

Abstract
Spontaneous tumor-initiated T cell priming is dependent on IFN-β production by tumor-resident dendritic cells. Based on recent observations indicating that IFN-β expression was dependent upon activation of the host STING pathway, we hypothesized that direct engagement of STING through intratumoral administration of specific agonists would result in effective antitumor therapy. After proof-of-principle studies using the mouse STING agonist DMXAA showed a potent therapeutic effect, we generated synthetic cyclic dinucleotide (CDN) derivatives that activated all human STING alleles as well as murine STING. Intratumoral injection of STING agonists induced profound regression of established tumors in mice and generated substantial systemic immune responses capable of rejecting distant metastases and providing long-lived immunologic memory. Synthetic CDNs have high translational potential as a cancer therapeutic.
Graphical abstract
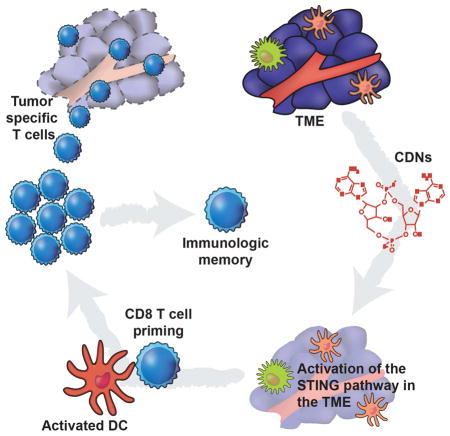
Introduction
The responsiveness of tumors to immunotherapy depends, at least in part, on the immunophenotype of the tumor microenvironment (TME) (Gajewski et al., 2013). Substantial evidence indicates that tumor-infiltrating lymphocytes (TILs) are correlated with favorable prognosis in diverse malignancies (Galon et al., 2012) and predicts a positive clinical outcome in response to several immunotherapy strategies (Postow et al., 2012; Wolchok et al., 2013). Understanding the underlying mechanisms that promote spontaneous T cell infiltration are critical towards developing new therapeutic strategies that can be used to effectively promote an immune-responsive TME.
Innate immune sensing in the TME is a critical step in promoting spontaneous tumor-initiated T cell priming and subsequent TIL infiltration (Fuertes et al., 2011). Transcriptional profiling analyses of melanoma patients has revealed that tumors containing infiltrating activated T cells are characterized by a type I IFN transcriptional signature (Harlin et al., 2009). Studies in mice have demonstrated that type I IFN signaling plays a critical role in tumor-initiated T cell priming (Diamond et al., 2011; Fuertes et al., 2011). Mice lacking the IFN-α/β receptor in DCs cannot reject immunogenic tumors, and CD8α+ DCs from these mice are defective in antigen cross-presentation to CD8+ T cells. Furthermore, Baft3−/− mice that lack the CD8α+ DC lineage lose the capacity to spontaneously prime tumor-specific CD8+ T cells (Fuertes et al., 2011; Hildner et al., 2008). These findings in humans and in mice indicate that the tumor-resident antigen presenting cell (APC) compartment is defective in non T cell-inflamed tumors. Thus, strategies to induce type I IFN signaling and APC activation in the TME to bridge the innate and adaptive immune responses may have therapeutic utility.
Recent work has demonstrated that activation of the STING pathway in tumor-resident host APCs is required for induction of a spontaneous CD8+ T cell response against tumor-derived antigens in vivo (Woo et al., 2014). In addition, activation of this pathway and the subsequent production of IFN-β contributes to the anti-tumor effect of radiation (Deng, 2014), which can be potentiated with co-administration of a natural STING agonist. STING (Stimulator of Interferon Genes, also known as TMEM173, MITA, ERIS, and MPYS) is a transmembrane protein localized to the endoplasmic reticulum that undergoes a conformational change in response to direct binding of cyclic dinucleotides (CDNs), resulting in a downstream signaling cascade involving TBK1 activation, IRF-3 phosphorylation, and production of IFN-β and other cytokines (Burdette et al., 2011; Burdette and Vance, 2013; Ishikawa and Barber, 2008). IFN-β is the signature cytokine induced in response to activating STING, by either exogenous CDNs produced by bacterial infection, or through binding of a structurally distinct endogenous CDN produced by a host cyclic GMP-AMP synthetase (cGAS) in response to sensing cytosolic double-stranded DNA (dsDNA) (Ablasser et al., 2013; Diner et al., 2013; McWhirter et al., 2009; Sun et al., 2013; Woodward et al., 2010; Zhang et al., 2013). These observations suggested that direct activation of the STING pathway in the TME by intratumoral injection of specific agonists might be an effective therapeutic strategy to promote broad tumor-initiated T cell priming against an individual’s tumor antigen repertoire.
To test this therapeutic approach, we began with 5,6-Dimethylxanthenone-4-acetic acid (DMXAA), a defined flavonoid compound known as a vascular disrupting agent that was shown to have anti-tumor activity in mouse models (Baguley and Ching, 1997). This drug ultimately failed in humans when combined with standard-of-care chemotherapy in a Phase 3 efficacy trial in non-small cell lung cancer (Lara et al., 2011). Interestingly, recent structure-function studies of mouse STING (mSTING) and human STING (hSTING) demonstrated that DMXAA is a direct ligand for mSTING (Conlon et al., 2013; Gao et al., 2013b; Kim et al., 2013; Prantner et al., 2012). However, detailed analysis revealed that polymorphisms in hSTING rendered it unable to bind DMXAA, therefore abrogating its activity in human cells. These findings provide a mechanistic insight for the lack of DMXAA efficacy in humans as well as the rationale for the development of new pharmacologic compounds that potently activate hSTING. While STING agonists are being developed as vaccine adjuvants (Dubesnky, 2013; Ebensen et al., 2011; Gray et al., 2012), whether STING agonists could have direct anti-tumor therapeutic effects has been under-explored, and the lack of defined agonists that could activate all known human STING alleles has been lacking.
In the current report, we confirm that DMXAA is a strong agonist of the mSTING pathway in vitro and in vivo. We show that intratumoral (IT) injection of DMXAA effectively primes CD8+ T cell responses to promote rejection of established tumors in a STING-dependent fashion. Based on these proof-of-concept results, we synthesized a large panel of CDNs and selected compounds capable of activating all known hSTING alleles. Unlike DMXAA, selected compounds indeed stimulated human PBMCs to produce IFN-β. Like DMXAA, these STING agonists exhibit significant antitumor efficacy in several mouse tumor models, without significant local or systemic toxicity. Strikingly, direct IT injection of selected CDNs into established B16 melanoma, CT26 colon, and 4T1 breast carcinomas resulted in rapid and profound tumor regression and promoted lasting systemic antigen-specific T cell immunity. These effects were entirely STING-dependent, and resulted in regression of non-injected tumors in the same hosts. We selected dithio-(RP, RP)-[cyclic[A(2′,5′)pA(3′,5′)p]], (ML RR-S2 CDA) as the lead molecule for continued development. This agent has high translational potential as a therapeutic intervention strategy to induce activation of the TME in multiple tumor types, with the mechanistic goal of generating effective tumor-initiated CD8+ T cell priming and lasting antitumor efficacy.
