engineering. Nat Commun. 2013;4:1762. doi: 10.1038/ncomms2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boch J, Scholze H, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Bulyk ML, Huang X, et al. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc Natl Acad Sci U S A. 2001;98(13):7158–7163. doi: 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CD, Warren CL, et al. Specificity landscapes of DNA binding molecules elucidate biological function. Proc Natl Acad Sci U S A. 2010;107(10):4544–4549. doi: 10.1073/pnas.0914023107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. Genome engineering with zinc-finger nucleases. Genetics. 2011;188(4):773–782. doi: 10.1534/genetics.111.131433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cermak T, Doyle EL, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12):e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Wen F, et al. Directed evolution of homing endonuclease I-SceI with altered sequence specificity. Protein Eng Des Sel. 2009;22(4):249–256. doi: 10.1093/protein/gzp001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Zhao H. A highly sensitive selection method for directed evolution of homing endonucleases. Nucleic Acids Res. 2005;33(18):e154. doi: 10.1093/nar/gni148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choo Y, Sanchez-Garcia I, et al. In vivo repression by a site-specific DNA-binding protein designed against an oncogenic sequence. Nature. 1994;372(6507):642–645. doi: 10.1038/372642a0. [DOI] [PubMed] [Google Scholar]
- Choulika A, Perrin A, et al. Induction of homologous recombination in mammalian chromosomes by using the I-SceI system of Saccharomyces cerevisiae. Mol Cell Biol. 1995;15(4):1968–1973. doi: 10.1128/mcb.15.4.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colleaux L, D'Auriol L, et al. Recognition and cleavage site of the intron-encoded omega transposase. Proc Natl Acad Sci U S A. 1988;85(16):6022–6026. doi: 10.1073/pnas.85.16.6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlem TJ, Hoshijima K, et al. Simple methods for generating and detecting locus-specific mutations induced with TALENs in the zebrafish genome. PLoS Genet. 2012;8(8):e1002861. doi: 10.1371/journal.pgen.1002861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Lee YK, et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell. 2013;12(2):238–251. doi: 10.1016/j.stem.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyon JB, Pattanayak V, et al. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. J Am Chem Soc. 2006;128(7):2477–2484. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
- Doyon Y, Vo TD, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8(1):74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- Dreier B, Beerli RR, et al. Development of zinc finger domains for recognition of the 5'-ANN-3' family of DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2001;276(31):29466–29478. doi: 10.1074/jbc.M102604200. [DOI] [PubMed] [Google Scholar]
- Dreier B, Fuller RP, et al. Development of zinc finger domains for recognition of the 5'-CNN-3' family DNA sequences and their use in the construction of artificial transcription factors. J Biol Chem. 2005;280(42):35588–35597. doi: 10.1074/jbc.M506654200. [DOI] [PubMed] [Google Scholar]
- Dreier B, Segal DJ, et al. Insights into the molecular recognition of the 5'-GNN-3' family of DNA sequences by zinc finger domains. J Mol Biol. 2000;303(4):489–502. doi: 10.1006/jmbi.2000.4133. [DOI] [PubMed] [Google Scholar]
- Fu Y, Foden JA, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, et al. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29(9):816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Gimble FS, Moure CM, et al. Assessing the plasticity of DNA target site recognition of the PI-SceI homing endonuclease using a bacterial two-hybrid selection system. J Mol Biol. 2003;334(5):993–1008. doi: 10.1016/j.jmb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- Greisman HA, Pabo CO. A general strategy for selecting high-affinity zinc finger proteins for diverse DNA target sites. Science. 1997;275(5300):657–661. doi: 10.1126/science.275.5300.657. [DOI] [PubMed] [Google Scholar]
- Gu K, Yang B, et al. R gene expression induced by a type-III effector triggers disease resistance in rice. Nature. 2005;435(7045):1122–1125. doi: 10.1038/nature03630. [DOI] [PubMed] [Google Scholar]
- Guilinger JP, Pattanayak V, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. 2014;11(4):429–435. doi: 10.1038/nmeth.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger JP, Thompson DB, et al. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014 doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirsch JA, Wah DA, et al. Crystallization and preliminary X-ray analysis of restriction endonuclease FokI bound to DNA. FEBS Lett. 1997;403(2):136–138. doi: 10.1016/s0014-5793(97)00039-2. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Wang H, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29(8):731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Paxton WA, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2(11):1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- Hwang WY, Fu Y, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat Biotechnol. 