

Abstract
Autoimmune carditis is associated with many human rheumatic conditions, including rheumatic fever, systemic lupus erythematosous, and rheumatoid arthritis. The immune mechanisms that mediate the cardiovascular pathology connected to these diseases are poorly defined. Several animal models are used to recapitulate human pathophysiology in order to better characterize the immunopathogenic mechanisms driving autoimmune endocardial inflammation. These animal models point toward common mechanisms mediating autoimmune endocarditis; in particular, CD4+ T cells and pro-inflammatory macrophages play critical roles in directing the disease process. The goals of this review are to discuss the prevailing animal models of autoimmune endocarditis and their underlying immunologic mechanisms, and to provide insight regarding potential therapeutic targets in humans.
Keywords: Valvular Carditis, Endocarditis, Autoimmune, Rheumatic, T cell, macrophage
Introduction
Endocarditis, inflammation of the inner lining and valves of the heart, can occur in both acute and chronic settings and can be caused by either infective or non-infective insults. Valvular damage, a serious complication resulting from chronic inflammation and scarring of the endothelium, can lead to congestive heart failure, which is often fatal [1, 2].
Infective endocarditis is caused by a microbial infection of the endocardium and accounts for approximately 10.8 hospitalizations per 100,000 people in the United States annually [3]. Lesions from infective endocarditis most commonly affect damaged or abnormal valves, prosthetic valves, or those of intravenous drug abusers. Staphylococcus aureus and Streptococcus spp. account for over 50% of infective endocarditis cases in the United States, but approximately 10% of cases are culture-negative [3]. Vegetations typically form on the left-sided heart valves, the mitral and aortic valves. Such vegetations can cause permanent valve damage or can embolize to the brain and cause complications [1]. Infective endocarditis has been reviewed recently elsewhere [1, 4]; this review focuses on autoimmune endocarditis.
Endocarditis is a serious manifestation of several autoimmune rheumatic diseases, including rheumatic fever, systemic lupus erythematosous (SLE), and rheumatoid arthritis. These forms of autoimmune endocarditis are characterized by antibody-initiated damage and activation of the endothelium, followed by inflammatory cell infiltration [5]. Rheumatic heart disease is initiated by an immune response to group A Streptococcal pharyngeal infection, in which T cells specific for group A Streptococcal M protein cross-react with cardiac myosin [5–7]. In acute rheumatic fever, all three layers of the heart become inflamed, including the endocardium, myocardium, and pericardium. Characteristic pathologic findings of rheumatic carditis include Aschoff nodules, focal granulomatous lesions of the myocardium, and Anitschkow cells, activated macrophages. In patients with SLE or anti-phospholipid syndrome, verrucous lesions form on the valvular and mural endocardium, a condition known as Libman-Sacks endocarditis [8–10]. Valve disease is also prevalent in patients with rheumatoid arthritis, with mitral regurgitation identified in 80% of patients who underwent echocardiographic evaluation [11, 12]. In each of these autoantibody-associated diseases, the mitral valve is the most commonly affected, although aortic valve insufficiency may arise [2, 10, 11].
Autoimmune endocarditis constitutes a major global health challenge. An estimated 15.6 to 17.9 million people have rheumatic heart disease worldwide and approximately 1.5% of people die each year due to the disease [13]. Rheumatic heart disease is most prevalent in developing countries is a major cause of cardiovascular death in children and young adults [13, 2]. Additionally, cardiovascular disease is a primary cause of death in patients with rheumatoid arthritis and SLE, primarily due to accelerated atherosclerosis [14–17]. Animal models have been used to recapitulate the human disease processes in order to define the immune mechanisms by which these diseases provoke endocardial inflammation. This review will focus on the existing animal models of autoimmune endocarditis and their proposed immunopathogenic mechanisms, and on how these models advance our understanding of the pathogenesis of autoimmune endocarditis in humans.
Animal Models of Autoimmune Endocarditis
Autoimmune endocarditis can be induced in experimental animals via several different means. These include inducing inflammation by injecting specific antibodies or pro-inflammatory agents or by using genetically-engineered animal models in which spontaneous production of autoantibodies occurs or in which an autoinflammatory state exists. These animal models aim to recapitulate the human disease processes, pathology, and immune mechanisms of common autoimmune diseases associated with valvular inflammation and endocarditis. Here we review the key animal models of autoimmune valvular carditis, focusing on post-Streptococcal rheumatic carditis, rheumatoid arthritis, and the spondyloarthropathies (Table 1).