Results
DMXAA stimulates the STING pathway in vitro
We first confirmed that DMXAA was a functional agonist of the STING pathway using mouse macrophages in vitro. STING aggregation was assessed using STING−/− macrophages expressing mSTING-HA. Control macrophages presented a diffuse pattern of STING in the cytoplasm, but after one hour of incubation with DMXAA, approximately 60% of cells displayed aggregates of STING in perinuclear sites (Figure 1A). Downstream phosphorylation of TBK1 and IRF3 was observed, which was abolished in STING−/− cells (Figure 1B). This correlated with an increase in the apparent molecular weight of STING, which has been reported to be due to its phosphorylation (Konno et al., 2013). STING−/− macrophages reconstituted with mSTING-HA showed restored phosphorylation of TBK1 and IRF3. IFN-β secretion was detected from wild-type (WT) but not from STING−/− macrophages in response to DMXAA (Figure 1C). Similar results were observed with bone marrow-derived DCs (BM-DC) from WT versus STING−/− mice (Figure S1A–B). We also used BM-DCs cells to study the expression of additional immunoregulatory molecules. IFN-β, IFN-α, TNF-α, IL-1β, IL-6 and IL12p40 were induced after stimulation with DMXAA in WT cells but not STING−/− BM-DCs (Figure S1C). Whereas LPS induced expression of CD40, PD-L1, CD86, and MHC class II in both WT and STING-deficient DCs, induction with DMXAA was observed only in WT cells (Figure S1D, S1E). Together, these data along with previous studies (Conlon et al., 2013; Gao et al., 2013b; Kim et al., 2013; Prantner et al., 2012) confirm that DMXAA is a strong agonist of mSTING, resulting in the production of IFN-β and other innate cytokines, and activation of DCs.
Figure 1. DMXAA activates the STING pathway and promotes the activation of APCs.

(A) STING−/− mouse bone marrow-derived macrophages (BMM) transduced to express STING-HA tag were stimulated for 1 hour with 50 μg/ml DMXAA, stained with specific antibodies against HA-tag, CD11b and DAPI. Single cell images were acquired in the ImageStream and data were analyzed with the IDEAS software (Amnis, Millipore). The data in the graph represent average of percentage of cells with aggregates from three independent experiments. (B) WT or STING−/− BMM were stimulated with 50 μg/ml of DMXAA for the indicated time points. The amount of pTBK1, total TBK1, pIRF3, total IRF3, STING and GAPDH was measured by Western blot. (C) WT or STING−/− BMM were stimulated with 50 μg/ml of DMXAA for 12 hours. The amount of secreted IFN-β was measured by ELISA.
DMXAA induces strong anti-tumor immunity in vivo
In order to evaluate whether stimulation of STING could augment anti-tumor immunity in vivo, we chose an intratumoral (IT) route of administration to focus activation on those APCs acquiring tumor antigens. To assess an antigen-specific immune response, we utilized the B16 melanoma cell line transduced to express the model antigen SIYRYYGL (B16.SIY) (Blank et al., 2004). B16.SIY tumor cells were inoculated into the flank of WT or STING deficient mice and injected IT with DMXAA at day 7. The dose of 500 μg of DMXAA was chosen after examining single doses ranging from 150 to 625 μg, with the highest dose of 625 μg showing unacceptable toxicity (data not shown). In WT animals the selected dosage induced potent tumor regression and complete tumor rejection in the majority of mice, however no reduction in tumor growth was observed in response to DMXAA in the absence of host STING (Figure 2A). Analysis of splenocytes five days after treatment showed a marked increase in the frequency of SIY-specific IFN-γ-producing T cells in WT mice, but not in STING−/− mice (Figure 2B). Similarly, treatment with DMXAA caused a high frequency of SIY-specific CD8+ T cells detected by SIY/Kb pentamer staining in WT animals, but not in STING-deficient mice (Figure 2C). Next, we examined whether DMXAA treatment had any effect in animals deficient in the type I IFN receptor (IFNAR). A significant portion of the anti-tumor effect of DMXAA was lost in IFNAR-deficient mice, and none of the deficient animals showed complete tumor rejection (Figure 2D).
Figure 2. Rejection of tumors in response to DMXAA is STING-dependent.

(A) WT or STING−/− C57BL/6 mice were inoculated with 106 B16.SIY cells in the left flank. When tumor volumes were 100–200 mm3 they received a single IT dose of 500 μg of DMXAA or saline. Tumor volume was measured at the indicated time points (n=5). (B–C) WT or STING−/− C57BL/6 mice (n=5) were treated as in (A) and 5 days later splenocytes were harvested and re-stimulated in vitro in the presence of culture medium or soluble SIY peptide for 16 hours. The frequency of tumor-specific IFN-γ–producing cells was assessed by ELISPOT (B), and the percentage of SIY-specific CD8+ T cells was assessed by staining splenocytes with antibodies against TCRβ, CD4, CD8 and SIY pentamer (C). Cells were acquired in the LSRII-Blue cytometer and analyzed with FlowJo software. Results are shown as mean ± s.e.m. * P < 0.05; ** P < 0.01. (D) WT or IFNAR−/− C57BL/6 mice were inoculated with 106 B16.SIY cells in the left flank (n=5). When tumor volumes were 100–200 mm3 they received a single IT dose of 500 μg of DMXAA or saline. Tumor volume was measured at the indicated time points. (E) WT mice that had rejected B16.SIY tumors were re-challenged with 106 B16.SIY in the contralateral flank. Naïve mice were used as controls. Tumor size was measured at the indicated time points. (F) WT mice were inoculated with 106 B16.SIY cells in the left and the right flanks (n=5). When tumor volumes were 100–200 mm3, 500 μg of DMXAA or saline was injected IT in the right flank only, and tumor volume was measured at the indicated time points. Data are representative of at least three independent experiments, or two independent experiments for the contralateral tumor model. Results are shown as mean tumor volume ± s.e.m. * P < 0.5; ** P < 0.01; *** P < 0.001. ns, not significant.
To determine whether immunologic memory was induced, WT mice that had rejected B16.SIY tumors were rechallenged 60 days after the initial inoculation with the same tumor cells. None of the rechallenged animals developed tumors (Figure 2E). We then investigated whether the anti-tumor immune response induced following DMXAA administration could be potent enough to reject non-injected secondary tumors. B16.SIY cells were injected in both flanks of mice but only one tumor was treated with DMXAA. Tumor regression was observed in both sites (Figure 2F), suggesting that IT DMXAA administration can have a therapeutic effect on distant tumors. This effect was unlikely secondary to systemic distribution of the drug, since deliberate systemic administration of DMXAA via intraperitoneal (IP) administration had an inferior therapeutic effect (data not shown). These results demonstrate that a STING agonist can activate tumor-specific immune response capable of eliminating distal tumors and protecting from tumor challenge.
To assess whether the potent anti-tumor efficacy resulting from IT administration of DMXAA could be broadly applied, we tested several additional syngeneic tumor models. Treatment with DMXAA significantly reduced the growth of B16.F10 (without expression of SIY) and TRAMP-C2 tumors in C57BL/6 mice; 4T-1 tumors in BALB/c mice; and Ag104L tumors in C3H mice, indicating that the therapeutic effect of DMXAA is not restricted to a specific tumor histology or mouse genetic background (Figure S2 A–D).
To determine whether the adaptive immune response was required for tumor control, B16.SIY cells were inoculated into RAG2−/− mice that lack mature T and B cells. DMXAA treatment lost most of its therapeutic effect in RAG2−/− hosts, although there was a partial control of tumor growth (Figure S2E). A similar loss of therapeutic effect was observed in TCRα−/− mice (Figure S2F) and in mice depleted of CD8+ T cells (Figure S2G), but not in mice depleted of CD4+ T cells or NK cells (Figure S2H and S2I). These results indicate that a significant component of the therapeutic effect of DMXAA is mediated by CD8+ T cells.
