

Abstract
CleavPredict (http://cleavpredict.sanfordburnham.org) is a Web server for substrate cleavage prediction for matrix metalloproteinases (MMPs). It is intended as a computational platform aiding the scientific community in reasoning about proteolytic events. CleavPredict offers in silico prediction of cleavage sites specific for 11 human MMPs. The prediction method employs the MMP specific position weight matrices (PWMs) derived from statistical analysis of high-throughput phage display experimental results. To augment the substrate cleavage prediction process, CleavPredict provides information about the structural features of potential cleavage sites that influence proteolysis. These include: secondary structure, disordered regions, transmembrane domains, and solvent accessibility. The server also provides information about subcellular location, co-localization, and co-expression of proteinase and potential substrates, along with experimentally determined positions of single nucleotide polymorphism (SNP), and posttranslational modification (PTM) sites in substrates. All this information will provide the user with perspectives in reasoning about proteolytic events. CleavPredict is freely accessible, and there is no login required.
Introduction
Proteolysis is an important posttranslational modification that involves irreversible hydrolysis of peptide bonds by proteinases. Proteolytic processing has a regulatory role in almost all biological pathways, including cell proliferation, cell death, and blood coagulation [1]. Proteinases identify their substrates with a high degree of specificity. Accurate identification of candidate substrates for proteinases has important implication for understanding the roles of these enzymes in physiological and pathological processes as well as for designing pharmacological intervention approaches. Identification of proteolytic substrates depends on a number of factors. One important factor is the primary substrate specificity, which is defined by a specific amino acid sequence in a substrate that is recognized by the active site of a given proteinase. The efficiency of a cleavage event is also related to the structural properties of the cleavage site. The cleavage site needs to be accessible at the protein surface. Recently, it has been shown that this property as measured by an absolute solvent accessibility index is essential for a proteolytic event to occur [2]. However, cleavage sites that are hidden in native proteins can become accessible as a result of unfolding, allosteric effects, and other proteolytic activity. The efficiency of a cleavage event is also related to the secondary structure of a cleaved amino acid sequence. However, recent statistical analysis of CutDB, the proteolytic event database [3], demonstrated that proteolytic events were uniformly distributed among three types of secondary structures, although with some enrichment in loops. Cleavages in α-helices were found to be relatively abundant in regions apparently prone to unfolding, while cleavages in β-structures tended to be located at the periphery of β-sheets [2]. Other obvious prerequisites for cleavage to occur are co-localization and co-expression. A proteolytic event is not possible if both substrate and proteinase are not in the same compartment of the cell and if they enter the same cell compartment at different times.
The majority of human proteinases have multiple (hundreds of) protein targets. For more than 550 known human proteinases, the potential range of normal and pathological proteolytic events is vast. Proteinases participate in a multitude of biological functions including cell cycle progression [4], cell differentiation [5], cell migration [6], tissue remodelling [7], cholesterol metabolism [8], blood coagulation [9] and apoptosis [10]. Given such widespread importance, it is not surprising that proteinases represent a significant fraction of all druggable targets [11], and that they are driving factors in diseases like emphysema [11], thrombosis [12], arthritis [13], Alzheimer’s [14] metastatic cancer [15], as well as those mediated by viral and bacterial pathogens [16–18].
Among all proteinases, extracellular proteinases matrix metalloproteinases, play a key role in degrading extracellular proteins that help the cell to communicate with its surroundings and function normally. They are important from the physiological, pathological, and pharmaceutical points of view [7, 19, 20]. Vertebrate MMPs have distinct but often overlapping substrate specificities. They can cleave essential extracellular matrix proteins, and, as such, they are highly regulated. The 23 human MMPs can be segregated into three groups; secreted proteinases, proteinases with a transmembrane domain, and proteinases anchored to the membrane with a GPI-linkage. Thus, every MMP is poised to modify interactions between cells, and between cells and the extracellular matrix. So, it is not surprising that MMPs are involved in tumor biology, synaptic plasticity, pulmonary disease, arthritis, atherosclerosis, and sepsis, along with many others [5, 21–27]. In pathological conditions MMPs can play a destructive role, e.g. in rheumatoid and osteoarthritis [28] by degrading key constituents of the extracellular matrix [29–33].
However, even in cases where considerable effort has been devoted to the study of an MMP, our understanding of the fundamental principles that determine their substrates and biological roles remains unclear. For example, MMPs are critical for angiogenesis [22]. Experimentally induced corneal angiogenesis is lacking in MMP-14 deficient mice [34] and is significantly diminished in the MMP-2 knockouts [35]. In a mouse model of retinal regeneration after injury, neovascularization is diminished in both MMP-2 and MMP-9 deficient mice, while it is almost absent in the double MMP-2/MMP-9 knockouts [36]. Inhibition of MMPs using synthetic and endogenous inhibitors has also been shown to down regulate tumor angiogenesis, which is indispensable for tumor development [37–42]. But what are the substrates for these proteinases in angiogenesis? Is a single substrate responsible for the effects of each MMP, or do they cleave distinct substrates?
Similarly, MMPs have a role at every stage of progression of atherosclerosis [43, 44]. MMPs promote matrix invasion by macrophages [45–47] and angiogenesis into vulnerable plaques [48]. These actions contribute to plaque formation and destabilization. On the other hand, MMP-2, 9, and 14 contribute to vascular smooth muscle cell migration and proliferation, thus stabilizing the plaques by increasing its thickness [49].
Recently, it was determined that MMPs (e.g. MMP 2, 3, 9, 14, 26) could be shuttled between cellular compartments. These “moonlighting” enzymes can target not only extracellular but also wide range of intracellular proteins [50, 51].
Understanding how MMP enzymes work and what their proteolytic networks are is of great importance to biologists. However, identification of their individual substrates is a more challenging task because of overlapping specificities of MMPs. Experimental discovery of proteinase targets is time- and resource- consuming. To facilitate this process, we have implemented an in silico method for predicting substrates for 11 out of the total 25 human MMPs. This method is available on-line via the publicly accessible CleavPredict Web server (http://cleavpredict.sanfordburnham.org). On this server, we used the enzyme-specific PWMs as a primary way to predict positions of scissile bonds in protein substrates. The PWMs have been derived based on the cleavage preferences determined from a high-throughput phage display experiment [52, 53]. An efficient and reliable tool for substrate prediction should take into account a number of factors that, if only considered together, define proper conditions for matching substrates with proteinases. We expect that screening the prediction hits using multiple lines of independent qualifiers for proteolytic events is less likely to return a false positive. To augment the predictive ability of the PWMs to recognize positions of scissile bonds, the server provides additional information and evidences (“yes” or “no” filters) for accepting the potential substrate candidate. These include secondary structural elements [54, 55], solvent exposure, presence of signal peptide, and co-expression and co-localization of the proteinase and the potential substrate. Likelihood of the proteolytic event is low if a proteinase and a substrate enter the same subcellular compartment at different times of the cell life cycle. The data on proteinase temporal behaviour can be assessed using the data of gene co-expression. If both proteinase and its substrate are localized in the same subcellular compartment, the substrate can be classified as a potential strong candidate for further experimental verification. In addition, the server reports the presence of SNPs, with available disease annotation, and PTMs in the substrate that could interfere with the proteolysis process. The analysis of SNPs is helpful in determining whether a new cleavage site was created in the disease protein or the existing (in norm) cleavage site was removed by mutation. Similarly, the presence of PTMs may fundamentally alter the function of the protein including availability of the cleavage site to proteolysis. The CleavPredict server also offers a comparison of our predictions with information contained in CutDB database [3], provides virtual mass spectrum based on predicted cleavage pattern, and finally displays predicted cleavage positions, together with SNPs and PTMs sites on the substrate’s structure, if it is only available in the PDB [56].