Table 1.
Models of autoimmune endocarditis and their associated immune mediators
| Model | Human Disease | Cellular Infiltrate | Immune Mediators |
|---|---|---|---|
| Streptococcus or Streptococal M protein-induced carditis | Rheumatic heart disease | CD4+ T cells of Th1 and Th17 effector lineage Macrophages |
TNF IFNγ IL-6 IL-10 IL-17 |
| K/BxN mouse | Rheumatoid arthritis and valvular carditis | CD4+ T cells Macrophages Tregs |
Fcγ receptors IFNγ IL-17 |
| IL-1ra-deficient mouse | Rheumatoid arthritis and aortitis Or DIRA |
Macrophages T cells |
TNF IL-1 |
| IL-23 Overexpression | Spondyloarthropathy and aortic root valve inflammation | CD4+ T cells of Th17 effector lineage Macrophages |
IL-6 IL-17 IL-22 |
Abbreviations: DIRA: deficiency of IL-1 receptor antagonist; IFNγ: interferon gamma; IL: interleukin; Th: helper T cell; TNF: tumor necrosis factor; Treg: regulatory T cell
Animal Models of Rheumatic Carditis
Cromartie and Craddock first demonstrated that rheumatic-like disease could be induced in mice via intraperitoneal injection of sonically-disrupted group A Streptococcal cells. This caused an inflammatory process in the endocardium, the mitral and aortic valves, myocardium, and pericardium. Within three days of injection, focal granulomatous lesions consisting of monocytes, lymphocytes, and Anitschkow cells with central necrosis similar to Aschoff bodies developed. Within eight weeks of injection, the acute inflammatory response subsided, and the aortic and mitral valve annuli demonstrated chronic inflammation and scarring. The acute and chronic pathologic findings in the mice showed striking similarity to the lesions of acute rheumatic fever and chronic rheumatic carditis in humans [18].
The theory that antibodies specific for Streptococcal epitopes are cross-reactive with cardiac antigens gained traction based on the finding that patients with acute rheumatic fever had circulating cardiac-reactive antibodies [19]. Specifically, it was suggested that following Streptococcal pharyngitis, antibodies generated against Streptococcal M cross-reacted with human cardiac tropomyosin. Extensive work from Cunningham’s group has demonstrated that monoclonal antibodies directed against Streptococcal M proteins or N-acetyl-βD-glucosamine (GlcNAc, the primary epitope of group A carbohydrate antigen) cross-react with cardiac myosin and laminin, thus establishing evidence for molecular mimicry [5–7, 19–21].
A rat model was developed based on this theory of molecular mimicry, in which Lewis rats were immunized with Streptococcal M protein and adjuvant. The rats developed valvular carditis and focal lesions of myocarditis with extensive T cell infiltration. Additionally, this model provided the first evidence that, in addition to antibody cross-reactivity, T-cell cross-reactivity also occurs. Specifically, T cells reactive to both Streptococcal M protein and cardiac myosin were present in the valvular lesions [22••]. Additional groups used this model to demonstrate that conserved regions of Streptococcal M protein [23] and individual peptides could also induce an immune response that mimics rheumatic heart disease [24, 25]. Furthermore, injecting rats with human or rat cardiac myosin plus adjuvant induced both myocarditis and valvular disease [26]. The lymphocytes in the myocardial infiltrate proliferated in response to Streptococcal M protein, providing further evidence that myosin-reactive T cells cross-react with Streptococcal M protein [26].
Immune Mechanisms of Rheumatic Carditis
The infiltrate in human rheumatic carditis is composed primarily of lymphocyte and mononuclear cells [27, 28]. Raizada et al first suggested a CD4+ T cell-mediated disease process in their examination of lymphocytic infiltrates in valvular tissue obtained from patients with rheumatic heart disease [27]. Similarly, both CD4+ T cells and CD68+ macrophages were identified in in the valves of Lewis rats immunized with recombinant Streptococcal M protein [24]. The expression of vascular cell adhesion molecule-1 (VCAM-1) was upregulated on the valvular endothelium of human patients with rheumatic heart disease; VCAM-1 enables extravasation of CD4+ and CD8+ T cells into the valvular tissue [29]. Mononuclear cells obtained from valve lesions of patients with rheumatic heart disease secreted tumor necrosis factor (TNF) and interferon gamma (IFNγ), suggesting that the CD4+ T cells were of the Th1 phenotype. Interleukin-10 (IL-10), a negative regulator of Th1 cells, was also secreted by the valve-infiltrating lymphocytes [30, 31].