Identification of synthetic human STING-activating molecules
Having shown that the STING pathway could be harnessed to promote tumor antigen-specific CD8+ T cell priming leading to significant therapeutic efficacy, we sought to identify compounds that could potently activate human STING and therefore be considered for clinical translation. Cyclic dinucleotides (CDNs) have been studied as small molecule second messengers synthesized by bacteria, which regulate diverse processes including motility and formation of biofilms (Romling et al., 2013). The immunogenicity of recombinant protein antigens can be augmented with CDNs used as an adjuvant, giving CDNs a potential application towards vaccine development (Dubesnky, 2013; Ebensen et al., 2011; Ebensen et al., 2007a; Ebensen et al., 2007b; Gray et al., 2012). We sought to develop synthetic CDN compounds with increased activity in human cells as well as the ability to engage all known polymorphic hSTING molecules. The availability of CDN-STING crystal structures, along with recent results describing hSTING allele/CDN-dependent signaling relationships, facilitated structure-based studies to design CDN compounds with increased activity. We synthesized compounds that varied in purine nucleotide base, structure of the phosphate bridge linkage, and substitution of the non-bridging oxygen atoms at the phosphate bridge with sulfur atoms. Native CDN molecules are sensitive to degradation by phosphodiesterases that are present in host cells or in the systemic circulation (Yan et al., 2008). We found that Rp, Rp (R,R) dithio-substituted diastereomer CDNs were both resistant to digestion with snake venom phosphodiesterase and induced higher expression of type I IFNs in human THP-1 cells, compared to the Rp, Rs (R,S) dithio-substituted diastereomers or unmodified CDNs (data not shown).
To increase their affinity for STING, CDNs were also synthesized with a phosphate bridge configuration containing both 2′-5′ and 3′-5′ linkages, termed “mixed linkage” (ML), as found in endogenous human CDNs produced by cGAS (Ablasser et al., 2013; Diner et al., 2013; Gao et al., 2013a; Wolchok et al., 2013). The synthesis of dithio mixed-linkage CDNs, via modifications of literature procedures (Gaffney et al., 2010), resulted in both R,R- and R,S dithio diastereomers which were purified and separated by a combination of silica gel and C18 reverse phase prep-HPLC chromatography, affording CDNs with ≥95% purity as shown for ML RR-S2 CDA (Figure S3A, upper panel). The spectra for both 1H NMR (data not shown) and the 31P NMR (y-axis of Figure S3A, upper panel) were consistent with ML RR-S2 CDA. Direct evidence for the regiochemistry of the phosphodiester linkages was obtained by 1H-1H COSY (correlation spectroscopy for assignment of ribose protons shown on x-axis of Figure S3A (lower panel), in combination with a 1H-31P HMBC (heteronuclear multiple-bond correlation spectroscopy) two-dimensional NMR (Figure S3A, lower panel). The three-dimensional X-ray crystal structure of ML RR-S2 CDA confirms the presence of the 2′-5′, 3′-5′ mixed phosphodiester linkage and a dithio [RP, RP] diastereomer configuration (Figure S3B).
Synthetic CDNs have enhanced binding affinity to STING and activate all known human STING alleles
We purified recombinant human STING and evaluated the relative binding affinity to various modified CDNs using differential scanning fluorimetry (DSF). DSF measures the stability of complex formation as a function of temperature as an indirect readout of protein-ligand association (Cavlar et al., 2013; Niesen et al., 2007). The increased shift in thermal stability of hSTING bound to ML RR-S2 CDA or ML RR-S2 cGAMP relative to unmodified CDNs indicates that R,R dithio and mixed linkage modifications enhance the binding affinity to STING (Figure 3A). Similar results were obtained using purified mouse STING (Figure S3C). It has been shown recently that the bisphosphothionate analog of endogenous cGAMP (ML cGAMP) is resistant to hydrolysis by ENPP1 phosphodiestersase, and thus is more potent at inducing IFN-β secretion in human THP1 cells (Li et al., 2014). Similarly, we found that R,R dithio-modified CDA compounds (ML RR-S2 CDA and RR-S2 CDA) showed enhanced type I IFN production over CDA in THP-1 human monocytes (Figure S3D).
Figure 3. Modified CDNs potently activate STING and signal through all human STING allelic variants.

(A) Purified human STING and murine STING binding to CDNs were analyzed by thermal shift assay. Temperature curves are the average from a representative experiment of three independent experiments performed in duplicate. Tm shift values are mean values + s.e.m. (B) Domain structure of hSTING is shown with the positions of the amino acid variations (bottom). The allelic frequencies of the hSTING isoforms shown on the left hand column were obtained from the 1000 Genome Project database as previously described (Yi et al., 2013). (C) HEK293T cells were stably transfected with the indicated STING alleles. Whole cell lysates from HEK293T cells stably expressing the indicated full length STING-HA proteins were analyzed by Western blot with anti-HA antibodies. (D) HEK293T cells expressing the indicated STING alleles were transfected with an IFN-β-luciferase reporter construct. After 24 hours, cells were stimulated for 6 hours with the indicated CDN compound (10 μM), and assessed for IFN-β-reporter activity. (E–F) Human PBMCs from donors with the indicated STING alleles were stimulated with 10 μM of the indicated CDN, or 100 μg/ml DMXAA (E), or human PBMCs from a donor homozygous for the reference variant (STINGREF/REF) was stimulated with 10 μM and 50 μM of the indicated CDN or 100 μg/ml DMXAA (F). After a 6 hour stimulation, fold induction of IFN-β was measured by q-RT-PCR and relative normalized expression was determined by comparison with untreated controls. Results are representative of at least two independent experiments.
Single nucleotide polymorphisms in the hSTING gene have been shown to affect the responsiveness to bacterial-derived canonical CDNs (Diner et al., 2013; Gao et al., 2013a). Five haplotypes of hSTING have been identified (WT, REF, HAQ, AQ and Q alleles), which vary at amino acid positions 71, 230, 232 and 293 (Figure 3B) (Jin et al., 2011; Yi et al., 2013). To test the responsiveness of the five hSTING variants to synthetic CDNs, we created stable HEK293T cell lines (defective in endogenous STING signaling) expressing each of the full length hSTING variants. Similar levels of STING protein were expressed in each of the cell lines (Figure 3C). DMXAA potently activated mSTING and failed to activate any of the five hSTING alleles (Figure S3E), consistent with previous studies evaluating one of the human STING alleles (Conlon et al., 2013). Cells expressing hSTINGREF responded poorly to stimulation with the bacterial CDN compounds cGAMP, CDA, and CDG, but were responsive to the endogenously produced cGAS product, ML cGAMP (Diner et al., 2013). Interestingly, the hSTINGQ allele was also refractory to the bacterial CDNs. Cells expressing mSTING were responsive to all of the CDNs tested (Figure 3D). Cells transformed with either an empty vector or expressing a non-functional mutant (I199N) STING protein (Goldenticket) (Sauer et al., 2011) were not responsive to any of the compounds (data not shown). In contrast, the dithio, mixed-linkage CDN derivatives (ML RR-CDA, ML RR-S2 CDG, and ML RR-S2 cGAMP) potently activated all five hSTING alleles, including the refractory hSTINGREF and hSTINGQ alleles (Figure 3D).
CDN derivatives potently induce STING-dependent signaling in murine and human immune cells
To determine whether modified CDNs activated downstream STING signaling, we assessed murine bone marrow macrophages (BMMs) isolated from WT C57BL/6 and STING−/− (Goldenticket) mice (Sauer, et al., 2011) for induction of IFN-β and other cytokines. Synthetic dithio mixed-linkage CDNs (ML RR-S2 CDA and ML RR-S2 CDG) induced the highest expression of IFN-β and the pro-inflammatory cytokines TNF-α, IL-6, and MCP-1 on a molar equivalent basis, as compared to endogenous ML cGAMP and the TLR3 agonist poly I:C (Figure S4A). The modified CDNs did not induce signaling in STING−/− BMMs, whereas, as expected, poly I:C agonists were still active. ML RR-S2 CDA was also found to induce aggregation of STING and induce phosphorylation of TBK1 and IRF3 in mouse BMM (Figure S4B and S4C). All of the modified CDNs tested also enhanced MHC class II and expression of co-stimulatory markers in a STING-dependent manner (Figure S4D).