In summary, the CleavPredict is a useful platform for reasoning about proteolytic events. It can be used as a discovery tool for formulating hypotheses that could be subsequently tested experimentally and conversely, it can be used for interpreting experimental findings, as has been already done in several projects [57–61]. Currently, CleavPredict is devoted to recognition of cleavage sites for MMPs, but it will be extended to other proteinases in the future. To our knowledge, none of the existing prediction methods incorporates all factors described here into the process of proteolytic substrate prediction for MMPs. The PROSPER webserver is another available prediction tool, which was developed by Pike et al. [62]. It offers the recognition of cleavage sites only for four MMPs (e.g. MMP-2, -9, -3, -7) and the batch mode functionality is not available.
Materials and Methods
Derivation of PWMs for predicting cleavage sites
The CleavPredict uses PWMs as a primary mechanism for cleavage prediction. We determined PWMs for the following enzymes: MMP-2, -3, -8, -9, -10, -14, -15, -16, -24, -17, and -25. We selected MMPs representing four main groups of enzymes according to the phylogenetic tree that was already published in our [52, 53] paper. Namely, we selected MMPs belonging to: a) a group containing simple hemopexin domain (MMP-3, MMP-8, MMP-10), b) gelatin binding MMPs (MMP-2, MMP-9), c) transmembrane MMPs (MMP-14, MMP-15, MMP-16, MMP-24), and d) GPI-linked MMPs (MMP-17, MMP-25). The PWMs were derived based on statistical analysis of enzyme specific substrates selected from phage display libraries. We used about 300 substrates for each MMP (S1 Table). Phage substrates are peptides containing constant flanking sequences (small letter) and variable six amino acid sequences (capital letters): ~ggsgPSA-LDAtasgaet~ (dash denominates the cleavage position) [53, 59].
The primary cleavage recognition method, derived for each individual MMP, has been obtained as follows. First, a set of phage substrates specific for a given MMP was selected. Then, the sequences of these substrates were aligned along the cleavage site and the frequencies (P(i AA, j)) of occurrence of each amino acid type, i AA, in each of the j-th position, ranging from P3 to P2’, were calculated. We use Schechter and Berger annotation for amino acid positions in substrates [63]. In Ref. [53] we demonstrated that the amino acids located at P3-P2’ positions are the most important in recognition of the substrates by MMP enzymes. Next, these frequencies at each position and amino acid were normalized by the distribution of amino acids in the set of background sequences. The background sequences comprised 766 peptides randomly selected from phage display library (S2 Table). Thus, the final PWM values for each amino acid iAA at the j-th position are calculated as:
| (1) |
We used log2 values of appropriate PWM(iAA,j) elements, log likelihood ratios, which allows for calculating scores by adding rather than multiplying the relevant values at each position in the PWM. The primary scoring function for substrate prediction is defined in Eqs 2 and 3 as a sum of log2 of PWM matrix elements for i AA amino acid type at the j-th position. Summation runs over P3-P2’ amino acid positions in the substrate:
| (2) |
where
| (3) |
If any element of the PWM is equal to zero, e.g. an amino acid type i AA was not observed at the j-th position in phage substrates, then the offset value is used instead. From numerical point of view this is done in order to avoid calculation of infinite value of log2(0) and yet add the sufficient penalty to the scoring function when an amino acid type i AA at j-th position is not observed in our learning set. The peptide bond is considered a cleavage site when the value of the score is above the threshold value. Both offset and threshold values are MMP specific and were derived using statistical analysis. This primary scoring function can be used to screen every peptide bond in test protein. The peptide bonds that have their score values above the threshold are considered to be potential cleavage sites.
The magnitudes of offset in Eq 3, and threshold are optimizable parameters of the method and are enzyme dependent. In order to establish their specific values for each MMP, we performed a series of 10-fold cross-validation calculations for a large set of offset and threshold values defined on two-dimensional grid. The optimal values of offset and threshold are those corresponding to the maximum value of F1-score. The F1 score is harmonic mean of precision and sensitivity in the machine learning theory. Our additional 10-fold cross-validation calculations demonstrated that extending the range of amino acid summation in Eq 2 beyond P3-P2’ positions does not change significantly the statistical evaluation metrics (results not shown).
We performed 10-fold cross validation calculations for each set of MMP specific substrates in two stages. In the first stage, each set was divided into two groups in approximately 2:1 ratio. The larger group of substrates was used for cross validation, e.g. involving training and validation, while the smaller set was used for independent testing of the performance of optimized scoring parameters. This smaller set is termed an “internal” test.
Next, we developed the final predictive model, in which for each MMP all available specific substrates were combined into the final training and validation sets, which were used for deriving the optimized values of thresholds and offsets parameters in 10-fold cross-validation. These are the values used for prediction cleavages in unknown targets incorporated in our web server. Combining all available data is a standard approach used to create the final prediction model for any “external” tests, as is described in Ref. [64]. The resultant log2 values of the MMP specific PWM matrices are provided in S3 Table. These PWMs have been applied to “external” test cases, such as evaluation of the set of substrates collected in CutDB (cutdb.burnham.org), and peptides determined as MMPs substrates by Overall et al. [65, 66].
Virtual Mass Spectroscopy
We implemented an automated script for calculating mass spectrum based on a predicted set of cleavage sites. All possible mass fragments are calculated with a monoisotopic set of masses for amino acids and displayed on a separate Web page after selecting the VMS (Virtual Mass Spectroscopy) button on the result page. The intensities of the mass fragments are defined as:
| (4) |
where the ws1 and ws2 characterize the cleavage efficiency of the cleavage at the N- and C-terminal sides of the molecular fragment, respectively. The ws1 and ws2 are normalized to [0–1]range values of their respective PWM scores defined by Eq 2. For example, Eq 5 defines normalization of the ws1 value:
| (5) |
where Scoremin_value and Scoremax_value are the minimum and maximum value of the score that can be obtained for a given PWM matrix. The first (N-terminal) and last (C-terminal) residues in the entire protein sequence have the ws1 and ws2 values assigned to 1.0.
External programs and databases used by CleavPredict Web server
There are two main types of query inputs for testing protein substrates in the CleavPredict server: an amino acid sequence or a structure in the PDB format. In the case when an amino acid sequence is an input, the server provides information about predicted secondary structure and disorder regions for a substrate calculated by Jnet (http://www.compbio.dundee.ac.uk/www-jpred/legacy/jnet/) [67] and Disopred (http://bioinf.cs.ucl.ac.uk/psipred/) [68] programs, respectively. If a query input is a PDB structure then the secondary structure elements and solvent accessibility is calculated with the DSSP program (http://swift.cmbi.ru.nl/gv/dssp/) [69, 70]. The SignalP v.4.0 program (http://www.cbs.dtu.dk/services/SignalP/) [71] is used to predict the presence or absence of a signal peptide. The TMHMM (http://www.cbs.dtu.dk/services/TMHMM/) [72] program is applied to predict the transmembrane domains.
In the CleavPredict server, we implemented a link to COXPRESdb (http://coxpresdb.jp) [73] to determine the co-expression of a proteinase and a substrate. The average rank of this event is calculated based on the correlation score and average co-expression score. The average correlation score between the substrate and the proteinase in a gene expression pattern is retrieved and presented. We linked the Mentha (http://mentha.uniroma2.it/about.php) [74] database to our Web server. It is used to determine whether the interaction between a proteinase and a substrate is reported in literature. Each interaction is assigned a reliability score (Mentha Score) that takes into account all the supporting evidence. The information about subcellular location and positions of SNPs is retrieved from the Uniprot resource portal and from the Humsavar database (http://www.uniprot.org/docs/humsavar) [75, 76]. The experimentally known posttranslational modifications of a substrate are determined based on information available from the curated dbPTM (http://dbptm.mbc.nctu.edu.tw) [77] database.