Th17 cells, a pro-inflammatory effector lineage of CD4+ T cells that secrete IL-17, IL-21, IL-22, and TNF have more recently been identified as key participants in autoimmune inflammatory states [32–34]. Guilherme and colleagues reported evidence of increased IL-17A expression in peripheral T lymphocytes from patients with chronic rheumatic heart disease compared to healthy donors [35]. Additionally, recurrent intranasal group A Streptococcal infection in mice shifted the antigen-specific T cell population toward an IL17A+IFN-γ+ phenotype [36]. A Th17-mediated inflammatory process has also been described in a mouse model of experimental autoimmune myocarditis in which BALB/c mice are injected with a synthesized peptide based on the cardiac myosin heavy chain α sequence. These mice developed CD4+ T cell-dependent myocarditis that progressed to dilated cardiomyopathy. It has been shown that the progression from myocarditis to dilated cardiomyopathy was dependent on the presence of IL-6 and IL-17 to drive a pro-inflammatory phenotype in monocytes and macrophages in the myocardium [37–39].
It is possible that the mechanisms that drive endocarditis in the Streptococcal model of rheumatic heart disease are similar to those described in this model of experimental autoimmune myocarditis. However, one group has suggested that the inflammatory mechanisms mediating endocarditis versus myocarditis may differ; specifically, in samples obtained from patients with rheumatic heart disease, the Th2 cytokine IL-4 was produced by many of the infiltrating cells in the myocardium, but by very few of the infiltrating cells in the mitral valve [30].
In sum, CD4+ T cells and pro-inflammatory macrophages seem to dominate the inflammatory infiltrates of human rheumatic carditis and its animals models [25]. Th1 and Th17 cells have been identified in both human patients and animal models, but the relative contributions of these two Th effector cell subsets remains undefined.
K/BxN Mouse Model of Inflammatory Arthritis
The K/BxN mouse was first described as an animal model of inflammatory arthritis with some similarities to rheumatoid arthritis in humans [40]. K/BxN mice express a transgene-encoded T cell receptor (TCR) termed “KRN” that recognizes a self-peptide derived from the ubiquitously-expressed enzyme glucose-6-phosphate isomerase (GPI), presented on the mouse major histocompatibility complex (MHC) class II molecule Ag7 derived from the NOD mouse strain. CD4+ T cells bearing the KRN TCR are activated by Ag7:GPI-expessing antigen presenting cells, allowing the CD4+ T cells in turn to stimulate GPI-reactive B cells to produce arthritis-inducing anti-GPI autoantibodies [40, 41]. Arthritis can be induced in naïve mice by injecting serum from K/BxN mice containing anti-GPI autoantibodies, a model system termed “serum transfer arthritis.”
In addition to developing spontaneous autoimmune arthritis, the TCR transgenic K/BxN mice also develop autoimmune valvular carditis, primarily affecting the mitral valve [42••]. Our group has used this model to study the immunopathogenic mechanisms mediating autoantibody-associated autoimmune valvular carditis.
Immune Mechanisms
Similar to murine models of rheumatic heart disease, CD4+ T cells and macrophages are the predominant cells infiltrating the inflamed mitral valves of K/BxN mice [42••]. Genetic absence of B cells or of CD40, a molecule required for immunoglobulin isotype switching, prevented valvular carditis in this model, strongly suggesting an autoantibody-dependent process. However, transfer of K/BxN serum to naïve mice produces only arthritis – not carditis – suggesting that autoantibodies alone are not sufficient to drive valvular carditis [42••]. Indeed, depletion of CD4+ T cells after the onset of arthritis prevented the development of valvular inflammation without affecting arthritis severity, suggesting that the sustained presence of CD4+ T cells is required to mediate valvular carditis [43].