To examine activation of STING signaling in primary human cells, we stimulated PBMCs from a panel of human donors harboring different STING alleles (hSTINGWT/WT, hSTINGWT/REF, hSTINGWT/HAQ) and measured induction of IFN-β. In contrast to a lack of activation by DMXAA, dithio-modified mixed linkage CDNs induced IFN-β expression across these donors (Figure 3E). Importantly, dithio-modified mixed linkage CDNs, as well as the non-canonical cGAS product ML cGAMP, induced IFN-β expression in a donor homozygous for the hSTINGREF allele, which was refractory to stimulation with canonical CDNs in HEK293T cells (Figure 3D and 3F). Looking at protein secretion by multiple donors that are homozygous for the hSTINGWT allele, ML RR-S2 CDA induced significantly higher levels of IFN-α when compared to ML cGAMP (Figure S4E). Thus, ML RR-S2 CDNs are viable clinical candidates capable of activating STING pathway in human cells harboring different STING alleles and genotypes.
Intratumoral delivery of synthetic CDN derivatives results in profound anti-tumor efficacy in established B16 melanoma
To evaluate whether modified dithio ML CDN compounds conferred increased anti-tumor activity, mice bearing established B16.F10 tumors were treated with three IT injections of CDN derivatives over a one-week period. While treatment with ML CDA and ML CDG had modestly reduced tumor growth, the R,R dithio derivatives profoundly inhibited tumor growth (Figure 4A). However, ML RR-S2 CDG was reactogenic, and these mice developed open wounds in the treated tumor that did not heal (data not shown). Lower dose levels of ML RR-S2 CDG were not efficacious (data not shown), indicating that this molecule had a narrow therapeutic index. In contrast, no injection site reactogenicity was observed with ML RR-S2 CDA, and several mice developed vitiligo upon fur regrowth following complete eradication of the treated tumor (data not shown). Importantly, these mice demonstrated significantly higher overall survival compared to mice treated with ML RR-S2 CDG (Figure S5A). ML RR-S2 CDA also showed higher anti-tumor control than the endogenous ML cGAMP (Figure 4B). Thus, ML RR-S2 CDA was selected for advancement to clinical development.
Figure 4. Synthetic CDN modifications significantly improve anti-tumor efficacy in established B16 tumors.

(A–B) WT C57BL/6 mice were inoculated with 5 × 104 B16.F10 cells in the left flank (n=8). When tumor volumes were 100 mm3 they received three 25 μg IT doses of ML CDA, ML CDG, ML RR-S2 CDG, or ML RR-S2 CDA or HBSS as control (A); or either three 50 μg doses of the endogenous cGAS product ML cGAMP, ML RR-S2 CDA, ML RR-S2 cGAMP or HBSS as control (n=8) (B). (C) WT C57BL/6 mice or STING−/− mice were treated with three IT doses of CDN ML RR-S2 CDA (50 μg), murine type B CpG ODN 1668 (50 μg) or HBSS vehicle control. Treatments were administered on the days indicated by the arrows and tumor measurements were taken twice weekly. Data are representative of at least two independent experiments. Results are shown as mean tumor volume ± s.e.m. ** P < 0.01, *** P < 0.001. ns, not significant.
To determine whether the CDN-induced anti-tumor efficacy was STING-dependent, we compared activity in B16 tumor-bearing WT (C57BL/6) and STING−/− mice. CDN therapeutic efficacy was completely lost in STING−/− mice. In contrast, a CpG TLR9 agonist (Kawarada et al., 2001) did modestly reduce tumor growth in STING−/− mice, demonstrating that this mouse strain is capable of mounting an immune-mediated antitumor response (Figure 4C). A dose response of the ML RR-S2 CDA compound was performed in B16 tumor-bearing mice, which identified an optimal antitumor dose level that also elicited maximum tumor antigen-specific CD8+ T cell responses (data not shown), and improved long-term survival to 50% (Figures S5B–C).
To evaluate which cell types are targeted by STING agonists in the TME, we analyzed the expression of IFN-β after ML RR-S2 CDA or DMXAA stimulation in BM-DC, bone marrow-derived macrophages, purified T cells from naïve mice, B16 tumor cells, mouse embryonic fibroblasts (MEFs) and mouse primary dermal fibroblasts. Except for tumor cells, all cell types tested were found to express IFN-β. However, expression in WT BM-DCs was ten times higher than in the other cell types (Figure S5D). To determine whether this was also the case in the TME, we sorted four different cell populations from pre-established B16 tumors: DCs (CD45+ CD11c+ MHCII+), macrophages (CD45+ CD11b+ F4/80+ MHC-II+), T cells (CD45+ CD3+), and endothelial cells (CD45− CD31+). Sorted cells were then stimulated ex vivo with ML RR-S2 CDA or DMXAA. All subsets expressed IFN-β upon stimulation with the STING agonists. Expression in macrophages was highest, followed by DCs, which were both higher as compared with lymphocytes and endothelial cells (Figure S5E). These data demonstrate that the main source of type I IFN in the TME is likely APCs, although stromal cells and T cells might also contribute. Interestingly and in agreement with these data, we observed that the CD8α+/CD103+ DCs play a critical role in vivo, as the therapeutic effect of DMXAA was significantly diminished in Batf3−/− mice (Figure S5F). Altogether, these findings suggest that CD8α+/CD103+ DCs are critical for curative anti-tumor therapy by STING agonists.
ML RR-S2 CDA induces lasting immune-mediated tumor rejection in multiple tumor types
To test anti-tumor efficacy in diverse tumor models, BALB/c mice bearing established 4T-1 colon or CT26 mammary carcinomas were treated with ML RR-S2 CDA. All treated animals showed significant and durable tumor regression. Mice that were cured of their primary tumor were completely resistant to re-challenge when the same tumor cell line was used (Figure 5A and 5B). However, animals cured from CT26 tumors after ML RR-S2 CDA treatment showed no protection when they were re-challenged with the 4T-1 tumor cells, demonstrating specificity (Figure 5B). Increased T cell responses were observed against the endogenous CT26 rejection antigen AH1 (Slansky et al., 2000) (Figure 5C). IT injection of ML RR-S2 CDA into one tumor in BALB/c mice bearing bilateral CT26 or 4T-1 tumors also demonstrated significant regression of the contralateral, untreated tumor (Figure 5D and Figure S5G). Using a different model to study the distal effect of ML RR-S2 CDA, we implanted B16.F10 melanoma in C57BL/6 mice, and seven days later intravenously infused B16.F10 melanoma cells to generate lung metastases. The two-week old established flank tumors were treated with ML RR-S2 CDA, DMXAA or HBSS control, and three weeks later lung metastases were enumerated. Mice treated in the flank tumor with ML RR-S2 CDA showed substantial control of distant lung metastases (Figure 5E). Together, these results demonstrate that IT injection with ML RR-S2 CDA eradicates multiple tumor types and primes an effective systemic CD8+ T cell immune response that significantly inhibits the growth of distal, untreated lesions.
Figure 5. ML RR-S2 CDA promotes immune-mediated tumor rejection.