CleavPredict Web server implementation
The CleavPredict Web server has been implemented using Python under the web2py (http://web2py.com/book) framework and running on an Apache server on a Linux machine. The cleavage-site predictions by PWM have been automated by implementing in-house Fortran programs, integrated with Python scripts for processing. Javascript and html were used to present the final results on the user interface. The server offers two options for querying the Web server: a single substrate query, e.g., Uniprot id, Fasta sequence, PDB id, or PDB file; and a batch mode, where multiple Fasta sequences or multiple PDB files can be submitted. When a PDB structure or a Fasta sequence is submitted, the server uses standalone BLASTp against Uniprot and PDB to determine the corresponding Uniprot id. This Uniprot id is used to retrieve appropriate information about co-expression, co-localization, SNPs, PTMs, and structure of the substrate. The PDB structures are displayed at the end of user interface using GLmol (http://webglmol.sourceforge.jp). In order to display the PDB structure with all P1 positions of predicted cleavage sites and amino acids modified by the presence of SNPs and PTMs the Uniprot and PDB structure sequence numbering were mapped using the PDBSWS server (http://www.bioinf.org.uk/pdbsws/) [78].
CleavPredict is currently configured on Apache/2.2.15 (CentOS) and Python2.6.6. The scripts are written in Python and Fortran, and the server uses a web2py framework. The web2py framework allows us to separate the components of our system into the model, the view, and the controller (MVC). The model represents the data of the application, the view specifies the user interface, and the controller handles the communication among all elements of the application. Computational time required for the cleavage site prediction depends on the size of a protein substrate but usually takes less then 30 seconds for a single case input.
Results
Validation of the prediction method
Phage display substrates—internal test
We performed 10-fold cross validation calculations for each set of MMP specific substrates. Each set was divided into two groups in approximately 2:1 ratio. The larger group of substrates, which was further divided into training and validating sets, was used for 10-fold cross validation for establishing the optimal values of offset and threshold for that set. The smaller sets, not seen by 10-fold cross validation, were used for independent testing of the performance of optimized scoring parameters. The results of these internal tests for all MMPs are summarized in Table 1, while the results for 10-fold cross-validation for larger set of substrates are presented in S4 Table. The accuracy of the method exceeds 85%, true positive rate is in the vicinity of 90% and false positive rate ranges from 2.9%, for MMP-16, to 12.7% for MMP-8.
Table 1. Recognition of MMPs cleavage positions in the subset of phage display peptides.
| Enzyme | Number of substrates | Sensitivity(TPR)(%) | Specificity (%) | Accuracy (%) | Precision(%) | MCC | FPR (%) | F1 | |
|---|---|---|---|---|---|---|---|---|---|
| 10F-CV set | Internal test (% of total) | ||||||||
| MMP-2 | 161 | 71 (31%) | 81.9 | 96.2 | 93.4 | 84.0 | 0.79 | 3.8 | 0.83 |
| MMP-9 | 164 | 98 (37%) | 84.4 | 96.7 | 94.4 | 85.2 | 0.81 | 3.3 | 0.85 |
| MMP-14 | 169 | 81 (32%) | 90.7 | 94.9 | 94.0 | 81.5 | 0.82 | 5.1 | 0.86 |
| MMP-15 | 159 | 71 (31%) | 80.8 | 94.2 | 91.1 | 80.8 | 0.75 | 5.8 | 0.81 |
| MMP-16 | 198 | 93 (32%) | 88.0 | 97.1 | 95.3 | 88.0 | 0.85 | 2.9 | 0.88 |
| MMP-24 | 177 | 97 (35%) | 93.6 | 95.1 | 94.8 | 81.7 | 0.84 | 4.9 | 0.87 |
| MMP-17 | 211 | 133 (39%) | 92.4 | 91.4 | 91.6 | 76.3 | 0.79 | 8.6 | 0.84 |
| MMP25 | 159 | 71 (31%) | 84.7 | 92.6 | 90.9 | 76.9 | 0.75 | 7.4 | 0.81 |
| MMP-3 | 304 | 87 (22%) | 91.1 | 93.2 | 92.7 | 78.5 | 0.80 | 6.8 | 0.84 |
| MMP-8 | 203 | 85 (30%) | 84.7 | 87.3 | 86.9 | 57.1 | 0.62 | 12.7 | 0.68 |
| MMP-10 | 170 | 42 (20%) | 97.8 | 91.7 | 92.9 | 72.6 | 0.80 | 8.3 | 0.83 |
Results for the internal statistical test of the scoring function obtained for the subset of substrates not seen by 10-fold cross-validation. TPR—true positive rate, MCC—Matthews correlation coefficient, FPR—false positive rate, F1 score—harmonic mean of precision and sensitivity.
Finally all substrates specific for each MMP have been combined. Each set was divided into 10 subsets and ten training and validating cycles were performed on them to generate the final model that is used for predicting cleavages in unknown targets. The results of this training are summarized in Table 2, where last column provides the optimized values of offset and threshold for each MMP. The corresponding log2 of PWM matrices are presented in S3 Table. For these optimal parameters, the cross-validation achieves high accuracy (>90%) with a true-positive rate above 78% and, in almost all cases, a false-positive rate less than 10% for phage substrates (Table 2).
Table 2. Average values for sensitivity, specificity, accuracy, precision, Matthews correlation coefficients, false positive rate, true positive rate and optimal values for threshold and offset from the 10-fold cross-validation using the entire sets of available substrates for every MMP.
| Sensitivity (TPR)(%) | Specificity (%) | Accuracy (%) | Precision (%) | MCC | FPR(%) | F1 | threshold/ offset | |
|---|---|---|---|---|---|---|---|---|
| MMP2 | 90.2±8.3 | 94.7±2.8 | 93.7±1.7 | 81.8±8.3 | 0.82±0.05 | 5.3±2.8 | 0.85±0.04 | 0.3 / -5.0 |
| MMP9 | 87.7±4.9 | 97.4±1.1 | 95.5±1.5 | 89.3±4.3 | 0.86±0.05 | 2.6±1.1 | 0.88±0.04 | 1.5 / -4.0 |
| MMP14 | 89.8±5.3 | 97.1±1.0 | 95.5±1.2 | 90.0±3.2 | 0.87±0.03 | 2.9±1.0 | 0.90±0.03 | 0.5 / -5.5 |
| MMP15 | 78.2±7.9 | 96.5±1.4 | 92.1±2.8 | 87.3±4.7 | 0.78±0.08 | 3.5±1.4 | 0.82±0.06 | 1.3 / -2.5 |
| MMP16 | 93.4±3.7 | 95.4±1.3 | 95.0±1.4 | 83.2±3.9 | 0.85±0.04 | 4.6±1.3 | 0.88±0.03 | 0.3 / -5.0 |
| MMP24 | 91.9±6.1 | 95.6±1.9 | 94.8±1.8 | 83.8±6.2 | 0.85±0.05 | 4.4±1.9 | 0.88±0.04 | 0.6 / -5.5 |
| MMP17 | 94.2±3.0 | 90.5±2.4 | 91.4±2.0 | 75.7±5.5 | 0.79±0.05 | 9.5±2.4 | 0.84±0.04 | 0.6 / -5.5 |
| MMP25 | 85.7±3.5 | 95.2±2.9 | 92.9±2.7 | 85.3±7.7 | 0.81±0.07 | 4.8±2.9 | 0.85±0.05 | -0.3 / -6.0 |
| MMP3 | 97.8±3.8 | 93.5±1.4 | 94.2±1.4 | 75.2±4.0 | 0.83±0.04 | 6.5±1.4 | 0.85±0.03 | 1.5 / -5.0 |
| MMP8 | 80.1±10.8 | 90.8±2.8 | 89.0±3.4 | 63.8±8.7 | 0.65±0.11 | 9.2±2.8 | 0.71±0.09 | 1.2 / -3.0 |
| MMP10 | 93.7±4.4 | 89.3±1.9 | 90.1±1.7 | 64.0±4.2 | 0.72±0.04 | 10.7±1.9 | 0.76±0.04 | 1.5 / -5.0 |
The calculations have been performed to establish the optimal values for threshold and offset parameters that are implemented in the CleavPredict web server for predicting cleavage sites in proteins. For each average value the sample standard deviations is provided. For abbreviations see Table 1.