2013;31(3):227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isalan M, Klug A, et al. A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat Biotechnol. 2001;19(7):656–660. doi: 10.1038/90264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, et al. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31(3):233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Jiang F, et al. Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science. 2014;343(6176):1247997. doi: 10.1126/science.1247997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013;14(1):49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juillerat A, Dubois G, et al. Comprehensive analysis of the specificity of transcription activator-like effector nucleases. Nucleic Acids Res. 2014;42(8):5390–5402. doi: 10.1093/nar/gku155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlon KS, Brown C, et al. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 2004;64(24):9160–9166. doi: 10.1158/0008-5472.CAN-04-0454. [DOI] [PubMed] [Google Scholar]
- Kay S, Hahn S, et al. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science. 2007;318(5850):648–651. doi: 10.1126/science.1144956. [DOI] [PubMed] [Google Scholar]
- Kim E, Kim S, et al. Precision genome engineering with programmable DNA-nicking enzymes. Genome Res. 2012;22(7):1327–1333. doi: 10.1101/gr.138792.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Kweon J, et al. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol. 2013;31(3):251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- Kim YG, Cha J, et al. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93(3):1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, et al. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- Kuscu C, Arslan S, et al. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol. 2014 doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- Lei Y, Guo X, et al. Efficient targeted gene disruption in Xenopus embryos using engineered transcription activator-like effector nucleases (TALENs) Proc Natl Acad Sci U S A. 2012;109(43):17484–17489. doi: 10.1073/pnas.1215421109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Huang S, et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011;39(1):359–372. doi: 10.1093/nar/gkq704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Huang S, et al. Modularly assembled designer TAL effector nucleases for targeted gene knockout and gene replacement in eukaryotes. Nucleic Acids Res. 2011;39(14):6315–6325. doi: 10.1093/nar/gkr188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Cradick TJ, et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014 doi: 10.1093/nar/gku402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Paxton WA, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell. 1996;86(3):367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Lukacsovich T, Yang D, et al. Repair of a specific double-strand break generated within a mammalian chromosome by yeast endonuclease I-SceI. Nucleic Acids Res. 1994;22(25):5649–5657. doi: 10.1093/nar/22.25.5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeder ML, Thibodeau-Beganny S, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31(2):294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Brodsky MH, et al. A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat Biotechnol. 2005;23(8):988–994. doi: 10.1038/nbt1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng X, Thibodeau-Beganny S, et al. Profiling the DNA-binding specificities of engineered Cys2His2 zinc finger domains using a rapid cell-based method. Nucleic Acids Res. 2007;35(11):e81. doi: 10.1093/nar/gkm385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Holmes MC, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25(7):778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- Miller JC, Tan S, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29(2):143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Moore FE, Reyon D, et al. Improved somatic mutagenesis in zebrafish using transcription activator-like effector nucleases (TALENs) PLoS One. 2012;7(5):e37877. doi: 10.1371/journal.pone.0037877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Mussolino C, Morbitzer R, et al. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39(21):9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, et al. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156(5):935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant AR, Brandl CJ, et al. Defining the sequence specificity of DNA-binding proteins by selecting binding sites from random-sequence oligonucleotides: analysis of yeast GCN4 protein. Mol Cell Biol. 1989;9(7):2944–2949. doi: 10.1128/mcb.9.7.2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn MJ, Starker CG, et al. TALEN-based Gene Correction for Epidermolysis Bullosa. Molecular Therapy. 2013 doi: 10.1038/mt.2013.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Lin S, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, Ramirez CL, et al. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8(9):765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petek LM, Russell DW, et al. Frequent endonuclease cleavage at off-target locations in vivo. Mol Ther. 2010;18(5):983–986. doi: 10.1038/mt.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis AA, Qureshi AM, et al. Design of compact, universal DNA microarrays for protein binding microarray experiments. J Comput Biol. 2008;15(7):655–665. doi: 10.1089/cmb.2007.0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez CL, Certo MT, et al. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res. 2012;40(12):5560–5568. doi: 10.1093/nar/gks179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebar EJ, Pabo CO. Zinc finger phage: affinity selection of fingers with new DNA-binding specificities. Science. 1994;263(5147):671–673. doi: 10.1126/science.8303274. [DOI] [PubMed] [Google Scholar]