(A–B) WT BALB/c mice were inoculated with 105 4T-1 breast cancer (A), or CT26 (B) colon carcinoma cells in the left flank. When tumor volumes were 100 mm3 they received three 50 μg doses IT of ML RR-S2 CDA, or HBSS vehicle control (left graph). Mice were re-implanted with 105 4T-1 (A and B); or CT26 (B) tumor cells on the opposite flank on day 55 post-initial tumor implantation. Naïve mice were used as controls (right graph) (n=8). (C) WT BALB/c mice were inoculated with 105 CT26 colon carcinoma cells in the left flank and treated on days 11, 14, and 18 with IT injections of ML RR-S2 CDA (25 μg each), or HBSS vehicle control (n= 4). 21 days post-implantation of CT26 tumors, PBMCs were stimulated with AH1 (gp70423–431) and assessed by IFN-γ ELISPOT assay. (D) WT BALB/c mice were implanted with 105 of CT26 tumor cells on both flanks. On the days indicated, mice were treated in one flank only with ML RR-S2 CDA (50 μg), or HBSS vehicle control (n=8). (E) WT C57BL/6 were inoculated with 5×104 B16.F10 melanoma cells on the right flank at day 0, and implanted IV with 105 cells on day 7. Naïve mice were implanted with cells IV only as a control. Flank tumors were treated on the days indicated with ML RR-S2 CDA (50 μg), DMXAA (150 μg) or HBSS control (n=8). On day 28, lungs were harvested and lung tumor nodules counted. The histogram depicts total numbers of nodules in the ML RR-S2 CDA, DMXAA or HBSS control treated mice, compared to the untreated IV-only tumor implanted mice. The images depict the ML RR-S2 CDA and HBSS control treated mice. Data are representative of at least two independent experiments. Results are shown as mean ± s.e.m. ** P < 0.01, *** P < 0.001.
Discussion
Our results indicate that IT administration of STING agonists generates a potent anti-tumor immune T cell response and striking durable disease regression in multiple mouse tumor models. Although DMXAA has a potent therapeutic effect in mice, it lacks the ability to activate hSTING. The chemically modified CDN compounds described here have the capacity to activate all human polymorphic STING molecules while retaining the ability to engage mSTING, and demonstrate a similarly impressive anti-tumor effect. The participation of the adaptive immune response for the observed antitumor responses is supported by the secondary rejection of non-injected tumors, the clearance of lung metastases, and long-term immunologic memory observed against autologous tumor re-challenge.
The mechanism of the therapeutic effect observed with the compounds tested in this study was absolutely dependent on host STING, and the majority of the antitumor effect was dependent upon T cells, specifically CD8+ T cells, as described (Jassar et al., 2005; Wallace et al., 2007). However, a partial therapeutic effect was observed in RAG−/− and TCRα−/− mice, indicating that innate immune cells are an important contributor to antitumor efficacy. Our in vitro data show that the various synthetic STING agonists tested induced IFN-β production by APCs via a mechanism that depended on the classical STING-TBK1-IRF3 signaling pathway (Ishikawa et al., 2009). In addition, STING agonists induced production of other cytokines, DC maturation, and also chemokine production in vitro. Previous work characterizing the vascular disrupting property of DMXAA demonstrated induction of TNF-α by stromal cells in vivo (Joseph et al., 1999), and attenuation of the therapeutic effect in TNFR−/− mice (Zhao et al., 2002). Thus, the T cell-independent component of the therapeutic effect of STING agonists in vivo may be mediated through early TNF-α mediated tumor vascular destruction. In addition, it seems likely that the induction of chemokines by STING agonists may also contribute to effective migration of activated T cells into the TME within the injected tumor site. This multi-faceted mechanism of action may explain the therapeutic potency of STING agonists against a range of cancers in vivo.
We developed synthetic CDN derivative molecules based on rationally-designed STING structure-function relationships. The lead molecule ML RR-S2 CDA has several features that improve both stability and lipophilicity, promoting significantly increased STING signaling as compared to endogenous and pathogen-derived CDNs. While canonical CDNs have been evaluated as vaccine adjuvants and were recently shown to inhibit growth of 4T-1 tumors when given by intraperitoneal injection (Chandra et al., 2014), those investigations used canonical CDNs which may not be appropriate for clinical development, since there are hSTING alleles at significant frequencies in the population that are refractory to these structures. In the present study we show the profound anti-tumor efficacy resulting from activation of the STING pathway with synthetic CDNs that not only activate all known hSTING alleles, but have significantly higher potency than the natural STING ligands generated by cGAS. Although occasional human donors showed higher responsiveness to cGAMP, such as the donor bearing the refractory reference allele represented in Figure 3F, overall ML RR-S2 CDA activated all known hSTING allelic variants. This compound will therefore be attractive for clinical development.
A possible limitation of the treatment approach described herein is the necessity for IT injection to achieve maximal therapeutic effect. However, a practical advantage of this strategy is that it has the potential to generate T cell responses against tumor-specific antigens expressed by a patient’s individual cancer. The attractiveness of IT injection approaches has been rekindled based on several recent clinical trial observations. IT injection of the oncolytic virus T-VEC has been shown in a randomized trial to provide improved clinical activity in melanoma patients compared with control (Goins et al., 2014). In addition, Levy and colleagues have shown that intratumoral injection of the TLR9 agonist CpG along with local low-dose radiation therapy had clinical activity in patients with non-Hodgkin’s lymphoma (Brody et al., 2010). Both of these studies demonstrated regression of non-injected lesions, consistent with the induction of tumor-specific T cells that could promote regression of tumors at distant sites. These observations, along with the impressive potency of STING agonists preclinically, support the development of clinical strategies for IT injection of STING agonists as a cancer therapeutic in patients.
Experimental Procedures
Cells and Cell Isolations
The cells used for the in vivo experiments were: the C57BL/6- derived melanoma cell lines B16.F10 and B16.F10.SIY (henceforth referred to as B16.SIY), the breast cancer 4T1 cell line, the colon cancer CT26 cell lines, all originally purchased from ATCC. All cells were maintained at 37°C with 7.5% CO2 in DMEM supplemented with 10% heat-inactivated FCS, penicillin, streptomycin, L-arginine, L-glutamine, folic acid, and L-asparagine.
Immortalized WT and STING−/− macrophages were obtained as described in Roberson et al.(Roberson and Walker, 1988). The WT macrophages were obtained from Dr. K Fitzgerald (U. Massachusetts). Non-immortalized macrophages were derived from the bone marrow of WT (C57BL/6) or STING−/− mice and cultured in BMM media (RPMI media with 5% CSF, 5% FBS, 1X L-glutamine, 1X Pen/Strep) for 7 days prior to use. Human PBMCs were isolated by density-gradient centrifugation using Ficoll-Paque Plus (GE Healthcare).
For stable overexpression of HA-STING in STING−/− macrophages, the full-length mSting-HA DNA sequence was generated by PCR. Sequence encoding full-length mSTING was amplified from pUNOI-mSTING plasmid (Invivogen) using a 5′ primer containing an EcoR1 site: gcagacGAATTCATGCCATACTCCAACCTGCATCCAGCCATCCCACGGCCCAGAGGTCA CCGCTCCAAATATGTAGCCCTCATCTTTCTGGTGGCCAG, and a 3′ primer containing the HA-tag nucleotide sequence, followed by a TGA stop codon and a NotI site: tcacatGCGGCCGCTCAGGCGTAGTCAGGCACGTCGTAAGGATAGATGAGGTCAGTGCG GAGTGGGAGAGGCTGATCC. The mSTING-HA PCR product was gel purified and double digested with EcoRI and NotI, then cloned into the multiple cloning site of pMXS-IRES-GFP with Quick ligation kit (NEB).
Stable HEK293T STING-expressing cell lines were generated with MSCV2.2 retroviral plasmids which contain STING cDNA cloned upstream of an IRES in frame with GFP. hSTING(REF)-HA, hSTING(WT)-HA, hSTING(HAQ)-HA, hSTING(Q)-HA and mSTING(WT)-HA retroviral plasmids were obtained from the Vance Laboratory at UC Berkeley. hSTING(AQ)-HA was derived from hSTING(Q)-HA using a QuickChange Site-Directed Mutagenesis kit (Stratagene). Retroviral vectors were transfected into the amphotropic Phoenix packaging cell line using Lipofectamine (Invitrogen). After two days viral supernatants were harvested and used for transduction of STING−/− macrophages or HEK293T cells. GFP+ cells were sorted in FACSAria (BD) or MoFlow cell sorters.
