

Abstract
Neurons in the brain must establish a balanced network of excitatory and inhibitory synapses during development for the brain to function properly. An imbalance between these synapses underlies various neurological and psychiatric disorders. The formation of excitatory and inhibitory synapses requires precise molecular control. In the hippocampus, the structure crucial for learning and memory, fibroblast growth factor 22 (FGF22) and FGF7 specifically promote excitatory or inhibitory synapse formation, respectively. Knockout of either Fgf gene leads to excitatory-inhibitory imbalance in the mouse hippocampus and manifests in an altered susceptibility to epileptic seizures, underscoring the importance of FGF-dependent synapse formation. However, the receptors and signaling mechanisms by which FGF22 and FGF7 induce excitatory and inhibitory synapse differentiation are unknown. Here, we show that distinct sets of overlapping FGF receptors (FGFRs), FGFR2b and FGFR1b, mediate excitatory or inhibitory presynaptic differentiation in response to FGF22 and FGF7. Excitatory presynaptic differentiation is impaired in Fgfr2b and Fgfr1b mutant mice; however, inhibitory presynaptic defects are only found in Fgfr2b mutants. FGFR2b and FGFR1b are required for an excitatory presynaptic response to FGF22, whereas only FGFR2b is required for an inhibitory presynaptic response to FGF7. We further find that FGFRs are required in the presynaptic neuron to respond to FGF22, and that FRS2 and PI3K, but not PLCγ, mediate FGF22-dependent presynaptic differentiation. Our results reveal the specific receptors and signaling pathways that mediate FGF-dependent presynaptic differentiation, and thereby provide a mechanistic understanding of precise excitatory and inhibitory synapse formation in the mammalian brain.
Keywords: Fibroblast growth factor receptor, Synapse formation, Excitatory-inhibitory balance, Hippocampus, Receptor tyrosine kinase signaling, Mouse
HIGHLIGHTED ARTICLE: Overlapping but distinct sets of FGF receptors mediate FGF22- and FGF7-dependent excitatory and inhibitory presynaptic differentiation in the mammalian hippocampus, in part through FRS2/PI3K signaling.
INTRODUCTION
Neurons of the developing brain must form a network of excitatory and inhibitory synapses in a highly organized fashion, which requires differential molecular control between each synapse type (Chia et al., 2013; Dabrowski and Umemori, 2011; Jin and Garner, 2008; Johnson-Venkatesh and Umemori, 2010; McAllister, 2007; Sanes and Yamagata, 2009; Shen and Scheiffele, 2010; Siddiqui and Craig, 2011; Williams and Umemori, 2014; Williams et al., 2010). An imbalance between excitatory and inhibitory drive in the brain may lead to various neurological and psychiatric diseases, including epilepsy, autism and schizophrenia (Lisman, 2012; Southwell et al., 2014). Thus, the components of molecular signaling mediating synapse formation are key factors in the vulnerability or resilience to such disorders. Hippocampal circuits are crucial for memory formation, emotional processing and social behavior. The hippocampal circuits consist of highly organized synaptic connections, in which excitatory projections relay from the entorhinal cortex (EC) to the dentate gyrus (DG) to CA3 to CA1 to the EC, and are balanced by inhibitory inputs from local interneurons. In the hippocampus, two fibroblast growth factors (FGFs), FGF22 and FGF7, are secreted from dendrites of CA3 pyramidal neurons, and promote differentiation of excitatory and inhibitory presynaptic terminals, respectively (Terauchi et al., 2010, 2015). That this FGF-dependent regulation of excitatory and inhibitory presynaptic differentiation is important for proper brain development is evidenced by altered seizure susceptibility of knockout (KO) animals: Fgf22-KO mice have decreased, and Fgf7-KO mice have increased susceptibility to epileptic seizures (Terauchi et al., 2010; Lee et al., 2012; Lee and Umemori, 2013). However, the signaling mechanisms underlying FGF-dependent synapse formation are currently not understood. Understanding the mechanisms through which FGF22 and FGF7 specifically promote excitatory and inhibitory presynaptic differentiation will give an important insight into how molecular signaling tunes discrete synapse formation.
FGFs regulate a variety of processes, including cell proliferation, migration, differentiation, tissue repair and response to injury in almost all organs (Thisse and Thisse, 2005; Turner and Grose, 2010; Ornitz and Itoh, 2001). In the nervous system, FGF signaling is important in various steps of development, including neural induction, patterning, proliferation, axon guidance and synapse formation (Guillemot and Zimmer, 2011; Jones and Basson, 2010; Mason, 2007; Stevens et al., 2010a; Umemori, 2009). For the 18 secreted FGFs, there are four FGF receptors (FGFRs), which, through b and c splice variants, encode seven distinct receptor tyrosine kinases (Chellaiah et al., 1994; Johnson et al., 1991; Ornitz et al., 1996; Belov and Mohammadi, 2013; Powers et al., 2000). Once FGFRs are activated, they recruit and/or phosphorylate a number of signaling molecules, including fibroblast growth factor receptor substrate 2 (FRS2), phosphoinositide 3-kinase (PI3K) and phospholipase C γ (PLCγ), which result in the activation of downstream pathways (Mohammadi et al., 1996; Furdui et al., 2006; Bae et al., 2009; Lemmon and Schlessinger, 2010). Therefore, the cellular responses to FGFs are regulated at multiple levels by a number of different factors. What is remarkable in FGF-dependent synapse formation is that FGF22 and FGF7 are secreted from the same CA3 pyramidal neurons but specifically organize excitatory or inhibitory presynaptic differentiation. Important questions regarding their specific effects include the identity of the receptors, location of their action and the signaling pathways involved. Understanding the precise FGF signaling mechanisms during excitatory and inhibitory synapse development will not only further our understanding of synaptogenesis, but will also yield more insight into how FGFs dictate cellular outcomes with such remarkable specificity.
Compared with what is known about signaling pathways involved in postsynaptic development (Ebert and Greenberg, 2013; Hagenston and Bading, 2011; Mabb and Ehlers, 2010; Shen and Cowan, 2010; Stamatakou and Salinas, 2014; Tolias et al., 2011), less is known about signaling pathways guiding presynaptic differentiation. The presynaptic contribution of individual signaling pathways has been studied using broad inhibition or overactivation: PI3K/AKT (Martin-Peña et al., 2006; Cuesto et al., 2011), extracellular signal-related kinase (ERK)/mitogen-activated protein kinase (MAPK) (Li et al., 2002; Kushner et al., 2005; Nakata et al., 2005; Wairkar et al., 2009; Giachello et al., 2010) and PLCγ (Yoshida et al., 2009) have been all proposed to be involved in presynaptic development. The contributions of different signaling pathways might reflect the molecular complexity of synapse formation, underscoring how crucial it is to understand the signaling pathways downstream of specific synaptogenic receptors to tease apart this complexity.
In this study, we asked the following important questions regarding the mechanisms of FGF-induced excitatory and inhibitory synapse formation: What are the receptors through which FGF22 and FGF7 mediate their synaptogenic effects? Where do the receptors act? What are the signaling pathways involved? Using multiple null and conditional KO mice and primary cultures prepared from them, we found that distinct but overlapping sets of FGFRs mediate excitatory versus inhibitory synapse differentiation through a presynaptic mechanism that involves PI3K and FRS2 signaling. Our work uncovers mechanisms of FGF-induced specific presynaptic differentiation and contributes to a profound understanding of how appropriate neural networks are established during development in the mammalian brain.
RESULTS
Excitatory presynaptic differentiation is impaired in Fgfr2b and Fgfr1b mutant mice
We first sought to identify the FGFRs involved in presynaptic differentiation by investigating whether KO mice of candidate FGFRs show similar synaptic deficits observed in Fgf22-KO or Fgf7-KO mice. We focused on FGFR2b and FGFR1b, because in mitogenesis assays with FGFR-expressing BaF3 cells, FGF22 activates FGFR2b and FGFR1b, and FGF7 activates FGFR2b (Zhang et al., 2006). Fgfr2 and Fgfr1b are expressed in excitatory neurons (Terauchi et al., 2010; Beer et al., 2000) and inhibitory interneurons in the hippocampus (supplementary material Fig. S1). To investigate excitatory presynaptic differentiation in mice lacking Fgfr2b or Fgfr1b, we stained brain sections for vesicular glutamate transporter 1 (VGLUT1), a marker of excitatory synaptic vesicles that cluster at nerve terminals as excitatory synapses develop (Fig. 1). Fgfr2b−/− mice (Fig. 1A) are not viable postnatally due to lung agenesis, whereas Fgfr2b+/− animals, although viable, have developmental defects such as stunted seminal vesicle development (Kuslak et al., 2007) and hypoplastic submandibular glands (Jaskoll et al., 2005). Hence, we investigated whether Fgfr2b haploinsufficiency affects excitatory presynaptic differentiation. We focused our analysis in CA3 of the hippocampus, because Fgf22-KO mice show specific defects in excitatory presynaptic differentiation in the CA3 (Terauchi et al., 2010). We found that Fgfr2b+/− mice have decreased intensity of VGLUT1 immunoreactivity in the stratum radiatum (SR) and stratum lucidum (SL) of hippocampal CA3 at postnatal day 8 (P8), an early stage of synapse formation (Fig. 1B,C). Further analysis revealed that VGLUT1 puncta are smaller and sparser in Fgfr2b+/− mice compared with wild-type (WT) littermates (Fig. 1D). We next investigated Fgfr1b−/− mice (Fig. 1E) for excitatory presynaptic differentiation. We found that VGLUT1 intensity is decreased both in the SL and SR layers of the hippocampal CA3 (Fig. 1F,G), and that VGLUT1 puncta are smaller and sparser in Fgfr1b−/− animals (Fig. 1H). We did not observe appreciable changes in postsynaptic differentiation, assessed by PSD95 immunostaining (an excitatory postsynaptic marker), in either Fgfr2b+/− or Fgfr1b−/− mice (supplementary material Fig. S2). Furthermore, VGLUT1 immunostaining in lateral septal nuclei does not perceptibly change in Fgfr mutant mice, suggesting that loss of Fgfr2b or Fgfr1b does not globally decrease VGLUT1 levels (supplementary material Fig. S3). These data suggest that both FGFR2b and FGFR1b are involved in excitatory presynaptic differentiation in the hippocampal CA3. To determine whether FGFR2b and FGFR1b compensate for the function of each other for excitatory presynaptic differentiation, we investigated the effect of Fgfr1b inactivation on an Fgfr2b+/− background. We found that Fgfr2b+/−Fgfr1b+/− (double heterozygotes) do not augment the loss of VGLUT1 staining in Fgfr2b+/− mice (Fig. 1I,J), suggesting that there is no apparent redundancy between FGFR2b and FGFR1b on excitatory presynaptic differentiation.