While autoantibodies appear to be required for the development of both arthritis and endocarditis in the K/BxN mice, distinct innate immune mechanisms direct the pathologic processes. Arthritis in K/BxN mice depends on complement C5 but not activating Fcg receptors [42••]. With respect to endocarditis, although complement C3 was deposited on the inflamed mitral valves (as in the human forms of autoimmune valvular carditis), genetic deficiency of C3 or C5 did not affect the severity of valve inflammation. Rather, in K/BxN mice, autoantibodies appear to be acting through activating Fcγ receptors to drive valvular carditis, specifically FcγRIII and FcγRIV on macrophages [42••, 44••]. Furthermore, depletion of macrophages reduced the severity of valve inflammation, suggesting that valvular carditis depends, at least in part, on the presence of macrophages [44••].
The nature of the CD4+ T cell cytokine profile driving valvular carditis in the K/BxN mouse model remains incompletely defined. IFNγ and IL-17 were both found in the valve infiltrate, whereas IL-4 was not [43]. However, genetic deletion of IFNγ had no effect on the severity of valvular carditis (BAB, unpublished results). A role for Foxp3+ regulatory T cells (Treg) in this model has also been demonstrated. Specifically, Treg cell development is impaired in mice lacking β2 integrin (CD18). In the K/BxN system, deficiency of CD18 resulted in fewer Treg cells and more severe cardiac inflammation without affecting arthritis severity [43]. Additionally, dual TCR T cells accelerated the development of autoimmunity in K/BxN mice by promoting positive selection, and the absence of dual TCR T cells prevented the development of valvular carditis [45]. Ongoing work by our group seeks to identify the key Th cytokines driving valvular carditis in this model and to characterize further the contribution of macrophages.
Arthritis and Carditis due to deficiency of IL-1 receptor antagonist (IL-1Ra)
IL-1 receptor antagonist (IL-1Ra) is a naturally occurring regulator of the pro-inflammatory IL-1 cytokine signaling system. BALB/c mice lacking the gene encoding IL-1Ra (Il1rn) developed spontaneous chronic inflammatory arthropathy similar to rheumatoid arthritis [46]. These mice produced autoantibodies directed against immunoglobulin, type II collagen, and double-stranded DNA. In addition to arthritis, these IL-1ra-deficient mice developed inflammatory thickening and stenosis of the aortic valve [47••]. Intriguingly, the development of arthritis depended on genetic background of the mice [48]; deficiency of IL-1Ra on the CL57B6/J background resulted in severe arteritis, but neither arthritis nor valvular disease [49]. Recently, genetic deficiency of IL-1Ra (DIRA) in humans has been described in a small number of patients. These patients develop inflammatory arthropathy and cutaneous inflammation; one patient had vasculitis, but no other cardiovascular phenotype has been reported [50, 51].
Immune Mechanism
As in the K/BxN model, the infiltrate in the inflamed aortic valve of IL-1Ra-deficient mice primarily consisted of monocytes/macrophages. Additionally, transfer of peripheral T cells from IL-1Ra-deficient mice into immunodeficient BALB/c hosts induced greater aortic valve inflammation than did transfer of peripheral T cells transferred from wild-type mice. Inflammation was associated with increased TNF levels in the plasma, and BALB/c mice genetically lacking both TNF and IL-1Ra (Tnf−/−/IL1rn−/−) were protected from valvular inflammation relative to their IL-1Ra-deficient but TNF-sufficient counterparts [47••]. Thus, this model too suggests that T cells and pro-inflammatory macrophages cooperate to drive autoimmune valvular carditis.
IL-23-Dependent Model of Spondyloarthropathy
Following injection with type II collagen-specific antibodies, B10.RIII mice develop severe enthesitis -- inflammation of tendons and ligaments, a characteristic clinical finding in human patients with spondyloarthropathy. The development of enthesitis in this mouse model was shown to depend on IL-23. In making this connection between type II collagen-specific antibodies and an IL-23 dependent mechanism, the authors investigated whether IL-23 alone could induce the same disease. Injection of IL-23 minicircle DNA induced systemic spondyloarthropathy, including aortic root and aortic valve inflammation, even in the absence of type II collagen-specific antibodies. Whether type II collagen-specific antibodies alone provoke aortic inflammation was not investigated [52••].
Immune Mechanism
In this model, IL-23 receptor-expressing CD4− CD8− T cells in the entheses responded to the overexpressed IL-23, resulting in the production of the cytokines typically associated with Th17 cells: IL-6, IL-17, and IL-22. Phenotypically similar cells were identified in the inflamed aortic root and aortic valve, along with increased expression of IL-22. The valve infiltrate included T cells, macrophages, and neutrophils [52••]. This model thus provides further evidence that T cells, particularly those making Th17-related cytokines, and macrophages can cooperate to drive autoimmune valvular carditis.