Protein Expression and Purification
STING ligand-binding domains (human amino acids(AA) 140-379, mouse AA 139-378) were cloned via ligation independent cloning into a custom pET based vector containing a 6xHIS-SUMO-tobacco etch virus protease (TEV) site. All plasmids were confirmed by sequencing. Proteins were expressed in BL21 DE3 Rosetta 2 cells (EMD Millipore). Cells were grown in LB media at 37°C until an OD600 of about 0.6. Cells were then shifted to 18°C and induced with 0.25 mM isopropyl-beta-D-thiogalactopyranoside and grown for 18–20 hrs. Fusion proteins were purified on Ni-NTA agarose (Qiagen). The 6xHIS-SUMO tag was removed by digestion with TEV protease (Sigma Aldrich) overnight during dialysis against buffer containing 20 mM Tris-HCL, 150 mM NaCl, 5mM Imidazole, 10% Glycerol, 0.5 mM Tris (2-carboxyethyl) phosphine, pH 7.5. After dialysis, the 6xHIS-SUMO tag and TEV protease were removed by Ni-NTA agarose. Proteins were concentrated to between 9 and 13 mg/ml. Aliquots were flash frozen in liquid nitrogen and stored at −80°C.
ImageStream analysis of STING aggregation in cells
STING−/− macrophages overexpressing STING-HA tag were stimulated for 1 hour with 50 μg/ml of DMXAA resuspended in 7.5% of NaHCO3, 50 μM of ML RR-S2 CDA resuspended in HBSS, or only the vehicles as control. After the incubation, cells were stained with anti-CD11b-APC (M1/70; BioLegend), rabbit anti-HA-tag (C29F4; Cell Signaling) and anti-Rabbit IgG-PE (Invitrogen), and DAPI (Invitrogen). Single cell images were acquired in the ImageStreamx Mark II (Amnis) and data were analyzed using IDEAS software.
Western blot analysis
WT, STING−/− macrophages, and STING−/− macrophages overexpressing STING-HA or an empty vector were stimulated with 50 μg/ml DMXAA for 0, 15, 60 or 180 minutes; BM-DCs from WT or STING−/− mice were stimulated with 25 μg/ml DMXAA for the same time-points. Proteins were extracted with Triton-X buffer (150 mM sodium chloride, 50 mM Tris, 1% Triton-X, pH 8.0) with proteinase inhibitors (Thermo scientific) and phosphatase inhibitors (Sigma). 30 μg of protein was electrophoresed in 10% SDS-PAGE gels and transferred onto Immobilon-FL membranes (Millipore). Blots were incubated with antibodies specific for phosphorylated TBK1 (Ser172), phosphorylated IRF3 (Ser396), total TBK1, STING and GAPDH (Cell Signaling) or total IRF3 (Invitrogen). Proteins from HEK293T lines stably expressing STING were extracted with M-PER (Thermo Scientific). 6 μg of protein was loaded onto a 4–12% MES NuPAGE gel (Life Technologies), transferred to nitrocellulose, and probed with anti-HA antibody (Santa Cruz). Anti-rabbit IRDye 680RD label secondary antibody was used for visualization of bands with the Odyssey Imaging system (LI-COR).
Differential Scanning Fluorimetry
Thermal shift assays were performed as (Cavlar et al., 2013). Assays were conducted with STING ligand binding domain at 1 mg/ml with or without various CDNs at 1 mM in 20mM Tris-HCL, 150 mM NaCl, pH 7.5 and 1:500 dilution of SYPRO Orange Dye (Life Technologies). The fluorescence as a function of temperature was recorded in a CFX 96 real time PCR machine (Bio-Rad) reading on the HEX channel EX 450–490 EM 560–580 nm. The temperature gradient was from 15–80°C ramping 0.5°C per 15 seconds. Curves were fit to a Boltzmann sigmoidal (Graph Pad Prism) to establish the midpoint of thermal unfolding (Tm).
Murine IFN-β ELISA
WT or STING−/− macrophages and BM-DCs from WT or STING−/− mice were stimulated with 50 μg/ml DMXAA. Conditioned media were collected after 4 hours. IFN-β concentration was assessed using VeriKine™ Mouse Interferon Beta ELISA Kit (PBL interferon source).
Quantitative RT-PCR analysis of cytokines
BM-DCs from WT or STING−/− mice were stimulated with 25 μg/ml DMXAA or 100 ng/ml LPS for 4 hours. Total RNA was isolated using the RNeasy® kit (Qiagen) and incubated with Deoxyribonuclease I, Amplification Grade (Invitrogen). cDNA was synthesized using High Capacity cDNA Reverse Transcription Kit (Applied Biosystem) and expression of cytokines was measured by real-time qRT-PCR using specific primers/probes for mouse INF-β, TNF-α, IL-6 and IL12p40, and pan-specific primers were to quantify expression of the IFN-α family. Primer sequences are listed in Table 1 in Supplementary Materials. PCR reactions were performed in the 7300 Real Time PCR system (Applied Biosystem). The results are expressed as 2−ΔCt using 18s as endogenous control.
WT BMM were stimulated with CDN at 5 μM in HBSS with the addition of Effectene (Qiagen) transfection reagent (per kit protocol). Human PBMCs were stimulated as indicated. Stimulated cells were and assessed by real-time qRT-PCR for gene expression of IFN-β1, MCP-1, TNF-α and IL-6 using the PrimePCR RNA purification and cDNA analysis system, and run on the CFX96 gene cycler (BioRad). Relative normalized expression was determined by comparing induced target gene expression to unstimulated controls, using the reference genes Gapdh and Ywhaz (mouse) and GusB and Pgk1 (human), genes confirmed to have a coefficient variable (CV) below 0.5 and M value below 1, and thus did not vary with different treatment conditions.
Mice
All animals were used according to protocols approved by Institutional Animal Use Committee of the University of Chicago and Aduro Biotech, Inc., and maintained in pathogen-free conditions in a barrier facility. C57BL/6, BALB/c, C3H/He and TCRα−/− mice were obtained from Jackson and Charles River. RAG2−/− mice were obtained from Taconic. Tmem173−/− (STING-deficient) mice were provided by Dr. G. Barber (University of Miami), and STING−/− (goldenticket) (Sauer JD, et al., 2011) and IFNAR−/− mice were purchased from Jackson. Batf3−/− mice were provided by Dr. Kenneth M. Murphy (Washington University School of Medicine, St. Louis, MO).
in vivo tumor experiments
106 of B16-SIY tumor cells, 5 × 104 B16.F10 tumor cells, 105 4T-1 and CT26, or 106 other tumor cells were injected s.c. in 100 μl DPBS or HBSS on the right flank of mice. Following tumor implantation, mice were randomized into treatment groups. When tumors were 100–200 mm3 in volume (5–7 mm wide), either one single or three doses of DMXAA resuspended in 7.5% of NaHCO3, or CDNs formulated in HBSS or vehicle control, were injected IT. Measurements of tumors were performed twice per week using calipers, and the tumor volume was calculated with the formula: V= (length × width2)/2. In some experiments, tumor-free survivors were rechallenged with tumor cells on the opposite flank several weeks after the injection of the primary tumor. Naïve mice were used as controls. For the contralateral experiments, mice were implanted on both flanks and only one tumor was treated. For the B16 melanoma lung metastasis experiments, mice were implanted on the flank with 5 × 104 cells B16.F10 on day 0, and then injected intravenously with 1 × 105 cells on day 7. Lungs were harvested on day 28. Administration of compounds, measurements of tumors and counting of lung tumors were performed in a blinded fashion.