Fig. 1.
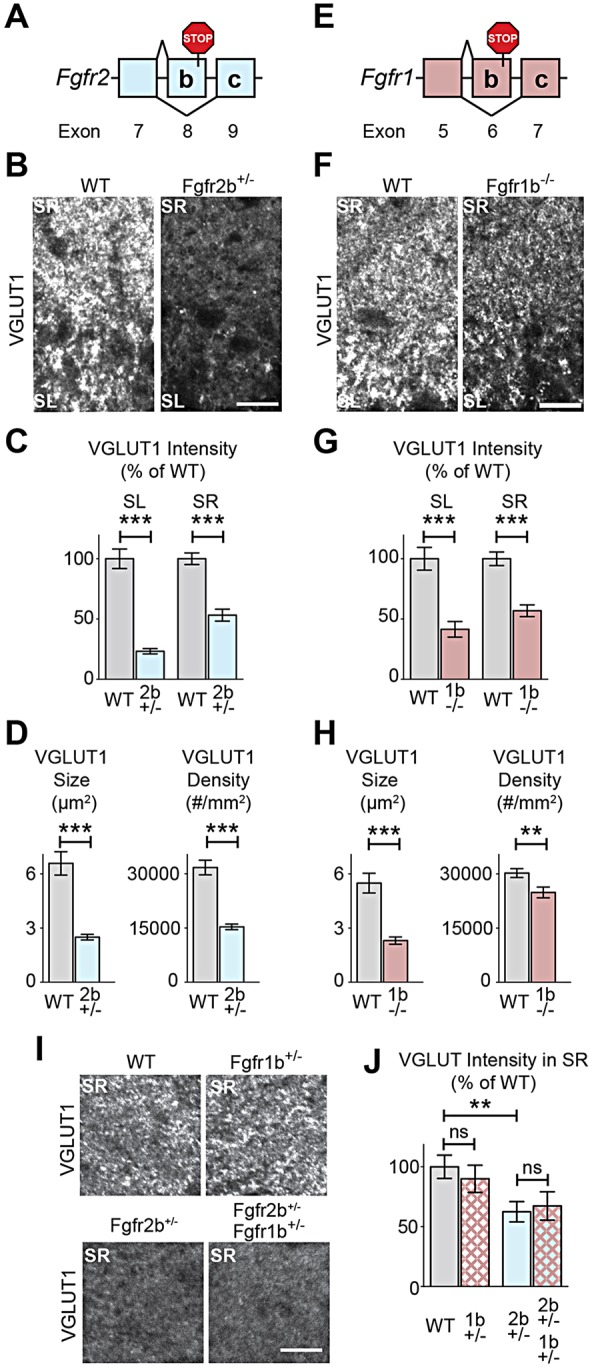
Loss of Fgfr2b or Fgfr1b results in decreased excitatory presynaptic differentiation in hippocampal CA3 in vivo. (A) Schematic of Fgfr2b isoform-specific deletion: stop codon introduced into Fgfr2 exon 8 leads to loss of Fgfr2b, but not -c splice isoforms. (B-D) Decreased excitatory presynaptic differentiation (VGLUT1 immunostaining) in hippocampal CA3 of Fgfr2b+/− mice compared with WT at P8. (B) Representative images. SL, stratum lucidum; SR, stratum radiatum. (C) Quantification of VGLUT1 intensity, normalized to WT littermates. (D) VGLUT1 puncta size and density in SL [n=(sections, animals) 77, 5 Fgfr2b+/−; 55, 3 WT.] (E) Schematic of Fgfr1b isoform-specific deletion: stop codon introduced into Fgfr1 exon 6 leads to loss of FGFR1b, but not -c splice isoforms. (F-H) Decreased excitatory presynaptic differentiation (VGLUT1 immunostaining) in hippocampal CA3 of Fgfr1b−/− mice compared with WT at P8. (F) Representative images. (G) Quantification of VGLUT1 intensity, normalized to WT littermates. (H) VGLUT1 puncta size and density in SL [n=(sections, animals) 76, 7 Fgfr1b−/−; 87, 7 WT.] (I,J) Loss of one copy of Fgfr1b does not exacerbate excitatory presynaptic defects in Fgfr2b+/− mice. (I) Representative images. (J) VGLUT1 intensity in SR, relative to WT littermates [n=(sections, animals) 15, 5 Fgfr2b+/−Fgfr1b+/−; 17, 5 Fgfr2b+/−; 20, 5 Fgfr1b+/−; 24, 6 WT.] ***P<0.001, **P<0.01, *P<0.05, ‘ns’ not significant by Student's t-test. Scale bars: 15 µm.
Inhibitory presynaptic differentiation is impaired in Fgfr2b but not Fgfr1b mutants
We next investigated inhibitory presynaptic differentiation in Fgfr2b+/− and Fgfr1b−/− mice by analyzing vesicular GABA transporter (VGAT) immunostaining to detect the clustering of inhibitory synaptic vesicles (Fig. 2). We found that Fgfr2b+/− mice have decreased VGAT intensity in SL and SR of hippocampal CA3 at P8 (Fig. 2A,B), resembling inhibitory presynaptic defects in Fgf7-KO mice (Terauchi et al., 2010). Further analysis showed that VGAT puncta in Fgfr2b+/− mice are smaller and sparser compared with WT littermates in SL (Fig. 2C). By contrast, Fgfr1b−/− animals do not have defects in inhibitory presynaptic differentiation measured by VGAT intensity, puncta size or density (Fig. 2D-F). Even on an Fgfr2b+/− background, which has quantitatively lower levels of FGFR-mediated signaling, additional loss of a copy of Fgfr1b (double heterozygotes) does not change the level of inhibitory presynaptic defects relative to Fgfr2b+/− (Fig. 2G,H). This is consistent with the notion that synapse formation is not simply regulated by the levels of FGFR signaling, but rather that FGFR2b specifically is required for inhibitory presynaptic differentiation. However, it is still possible that loss of both copies of Fgfr1b on an Fgfr2b+/− background might affect the level of inhibitory presynaptic defects, which might pose an interesting future study. We did not observe appreciable changes in gephyrin immunostaining (an inhibitory postsynaptic marker) in Fgfr2b+/− mice (supplementary material Fig. S2). VGAT immunostaining in lateral septal nuclei does not perceptibly change in Fgfr2b+/− mice (supplementary material Fig. S3). These results suggest that FGFR2b, but not FGFR1b, contributes to inhibitory presynaptic differentiation in hippocampal CA3.
Fig. 2.
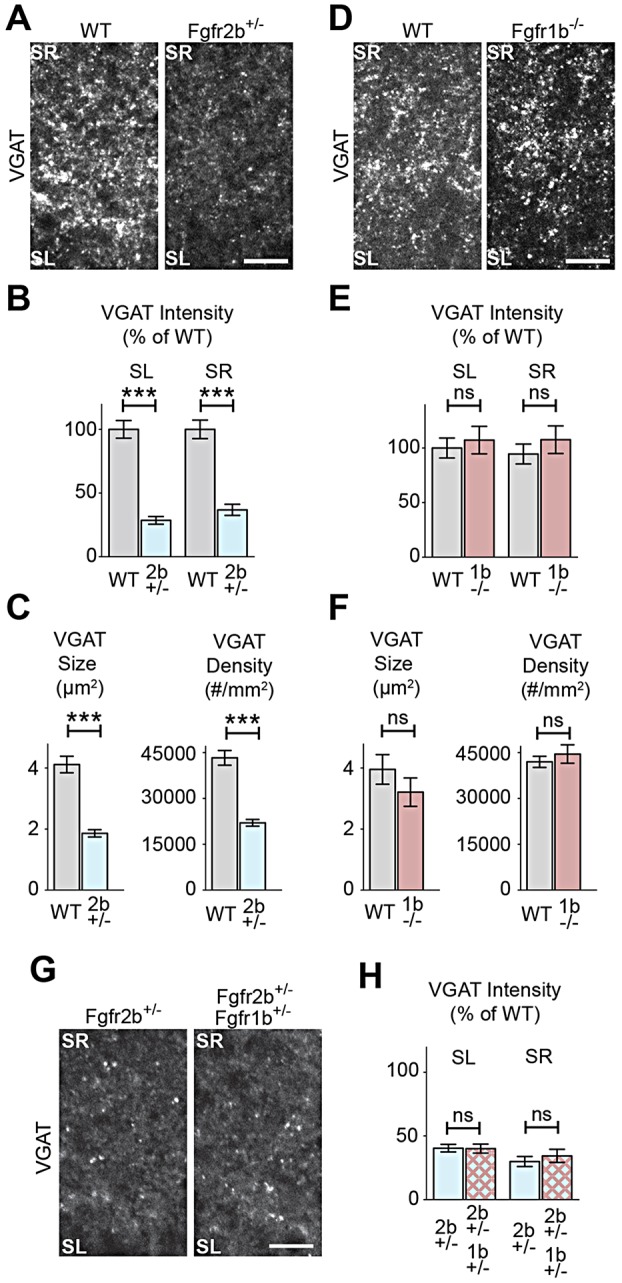
Loss of Fgfr2b, but not Fgfr1b, results in decreased inhibitory presynaptic differentiation in hippocampal CA3 in vivo. (A-C) Decreased inhibitory presynaptic differentiation (VGAT immunostaining) in CA3 of Fgfr2b+/− mice compared with WT at P8. (A) Representative images. (B) Quantification of VGAT intensity, normalized to WT littermates. (C) VGAT puncta size and density in SL [n=(sections, animals) 77, 5 Fgfr2b+/−; 52, 3 WT.] (D-F) No change in inhibitory presynaptic differentiation (VGAT immunostaining) in hippocampal CA3 of Fgfr1b−/− mice compared with WT at P8. (D) Representative images. (E) Quantification of VGAT intensity, normalized to WT littermates. (F) VGAT size and density in SL [n=(sections, animals) 71, 6 Fgfr1b−/−; 71, 6 WT.] (G,H) Loss of one copy of Fgfr1b does not exacerbate inhibitory presynaptic defects in Fgfr2b+/− mice. (G) Representative images. (H) VGAT intensity, relative to WT littermates [n=(sections, animals) 15, 5 Fgfr2b+/−Fgfr1b+/−; 14, 5 Fgfr2b+/−; 12, 4 WT.] Scale bars: 15 µm.