The worst results in the above tests, e.g. characterized by the lowest F1 score, have been obtained for MMP-8 enzyme. According to our analysis the MMP-8 is characterized by the widest specificity. In S1 Fig, in Supporting Material we present the sequence logos for the substrate recognition motifs for all 11 MMPs obtained from the analysis of phage display substrates. According to the frequency of occurrence, S1 Fig, almost none of the positions in MMP-8 substrates, contributes significantly to substrate specificity contrary to what was observed for other MMPs. This makes the development of statistically robust prediction model a difficult task.
Analysis of proteome samples—external test
As for the first external we choose two sets of peptide substrates identified by Overall et al. in proteome samples for MMP-2 and MMP-9 enzymes [65, 66]. The first set of 1775 substrates for MMP-2 [66] reported in 2008, has been obtained by the proteomic identification of proteinases cleavage sites (PICS) method combined with liquid chromatography-tandem mass spectrometry (LC-MS/MS). This approach when combined with bioinformatical analysis allows for identification of the prime side sequences of the cleaved peptides. The separate bioinformatical analysis is used to establish the non-prime sequences and then to deduct the position of the cleavage sites. The duration of the enzymatic reaction was chosen to vary between 1–16 h. In 2010 Prudova and Overall [65] proposed more advanced technique called iTRAQ-TAILS, that involves isotopic labelling of substrates. This technique was applied to study specificity of several enzymes, including MMP-2 and MMP-9, for which the authors identified 201 and 19 substrates, respectively. The sensitivity of the method depends on the statistically defined reporter ion ratio cutoff for MS/MS fragmentation in samples with and without the enzyme treatment. This cutoff was uniformly established using GluC enzyme, because its specificity is well known, and cutoff ratio can be validated. Thus, both methods produced the most comprehensive to date set of well-defined peptide substrates.
We applied our prediction method to all reported peptides. The experimentally identified cleavage sites were considered to be a positive set while all other peptides bonds were negative set. We used CleavPredict to evaluate all peptide bonds in substrates and calculated the distribution of PWM score values for the positive and negative sets. The results are presented in Figs 1 and 2. In each case the separation between the scores for negative and positive sets is significant. In the two-sample Kolmogorov-Smirnov (KS) test the value of D statistic is equal to 0.6.
Fig 1. Distribution of PWM scores for peptide substrates of MMP-2 from Schilling et al. [66].

Red line—distribution of PWM score values for experimentally identified cleaved peptide bonds, black line—distribution of scores for all other peptide bonds. Red—dashed line represents distribution of scores for set of cleaved peptide bonds corrected by replacing poorly scored peptide bonds by those that have their scores above the threshold and were located in the vicinity of experimentally predicted positions. The separation between the cleavage site scores and the scores for other peptide bonds was subject to Kolmogorov-Smirnov test yielding D = 0.60 and D = 0.66 for red and red-dashed distributions, respectively, when tested against the black one.
Fig 2. Distribution of PWM scores for peptide substrates of MMP-2 and MMP-9 from Prudova et al. [65].
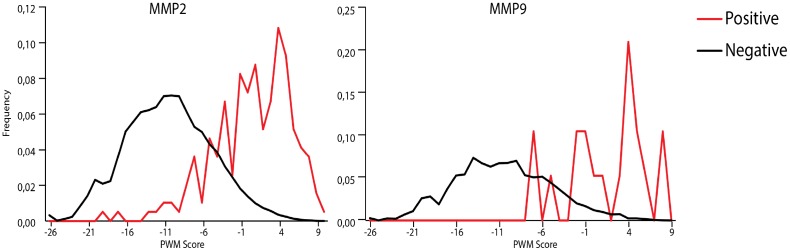
Red and black lines are distributions of PWM score values for experimentally identified cleaved peptide bonds and for all other peptide bonds, respectively. For both MMP-2 and MMP-9 the Kolmogorov-Smirnov test yields D = 0.60.
Experimental identification of cleavage positions in Ref. [66] depends on the accuracy of mass spectroscopy method and there is some level of ambiguity introduced by two separate bioinformatical procedures used for analysis of prime and non-prime product sequences. We hypothesize that the actual cleavage sites could be found within 1 to 4 peptides bonds next to the reported ones. Analysis of our prediction data showed it may be the case. We only include those peptide bonds that scored above the method threshold. We found 184 cleaved peptides bonds in the vicinity of experimentally identified ones that have PWM scores higher than the threshold and better match sequence patterns observed in our phage display experiment. When taking into account those new cleavage positions the discrimination between non-cleaved and cleaved peptide bonds becomes more pronounced (D value in KS test is 0.66), see Fig 1 (“corrected” curve) and Table 3. This result, of course, does not preclude the biases in our approach. Understanding them would lead to the improvement of our algorithm. Nevertheless, our results demonstrate that identification of the proper cleavage position in some substrates reported by Overall et al. could be revisited.
Table 3. Results for the external statistical test of the scoring function for the MMP-2 and MMP-9 cleavages in peptide substrates identified by Overall et al.
| Exper. cleavage sites | Predicted cleavage sites | TPR(%) | FPR(%) | Precision(%) | Accuracy(%) | Specificity(%) | |
|---|---|---|---|---|---|---|---|
| 1775 | 864 | 49.0 | 4.0 | 42.3 | 93.0 | 96.0 | MMP2 Schilling, Overall, 2008 [66] |
| 1775 | 1048 | 59.0 | 4.0 | 51.3 | 94.0 | 96.0 | MMP2 Schilling, Overall, 2008 [66] (corrected) |
| 201 | 120 | 60.0 | 3.0 | 57.1 | 94.0 | 97.0 | MMP2 Prudova, Overall, 2010 [65] |
| 19 | 13 | 68.0 | 2.0 | 72.2 | 96.0 | 98.0 | MMP9 Prudova, Overall, 2010 [65] |
CutDB—external test
Further on we validated our PWM—based scoring method in another “external” test performed on substrates collected in CutDB [3]. We selected only those MMPs for which sufficient number of cleavage events and protein substrates is available. Thus, we applied our prediction algorithm to calculate scores for cleavage sites in substrates of five MMPs including: MMP-9 (334 cleavages in 88 unique substrates), MMP-2 (135 cleavages in 50 substrates), MMP-14 (89 cleavages in 38 substrates), MMP-3 (186 cleavages in 67 proteins) and MMP-8 (85 cleavages in 26 proteins). In these calculations, the positive set constitutes the cleavage sites reported in the literature (CutDB) for appropriate protein substrates for each MMP, while for the negative set we choose peptide bonds randomly selected from the same protein substrates that are not cleaved by MMP. The ratio of positive to negative cases is 1:100. The results of our prediction calculations are collected in Table 4. It demonstrates that for experimental protein substrates the PWM approach yields the accuracy reaching the level of 70% for most MMPs, while the false-positive rate is in the range of 30%, with the exception of MMP-8 and MMP-9 for which false-positive rate is 61 and 47%, respectively. The appropriate ROC curves are presented in Fig 3. Area under the curve (AUC) (Table 4), in most cases is well above 0.8, which demonstrate a good ability of our method to discriminate between cleavable and non-cleavable peptide bonds for MMP hydrolysis. The high level of false-positive rate is not satisfactory here and substantially higher than for uniformly identified substrates by Overall et al., as discussed above. However, we are aware that the reported cleavage sites come from highly heterogeneous sources and may not all be entirely accurate, either because denatured proteins were used as substrates, or because when the study was performed the methods for determining the position of the cleavage sites, including mass spectroscopy, were not as robust as methods available today. What is more important, the conditions used for studying cleavage events reported in the literature could differ substantially from those used in our phage display experiment. The conditions used in our high throughput phage display experiment allow measuring important cleavage events with observed kcat/KM values above 3000 sec-1M-1 [53]. Thus, if our predictive algorithm is sufficiently accurate, we may be able to identify the reported cleavage sites that are “suspect.”