Conclusion
Autoimmune valvular carditis accompanies many human rheumatic conditions, including rheumatic fever, systemic lupus erythematosus and the related antiphospholipid syndrome, as well as rheumatoid arthritis. We have reviewed the animal models that are currently being used to understand the immunopathogenic mechanisms mediating cardiovascular pathology. Autoantibodies appear to be critical in some of the models, but not all. Common threads do emerge, however, with predominant roles for CD4+ T cells of the Th1 and Th17 effector lineages as well as pro-inflammatory macrophages (Fig. 1). Ongoing work is directed at understanding how these components of the immune response -- autoantibodies, effector T cells, and macrophages -- interact to drive cardiac valve inflammation in the setting of systemic autoimmune disease. Identification of the key pro-inflammatory pathways involved, likely including IL-17 and related molecules, TNF, IL-1, and IL-6, is expected to lead to improved therapies for patients with autoimmune valvular carditis.
Figure 1. Model of the pathogenesis of autoimmune valvular carditis.
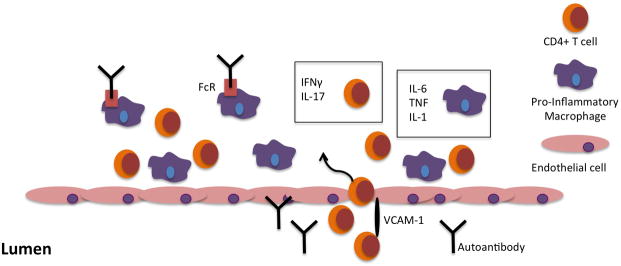
CD4+ T cells, with the help of VCAM-1, and macrophages extravasate across the endothelium into the valve interstitium. Th1 and Th17 effector CD4+ T cell lineages are characterized by the production of IFNγ and IL-17, respectively. Macrophages, in some cases stimulated by antibodies engaging activating Fc receptors, release the pro-inflammatory cytokines IL-6, TNF, and IL-1.
Acknowledgments
Research in Dr. Binstadt’s laboratory was funded by the National Institutes of Health National Heart, Lung, and Blood Institute R01 HL121093, National Institute of Arthritis and Musculoskeletal and Skin Diseases R03 AR057101, an Innovative Research Grant from the Rheumatology Research Foundation, and by the University of Minnesota Department of Pediatrics.
References
Papers of particular interest have been highlighted as:
• Of importance
•• Of major importance
- 1.Mylonakis E, Calderwood SB. Infective endocarditis in adults. N Engl J Med. 2001;345(18):1318–30. doi: 10.1056/NEJMra010082. [DOI] [PubMed] [Google Scholar]
- 2.Marijon E, Mirabel M, Celermajer DS, Jouven X. Rheumatic heart disease. Lancet. 2012;379(9819):953–64. doi: 10.1016/S0140-6736(11)61171-9. [DOI] [PubMed] [Google Scholar]
- 3.Bor DH, Woolhandler S, Nardin R, Brusch J, Himmelstein DU. Infective endocarditis in the U.S., 1998–2009: a nationwide study. PloS one. 2013;8(3):e60033. doi: 10.1371/journal.pone.0060033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Werdan K, Dietz S, Loffler B, Niemann S, Bushnaq H, Silber RE, et al. Mechanisms of infective endocarditis: pathogen-host interaction and risk states. Nat Rev Cardiol. 2014;11(1):35–50. doi: 10.1038/nrcardio.2013.174. [DOI] [PubMed] [Google Scholar]
- 5.Galvin JE, Hemric ME, Ward K, Cunningham MW. Cytotoxic mAb from rheumatic carditis recognizes heart valves and laminin. J Clin Invest. 2000;106(2):217–24. doi: 10.1172/JCI7132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cunningham MW, Antone SM, Gulizia JM, McManus BM, Fischetti VA, Gauntt CJ. Cytotoxic and viral neutralizing antibodies crossreact with streptococcal M protein, enteroviruses, and human cardiac myosin. Proc Natl Acad Sci U S A. 1992;89(4):1320–4. doi: 10.1073/pnas.89.4.1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krisher K, Cunningham MW. Myosin: a link between streptococci and heart. Science. 1985;227(4685):413–5. doi: 10.1126/science.2578225. [DOI] [PubMed] [Google Scholar]
- 8.Tincani A, Rebaioli CB, Taglietti M, Shoenfeld Y. Heart involvement in systemic lupus erythematosus, anti-phospholipid syndrome and neonatal lupus. Rheumatology. 2006;45(Suppl 4):iv8–13. doi: 10.1093/rheumatology/kel308. [DOI] [PubMed] [Google Scholar]