FGFRs are required during the postnatal stage of presynaptic differentiation in vivo
FGFR2 and FGFR1 also regulate embryonic neuronal development, including neuronal precursor specification and proliferation (Paek et al., 2009; Gutin et al., 2006; Stevens et al., 2010b). To exclude the influence of embryonic events to presynaptic defects in Fgfr2b+/− and Fgfr1b−/− mice, we utilized conditional Fgfr-KO mice and tested whether the receptors are necessary during the postnatal synaptogenic stage. We selectively deleted Fgfr2 or Fgfr1 in a temporally controlled manner by crossing mice possessing tamoxifen-inducible Cre (CreER) under the actin promoter (Guo et al., 2002) with mice with a conditional allele of Fgfr2 (Fgfr2flox) (Fig. 3A; Yu et al., 2003) or Fgfr1 (Fgfr1flox) (Fig. 3B; Hoch and Soriano, 2006). Tamoxifen injection (100 µg) efficiently induces CreER-mediated excision of floxed genes (supplementary material Fig. S4). We injected tamoxifen into actin-CreER::Fgfr2flox/flox or actin-CreER::Fgfr1flox/flox pups at P0, resulting in Fgfr2-KO or Fgfr1-KO animals, in which both b and c receptor splice forms are deleted postnatally. Both Fgfr2-KO and Fgfr1-KO animals have decreased excitatory presynaptic differentiation in hippocampal CA3 at P8, as measured by VGLUT1 immunoreactivity, compared with their respective controls (Fig. 3C-E). VGLUT1 puncta are smaller in Fgfr2-KO animals, and both are smaller and sparser in Fgfr1-KO animals (Fig. 3E). However, only Fgfr2-KO, but not Fgfr1-KO animals, have decreased inhibitory presynaptic differentiation at P8, measured by VGAT immunoreactivity (Fig. 3F-H). In Fgfr2-KO, but not Fgfr1-KO animals, VGAT puncta are smaller and sparser (Fig. 3H). Both the excitatory and inhibitory presynaptic defects in Fgfr-KO mice persisted at P14 (supplementary material Fig. S5), the end of initial synapse formation in the hippocampus, suggesting that Fgfr inactivation does not simply delay presynaptic differentiation. Thus, during the postnatal synaptogenic stage, excitatory presynaptic differentiation in hippocampal CA3 requires both FGFR2 and FGFR1, and inhibitory presynaptic differentiation requires FGFR2 but not FGFR1.
Fig. 3.
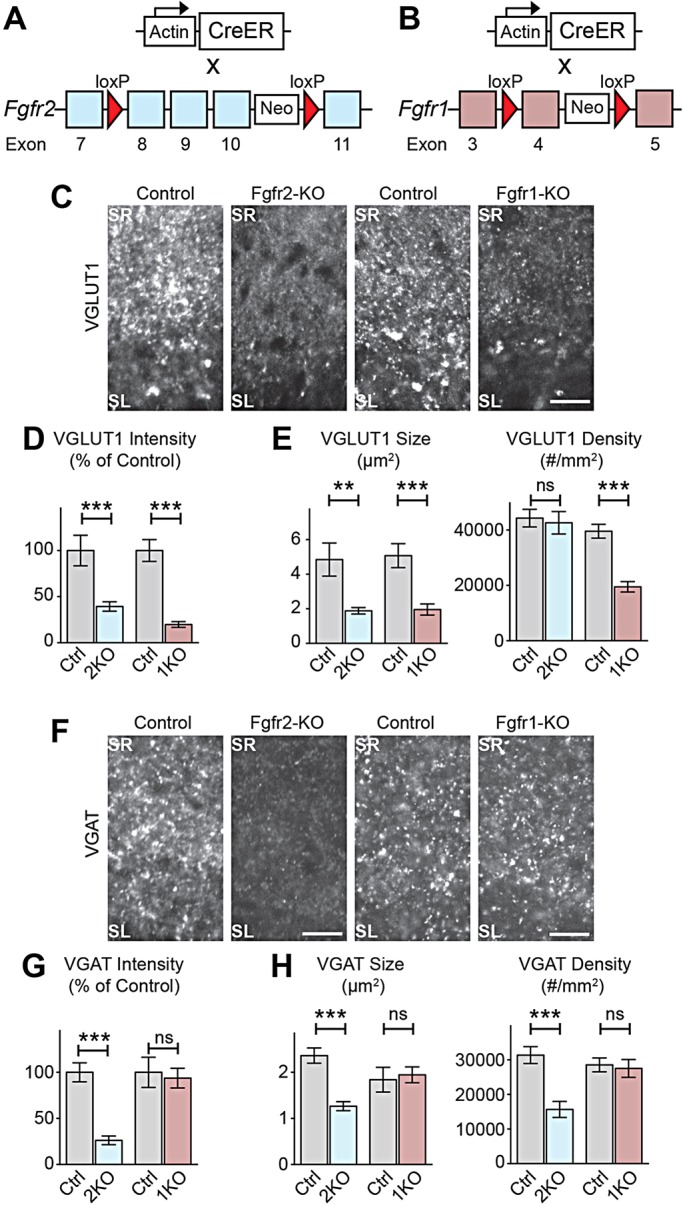
Temporally restricted loss of Fgfr2 and Fgfr1 during the synaptogenic period results in deficiencies in presynaptic differentiation in hippocampal CA3 in vivo. (A,B) Conditional Fgfr-KO strategy: mice carrying actin-promoter-driven CreER were crossed with Fgfr2flox/flox or Fgfr1flox/flox mice. Tamoxifen injection at P0 induced Fgfr deletion (Fgfr2-KO and Ffgr1-KO, respectively) postnatally. Controls: Cre-negative, tamoxifen-injected littermates. (C-E) Decreased excitatory presynaptic differentiation (VGLUT1 immunostaining) in CA3 of Fgfr2-KO and Fgfr1-KO at P8. (C) Representative images. (D) VGLUT1 intensity in SL, normalized to littermate controls. (E) VGLUT1 puncta size and density in SL [n=(sections, animals) 28, 4 Fgfr2-KO; 21, 3 Fgfr2-control; 24, 4 Fgfr1-KO; 24, 4 Fgfr1-control.] (F-H) Decreased inhibitory presynaptic differentiation (VGAT immunostaining) in CA3 of Fgfr2-KO but not Fgfr1-KO at P8. (F) Representative images. (G) VGAT intensity in SL, normalized to littermate controls. (H) VGAT puncta size and density in SL [n=(sections, animals) 34, 5 Fgfr2-KO; 35, 5 Fgfr2-control; 18, 3 Fgfr1-KO; 18, 3 Fgfr1-control.] Scale bars: 15 µm.
FGF22 requires FGFR2b and FGFR1b to induce excitatory presynaptic differentiation
We have shown that FGFR2b and FGFR1b are necessary for excitatory presynaptic differentiation in vivo (Figs 1 and 3). We next tested whether FGFR2b and FGFR1b are receptors for FGF22 to organize excitatory presynaptic terminals in neurons. We cultured hippocampal neurons from Fgfr2b−/− embryos, Fgfr1b−/− P0 pups and their respective WT littermates, treated with FGF22 on day two in vitro (DIV2), and examined response to FGF22 by VGLUT1 staining on DIV14 (Fig. 4A). FGF22 acts on inputs synapsing onto CA3 pyramidal neurons (Terauchi et al., 2010), so we focused our analysis on synapses forming onto CA3 pyramidal neurons. CA3 neurons were identified by staining with the antibody Py (Woodhams et al., 1989), and VGLUT1 puncta contacting Py+ dendrites were quantified. WT neurons respond to FGF22 with a twofold increase in VGLUT1 signal along CA3 dendrites; however, Fgfr2b−/− neurons do not respond to FGF22 (Fig. 4B,C). Closer analysis revealed that the response to FGF22 in WT neurons, but not in Fgfr2b−/− neurons, comprises increases in VGLUT1 puncta density along CA3 dendrites (Fig. 4D) and VGLUT1 puncta size (Fig. 4E). Fgfr1b−/− neurons also do not respond to FGF22 (Fig. 4F-I): there was no FGF22-induced increase in density (Fig. 4H) or size (Fig. 4I) of VGLUT1 puncta. In control experiments examining neuronal cell fate, dendritic morphology of CA3 neurons and their excitatory postsynaptic differentiation, we found no apparent differences between WT neurons, Fgfr2b−/− neurons or Fgfr1b−/− neurons in the presence or absence of FGF22 (supplementary material Figs S6 and S7), suggesting that FGFR2b and FGFR1b primarily affect presynaptic differentiation. Together, these results indicate that both FGFR2b and FGFR1b are required for FGF22-dependent induction of excitatory presynaptic differentiation.
Fig. 4.