IFN-γ ELISPOT and SIY-pentamer staining
Splenocytes were analyzed 5 days after the first IT injection of DMXAA or ML RR-S2 CDA. For the ELISPOTs, 106 splenocytes were plated per well and stimulated overnight with SIY peptide (160 nM) or AH1 (1 μM) peptide, with PMA (50 ng/ml) plus ionomycin (0.5 μM) as a positive control, or medium as negative control. Spots were developed using the BD mouse IFN-γ kit according to the manufacturer’s instructions and the number of spots was measured using an Immunospot Series 3 Analyzer and analyzed using ImmunoSpot software (Cellular Technology Ltd). For SIY-pentamer staining, splenocytes were preincubated for 15 min with anti-CD16/32 monoclonal antibody (93) to block potential nonspecific binding, and labeled with PE-MHC class I pentamer (Proimmune) consisting of murine H-2Kb complexed to SIYRYYGL (SIY) peptide, anti–TCRβ-AF700 (H57-597), anti–CD8-Pacific Blue (53-6.7), anti–CD4-Pacific Orange (RM4-5) (all antibodies from BioLegend) and the Fixable Viability Dye eFluor 450 (eBioscience). Stained cells were analyzed using LSR II cytometer with FACSDiva software (BD). Data analysis was conducted with FlowJo software (Tree Star).
Luciferase Assay
104 HEK293T cells were seeded in 96-well plates and transiently transfected (Lipofectamine 2000) with human IFN-β firefly reporter plasmid(Fitzgerald et al., 2003) and TK-Renilla luciferase reporter for normalization. The following day, cells were stimulated with 10 μM of each CDN or 100 μg/ml DMXAA using digitonin permeabilization (50 mM HEPES, 100 mM KCL, 3 mM MgCl2, 0.1 mM DTT, 85 mM Sucrose, 0.2% BSA, 1 mM ATP, 0.1 mM GTP, 10 ug/ml digitonin) to ensure uniform uptake. After 20 min, stimulation mixtures were removed and normal media was added. After a total of 6 hours, cell lysates were prepared and reporter gene activity measured using the Dual Luciferase Assay System (Promega) on a Spectramax M3 luminometer.
Statistical analysis
Student’s paired t-test was used to calculate two-tailed p values to estimate statistical significance of differences between two treatment groups using Prism 6 software. The number of mice per group and the statistically significant P values are labeled in the figures and/or legends with asterisks.
Supplementary Material
Acknowledgments
We thank Meredith Leong and Pete Lauer and Russell Vance for discussions, Jake Bruml, Ryan Duggan, Michael Y.K. Leung and Diana Ranoa for technical assistance and Elie Diner for retroviral constructs. We thank Hector Nolla at the UC Berkeley Cancer Research Laboratory (CRL) for help with cell sorting. L.C was supported by a post-doctoral fellowship from the Ramon Areces Foundation. This work was supported by P01 CA97296 and R01CA181160 from the National Cancer Institute.
Footnotes
AUTHOR CONTRIBUTIONS
L.C., L.H.G., T.W.D. and T.F.G. conceived the research and conducted the experiments with S.M.M., D.B.K., K.E.S., E.L., J.J.L., G.E.K., T.B., S.W., and K.M. T.W.D. and T.F.G. conceived and supervised the entire project and wrote and revised the manuscript with L.C., L.H.G., K.E.G, and S.M.M.
COMPETING FINANCIAL INTERESTS
L.H.G., S.M.M., D.B.K., K.E.S., G.E.K., E.L, T.B., J.J.L., K.M. and T.W.D are all paid employees of Aduro BioTech, hold stock in the company, and may be inventors on patent applications that apply to the CDN molecules described in the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Rohl I, Hopfner KP, Ludwig J, Hornung V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498:380–384. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baguley BC, Ching LM. Immunomodulatory actions of xanthenone anticancer agents. BioDrugs: clinical immunotherapeutics, biopharmaceuticals and gene therapy. 1997;8:119–127. doi: 10.2165/00063030-199708020-00005. [DOI] [PubMed] [Google Scholar]
- Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, Gajewski TF. PD-L1/B7H-1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64:1140–1145. doi: 10.1158/0008-5472.can-03-3259. [DOI] [PubMed] [Google Scholar]
- Brody JD, Ai WZ, Czerwinski DK, Torchia JA, Levy M, Advani RH, Kim YH, Hoppe RT, Knox SJ, Shin LK, et al. In situ vaccination with a TLR9 agonist induces systemic lymphoma regression: a phase I/II study. J Clin Oncol. 2010;28:4324–4332. doi: 10.1200/JCO.2010.28.9793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE. STING is a direct innate immune sensor of cyclic di-GMP. Nature. 2011;478:515–518. doi: 10.1038/nature10429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nature immunology. 2013;14:19–26. doi: 10.1038/ni.2491. [DOI] [PubMed] [Google Scholar]
- Cavlar T, Deimling T, Ablasser A, Hopfner KP, Hornung V. Species-specific detection of the antiviral small-molecule compound CMA by STING. The EMBO journal. 2013;32:1440–1450. doi: 10.1038/emboj.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Quispe-Tintaya W, Jahangir A, Asafu-Adjei D, Ramos I, Sintim HO, Zhou J, Hayakawa Y, Karaolis DK, Gravekamp C. STING ligand c-di-GMP improves cancer vaccination against metastatic breast cancer. Cancer immunology research. 2014 doi: 10.1158/2326-6066.CIR-13-0123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon J, Burdette DL, Sharma S, Bhat N, Thompson M, Jiang Z, Rathinam VAK, Monks B, Jin T, Xiao TS, et al. Mouse, but not Human STING, Binds and Signals in Response to the Vascular Disrupting Agent 5,6-Dimethylxanthenone-4-Acetic Acid. Journal of Immunology. 2013 doi: 10.4049/jimmunol.1300097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond MS, Kinder M, Matsushita H, Mashayekhi M, Dunn GP, Archambault JM, Lee H, Arthur CD, White JM, Kalinke U, et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J Exp Med. 2011;208:1989–2003. doi: 10.1084/jem.20101158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell reports. 2013;3:1355–1361. doi: 10.1016/j.celrep.2013.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubesnky TW, Kanne DB, Leong ML. Rationale, Progress and Development of Vaccines Utilizing STING-Activating Cyclic Dinucleotide Adjuvants. Therapeutic Advances in Vaccines. 2013 doi: 10.1177/2051013613501988. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebensen T, Libanova R, Schulze K, Yevsa T, Morr M, Guzman CA. Bis-(3′,5′)-cyclic dimeric adenosine monophosphate: strong Th1/Th2/Th17 promoting mucosal adjuvant. Vaccine. 2011;29:5210–5220. doi: 10.1016/j.vaccine.2011.05.026. [DOI] [PubMed] [Google Scholar]
- Ebensen T, Schulze K, Riese P, Link C, Morr M, Guzman CA. The bacterial second messenger cyclic diGMP exhibits potent adjuvant properties. Vaccine. 2007a;25:1464–1469. doi: 10.1016/j.vaccine.2006.10.033. [DOI] [PubMed] [Google Scholar]