Table 4. Results for the external statistical test of the scoring function for the MMPs protein substrates collected in CutDB database.
| Enzyme | Cleavage sites in CutDB | Predicted cleavage sites | TPR(%) | FPR (%) | Accuracy(%) | Area Under theCurve (AUC) |
|---|---|---|---|---|---|---|
| MMP-2 | 135 | 115 | 85.2 | 29.4 | 70.7 | 0.862 |
| MMP-9 | 344 | 296 | 88.6 | 46.6 | 53.6 | 0.836 |
| MMP-14 | 89 | 61 | 68.5 | 29.0 | 71.0 | 0.760 |
| MMP-3 | 186 | 155 | 83.3 | 28.2 | 71.8 | 0.849 |
| MMP-8 | 85 | 83 | 97.6 | 61.0 | 39.0 | 0.890 |
Fig 3. ROC curves for prediction cleavage sites in proteins collected in CutDB for MMP-2, MMP-3, MMP-8, MMP-9 and MMP-14.

Description of an input and output for CleavPredict server
Workflow of the CleavPredict web server is presented in Fig 4.
Fig 4. Workflow of the CleavPredict Web server.

Top: the types of input queries; middle: the first tier of the output data; bottom: the second tier of the results obtained using a Uniprot id as for the input protein substrate. Blast program and mapping is used for determining the Uniprot id.
User input
The user can submit either a single protein query in the interactive mode or a multi-protein query in the batch mode. In a single-protein-query mode, the input for a potential protein substrate can be provided in the form of a Uniprot accession number, a Fasta sequence, an uploaded PDB file, or a PDB id (Fig 4: Query Type). In the batch mode, the server accepts either a file containing multiple Fasta sequences, a list of multiple PDB ids (with a single space between each id), or multiple PDB files uploaded from a local computer. Once the input protein substrate(s) is/are defined, the user selects the MMP type from the list for which cleavage predictions will be calculated. The “Submit” button submits the input query for cleavage prediction calculations.
Outputs
The two sections/tiers of the output data are summarized in Fig 4. All the results are shown in tabular form (see example in Fig 5). The form of the first section of the output depends on the type of input query. When a query input is in the form of a protein sequence, then the first table contains a list of predicted P1 cleavage positions, 10 amino acid sequences around the cleavage site, a PWM score, the predicted secondary structure (alphahelix; ‘H’, beta-sheet; ‘E’. loop; ‘_’) and the predicted disorder (order: ‘.’; disorder ‘*’) characterizing this region of 10 amino acids. In addition, for each of these 10 amino acid positions, the server reports confidence scores of prediction in the range of 0–9, calculated by the Jnet and the Disopred programs. The confidence score is computed for every amino acid position, as a separate number. Thus, for ten amino acids fragment the server reports ten numbers in the form of a chain of consecutive values. The table also provides information about the presence of transmembrane domain for 10 amino acids region around the cleavage site, as predicted by the TMHMM program; and N-terminal and C-terminal mass fragments resultant from each cleavage event (Fig 5, label: A).
Fig 5. Snapshots of the result pages.

As an example the prediction of the cleavage positions in Q15848 protein for MMP2 enzyme is demonstrated. This section contains information about signal peptide prediction, subcellular location, co-expression and co-localization information, known cleavages in CutDB, data on experimentally identified SNPs and PTMs, congregated into tables.
When the PDB structure is a query input, the above-described table is partially modified. In this case, the server provides information about solvent accessibility of the cleavage site and actual secondary structure assignment, instead of predicted parameters. These properties are calculated by the DSSP program and provided in the CleavPredict result page for the region of 10 amino acids around the predicted cleavage sites.
When the Uniprot id is not provided explicitly in the input query, the BLASTp is run internally against the Swissprot database to determine the Uniprot accession number. This number is necessary to retrieve other information about the query substrate, such as co-expression, co-localization, SNPs and PTMs from appropriate databases. In the case when the PDB id is provided as an input, PDB—Uniprot id mapping is used instead of BLASTp.
The second section of the result page (Fig 5) contains information about: B) the distribution of masses after the cleavage via link to virtual mass spectroscopy results (VMS button. See Supporting Material Figure A in S1 File for example of an output from VMS); C) the presence of a signal peptide in the substrate, indicating whether it belongs to the set of secreted proteins; D) the subcellular localization of the substrate and the proteinase; E) the co-expression of the substrate and the proteinase retrieved from COXPRESSdb; F) the physical interaction between the substrate and the enzyme retrieved from the Mentha database; G) known cleavages in the query substrate retrieved from CutDB, that can be used for making comparison with CleavPredict predictions, and H) known SNPs, with disease annotation, when available, and PTMs in the substrate. Additionally, for user convenience, the server displays sequences of the query substrate in the Fasta format and color-code the predicted cleavages, SNPs, and PTMs (Supporting Material Figure B in S1 File). This information is also displayed in the PDB structure, if it is available, using the GLmol viewer.
In the batch mode, results of the calculations can be downloaded from the server or may be sent to the user’s e-mail address that is optionally provided at the time of query input.
Conclusions and Future Developments
We will continue to integrate more proteinases into our CleavPredict Web server. This includes thrombin, furin, caspases, and others for which large set of substrates could be extracted from the literature, from the MEROPS database [79], or from our own effort aimed at high-throughput profiling of proteinases [53, 80–82]. We will work toward integrating PWMs with structural elements information into a single unified scoring function that will be used for discrimination between cleaved and non-cleaved peptide bonds. Initial work toward this goal has been already published recently [2, 54]. For many proteins, the 3D structure is not available. Instead of relying on prediction of secondary structure elements, we will incorporate a mechanism that can be used to build a homology model for the potential substrate. Homology modelling will be performed using the FFAS server (ffas.burnham.org). Our computational platform can be further extended by connecting predicted cleavage events to the chain of other events using a library of pathways and networks. The combined knowledge of the position of cleavage sites, SNPs and PTMs in the vicinity of cleavage sites, as well as knowledge of pathways and networks could be used to discover relationships between aberrant proteolytic events and potential disease or syndromes. The main problem with most, if not all, prediction methods is over-prediction of the substrates. In the case of MMPs—this problem is partially related to their broad and overlapping specificity.
We believe that CleavPredict can become a versatile hypothesis generator guiding future experiments in basic and transitional medical research. The CleavPredict has been already successfully applied to several practical scientific projects related to discovery of new MMPs substrates and helped in interpreting experimental findings [57–61].
Supporting Information
Left column—frequency logos, right column—information content logos. The logos have been created using WebLogo on-line web server: weblogo.berkeley.edu [83].
(JPEG)
{kind=link}
(A) Snapshot of the web page demonstrating the top of the scrollable table containing virtual mass spectrum data displayed after selecting the VMS button on the first result page. (B) Graphical display of the cleavage P1 positions (red), SNPs (green), and PTMs (blue).
(DOC)
(DOC)
(DOC)
The header of each matrix contains the values of offset and threshold. The offset values are already incorporated into the log2 PWM matrices in appropriate positions where given amino acid is not observed in phage display substrates.
(DOC)
The calculations have been performed to establish the optimal values for threshold and offset parameters that are implemented in the CleavPredict web server for predicting cleavage sites in proteins. For each average value the sample standard deviations is provided. For abbreviations see Table 1.