Fgfr2b−/− or Fgfr1b−/− neurons do not respond to FGF22 in culture. (A) Experimental scheme. Hippocampal neurons were cultured from Fgfr2b−/− embryos or Fgfr1b−/− pups and WT littermates. FGF22 (2 nM) was applied on DIV2. Neurons were stained at DIV14 against VGLUT1 and Py, a marker of CA3 pyramidal neurons. VGLUT1 puncta contacting CA3 dendrites were analyzed. (B-E) Fgfr2b−/− neurons do not respond to FGF22. (B) Representative images of VGLUT1 puncta (green) contacting CA3 dendrites (red). Contacting puncta are indicated in WT condition with arrowheads. (C) Quantification of FGF22 responsiveness. For each genotype, total intensity of VGLUT1 on CA3 dendrite of FGF22-treated neurons was divided by that of untreated neurons. (D) VGLUT1 density and (E) size along CA3 dendrites [n=(neurons, experiments) 25, 3 WT; 30, 3 WT+FGF22; 34, 3 Fgfr2b−/−; 30, 3 Fgfr2b−/−+FGF22.] (F-I) Fgfr1b−/− neurons do not respond to FGF22. (F) Representative images. (G) VGLUT1 response to FGF22, quantified as in C. (H) VGLUT1 density and (I) size along CA3 dendrites [n=(neurons, experiments) 45, 6 WT; 45, 6 WT+FGF22; 44, 6 Fgfr1b−/−; 44, 6 Fgfr1b−/−+FGF22.] In scatter plots, each dot represents average value from an individual neuron analyzed. Scale bars: 5 µm.
FGFRs are necessary during the synaptogenic stage for FGF22-dependent excitatory presynaptic differentiation
To determine whether FGFRs are required specifically during the synaptogenic stage in vitro, we temporally restricted Fgfr2 and Fgfr1 deletion. Hippocampal neurons were cultured from actin-CreER::Fgfr2flox/flox or actin-CreER::Fgfr1flox/flox mice. On DIV1, 10 nM 4-hydroxytamoxifen (4-OHT) was applied to cultures, generating Fgfr2-KO or Fgfr1-KO neurons. We found that this dose efficiently inactivates Fgfrs (supplementary material Fig. S8). FGF22 was applied on DIV2, and the neurons were assessed on DIV14 for VGLUT1 clustering on Py+ dendrites (Fig. 5A). Control neurons respond to FGF22 with a threefold increase in VGLUT1 immunoreactivity along CA3 dendrites, whereas Fgfr2-KO neurons do not respond (Fig. 5B,C). There were no FGF22 responses in VGLUT1 puncta density (Fig. 5D) and size (Fig. 5E) in Fgfr2-KO neurons. Conversely, Fgfr1-KO neurons have an attenuated, but still present response to FGF22 (Fig. 5F,G). Where control neurons respond to FGF22 with a threefold increase in VGLUT1 signal along CA3 dendrites, Fgfr1-KO neurons respond with a twofold increase (Fig. 5G), with significant increases in VGLUT1 density (Fig. 5H) and size (Fig. 5I). Therefore, we conclude that FGFR2 is required during the synaptogenic stage to respond to FGF22 to induce excitatory presynaptic differentiation, whereas FGFR1 is not absolutely required, but is involved during this stage.
Fig. 5.

Fgfr2-KO neurons, in which receptor deletion is temporally restricted to synaptogenic period, do not respond to FGF22 in culture. (A) Experimental scheme. Hippocampal neurons were cultured from actin-CreER::Fgfr2flox/flox or actin-CreER::Fgfr1flox/flox pups and littermate controls lacking CreER. 10 nM 4-OHT was applied at DIV1 to induce receptor deletion, and FGF22 was applied at DIV2. Neurons were stained at DIV14 against VGLUT1 and Py. VGLUT1 puncta contacting CA3 dendrites were analyzed. (B-E) Fgfr2-KO neurons do not respond to FGF22. (B) Representative images of VGLUT1 puncta (green) contacting CA3 dendrites (red). (C) VGLUT1 response to FGF22, quantified as in Fig. 4C. (D) VGLUT1 density and (E) size along CA3 dendrites [n=(neurons, experiments) 25, 3 control; 25, 3 control+FGF22; 29, 3 Fgfr2-KO; 29, 3 Fgfr2-KO+FGF22.] (F-I) Fgfr1-KO neurons partially respond to FGF22. (F) Representative images. (G) VGLUT1 response to FGF22, quantified as in Fig. 4C. (H) VGLUT1 density and (I) size along CA3 dendrites [n=(neurons, experiments) 35, 4 control; 35, 4 control+FGF22; 40, 4 Fgfr1-KO; 35, 4 Fgfr1-KO+FGF22.] Scale bars: 5 µm.
FGF7 requires FGFR2b but not FGFR1b to induce inhibitory presynaptic differentiation
Fgfr2b+/− mice exhibit defects in inhibitory presynaptic differentiation (Figs 2 and 3), similar to Fgf7-KO mice (Terauchi et al., 2010). We next tested whether FGFR2b is a receptor for FGF7 to organize inhibitory presynaptic terminals. Hippocampal neurons from Fgfr2b−/− embryos, Fgfr1b−/− P0 pups and WT littermates were treated on DIV2 with FGF7, and assessed at DIV14 for VGAT clustering on Py+ dendrites (Fig. 6A). WT neurons respond to FGF7 with a twofold increase in VGAT staining along CA3 dendrites, whereas Fgfr2b−/− neurons do not respond (Fig. 6B-E). In Fgfr2b−/− neurons, FGF7 does not increase VGAT puncta density (Fig. 6D) or size (Fig. 6E). Meanwhile, Fgfr1b−/− neurons respond equally to WT controls with a twofold increase in VGAT intensity along CA3 dendrites (Fig. 6F,G), as well as increases in VGAT density (Fig. 6H) and size (Fig. 6I). We observed no apparent changes in dendritic morphology of CA3 neurons and inhibitory postsynaptic differentiation on CA3 dendrites between WT cultures, Fgfr2b−/− cultures or Fgfr1b−/− cultures, with or without FGF7 (supplementary material Fig. S9). Together, these data suggest that FGFR2b, but not FGFR1b, is required for FGF7-induced inhibitory presynaptic differentiation.
Fig. 6.

Fgfr2b−/− neurons do not, but Fgfr1b−/− neurons do respond to FGF7 in culture. (A) Experimental scheme. Hippocampal neurons were cultured from Fgfr2b−/− embryos or Fgfr1b−/− pups and WT littermates. FGF7 (2 nM) was applied on DIV2. Neurons were stained at DIV14 against VGAT and Py. VGAT puncta contacting CA3 dendrites were analyzed. (B-E) Fgfr2b−/− neurons do not respond to FGF7. (B) Representative images of VGAT puncta (green) contacting CA3 dendrites (red). (C) Quantification of FGF7 responsiveness. For each genotype, total intensity of VGAT on CA3 dendrite of FGF7-treated neurons was divided by that of untreated neurons. (D) VGAT density and (E) size along CA3 dendrites [n=(neurons, experiments) 30, 3 WT; 38, 3 WT+FGF7; 25, 3 Fgfr2b−/−; 25, 3 Fgfr2b−/−+FGF7.] (F-I) Fgfr1b−/− neurons respond to FGF7. (F) Representative images. (G) VGAT response to FGF7, quantified as in C. (H) VGAT density and (I) size along CA3 dendrites [n=(neurons, experiments) 23, 3 WT; 26, 3 WT+FGF7; 24, 3 Fgfr1b−/−; 23, 3 Fgfr1b−/−+FGF7.] Scale bars: 5 µm.
FGFR2 is necessary during the synaptogenic stage in FGF7-dependent inhibitory presynaptic differentiation
To confirm that FGFR2 is required during the synaptogenic stage to respond to FGF7, we temporally restricted Fgfr2 deletion. Actin-CreER::Fgfr2flox/flox hippocampal cultures were treated with 10 nM 4-OHT on DIV1 to induce receptor deletion, FGF7 on DIV2, and assessed on DIV14 for VGAT clustering along Py+ dendrites (Fig. 7A). In response to FGF7, control neurons have a threefold increase in VGAT intensity along CA3 dendrites, whereas Fgfr2-KO neurons do not respond (Fig. 7B,C). Where control neurons respond to FGF7 with increases in VGAT puncta density (Fig. 7D) and size (Fig. 7E), Fgfr2-KO neurons do not. Thus, we conclude that Fgfr2 is required during the synaptogenic stage to respond to FGF7.
Fig. 7.
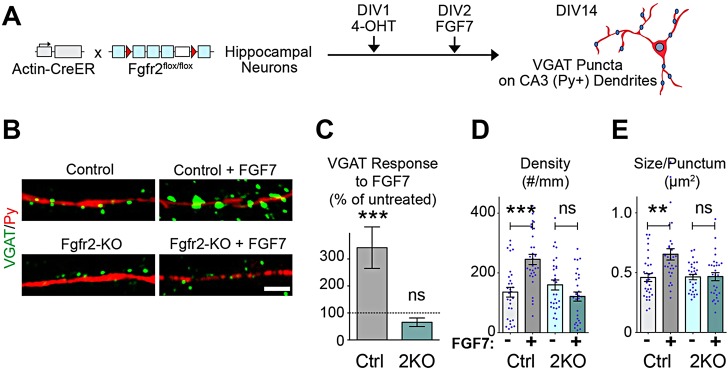
Fgfr2-KO neurons, in which receptor deletion is temporally restricted to synaptogenic period, do not respond to FGF7 in culture. (A) Experimental scheme. Hippocampal neurons were cultured from actin-CreER::Fgfr2flox/flox pups and littermate controls lacking CreER. 4-OHT was applied at DIV1, and FGF7 was applied at DIV2. Neurons were stained for VGAT and Py at DIV14. VGAT puncta contacting CA3 dendrites were analyzed. (B-E) Fgfr2-KO neurons do not respond to FGF7. (B) Representative images of VGAT puncta (green) contacting CA3 dendrites (red). (C) VGAT response to FGF7, quantified as in Fig. 6C. (D) VGAT density and (E) size along CA3 dendrites [n=(neurons, experiments) 30, 3 control; 30, 3 control+FGF7; 29, 3 Fgfr2-KO; 29, 3 Fgfr2-KO+FGF7.] Scale bar: 5 µm.