- Reik A, Zhou Y, et al. Zinc finger nucleases targeting the glucocorticoid receptor allow IL-13 zetakine transgenic CTLs to kill glioblastoma cells in vivo in the presence of immunosuppressing glucocorticoids. Mol Ther. 2008;16(Suppl 1):S13–S14. [Google Scholar]
- Reyon D, Tsai SQ, et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30(5):460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouet P, Smih F, et al. Introduction of double-strand breaks into the genome of mouse cells by expression of a rare-cutting endonuclease. Mol Cell Biol. 1994;14(12):8096–8106. doi: 10.1128/mcb.14.12.8096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samson M, Libert F, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382(6593):722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- Sander JD, Dahlborg EJ, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA) Nat Methods. 2011;8(1):67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Joung JK. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat Biotechnol. 2014;32(4):347–355. doi: 10.1038/nbt.2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander JD, Ramirez CL, et al. In silico abstraction of zinc finger nuclease cleavage profiles reveals an expanded landscape of off-target sites. Nucleic Acids Res. 2013;41(19):e181. doi: 10.1093/nar/gkt716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapranauskas R, Gasiunas G, et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011;39(21):9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlatti G, Tresoldi E, et al. In vivo evolution of HIV-1 co-receptor usage and sensitivity to chemokine-mediated suppression. Nat Med. 1997;3(11):1259–1265. doi: 10.1038/nm1197-1259. [DOI] [PubMed] [Google Scholar]
- Segal DJ, Dreier B, et al. Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5'-GNN-3' DNA target sequences. Proc Natl Acad Sci U S A. 1999;96(6):2758–2763. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proc Natl Acad Sci U S A. 2011;108(25):10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, et al. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507(7490):62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szczepek M, Brondani V, et al. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25(7):786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- Tebas P, Stein D, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson L, Usal C, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29(8):695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- Thiesen HJ, Bach C. Target Detection Assay (TDA): a versatile procedure to determine DNA binding sites as demonstrated on SP1 protein. Nucleic Acids Res. 1990;18(11):3203–3209. doi: 10.1093/nar/18.11.3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Wyvekens N, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014 doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanamee ES, Santagata S, et al. FokI requires two specific DNA sites for cleavage. J Mol Biol. 2001;309(1):69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- Wang J, Friedman G, et al. Targeted gene addition to a predetermined site in the human genome using a ZFN-based nicking enzyme. Genome Res. 2012;22(7):1316–1326. doi: 10.1101/gr.122879.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, et al. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood AJ, Lo TW, et al. Targeted genome editing across species using ZFNs and TALENs. Science. 2011;333(6040):307. doi: 10.1126/science.1207773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Yang WP, et al. Building zinc fingers by selection: toward a therapeutic application. Proc Natl Acad Sci U S A. 1995;92(2):344–348. doi: 10.1073/pnas.92.2.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Scott DA, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol. 2014 doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B, Sugio A, et al. Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc Natl Acad Sci U S A. 2006;103(27):10503–10508. doi: 10.1073/pnas.0604088103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanover C, Bradley P. Extensive protein and DNA backbone sampling improves structure-based specificity prediction for C2H2 zinc fingers. Nucleic Acids Res. 2011;39(11):4564–4576. doi: 10.1093/nar/gkr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhu S, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509(7501):487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- Zykovich A, Korf I, et al. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Res. 2009;37(22):e151. doi: 10.1093/nar/gkp802. [DOI] [PMC free article] [PubMed] [Google Scholar]