(DOC)
Acknowledgments
The authors would like to thank David Huhta from Sanford-Burnham Medical Research Institute for his help in solving technical problems associated with setting up the CleavPredict server.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This work was supported by the National Institutes of Health (NIH) grant R01GM098835 (PC) and by the Russian Foundation grant: 15-04-06861a and Russian Academy of Sciences via program "Molecular and Cellular Biology" (MK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lopez-Otin C, Bond JS. Proteases: multifunctional enzymes in life and disease. J Biol Chem. 2008;283(45):30433–7. Epub 2008/07/25. 10.1074/jbc.R800035200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Belushkin AA, Vinogradov DV, Gelfand MS, Osterman AL, Cieplak P, Kazanov MD. Sequence-derived structural features driving proteolytic processing. Proteomics. 2014;14(1):42–50. Epub 2013/11/15. 10.1002/pmic.201300416 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Igarashi Y, Eroshkin A, Gramatikova S, Gramatikoff K, Zhang Y, Smith JW, et al. CutDB: a proteolytic event database. Nucleic acids research. 2007;35(Database issue):D546–9. Epub 2006/12/05. 10.1093/nar/gkl813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lopez-Aviles S, Uhlmann F. Cell cycle: the art of multi-tasking. Current biology: CB. 2010;20(3):R101–3. Epub 2010/02/11. 10.1016/j.cub.2010.01.001 . [DOI] [PubMed] [Google Scholar]
- 5. Churg A, Zhou S, Wright JL. Series "matrix metalloproteinases in lung health and disease": Matrix metalloproteinases in COPD. The European respiratory journal: official journal of the European Society for Clinical Respiratory Physiology. 2012;39(1):197–209. Epub 2011/09/17. 10.1183/09031936.00121611 . [DOI] [PubMed] [Google Scholar]
- 6. Critchley DR. Smurf1 zaps the talin head. Nature cell biology. 2009;11(5):538–40. Epub 2009/05/01. 10.1038/ncb0509-538 . [DOI] [PubMed] [Google Scholar]
- 7. Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat Rev Mol Cell Biol. 2007;8(3):221–33. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brown MS, Goldstein JL. The SREBP pathway: regulation of cholesterol metabolism by proteolysis of a membrane-bound transcription factor. Cell. 1997;89(3):331–40. . [DOI] [PubMed] [Google Scholar]
- 9. Coughlin SR. Thrombin signalling and protease-activated receptors. Nature. 2000;407(6801):258–64. . [DOI] [PubMed] [Google Scholar]
- 10. Salvesen GS, Dixit VM. Caspases: intracellular signaling by proteolysis. Cell. 1997;91(4):443–6. Epub 1997/12/09. . [DOI] [PubMed] [Google Scholar]
- 11. Overall CM, Kleifeld O. Tumour microenvironment—opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nature reviews Cancer. 2006;6(3):227–39. Epub 2006/02/25. 10.1038/nrc1821 . [DOI] [PubMed] [Google Scholar]
- 12. Carrell RW, Owen MC. Plakalbumin, alpha 1-antitrypsin, antithrombin and the mechanism of inflammatory thrombosis. Nature. 1985;317(6039):730–2. . [DOI] [PubMed] [Google Scholar]
- 13. Holmbeck K, Bianco P, Caterina J, Yamada S, Kromer M, Kuznetsov SA, et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell. 1999;99(1):81–92. . [DOI] [PubMed] [Google Scholar]
- 14. Haass C, De Strooper B. The presenilins in Alzheimer's disease—proteolysis holds the key. Science. 1999;286(5441):916–9. . [DOI] [PubMed] [Google Scholar]
- 15. Wuarin J, Nurse P. Regulating S phase: CDKs, licensing and proteolysis. Cell. 1996;85(6):785–7. . [DOI] [PubMed] [Google Scholar]
- 16. Buller RM, Palumbo GJ. Poxvirus pathogenesis. Microbiol Rev. 1991;55(1):80–122. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rossetto O, de Bernard M, Pellizzari R, Vitale G, Caccin P, Schiavo G, et al. Bacterial toxins with intracellular protease activity. Clin Chim Acta. 2000;291(2):189–99. . [DOI] [PubMed] [Google Scholar]
- 18. Lorenz IC, Marcotrigiano J, Dentzer TG, Rice CM. Structure of the catalytic domain of the hepatitis C virus NS2-3 protease. Nature. 2006;442(7104):831–5. . [DOI] [PubMed] [Google Scholar]
- 19. Coussens LM, Fingleton B, Matrisian LM. Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science. 2002;295(5564):2387–92. Epub 2002/03/30. 10.1126/science.1067100 . [DOI] [PubMed] [Google Scholar]
- 20. Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74. . [DOI] [PubMed] [Google Scholar]
- 21. Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. Epub 2010/04/08. 10.1016/j.cell.2010.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Page-McCaw A, Ewald AJ, Werb Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nature reviews Molecular cell biology. 2007;8(3):221–33. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dziembowska M, Wlodarczyk J. MMP9: a novel function in synaptic plasticity. The international journal of biochemistry & cell biology. 2012;44(5):709–13. Epub 2012/02/14. 10.1016/j.biocel.2012.01.023 . [DOI] [PubMed] [Google Scholar]
- 24. Troeberg L, Nagase H. Proteases involved in cartilage matrix degradation in osteoarthritis. Biochimica et biophysica acta. 2012;1824(1):133–45. Epub 2011/07/23. 10.1016/j.bbapap.2011.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Burrage PS, Mix KS, Brinckerhoff CE. Matrix metalloproteinases: role in arthritis. Frontiers in bioscience: a journal and virtual library. 2006;11:529–43. Epub 2005/09/09. . [DOI] [PubMed] [Google Scholar]
- 26. Hwang IK, Park SM, Kim SY, Lee ST. A proteomic approach to identify substrates of matrix metalloproteinase-14 in human plasma. Biochimica et biophysica acta. 2004;1702(1):79–87. . [DOI] [PubMed] [Google Scholar]
- 27. Vanlaere I, Libert C. Matrix metalloproteinases as drug targets in infections caused by gram-negative bacteria and in septic shock. Clinical microbiology reviews. 2009;22(2):224–39, Table of Contents. Epub 2009/04/16. 10.1128/CMR.00047-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Murphy G, Nagase H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: destruction or repair? Nat Clin Pract Rheumatol. 2008;4(3):128–35. Epub 2008/02/07. ncprheum0727 [pii] 10.1038/ncprheum0727 . [DOI] [PubMed] [Google Scholar]
- 29. Wu W, Billinghurst RC, Pidoux I, Antoniou J, Zukor D, Tanzer M, et al. Sites of collagenase cleavage and denaturation of type II collagen in aging and osteoarthritic articular cartilage and their relationship to the distribution of matrix metalloproteinase 1 and matrix metalloproteinase 13. Arthritis Rheum. 2002;46(8):2087–94. Epub 2002/09/05. 10.1002/art.10428 . [DOI] [PubMed] [Google Scholar]
- 30. Fraser A, Fearon U, Billinghurst RC, Ionescu M, Reece R, Barwick T, et al. Turnover of type II collagen and aggrecan in cartilage matrix at the onset of inflammatory arthritis in humans: relationship to mediators of systemic and local inflammation. Arthritis Rheum. 2003;48(11):3085–95. Epub 2003/11/13. 10.1002/art.11331 . [DOI] [PubMed] [Google Scholar]