- 9.Blank M, Shani A, Goldberg I, Kopolovic J, Amigo MC, Magrini L, et al. Libman-Sacks endocarditis associated with antiphospholipid syndrome and infection. Thromb Res. 2004;114(5–6):589–92. doi: 10.1016/j.thromres.2004.06.039. [DOI] [PubMed] [Google Scholar]
- 10.Hojnik M, George J, Ziporen L, Shoenfeld Y. Heart valve involvement (Libman-Sacks endocarditis) in the antiphospholipid syndrome. Circulation. 1996;93(8):1579–87. doi: 10.1161/01.cir.93.8.1579. [DOI] [PubMed] [Google Scholar]
- 11.Voskuyl A. The heart and cardiovascular manifestations in rheumatoid arthritis. Rheumatology. 2006;45(suppl 4):iv4–iv7. doi: 10.1093/rheumatology/kel313. [DOI] [PubMed] [Google Scholar]
- 12.Guedes C, Bianchi-Fior P, Cormier B, Barthelemy B, Rat AC, Boissier MC. Cardiac manifestations of rheumatoid arthritis: A case–control transesophageal echocardiography study in 30 patients. Arthritis Care Res. 2001;45(2):129–35. doi: 10.1002/1529-0131(200104)45:2<129::AID-ANR164>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 13.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5(11):685–94. doi: 10.1016/s1473-3099(05)70267-x. [DOI] [PubMed] [Google Scholar]
- 14.Wolfe F, Mitchell DM, Sibley JT, Fries JF, Bloch DA, Williams CA, et al. The mortality of rheumatoid arthritis. Arthritis Rheum. 1994;37(4):481–94. doi: 10.1002/art.1780370408. [DOI] [PubMed] [Google Scholar]
- 15.Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. 2005;52(3):722–32. doi: 10.1002/art.20878. [DOI] [PubMed] [Google Scholar]
- 16.Jacobsen S, Petersen J, Ullman S, Junker P, Voss A, Rasmussen J, et al. Mortality and causes of death of 513 Danish patients with systemic lupus erythematosus. Scand J Rheumatol. 1999;28(2):75–80. doi: 10.1080/030097499442522. [DOI] [PubMed] [Google Scholar]
- 17.Bartels CM, Buhr KA, Goldberg JW, Bell CL, Visekruna M, Nekkanti S, et al. Mortality and cardiovascular burden of systemic lupus erythematosus in a US population-based cohort. J Rheumatol. 2014;41(4):680–7. doi: 10.3899/jrheum.130874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cromartie WJ, Craddock JG. Rheumatic-like cardiac lesions in mice. Science. 1966;154(3746):285–7. doi: 10.1126/science.154.3746.285. [DOI] [PubMed] [Google Scholar]
- 19.Cunningham MW, Swerlick RA. Polyspecificity of antistreptococcal murine monoclonal antibodies and their implications in autoimmunity. J Exp Med. 1986;164(4):998–1012. doi: 10.1084/jem.164.4.998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenderson PG, Fischetti VA, Cunningham MW. Tropomyosin shares immunologic epitopes with group A streptococcal M proteins. J Immunol. 1989;142(7):2475–81. [PubMed] [Google Scholar]
- 21.Shikhman AR, Greenspan N, Cunningham MW. A subset of mouse monoclonal antibodies cross-reactive with cytoskeletal proteins and group A streptococcal M proteins recognizes N-acetyl-beta-D-glucosamine. J Immunol. 1993;151(7):3902–13. [PubMed] [Google Scholar]
- ••22.Quinn A, Kosanke S, Fischetti VA, Factor SM, Cunningham MW. Induction of autoimmune valvular heart disease by recombinant streptococcal m protein. Infect Immun. 2001;69(6):4072–8. doi: 10.1128/iai.69.6.4072-4078.2001. This paper describes the method for inducing autoimmune valvular carditis in Lewis rats by immunization with Streptococcal M protein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lymbury RS, Olive C, Powell KA, Good MF, Hirst RG, LaBrooy JT, et al. Induction of autoimmune valvulitis in Lewis rats following immunization with peptides from the conserved region of group A streptococcal M protein. J Autoimmun. 2003;20(3):211–7. doi: 10.1016/s0896-8411(03)00026-x. [DOI] [PubMed] [Google Scholar]