FGFR2 and FGFR1 localize and function presynaptically to respond to FGF22
As FGF22 and FGF7 are secreted from postsynaptic sites and retrogradely induce differentiation at presynaptic terminals, we hypothesize that FGFR2b and FGFR1b are localized and function presynaptically in the axon. Previous studies reported that FGFR1 and FGFR2 are expressed in axons, dendrites and soma of cultured hippocampal neurons (Li et al., 2002); however, there are no adequate antibodies against FGFR2b or FGFR1b to directly test their endogenous localization. Thus, we used an alternative strategy of overexpressing fluorescently tagged FGFR2b and FGFR1b in cultured neurons to follow their localization (Fig. 8). FGFR2b-GFP and FGFR1b-GFP both localized throughout the cell including the axon, along which FGFRs accumulate in a punctate manner (Fig. 8A,B). In order to demonstrate their localization to presynaptic terminals within the axon, we co-transfected fluorescently tagged synaptophysin, which marks synaptic vesicles (Li and Murthy, 2001; De Paola et al., 2003; Umemori et al., 2004). The majority of FGFR2b-GFP colocalized with synaptophysin-mCherry (Fig. 8C), and the majority of FGFR1b-GFP colocalized with synaptophysin-mCherry (Fig. 8D). This indicates that FGFR2b and FGFR1b can indeed localize to axon terminals, consistent with a presynaptic role.
Fig. 8.
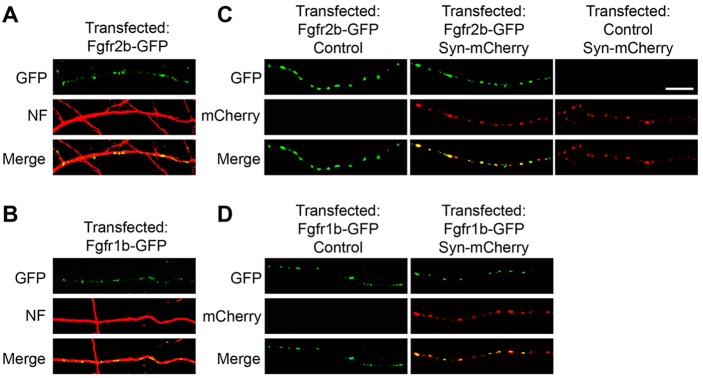
FGFR2b and FGFR1b localize to presynaptic terminals. WT neurons in culture were transfected with GFP-tagged Fgfr constructs. (A,B) FGFR2b-GFP (A) and FGFR1b-GFP (B) localize to neurofilament (NF)+ axons. (C,D) When co-transfected with synaptophysin-mCherry, the majority of FGFR2b-GFP (C) and FGFR1b-GFP (D) puncta colocalize with synaptophysin-mCherry. Scale bar: 10 µm.
We then examined whether the receptors function presynaptically. FGF22 acts on excitatory axon terminals contacting CA3 pyramidal neurons, including those from dentate granule cells (DGCs). Therefore, we first focused on FGF22-induced excitatory presynaptic differentiation within the axons of DGCs. Neuronal cultures prepared from DG and CA3 from Fgfr2flox/flox or Fgfr1flox/flox animals were sparsely co-transfected at DIV1 with Cre, to induce cell-autonomous receptor deletion, and synaptophysin-YFP, to visualize synaptic vesicles. Co-transfection efficiency was nearly 100%, and efficiency of Cre-mediated excision of floxed genes was estimated to be ∼90% (supplementary material Fig. S4). Thus, the Fgfr was deleted in transfected, synaptophysin-YFP+ cells, whereas untransfected cells remained WT. The transfection was very sparse to analyze single cells not contacting other transfected cells (note that synaptophysin-YFP is under the CMV promoter so that we could identify all transfected cells, including astrocytes; see supplementary material Fig. S10), ensuring a cell-autonomous effect. DGCs were identified by staining for the DGC marker Prox1 (Oliver et al., 1993; Williams et al., 2011). Fgfr deletion did not obviously affect DGC axon length (supplementary material Fig. S11). The neurons were treated with FGF22 at DIV2 and fixed for visualization at DIV11 (Fig. 9A,B). Control DGCs (transfected with non-Cre plasmid) responded to FGF22 to induce synaptophysin-YFP clustering, whereas both Fgfr2-deleted and Fgfr1-deleted DGCs failed to respond (Fig. 9C,D), both in terms of synaptophysin-YFP puncta density within the axon (Fig. 9E) or size (Fig. 9F). These data suggest that both FGFR2 and FGFR1 are essential and function in the presynaptic neuron to respond to FGF22.
Fig. 9.

Selective deletion of Fgfr2 or Fgfr1 in dentate granule cells eliminates response to FGF22, and that of Fgfr2 in interneurons eliminates response to FGF7. (A) Experimental scheme. DG and CA3 neurons were cultured from Fgfr2flox/flox or Fgfr1flox/flox pups. At DIV1, neurons were sparsely transfected with Cre to induce Fgfr deletion (Fgfr2-deleted or Fgfr1-deleted) and with synaptophysin-YFP to label synaptic vesicles. Control transfections consisted of synaptophysin-YFP and empty vector without Cre. Neurons were treated with 3 nM FGF22 on DIV2. At DIV11, neurons were stained for Prox1 to identify DGCs. Synaptophysin-YFP puncta in axons from discretely transfected DGCs were analyzed. (B) Example of synaptophysin-YFP-transfected DGC. (C-F) Fgfr2-deleted and Fgfr1-deleted DGCs do not respond to FGF22. (C) Representative images of synaptophysin-YFP in DGC axons (D) Quantification of FGF22 responsiveness, calculated as total synaptophysin-YFP signal per mm of axon in FGF22-treated DGCs divided by that of untreated DGCs. (E) Synaptophysin-YFP density and (F) size within DGC axons [n=(neurons, experiments) 25, 7 control; 40, 7 control+FGF22; 13, 3 Fgfr2-deleted; 18, 3 Fgfr2-deleted+FGF22; 16, 4 Fgfr1-deleted; 17, 4 Fgfr1-deleted+FGF22.] (G) Experimental scheme. DG and CA3 neurons were cultured from Fgfr2flox/flox pups. At DIV1, neurons were sparsely transfected with Cre and synaptophysin-YFP. FGF7 (3 nM) was applied on DIV2. At DIV11, neurons were stained for GABA to identify interneurons. Synaptophysin-YFP puncta in axons from discretely transfected interneurons were analyzed. (H) Example of synaptophysin-YFP-transfected interneuron. (I-L) Fgfr2-deleted interneurons do not respond to FGF7. (I) Representative images of synaptophysin-YFP in interneuron axons. (J) Quantification of FGF7 responsiveness, calculated as total synaptophysin-YFP signal per mm of axon in FGF7-treated interneurons divided by untreated interneurons. (K) Synaptophysin-YFP density and (L) size within interneuron axons [n=(neurons, experiments) 10, 3 control; 15, 3 control+FGF7; 12, 3 Fgfr2-deleted; 16, 3 Fgfr2-deleted+FGF22.] Scale bars: 100 µm (in B,H); 10 µm (in C,I).
Next, we studied the functional presynaptic requirement of FGFR2 at inhibitory synapses in response to FGF7. To do this, we performed a similar experiment as described above, but treated with FGF7 and identified transfected interneurons using GABA immunoreactivity (Fig. 9G,H). We found that control interneurons respond to FGF7 to induce synaptophysin-YFP clustering both in terms of density and size, whereas Fgfr2-KO interneurons failed to respond (Fig. 9I-L). These data suggest that Fgfr2 functions in the presynaptic interneuron to respond to FGF7.
Because each coverslip in our experiment contained more than one Cre/synaptophysin-YFP-transfected (i.e. Fgfr-KO) cell, we cannot completely exclude the possibility that other Fgfr-KO cells on the coverslip indirectly affected synaptophysin-YFP clustering in the neuron we examined. However, because only ∼0.1% of cells were transfected (supplementary material Fig. S10), untransfected WT cells would probably mask the indirect effect of sparse receptor deletion. Thus, our data suggest that FGFRs function directly in presynaptic neurons.
FGFR2b requires kinase activity, and FRS2 and PI3K binding to respond to FGF22
Our experiments show that FGFR2 is required in the presynaptic neuron to respond to FGF22. We next asked whether kinase activity is required for and which intracellular signaling pathways are involved in FGF22-dependent presynaptic differentiation. FGFRs belong to the receptor tyrosine kinase family of proteins and have an intracellular kinase domain, which becomes activated after ligand-induced receptor dimerization, triggering a multi-step autophosphorylation cascade (Mohammadi et al., 1996; Furdui et al., 2006; Bae et al., 2009; Lemmon and Schlessinger, 2010). Phosphorylation sites within the intracellular domain also provide docking sites for adaptor proteins, such as FRS2, PI3K and PLCγ, which activate downstream signaling pathways, such as the MAPK/ERK and AKT by FRS2, AKT by PI3K, or IP3/DAG/Ca2+ by PLCγ (Mohammadi et al., 1992, 1996; Burgar et al., 2002; Hart et al., 2001; Hatch et al., 2006; Songyang et al., 1993; Hadari et al., 1998; Ong et al., 2001). In order to study signaling pathways specifically downstream of FGFR2b, we created Fgfr2b mutant constructs to perturb specific downstream signaling (Fig. 10A): a kinase dead (KD) mutant and mutants lacking the FRS2, the PI3K or the PLCγ binding site. We examined whether the mutated FGFR2b constructs can restore FGF22-induced synaptophysin-YFP response in Fgfr2-deleted DGCs. We co-transfected Fgfr2b constructs with Cre and synaptophysin-YFP into DG + CA3 Fgfr2flox/flox neuronal cultures at DIV1, treated with FGF22 at DIV2, and assessed synaptophysin-YFP clustering at DIV11 (Fig. 10B). As in Fig. 9, DGCs transfected with non-Cre plasmid (control) responded to FGF22, whereas Fgfr2-deleted DGCs did not (Fig. 10C,D). Expression of FGFR2b-WT in Fgfr2-deleted DGCs rescued FGF22-responsiveness, but expression of FGFR2b-KD, FGFR2b-FRS2-deficient or FGFR2b-PI3K-deficient mutants did not. Interestingly, FGFR2b-PLCγ-deficient rescued FGF22-responsiveness (Fig. 10D), although only the rescue of synaptophysin-YFP density (Fig. 10E) and not size (Fig. 10F) was statistically significant. These data indicate that kinase activity of FGFR2b is required to respond to FGF22, and that FGFR2b requires signaling downstream of FRS2 and PI3K to induce excitatory presynaptic differentiation in response to FGF22.