- Ebensen T, Schulze K, Riese P, Morr M, Guzman CA. The bacterial second messenger cdiGMP exhibits promising activity as a mucosal adjuvant. Clinical and vaccine immunology: CVI. 2007b;14:952–958. doi: 10.1128/CVI.00119-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- Fuertes MB, Kacha AK, Kline J, Woo SR, Kranz DM, Murphy KM, Gajewski TF. Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J Exp Med. 2011;208:2005–2016. doi: 10.1084/jem.20101159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffney BL, Veliath E, Zhao J, Jones RA. One-flask syntheses of c-di-GMP and the [Rp,Rp] and [Rp,Sp] thiophosphate analogues. Organic letters. 2010;12:3269–3271. doi: 10.1021/ol101236b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, Spranger S. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol. 2013 doi: 10.1016/j.coi.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Galon J, Pages F, Marincola FM, Angell HK, Thurin M, Lugli A, Zlobec I, Berger A, Bifulco C, Botti G, et al. Cancer classification using the Immunoscore: a worldwide task force. Journal of translational medicine. 2012;10:205. doi: 10.1186/1479-5876-10-205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, et al. Cyclic [G(20,50)pA(30,50)p] Is the Metazoan Second Messenger Produced by DNA-Activated Cyclic GMP-AMP Synthase. Cell. 2013a:1–14. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, et al. Structure-function analysis of STING activation by c[G(2′,5′)pA(3′,5′)p] and targeting by antiviral DMXAA. Cell. 2013b;154:748–762. doi: 10.1016/j.cell.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goins WF, Huang S, Cohen JB, Glorioso JC. Engineering HSV-1 vectors for gene therapy. Methods in molecular biology. 2014;1144:63–79. doi: 10.1007/978-1-4939-0428-0_5. [DOI] [PubMed] [Google Scholar]
- Gray PM, Forrest G, Wisniewski T, Porter G, Freed DC, DeMartino JA, Zaller DM, Guo Z, Leone J, Fu TM, et al. Evidence for cyclic diguanylate as a vaccine adjuvant with novel immunostimulatory activities. Cellular immunology. 2012;278:113–119. doi: 10.1016/j.cellimm.2012.07.006. [DOI] [PubMed] [Google Scholar]
- Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–3085. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322:1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455:674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassar AS, Suzuki E, Kapoor V, Sun J, Silverberg MB, Cheung L, Burdick MD, Strieter RM, Ching LM, Kaiser LR, et al. Activation of tumor-associated macrophages by the vascular disrupting agent 5,6-dimethylxanthenone-4-acetic acid induces an effective CD8+ T-cell-mediated antitumor immune response in murine models of lung cancer and mesothelioma. Cancer Res. 2005;65:11752–11761. doi: 10.1158/0008-5472.CAN-05-1658. [DOI] [PubMed] [Google Scholar]
- Jin L, Xu LG, Yang IV, Davidson EJ, Schwartz DA, Wurfel MM, Cambier JC. Identification and characterization of a loss-of-function human MPYS variant. Genes and immunity. 2011;12:263–269. doi: 10.1038/gene.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph WR, Cao Z, Mountjoy KG, Marshall ES, Baguley BC, Ching LM. Stimulation of tumors to synthesize tumor necrosis factor-alpha in situ using 5,6-dimethylxanthenone-4-acetic acid: a novel approach to cancer therapy. Cancer Res. 1999;59:633–638. [PubMed] [Google Scholar]
- Kawarada Y, Ganss R, Garbi N, Sacher T, Arnold B, Hammerling GJ. NK- and CD8(+) T cell-mediated eradication of established tumors by peritumoral injection of CpG-containing oligodeoxynucleotides. J Immunol. 2001;167:5247–5253. doi: 10.4049/jimmunol.167.9.5247. [DOI] [PubMed] [Google Scholar]
- Kim S, Li L, Maliga Z, Yin Q, Wu H, Mitchison TJ. Anticancer flavonoids are mouse-selective STING agonists. ACS chemical biology. 2013;8:1396–1401. doi: 10.1021/cb400264n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno H, Konno K, Barber GN. Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell. 2013;155:688–698. doi: 10.1016/j.cell.2013.09.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara PN, Jr, Douillard JY, Nakagawa K, von Pawel J, McKeage MJ, Albert I, Losonczy G, Reck M, Heo DS, Fan X, et al. Randomized phase III placebo-controlled trial of carboplatin and paclitaxel with or without the vascular disrupting agent vadimezan (ASA404) in advanced non-small-cell lung cancer. J Clin Oncol. 2011;29:2965–2971. doi: 10.1200/JCO.2011.35.0660. [DOI] [PubMed] [Google Scholar]
- Li L, Yin Q, Kuss P, Maliga Z, Millan JL, Wu H, Mitchison TJ. Hydrolysis of 2′3′-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nature chemical biology. 2014;10:1043–1048. doi: 10.1038/nchembio.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, et al. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J Exp Med. 2009;206:1899–1911. doi: 10.1084/jem.20082874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niesen FH, Berglund H, Vedadi M. The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nature protocols. 2007;2:2212–2221. doi: 10.1038/nprot.2007.321. [DOI] [PubMed] [Google Scholar]
- Postow MA, Harding J, Wolchok JD. Targeting immune checkpoints: releasing the restraints on anti-tumor immunity for patients with melanoma. Cancer J. 2012;18:153–159. [PMC free article] [PubMed] [Google Scholar]
- Prantner D, Perkins DJ, Lai W, Williams MS, Sharma S, Fitzgerald KA, Vogel SN. 5,6-Dimethylxanthenone-4-acetic acid (DMXAA) activates stimulator of interferon gene (STING)-dependent innate immune pathways and is regulated by mitochondrial membrane potential. The Journal of biological chemistry. 2012;287:39776–39788. doi: 10.1074/jbc.M112.382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson SM, Walker WS. Immortalization of cloned mouse splenic macrophages with a retrovirus containing the v-raf/mil and v-myc oncogenes. Cellular immunology. 1988;116:341–351. doi: 10.1016/0008-8749(88)90236-5. [DOI] [PubMed] [Google Scholar]
- Romling U, Galperin MY, Gomelsky M. Cyclic di-GMP: the first 25 years of a universal bacterial second messenger. Microbiology and molecular biology reviews: MMBR. 2013;77:1–52. doi: 10.1128/MMBR.00043-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer JD, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, et al. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of Sting in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infection and immunity. 2011;79:688–694. doi: 10.1128/IAI.00999-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slansky JE, Rattis FM, Boyd LF, Fahmy T, Jaffee EM, Schneck JP, Margulies DH, Pardoll DM. Enhanced antigen-specific antitumor immunity with altered peptide ligands that stabilize the MHC-peptide-TCR complex. Immunity. 2000;13:529–538. doi: 10.1016/s1074-7613(00)00052-2. [DOI] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science. 2013;339:786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace A, LaRosa DF, Kapoor V, Sun J, Cheng G, Jassar A, Blouin A, Ching LM, Albelda SM. The vascular disrupting agent, DMXAA, directly activates dendritic cells through a MyD88-independent mechanism and generates antitumor cytotoxic T lymphocytes. Cancer Res. 2007;67:7011–7019. doi: 10.1158/0008-5472.CAN-06-3757. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo S, Fuertes BM, Corrales L, SS, MF, Leung M, RD, YW, GN, BK, AF, et al. STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity. 2014;41:830–842. doi: 10.1016/j.immuni.2014.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science. 2010;328:1703–1705. doi: 10.1126/science.1189801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Wang X, KuoLee R, Chen W. Synthesis and immunostimulatory properties of the phosphorothioate analogues of cdiGMP. Bioorganic & medicinal chemistry letters. 2008;18:5631–5634. doi: 10.1016/j.bmcl.2008.08.088. [DOI] [PubMed] [Google Scholar]
- Yi G, Brendel VP, Shu C, Li P, Palanathan S, Cheng Kao C. Single Nucleotide Polymorphisms of Human STING Can Affect Innate Immune Response to Cyclic Dinucleotides. PloS one. 2013;8:e77846. doi: 10.1371/journal.pone.0077846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Molecular cell. 2013;51:226–235. doi: 10.1016/j.molcel.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Ching LM, Kestell P, Baguley BC. The antitumour activity of 5,6-dimethylxanthenone-4-acetic acid (DMXAA) in TNF receptor-1 knockout mice. British journal of cancer. 2002;87:465–470. doi: 10.1038/sj.bjc.6600479. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.