- 24.Gorton D, Blyth S, Gorton J, Govan B, Ketheesan N. An alternative technique for the induction of autoimmune valvulitis in a rat model of rheumatic heart disease. J Immunol Methods. 2010;355(1):80–5. doi: 10.1016/j.jim.2010.02.013. [DOI] [PubMed] [Google Scholar]
- 25.Gorton D, Govan B, Olive C, Ketheesan N. B-and T-cell responses in group a streptococcus M-protein-or Peptide-induced experimental carditis. Infect Immun. 2009;77(5):2177–83. doi: 10.1128/IAI.01514-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galvin JE, Hemric ME, Kosanke SD, Factor SM, Quinn A, Cunningham MW. Induction of myocarditis and valvulitis in lewis rats by different epitopes of cardiac myosin and its implications in rheumatic carditis. Am J Pathol. 2002;160(1):297–306. doi: 10.1016/s0002-9440(10)64373-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raizada V, Williams RC, Jr, Chopra P, Gopinath N, Prakash K, Sharma K, et al. Tissue distribution of lymphocytes in rheumatic heart valves as defined by monoclonal anti-T cell antibodies. Am J Med. 1983;74(1):90–6. doi: 10.1016/0002-9343(83)91124-5. [DOI] [PubMed] [Google Scholar]
- 28.Kemeny E, Grieve T, Marcus R, Sareli P, Zabriskie JB. Identification of mononuclear cells and T cell subsets in rheumatic valvulitis. Clin Immunol Immunopathol. 1989;52(2):225–37. doi: 10.1016/0090-1229(89)90174-8. [DOI] [PubMed] [Google Scholar]
- 29.Roberts S, Kosanke S, Dunn ST, Jankelow D, Duran CM, Cunningham MW. Pathogenic mechanisms in rheumatic carditis: focus on valvular endothelium. J Infect Dis. 2001;183(3):507–11. doi: 10.1086/318076. [DOI] [PubMed] [Google Scholar]
- 30.Guilherme L, Cury P, Demarchi LM, Coelho V, Abel L, Lopez AP, et al. Rheumatic heart disease: proinflammatory cytokines play a role in the progression and maintenance of valvular lesions. Am J Pathol. 2004;165(5):1583–91. doi: 10.1016/s0002-9440(10)63415-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fae K, Oshiro S, Toubert A, Charron D, Kalil J, Guilherme L. How an autoimmune reaction triggered by molecular mimicry between streptococcal M protein and cardiac tissue proteins leads to heart lesions in rheumatic heart disease. J Autoimmun. 2005;24(2):101–9. doi: 10.1016/j.jaut.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 32.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 33.Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y, et al. Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med. 2008;205(4):799–810. doi: 10.1084/jem.20071258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Afzali B, Lombardi G, Lechler R, Lord G. The role of T helper 17 (Th17) and regulatory T cells (Treg) in human organ transplantation and autoimmune disease. Clin Exp Immunol. 2007;148(1):32–46. doi: 10.1111/j.1365-2249.2007.03356.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guilherme L, Kalil J. Rheumatic fever and rheumatic heart disease: cellular mechanisms leading autoimmune reactivity and disease. J Clin Immunol. 2010;30(1):17–23. doi: 10.1007/s10875-009-9332-6. [DOI] [PubMed] [Google Scholar]
- 36.Dileepan T, Linehan JL, Moon JJ, Pepper M, Jenkins MK, Cleary PP. Robust antigen specific th17 T cell response to group A Streptococcus is dependent on IL-6 and intranasal route of infection. PLoS Pathog. 2011;7(9):e1002252. doi: 10.1371/journal.ppat.1002252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu L, Ong S, Talor MV, Barin JG, Baldeviano GC, Kass DA, et al. Cardiac fibroblasts mediate IL-17A-driven inflammatory dilated cardiomyopathy. J Exp Med. 2014;211(7):1449–64. doi: 10.1084/jem.20132126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6–dependent Th17 cell development and survival. J Exp Med. 2008;205(10):2281–94. doi: 10.1084/jem.20071119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106(10):1646–55. doi: 10.1161/circresaha.109.213157. [DOI] [PubMed] [Google Scholar]