Fig. 10.

FRS2 and PI3K signaling is required downstream of FGFR2b for presynaptic response to FGF22. (A) Schematic of FGFR2b mutants. FGFR2b-KD: Y561E and Y562E mutations to inactivate kinase activity. FGFR2b-FRS2-deficient: V332 and T333 deletion to remove FRS2 binding site. FGFR2b-PI3K-deficient: Y638A mutation to remove PI3K binding site. FGFR2b-PLCγ-deficient: Y674A mutation to remove PLCγ binding site. (B) Experimental scheme. DG and CA3 neurons were cultured from Fgfr2flox/flox pups. At DIV1, neurons were sparsely transfected with Cre, Fgfr2b constructs and synaptophysin-YFP. Empty vector was used as a control. Neurons were treated with FGF22 at DIV2 and stained at DIV11 for Prox1 to identify DGCs. Synaptophysin-YFP puncta in axons from discretely transfected DGCs were analyzed. (C-F) Expression of FGFR2b-WT or FGFR2b-PLCγ-deficient, but not FGFR2b-KD, FGFR2b-FRS2-deficient or FGFR2b-PI3K-deficient restores FGF22 responsiveness in Fgfr2-deleted DGCs. (C) Representative images of synaptophysin-YFP in DGC axons. (D) Synaptophysin-YFP response to FGF22, quantified as in Fig. 9D. (E) Synaptophysin-YFP density and (F) size within DGC axons. [n=(neurons, experiments) 7, 3 control; 7, 3 control+FGF22; 6, 3 Fgfr2-deleted; 10, 3 Fgfr2-deleted+FGF22; 13, 3 FGFR2b-WT; 18, 3 FGFR2b-WT+FGF22; 8, 3 FGFR2b-KD; 7, 3 FGFR2b-KD+FGF22; 9, 3 FGFR2b-FRS2-deficient; 13, 3 FGFR2b-FRS2-deficient+FGF22; 12, 3 FGFR2b-PI3K-deficient; 13, 3 FGFR2b-PI3K-deficient+FGF22; 13, 3 FGFR2b-PLCγ-deficient; 13, 3 FGFR2b-PLCγ-deficient+FGF22]. Scale bar: 10 µm.
DISCUSSION
Numerous synaptogenic molecules, such as FGFs, Wnts, neurotrophins, Eph/ephrins, neurexins/neuroligins and leucine-rich repeat transmembrane proteins (LRRTMs), have been identified (Dai and Peng, 1995; Umemori et al., 2004; Terauchi et al., 2010; de Wit et al., 2011; Dickins and Salinas, 2013; Park and Poo, 2013; Siddiqui and Craig, 2011; Xu and Henkemeyer, 2012), but the underlying mechanisms through which they organize presynaptic differentiation are largely unknown. In this study, we demonstrated, using both in vivo and in vitro evidence, that differential use of FGFRs by FGF22 and FGF7 contributes to their presynaptic effects on excitatory and inhibitory synapses in the CA3 of the hippocampus, where the receptors are localized presynaptically in DGCs and interneurons, and that FGFR2b utilizes FRS2 and PI3K signaling to promote synaptic vesicle accumulation. Together, we provide novel insights into mechanisms of excitatory and inhibitory presynaptic differentiation in the mammalian hippocampus that are crucial for proper brain function.
The coordinated development of excitatory and inhibitory synapses in the hippocampal CA3 is guided by FGF22 and FGF7 with remarkable specificity. After the fate of excitatory and inhibitory neurons has been specified, and as their axons reach CA3, FGF22 and FGF7 are secreted from CA3 dendrites at discrete excitatory and inhibitory postsynaptic sites, respectively, to promote presynaptic differentiation of each type of synapses (Terauchi et al., 2010, 2015). We found that, in addition to specific localization of ligand secretion, overlapping but distinct sets of FGFRs are required for the differentiation of excitatory or inhibitory presynaptic terminals, providing robustness to the system. FGF22 and FGF7 both activate FGFR2b, but we propose that FGFR1b plays an important role in coordinating specificity. FGF22, but not FGF7, activates FGFR1b in in vitro suspension cell proliferation assays (Zhang et al., 2006), and we found that, in the hippocampus, FGFR1b is required for excitatory synapse formation in response to FGF22 (Fig. 4), but not for inhibitory synapse formation in response to FGF7 (Fig. 6). Consistent with this specificity, Fgfr2b-lacking mice have excitatory and inhibitory presynaptic deficits, and Fgfr1b-lacking mice have only excitatory presynaptic deficits (Figs 1-3). The receptor KO mice do not completely lose excitatory or inhibitory presynaptic differentiation, which suggests that presynaptic differentiation is not fully dependent on FGF7 and FGF22. It is likely that other FGFs, FGFRs and/or other synaptogenic molecules contribute to and coordinate presynaptic differentiation. What is the relationship between FGFR2b and FGFR1b in promoting excitatory presynaptic differentiation in response to FGF22? Both FGFR2b and FGFR1b appear to be necessary presynaptically for responsiveness to FGF22 in vitro (Fig. 9); however, they do not appear to be redundant in terms of their effect on excitatory presynaptic differentiation (Fig. 1I,J). One possibility is that FGFR2b and FGFR1b form heterodimers. Under conditions of complete ligand absence, FGFR2 and FGFR1 can form heterodimers (Wang et al., 1997) and the ligand can induce transphosphorylation (Bellot et al., 1991), although it has not yet been demonstrated whether the native forms do so during development. Alternatively, they cooperate as independent homodimers to promote presynaptic differentiation, in which FGFR2b is the dominant receptor required for both excitatory and inhibitory presynaptic differentiation, whereas FGFR1b specifically modulates excitatory presynaptic differentiation in response to FGF22. Interestingly, FGFR1b appears to have a modulatory role in relation to FGFR2b in other systems. For example, FGF10, which, like FGF22, activates both FGFR2b and FGFR1b (Zhang et al., 2006), plays an important role in lung and submandibular gland development. In both systems, FGFR2b promotes glandular proliferation and elongation and induces FGFR1b expression at the end buds, whereas FGFR1b is important specifically for end bud expansion (Steinberg et al., 2005; Patel et al., 2008). Identification of the precise roles of FGFR1b in excitatory presynaptic differentiation is an important next question.
We found that FGFR2b requires kinase activity as well as binding of FRS2 and PI3K for synaptic vesicle recruitment in response to FGF22 (Fig. 10). All three are upstream of AKT signaling, and because the effect of mutating any of these three pathways had a similar effect in our experiments, it is reasonable to speculate that AKT signaling is involved in excitatory presynaptic differentiation downstream of FGFR2b. However, phosphorylation of FRS2 by FGFRs activates not only the PI3K/AKT pathway (through Grb2/Gab1 binding; Ong et al., 2001), but also the MAPK/ERK pathway (through Grb2/Shp2 binding; Hadari et al., 1998). Indeed, deletion of FGFR1 and/or FGFR2 leads to decreases in phospho-MAPK/ERK and phospho-AKT levels (Emmenegger et al., 2013; Zhao et al., 2008; Cai et al., 2013; Hoch and Soriano, 2006; Loilome et al., 2009; Kondo et al., 2007), and overexpression of FGFR2b or FGFR1 increases both phospho-AKT and phospho-ERK levels (Cha et al., 2008; Freeman et al., 2003; Acevedo et al., 2007). It is known that crosstalk can occur between the PI3K/AKT and MAPK/ERK pathways (Moelling et al., 2002), thus, possibly, the MAPK/ERK pathway is being triggered congruently. Indeed, a previous study linked FGF2-induced synaptogenic activity to the MAPK/ERK pathway: application of MAPK/ERK-specific inhibitors blocked the synaptogenic effect of FGF2 (Li et al., 2002). Finally, recent evidence suggests a role for WAVE regulatory complex (WRC) in the regulation of actin cytoskeletal dynamics downstream of synaptic cell adhesion molecules (Chia et al., 2014; Chen et al., 2014). Interestingly, FRS2 contains a putative WRC-binding consensus sequence (Chen et al., 2014), and PI3K can contribute to actin cytoskeletal dynamics (Cain and Ridley, 2009). An interesting possibility is that FGFR2b-FRS2/PI3K signaling might be controlling the local actin environment at the presynaptic terminal for presynaptic differentiation at the site of activation. Finally, two interesting questions remain: Does FGFR1b act through the same or distinct signaling pathways to control excitatory presynaptic differentiation? And do distinct signaling pathways contribute to FGFR-dependent excitatory and inhibitory presynaptic differentiation?