- 31. Lark MW, Bayne EK, Flanagan J, Harper CF, Hoerrner LA, Hutchinson NI, et al. Aggrecan degradation in human cartilage. Evidence for both matrix metalloproteinase and aggrecanase activity in normal, osteoarthritic, and rheumatoid joints. J Clin Invest. 1997;100(1):93–106. Epub 1997/07/01. 10.1172/JCI119526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Andersen TL, del Carmen Ovejero M, Kirkegaard T, Lenhard T, Foged NT, Delaisse JM. A scrutiny of matrix metalloproteinases in osteoclasts: evidence for heterogeneity and for the presence of MMPs synthesized by other cells. Bone. 2004;35(5):1107–19. Epub 2004/11/16. S8756-3282(04)00261-3 [pii] 10.1016/j.bone.2004.06.019 . [DOI] [PubMed] [Google Scholar]
- 33. Benito MJ, Veale DJ, FitzGerald O, van den Berg WB, Bresnihan B. Synovial tissue inflammation in early and late osteoarthritis. Ann Rheum Dis. 2005;64(9):1263–7. Epub 2005/02/26. ard.2004.025270 [pii] 10.1136/ard.2004.025270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhou Z, Apte SS, Soininen R, Cao R, Baaklini GY, Rauser RW, et al. Impaired endochondral ossification and angiogenesis in mice deficient in membrane-type matrix metalloproteinase I. Proc Natl Acad Sci U S A. 2000;97(8):4052–7. Epub 2000/03/29. 10.1073/pnas.060037197060037197 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kato T, Kure T, Chang JH, Gabison EE, Itoh T, Itohara S, et al. Diminished corneal angiogenesis in gelatinase A-deficient mice. FEBS Lett. 2001;508(2):187–90. Epub 2001/11/24. S0014-5793(01)02897-6 [pii]. . [DOI] [PubMed] [Google Scholar]
- 36. Lambert V, Wielockx B, Munaut C, Galopin C, Jost M, Itoh T, et al. MMP-2 and MMP-9 synergize in promoting choroidal neovascularization. FASEB J. 2003;17(15):2290–2. Epub 2003/10/18. doi: 10.1096/fj.03-0113fje 03-0113fje [pii]. . [DOI] [PubMed] [Google Scholar]
- 37. Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86(3):353–64. Epub 1996/08/09. S0092-8674(00)80108-7 [pii]. . [DOI] [PubMed] [Google Scholar]
- 38. Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci U S A. 2001;98(22):12485–90. Epub 2001/10/19. 10.1073/pnas.171460498171460498 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oh J, Takahashi R, Kondo S, Mizoguchi A, Adachi E, Sasahara RM, et al. The membrane-anchored MMP inhibitor RECK is a key regulator of extracellular matrix integrity and angiogenesis. Cell. 2001;107(6):789–800. Epub 2001/12/19. S0092-8674(01)00597-9 [pii]. . [DOI] [PubMed] [Google Scholar]
- 40. Martin DC, Sanchez-Sweatman OH, Ho AT, Inderdeo DS, Tsao MS, Khokha R. Transgenic TIMP-1 inhibits simian virus 40 T antigen-induced hepatocarcinogenesis by impairment of hepatocellular proliferation and tumor angiogenesis. Lab Invest. 1999;79(2):225–34. Epub 1999/03/06. . [PubMed] [Google Scholar]
- 41. Li H, Lindenmeyer F, Grenet C, Opolon P, Menashi S, Soria C, et al. AdTIMP-2 inhibits tumor growth, angiogenesis, and metastasis, and prolongs survival in mice. Hum Gene Ther. 2001;12(5):515–26. Epub 2001/03/27. 10.1089/104303401300042429 . [DOI] [PubMed] [Google Scholar]
- 42. Gatto C, Rieppi M, Borsotti P, Innocenti S, Ceruti R, Drudis T, et al. BAY 12–9566, a novel inhibitor of matrix metalloproteinases with antiangiogenic activity. Clin Cancer Res. 1999;5(11):3603–7. Epub 1999/12/10. . [PubMed] [Google Scholar]
- 43. Newby AC. Dual role of matrix metalloproteinases (matrixins) in intimal thickening and atherosclerotic plaque rupture. Physiol Rev. 2005;85(1):1–31. Epub 2004/12/25. 85/1/1 [pii] 10.1152/physrev.00048.2003 . [DOI] [PubMed] [Google Scholar]
- 44. Newby AC. Metalloproteinases and vulnerable atherosclerotic plaques. Trends Cardiovasc Med. 2007;17(8):253–8. Epub 2007/11/21. S1050-1738(07)00178-8 [pii] 10.1016/j.tcm.2007.09.001 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci U S A. 1996;93(9):3942–6. Epub 1996/04/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Johnson JL, Baker AH, Oka K, Chan L, Newby AC, Jackson CL, et al. Suppression of atherosclerotic plaque progression and instability by tissue inhibitor of metalloproteinase-2: involvement of macrophage migration and apoptosis. Circulation. 2006;113(20):2435–44. Epub 2006/05/17. CIRCULATIONAHA.106.613281 [pii] 10.1161/CIRCULATIONAHA.106.613281 . [DOI] [PubMed] [Google Scholar]
- 47. Johnson JL, Fritsche-Danielson R, Behrendt M, Westin-Eriksson A, Wennbo H, Herslof M, et al. Effect of broad-spectrum matrix metalloproteinase inhibition on atherosclerotic plaque stability. Cardiovasc Res. 2006;71(3):586–95. Epub 2006/06/09. S0008-6363(06)00192-1 [pii] 10.1016/j.cardiores.2006.05.009 . [DOI] [PubMed] [Google Scholar]
- 48. Pepper MS. Role of the matrix metalloproteinase and plasminogen activator-plasmin systems in angiogenesis. Arterioscler Thromb Vasc Biol. 2001;21(7):1104–17. Epub 2001/07/14. . [DOI] [PubMed] [Google Scholar]
- 49. Newby AC. Matrix metalloproteinases regulate migration, proliferation, and death of vascular smooth muscle cells by degrading matrix and non-matrix substrates. Cardiovasc Res. 2006;69(3):614–24. Epub 2005/11/04. S0008-6363(05)00407-4 [pii] 10.1016/j.cardiores.2005.08.002 . [DOI] [PubMed] [Google Scholar]
- 50. Butler GS, Overall CM. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat Rev Drug Discov. 2009;8(12):935–48. Epub 2009/12/02. 10.1038/nrd2945 . [DOI] [PubMed] [Google Scholar]
- 51. Butler GS, Overall CM. Updated biological roles for matrix metalloproteinases and new "intracellular" substrates revealed by degradomics. Biochemistry. 2009;48(46):10830–45. Epub 2009/10/13. 10.1021/bi901656f . [DOI] [PubMed] [Google Scholar]
- 52. Ratnikov B, Cieplak P, Smith JW. High throughput substrate phage display for protease profiling. Methods Mol Biol. 2009;539:93–114. Epub 2009/04/21. 10.1007/978-1-60327-003-8_6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ratnikov BI, Cieplak P, Gramatikoff K, Pierce J, Eroshkin A, Igarashi Y, et al. Basis for substrate recognition and distinction by matrix metalloproteinases. Proc Natl Acad Sci U S A. 2014;111(40):E4148–55. 10.1073/pnas.1406134111 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kazanov MD, Igarashi Y, Eroshkin AM, Cieplak P, Ratnikov B, Zhang Y, et al. Structural determinants of limited proteolysis. Journal of proteome research. 2011;10(8):3642–51. Epub 2011/06/21. 10.1021/pr200271w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochimica et biophysica acta. 1998;1382(2):191–206. Epub 1998/04/16. S0167-4838(97)00175-1 [pii]. . [DOI] [PubMed] [Google Scholar]