- 40.Kouskoff V, Korganow AS, Duchatelle V, Degott C, Benoist C, Mathis D. Organ-specific disease provoked by systemic autoimmunity. Cell. 1996;87(5):811–22. doi: 10.1016/s0092-8674(00)81989-3. [DOI] [PubMed] [Google Scholar]
- 41.Matsumoto I, Staub A, Benoist C, Mathis D. Arthritis provoked by linked T and B cell recognition of a glycolytic enzyme. Science. 1999;286(5445):1732–5. doi: 10.1126/science.286.5445.1732. [DOI] [PubMed] [Google Scholar]
- ••42.Binstadt BA, Hebert JL, Ortiz-Lopez A, Bronson R, Benoist C, Mathis D. The same systemic autoimmune disease provokes arthritis and endocarditis via distinct mechanisms. Proc Natl Acad Sci U S A. 2009;106(39):16758–63. doi: 10.1073/pnas.0909132106. This is the initial description of valvular carditis in K/BxN mice, and demonstrates that carditis depends on activating Fcγ receptors but not complement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Haasken S, Auger JL, Binstadt BA. Absence of β2 Integrins Impairs Regulatory T Cells and Exacerbates CD4+ T Cell-Dependent Autoimmune Carditis. J Immunol. 2011;187(5):2702–10. doi: 10.4049/jimmunol.1000967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••44.Hobday PM, Auger JL, Schuneman GR, Haasken S, Verbeek JS, Binstadt BA. Fcgamma receptor III and Fcgamma receptor IV on macrophages drive autoimmune valvular carditis in mice. Arthritis Rheumatol. 2014;66(4):852–62. doi: 10.1002/art.38311. This paper demonstrates the importance of macrophages and particular activating Fcγ receptors in the K/BxN mouse model of valvular carditis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Auger JL, Haasken S, Steinert EM, Binstadt BA. Incomplete TCR-β allelic exclusion accelerates spontaneous autoimmune arthritis in K/BxN TCR transgenic mice. Eur J Immunol. 2012;42(9):2354–62. doi: 10.1002/eji.201242520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Horai R, Saijo S, Tanioka H, Nakae S, Sudo K, Okahara A, et al. Development of chronic inflammatory arthropathy resembling rheumatoid arthritis in interleukin 1 receptor antagonist-deficient mice. J Exp Med. 2000;191(2):313–20. doi: 10.1084/jem.191.2.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••47.Isoda K, Matsuki T, Kondo H, Iwakura Y, Ohsuzu F. Deficiency of interleukin-1 receptor antagonist induces aortic valve disease in BALB/c mice. Arterioscler Thromb Vasc Biol. 2010;30(4):708–15. doi: 10.1161/atvbaha.109.201749. This paper demonstrates the occurrence of TNF-dependent aortic valve inflammation in IL-1ra-deficient BALB/c mice. [DOI] [PubMed] [Google Scholar]
- 48.Zhou F, He X, Iwakura Y, Horai R, Stuart JM. Arthritis in mice that are deficient in interleukin-1 receptor antagonist is dependent on genetic background. Arthritis Rheum. 2005;52(12):3731–8. doi: 10.1002/art.21481. [DOI] [PubMed] [Google Scholar]
- 49.Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191(2):303–12. doi: 10.1084/jem.191.2.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Aksentijevich I, Masters SL, Ferguson PJ, Dancey P, Frenkel J, van Royen-Kerkhoff A, et al. An autoinflammatory disease with deficiency of the interleukin-1–receptor antagonist. N Engl J Med. 2009;360(23):2426–37. doi: 10.1056/NEJMoa0807865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reddy S, Jia S, Geoffrey R, Lorier R, Suchi M, Broeckel U, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med. 2009;360(23):2438–44. doi: 10.1056/NEJMoa0809568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••52.Sherlock JP, Joyce-Shaikh B, Turner SP, Chao C-C, Sathe M, Grein J, et al. IL-23 induces spondyloarthropathy by acting on ROR-[gamma] t+ CD3+ CD4-CD8-entheseal resident T cells. Nat Med. 2012;18(7):1069–76. doi: 10.1038/nm.2817. This study describes the occurrence of aortic root inflammation in a mouse model of IL-23-driven spondyloarthropathy. [DOI] [PubMed] [Google Scholar]