Unbalanced excitation-inhibition has behavioral consequences: Fgf22-KO and Fgf7-KO mice have altered epileptic seizure susceptibility (Terauchi et al., 2010; Lee et al., 2012; Lee and Umemori, 2013). Additionally, other FGFs have also been implicated in epilepsy (Paradiso et al., 2013). What are the behavioral, neurological or psychiatric consequences of losing FGFR2b or FGFR1b signaling in humans? Dominant negative mutations and mutations leading to overactivation in FGFR2 and FGFR1 cause craniosynostosis syndromes, dominated by cranial defects, which also include seizures and intellectual disability (Agochukwu et al., 2012; Melville et al., 2010; Stevens et al., 2010a; Williams and Umemori, 2014). Analysis of single-nucleotide polymorphisms in human patients has linked mutations in FGFR2 to susceptibility to schizophrenia (O'Donovan et al., 2009) and bipolar disorder (Wang et al., 2012), and FGFR1 mutations to susceptibility to depression (Gaughran et al., 2006) and schizophrenia (Shi et al., 2011). These analyses do not differentiate between the isoform-specific contributions of the receptors; however, all of this evidence supports the idea that FGFR signaling is crucial for proper neural development, and FGFRs might prove to be important druggable therapeutic targets in neurological diseases.
MATERIALS AND METHODS
Mouse strains
Fgfr1b mutant mice were from Juha Partanen (University of Helsinki, Finland) (Partanen et al., 1998). Fgfr2b mutant mice (Fox et al., 2007), Fgfr2flox/flox mice (Yu et al., 2003; Umemori et al., 2004), Fgfr1flox/flox mice (Hoch and Soriano, 2006) and actin-CreER mice (Guo et al., 2002; Umemori et al., 2004) were described previously. All mice were on a C57BL/6 background, except Fgfr1b−/− mice, which were on a mixed 129sv/CD-1 background. WT mice used were either C57BL/6 or ICR/CD-1. All animal care and use was in accordance with the institutional guidelines and approved by the Institutional Animal Care and Use Committees at Boston Children's Hospital and University of Michigan.
Neuronal culture
For FGF responsiveness assays (Figs 4-7), hippocampi were dissected from P0 pups or E18-E19 embryos, dissociated with 0.5% trypsin and grown in culture media [B27 (Gibco), 2 mM L-glutamine, 1× penicillin-streptomycin in neurobasal medium (Gibco)] at a density of 36,000 cells/poly-D-lysine-coated coverslip. For DGC transfections (Figs 8-10), dissociated DG and CA3 cells were plated at a density of 50,000 cells/coverslip, and culture media was supplemented with 5 mM KCl. 1 μg of plasmids per coverslip were transfected using a CalPhos transfection kit (Clontech). FGF22 (R&D Systems) and FGF7 (PeproTech) were dissolved to 2 nM for responsiveness experiments and 3 nM for synaptophysin-YFP experiments.
Plasmids
To generate Fgfr1b-GFP, the coding region of Fgfr1b cDNA was PCR-amplified from neonatal mouse skin cDNA and fused with GFP in pEGFP-N1 (Clontech). To generate Fgfr2b-EGFP, the coding region of Fgfr2b cDNA from IMAGE clone 5349249 (ATCC) was inserted into pcDNA3.1 with EGFP cDNA.
Fgfr2b mutant constructs were generated using PCR amplification and insertion into the pAP-TAG5 plasmid (GenHunter) using the following combinations of primers: 5′-ATACTAGTCATGGGATTACCGTCC-3′ (primer-N) with 5′-ATGGGCCCTCATGTTTTAACACTGC-CG3′ (primer-C) for WT Fgfr2b; primer-N with 5′-ATGTCGACTTC-CAGTCAAGTGGATGGCTCC-3′ and 5′-ATGTCGACCATTTGTGGTC-TTTTTTTCTTCGTCTATGTTGTTG-3′ with primer-C for Fgfr2b-KD; primer-N with 5′-ATCTGCAGAGTCCAGCTCCTCCA-TGAAC-3′ and 5′-ATCTGCAGAAACCTGTCTCCGCAGGGGG-3′ with primer-C for Fgfr2b-FRS2; primer-N with 5′-ATGAGCTAGCCATGATG-ATGAG-3′ and 5′-TCATGGCTAGCTCATTGGTGC-3′ with primer-C for Fgfr2b-PI3K; and primer-N with 5′-ATAAGCTTTGGATCTCACCC-AGCCTC-3′ and 5′-ATAAAGCTTCC TCATTGGTTGTGAGAG-3′ with primer-C for Fgfr2b-PLCγ. All PCR products were verified by sequencing.
Immunostaining
P8 animals were sacrificed by decapitation (Figs 1 and 2) or perfusion with 4% PFA (Fig. 3). Brains were fixed in 4% PFA overnight. Sagittal sections (20 μm) were cryosectioned. Cultured neurons were fixed with 100% methanol, at −20°C for 5 min. The following antibodies and dilutions were used: anti-VGLUT1 (Millipore, AB5905; 1:4000), anti-VGAT (Synaptic Systems, 131003; 1:4000), anti-GFP (Aves, 1020; 1:5000), anti-Prox1 (Millipore, MAB5652; 1:500) and antibody Py [1:25, a gift from M. Webb (Johnson & Johnson) and P. L. Woodhams (GlaxoSmithKline)]. Glycerol with p-phenylenediamine or n-propyl gallate was used as mounting medium.
Image acquisition and analysis
Twelve-bit images were acquired with epifluorescence microscopes (Olympus) using 20× (Figs 1-3) and 40× lenses (Figs 4-10) with F-View II CCD (Soft Imaging System) and XM10 (Olympus) cameras at 1376×1032 pixels resolution.
Section data were analyzed using MetaMorph software. For each image, a threshold was chosen to exclude signal from background, based on intensity of the fimbria, a myelinated tract of axons exiting CA3 medially.
For analysis of VGLUT1 or VGAT puncta on Py+ dendrites (Figs 4-7), images were merged using ImageJ (NIH) and Adobe Photoshop in 16-bit, and masks were drawn in Adobe Photoshop over Py+ dendrites. Image thresholds were chosen to subtract background staining and separate each punctum. The threshold was then subtracted from the intensity measurement. Dendritic lengths were measured manually by tracing the length in MetaMorph.
Synaptophysin-YFP images (Figs 9 and 10) were analyzed using Fiji software package. The entire axonal arbor was imaged and analyzed. Images were processed to exclude dendrites and converted to 8-bit. For each neuron, a threshold was chosen separately to capture the dimmest punctum and remove diffuse background axonal staining. Masks were applied to images, and puncta size and count were calculated in Fiji. Axon length was traced manually in Fiji.
The statistical tests performed were two-tailed Student's t-tests. All data are expressed as mean±s.e.m.
Supplementary Material
Acknowledgements
We thank Masahiro Yasuda, Erin Johnson-Venkatesh and Sivapratha Nagappan Chettiar for critical reading of the manuscript; Mei Zhang, Andrew McNamara, Patricia Yee, Hsin-Lan Wen and Noreen Francis for technical assistance.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
H.U. and A.D. designed the experiments and prepared the manuscript; A.D., A.T. and C.S. performed the experiments; H.U. supervised the project; all authors analyzed data and commented on the manuscript.
Funding
This work was supported by the National Institutes of Health [grant NS070005 to H.U.]. A.D. was also supported by the University of Michigan Organogenesis Program Training Grant T32 HD007505 and Medical Scientist Training Program Training Grant T32 GM07863. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.115568/-/DC1
References
- Acevedo V. D., Gangula R. D., Freeman K. W., Li R., Zhang Y., Wang F., Ayala G. E., Peterson L. E., Ittmann M. and Spencer D. M. (2007). Inducible FGFR-1 activation leads to irreversible prostate adenocarcinoma and an epithelial-to-mesenchymal transition. Cancer Cell 12, 559-571. 10.1016/j.ccr.2007.11.004 [DOI] [PubMed] [Google Scholar]
- Agochukwu N. B., Solomon B. D. and Muenke M. (2012). Impact of genetics on the diagnosis and clinical management of syndromic craniosynostoses. Childs Nerv. Syst. 28, 1447-1463. 10.1007/s00381-012-1756-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae J. H., Lew E. D., Yuzawa S., Tomé F., Lax I. and Schlessinger J. (2009). The selectivity of receptor tyrosine kinase signaling is controlled by a secondary SH2 domain binding site. Cell 138, 514-524. 10.1016/j.cell.2009.05.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beer H.-D., Vindevoghel L., Gait M. J., Revest J.-M., Duan D. R., Mason I., Dickson C. and Werner S. (2000). Fibroblast growth factor (FGF) receptor 1-IIIb is a naturally occurring functional receptor for FGFs that is preferentially expressed in the skin and the brain. J. Biol. Chem. 275, 16091-16097. 10.1074/jbc.275.21.16091 [DOI] [PubMed] [Google Scholar]
- Bellot F., Crumley G., Kaplow J. M., Schlessinger J., Jaye M. and Dionne C. A. (1991). Ligand-induced transphosphorylation between different FGF receptors. EMBO J. 10, 2849-2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belov A. A. and Mohammadi M. (2013). Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb. Perspect. Biol. 5, a015958 10.1101/cshperspect.a015958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgar H. R., Burns H. D., Elsden J. L., Lalioti M. D. and Heath J. K. (2002). Association of the signaling adaptor FRS2 with fibroblast growth factor receptor 1 (Fgfr1) is mediated by alternative splicing of the juxtamembrane domain. J. Biol. Chem. 277, 4018-4023. 10.1074/jbc.M107785200 [DOI] [PubMed] [Google Scholar]
- Cai Z., Tao C., Li H., Ladher R., Gotoh N., Feng G.-S., Wang F. and Zhang X. (2013). Deficient FGF signaling causes optic nerve dysgenesis and ocular coloboma. Development 140, 2711-2723. 10.1242/dev.089987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cain R. J. and Ridley A. J. (2009). Phosphoinositide 3-kinases in cell migration. Biol. Cell. 101, 13-29. 10.1042/BC20080079 [DOI] [PubMed] [Google Scholar]
- Cha J. Y., Lambert Q. T., Reuther G. W. and Der C. J. (2008). Involvement of fibroblast growth factor receptor 2 isoform switching in mammary oncogenesis. Mol. Cancer Res. 6, 435-445. 10.1158/1541-7786.MCR-07-0187 [DOI] [PubMed] [Google Scholar]