- 56. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, et al. The Protein Data Bank. Nucleic Acids Res. 2000;28(1):235–42. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Golubkov VS, Chekanov AV, Cieplak P, Aleshin AE, Chernov AV, Zhu W, et al. The Wnt/planar cell polarity protein-tyrosine kinase-7 (PTK7) is a highly efficient proteolytic target of membrane type-1 matrix metalloproteinase: implications in cancer and embryogenesis. J Biol Chem. 2010;285(46):35740–9. Epub 2010/09/15. 10.1074/jbc.M110.165159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Golubkov VS, Cieplak P, Chekanov AV, Ratnikov BI, Aleshin AE, Golubkova NV, et al. Internal cleavages of the autoinhibitory prodomain are required for membrane type 1 matrix metalloproteinase activation, although furin cleavage alone generates inactive proteinase. The Journal of biological chemistry. 2010;285(36):27726–36. Epub 2010/07/08. 10.1074/jbc.M110.135442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shiryaev SA, Savinov AY, Cieplak P, Ratnikov BI, Motamedchaboki K, Smith JW, et al. Matrix metalloproteinase proteolysis of the myelin basic protein isoforms is a source of immunogenic peptides in autoimmune multiple sclerosis. PloS one. 2009;4(3):e4952 Epub 2009/03/21. 10.1371/journal.pone.0004952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shiryaev SA, Cieplak P, Aleshin AE, Sun Q, Zhu W, Motamedchaboki K, et al. Matrix metalloproteinase proteolysis of the mycobacterial HSP65 protein as a potential source of immunogenic peptides in human tuberculosis. Febs J. 2011;278(18):3277–86. Epub 2011/07/15. 10.1111/j.1742-4658.2011.08244.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Shiryaev SA, Remacle AG, Savinov AY, Chernov AV, Cieplak P, Radichev IA, et al. Inflammatory proprotein convertase-matrix metalloproteinase proteolytic pathway in antigen-presenting cells as a step to autoimmune multiple sclerosis. The Journal of biological chemistry. 2009;284(44):30615–26. Epub 2009/09/04. 10.1074/jbc.M109.041244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Song J, Tan H, Perry AJ, Akutsu T, Webb GI, Whisstock JC, et al. PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PloS one. 2012;7(11):e50300 Epub 2012/12/05. 10.1371/journal.pone.0050300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. 1967. Biochemical and biophysical research communications. 1967;425(3):497–502. 10.1016/j.bbrc.2012.08.015 . [DOI] [PubMed] [Google Scholar]
- 64. Witten IH, Frank E, Hall MA. Data Mining: Practical Machines Learning Tools and Techniques. third edition ed. Amsterdam: Elsevier; 2011. 149 p. [Google Scholar]
- 65. Prudova A, auf dem Keller U, Butler GS, Overall CM. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Molecular & cellular proteomics: MCP. 2010;9(5):894–911. Epub 2010/03/23. 10.1074/mcp.M000050-MCP201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Schilling O, Overall CM. Proteome-derived, database-searchable peptide libraries for identifying protease cleavage sites. Nat Biotechnol. 2008;26(6):685–94. . [DOI] [PubMed] [Google Scholar]
- 67. Cuff JA, Barton GJ. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins. 2000;40(3):502–11. Epub 2000/06/22. . [DOI] [PubMed] [Google Scholar]
- 68. Ward JJ, McGuffin LJ, Bryson K, Buxton BF, Jones DT. The DISOPRED server for the prediction of protein disorder. Bioinformatics. 2004;20(13):2138–9. Epub 2004/03/27. 10.1093/bioinformatics/bth195bth195 [pii]. . [DOI] [PubMed] [Google Scholar]
- 69. Joosten RP, te Beek TA, Krieger E, Hekkelman ML, Hooft RW, Schneider R, et al. A series of PDB related databases for everyday needs. Nucleic Acids Res. 2011;39(Database issue):D411–9. Epub 2010/11/13. 10.1093/nar/gkq1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kabsch W, Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22(12):2577–637. Epub 1983/12/01. 10.1002/bip.360221211 . [DOI] [PubMed] [Google Scholar]
- 71. Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods. 2011;8(10):785–6. Epub 2011/10/01. 10.1038/nmeth.1701 . [DOI] [PubMed] [Google Scholar]
- 72. Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol. 2001;305(3):567–80. Epub 2001/01/12. 10.1006/jmbi.2000.4315 . [DOI] [PubMed] [Google Scholar]
- 73. Obayashi T, Okamura Y, Ito S, Tadaka S, Motoike IN, Kinoshita K. COXPRESdb: a database of comparative gene coexpression networks of eleven species for mammals. Nucleic Acids Res. 2013;41(Database issue):D1014–20. Epub 2012/12/04. 10.1093/nar/gks1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Calderone A, Castagnoli L, Cesareni G. mentha: a resource for browsing integrated protein-interaction networks. Nat Methods. 2013;10(8):690–1. Epub 2013/08/01. 10.1038/nmeth.2561 . [DOI] [PubMed] [Google Scholar]
- 75. Magrane M, Consortium U. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford). 2011;2011:bar009 Epub 2011/03/31. 10.1093/database/bar009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Yip YL, Scheib H, Diemand AV, Gattiker A, Famiglietti LM, Gasteiger E, et al. The Swiss-Prot variant page and the ModSNP database: a resource for sequence and structure information on human protein variants. Hum Mutat. 2004;23(5):464–70. Epub 2004/04/27. 10.1002/humu.20021 . [DOI] [PubMed] [Google Scholar]
- 77. Lu CT, Huang KY, Su MG, Lee TY, Bretana NA, Chang WC, et al. DbPTM 3.0: an informative resource for investigating substrate site specificity and functional association of protein post-translational modifications. Nucleic Acids Res. 2013;41(Database issue):D295–305. Epub 2012/11/30. 10.1093/nar/gks1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Martin AC. Mapping PDB chains to UniProtKB entries. Bioinformatics. 2005;21(23):4297–301. Epub 2005/09/29. 10.1093/bioinformatics/bti694 . [DOI] [PubMed] [Google Scholar]
- 79. Rawlings ND, Waller M, Barrett AJ, Bateman A. MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic acids research. 2014;42(Database issue):D503–9. Epub 2013/10/26. 10.1093/nar/gkt953 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Shiryaev SA, Chernov AV, Golubkov VS, Thomsen ER, Chudin E, Chee MS, et al. High-resolution analysis and functional mapping of cleavage sites and substrate proteins of furin in the human proteome. PloS one. 2013;8(1):e54290 Epub 2013/01/22. 10.1371/journal.pone.0054290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shiryaev SA, Remacle AG, Chernov AV, Golubkov VS, Motamedchaboki K, Muranaka N, et al. Substrate cleavage profiling suggests a distinct function of Bacteroides fragilis metalloproteinases (fragilysin and metalloproteinase II) at the microbiome-inflammation-cancer interface. The Journal of biological chemistry. 2013;288(48):34956–67. Epub 2013/10/23. 10.1074/jbc.M113.516153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Shiryaev SA, Thomsen ER, Cieplak P, Chudin E, Cheltsov AV, Chee MS, et al. New details of HCV NS3/4A proteinase functionality revealed by a high-throughput cleavage assay. PloS one. 2012;7(4):e35759 Epub 2012/05/05. 10.1371/journal.pone.0035759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Crooks GE, Hon G, Chandonia JM, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14(6):1188–90. . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Left column—frequency logos, right column—information content logos. The logos have been created using WebLogo on-line web server: weblogo.berkeley.edu [83].
(JPEG)
(A) Snapshot of the web page demonstrating the top of the scrollable table containing virtual mass spectrum data displayed after selecting the VMS button on the first result page. (B) Graphical display of the cleavage P1 positions (red), SNPs (green), and PTMs (blue).
(DOC)
(DOC)
(DOC)
The header of each matrix contains the values of offset and threshold. The offset values are already incorporated into the log2 PWM matrices in appropriate positions where given amino acid is not observed in phage display substrates.
(DOC)
The calculations have been performed to establish the optimal values for threshold and offset parameters that are implemented in the CleavPredict web server for predicting cleavage sites in proteins. For each average value the sample standard deviations is provided. For abbreviations see Table 1.
(DOC)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.