- Chellaiah A. T., McEwen D. G., Werner S., Xu J. and Ornitz D. M. (1994). Fibroblast growth factor receptor (FGFR) 3. Alternative splicing in immunoglobulin-like domain III creates a receptor highly specific for acidic FGF/FGF-1. J. Biol. Chem. 269, 11620-11627. [PubMed] [Google Scholar]
- Chen B., Brinkmann K., Chen Z., Pak C. W., Liao Y., Shi S., Henry L., Grishin N. V., Bogdan S. and Rosen M. K. (2014). The WAVE regulatory complex links diverse receptors to the actin cytoskeleton. Cell 156, 195-207. 10.1016/j.cell.2013.11.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia P. H., Li P. and Shen K. (2013). Cell biology in neuroscience: cellular and molecular mechanisms underlying presynapse formation. J. Cell Biol. 203, 11-22. 10.1083/jcb.201307020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia P. H., Chen B., Li P., Rosen M. K. and Shen K. (2014). Local f-actin network links synapse formation and axon branching. Cell 156, 208-220. 10.1016/j.cell.2013.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuesto G., Enriquez-Barreto L., Caramés C., Cantarero M., Gasull X., Sandi C., Ferrús A., Acebes Á. and Morales M. (2011). Phosphoinositide-3-kinase activation controls synaptogenesis and spinogenesis in hippocampal neurons. J. Neurosci. 31, 2721-2733. 10.1523/JNEUROSCI.4477-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dabrowski A. and Umemori H. (2011). Orchestrating the synaptic network by tyrosine phosphorylation signalling. J. Biochem. 149, 641-653. 10.1093/jb/mvr047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z. and Peng H. B. (1995). Presynaptic differentiation induced in cultured neurons by local application of basic fibroblast growth factor. J. Neurosci. 15, 5466-5475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paola V., Arber S. and Caroni P. (2003). AMPA receptors regulate dynamic equilibrium of presynaptic terminals in mature hippocampal networks. Nat. Neurosci. 6, 491-500. [DOI] [PubMed] [Google Scholar]
- de Wit J., Hong W., Luo L. and Ghosh A. (2011). Role of leucine-rich repeat proteins in the development and function of neural circuits. Annu. Rev. Cell Dev. Biol. 27, 697-729. 10.1146/annurev-cellbio-092910-154111 [DOI] [PubMed] [Google Scholar]
- Dickins E. M. and Salinas P. C. (2013). Wnts in action: from synapse formation to synaptic maintenance. Front. Cell. Neurosci. 7, 162 10.3389/fncel.2013.00162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert D. H. and Greenberg M. E. (2013). Activity-dependent neuronal signalling and autism spectrum disorder. Nature 493, 327-337. 10.1038/nature11860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmenegger B. A., Hwang E. I., Moore C., Markant S. L., Brun S. N., Dutton J. W., Read T.-A., Fogarty M. P., Singh A. R., Durden D. L. et al. (2013). Distinct roles for fibroblast growth factor signaling in cerebellar development and medulloblastoma. Oncogene 32, 4181-4188. 10.1038/onc.2012.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox M. A., Sanes J. R., Borza D.-B., Eswarakumar V. P., Fässler R., Hudson B. G., John S. W. M., Ninomiya Y., Pedchenko V., Pfaff S. L. et al. (2007). Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 129, 179-193. 10.1016/j.cell.2007.02.035 [DOI] [PubMed] [Google Scholar]
- Freeman K. W., Welm B. E., Gangula R. D., Rosen J. M., Ittamnn M., Greenberg N. M. and Spencer D. M. (2003). Inducible prostate intraepithelial neoplasia with reversible hyperplasia in conditional FGFR1-expressing mice. Cancer Res. 63, 8256-8263. [PubMed] [Google Scholar]
- Furdui C. M., Lew E. D., Schlessinger J. and Anderson K. S. (2006). Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol. Cell. 21, 711-717. 10.1016/j.molcel.2006.01.022 [DOI] [PubMed] [Google Scholar]
- Gaughran F., Payne J., Sedgwick P. M., Cotter D. and Berry M. (2006). Hippocampal FGF-2 and FGFR1 mRNA expression in major depression, schizophrenia and bipolar disorder. Brain Res. Bull. 70, 221-227. 10.1016/j.brainresbull.2006.04.008 [DOI] [PubMed] [Google Scholar]
- Giachello C. N. G., Fiumara F., Giacomini C., Corradi A., Milanese C., Ghirardi M., Benfenati F. and Montarolo P. G. (2010). MAPK/Erk-dependent phosphorylation of synapsin mediates formation of functional synapses and short-term homosynaptic plasticity. J. Cell. Sci. 123, 881-893. 10.1242/jcs.056846 [DOI] [PubMed] [Google Scholar]
- Guillemot F. and Zimmer C. (2011). From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71, 574-588. 10.1016/j.neuron.2011.08.002 [DOI] [PubMed] [Google Scholar]
- Guo C., Yang W. and Lobe C. G. (2002). A Cre recombinase transgene with mosaic, widespread tamoxifen-inducible action. Genesis 32, 8-18. 10.1002/gene.10021 [DOI] [PubMed] [Google Scholar]
- Gutin G., Fernandes M., Palazzolo L., Paek H., Yu K., Ornitz D. M., McConnell S. K. and Hébert J. M. (2006). FGF signalling generates ventral telencephalic cells independently of SHH. Development 133, 2937-2946. 10.1242/dev.02465 [DOI] [PubMed] [Google Scholar]
- Hadari Y. R., Kouhara H., Lax I. and Schlessinger J. (1998). Binding of Shp2 tyrosine phosphatase to FRS2 is essential for fibroblast growth factor-induced PC12 cell differentiation. Mol. Cell. Biol. 18, 3966-3973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenston A. M. and Bading H. (2011). Calcium signaling in synapse-to-nucleus communication. Cold Spring Harb. Perspect. Biol. 3, a004564 10.1101/cshperspect.a004564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart K. C., Robertson S. C. and Donoghue D. J. (2001). Identification of tyrosine residues in constitutively activated fibroblast growth factor receptor 3 involved in mitogenesis, Stat activation, and phosphatidylinositol 3-kinase activation. Mol. Biol. Cell 12, 931-942. 10.1091/mbc.12.4.931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatch N. E., Hudson M., Seto M. L., Cunningham M. L. and Bothwell M. (2006). Intracellular retention, degradation, and signaling of glycosylation-deficient FGFR2 and craniosynostosis syndrome-associated FGFR2C278F. J. Biol. Chem. 281, 27292-27305. 10.1074/jbc.M600448200 [DOI] [PubMed] [Google Scholar]
- Hoch R. V. and Soriano P. (2006). Context-specific requirements for Fgfr1 signaling through Frs2 and Frs3 during mouse development. Development 133, 663-673. 10.1242/dev.02242 [DOI] [PubMed] [Google Scholar]
- Jaskoll T., Abichaker G., Witcher D., Sala F. G., Bellusci S., Hajihosseini M. K. and Melnick M. (2005). FGF10/FGFR2b signaling plays essential roles during in vivo embryonic submandibular salivary gland morphogenesis. BMC Dev. Biol. 5, 11 10.1186/1471-213X-5-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y. and Garner C. C. (2008). Molecular mechanisms of presynaptic differentiation. Annu. Rev. Cell Dev. Biol. 24, 237-262. 10.1146/annurev.cellbio.23.090506.123417 [DOI] [PubMed] [Google Scholar]
- Johnson D. E., Lu J., Chen H., Werner S. and Williams L. T. (1991). The human fibroblast growth factor receptor genes: a common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol. Cell. Biol. 11, 4627-4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson-Venkatesh E. M. and Umemori H. (2010). Secreted factors as synaptic organizers. Eur. J. Neurosci. 32, 181-190. 10.1111/j.1460-9568.2010.07338.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K. and Basson M. A. (2010). FGF ligands emerge as potential specifiers of synaptic identity. Cellscience 7, 33-42. 10.1111/j.1460-9568.2010.07338.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T., Zheng L., Liu W., Kurebayashi J., Asa S. L. and Ezzat S. (2007). Epigenetically controlled fibroblast growth factor receptor 2 signaling imposes on the RAS/BRAF/Mitogen-activated protein kinase pathway to modulate thyroid cancer progression. Cancer Res. 67, 5461-5470. 10.1158/0008-5472.CAN-06-4477 [DOI] [PubMed] [Google Scholar]
- Kushner S. A., Elgersma Y., Murphy G. G., Jaarsma D., van Woerden G. M., Hojjati M. R., Cui Y., LeBoutillier J. C., Marrone D. F., Choi E. S. et al. (2005). Modulation of presynaptic plasticity and learning by the H-ras/extracellular signal-regulated kinase/synapsin I signaling pathway. J. Neurosci. 25, 9721-9734. 10.1523/JNEUROSCI.2836-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuslak S. L., Thielen J. L. and Marker P. C. (2007). The mouse seminal vesicle shape mutation is allelic with Fgfr2. Development 134, 557-565. 10.1242/dev.02741 [DOI] [PubMed] [Google Scholar]
- Lee C. H. and Umemori H. (2013). Suppression of epileptogenesis-associated changes in response to seizures in FGF22-deficient mice. Front. Cell. Neurosci. 7, 43 10.3389/fncel.2013.00043 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Lee C. H., Javed D., Althaus A. L., Parent J. M. and Umemori H. (2012). Neurogenesis is enhanced and mossy fiber sprouting arises in FGF7-deficient mice during development. Mol. Cell. Neurosci. 51, 61-67. 10.1016/j.mcn.2012.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon M. A. and Schlessinger J. (2010). Cell signaling by receptor tyrosine kinases. Cell 141, 1117-1134. 10.1016/j.cell.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z. and Murthy V. N. (2001). Visualizing postendocytic traffic of synaptic vesicles at hippocampal synapses. Neuron 31, 593-605. 10.1016/S0896-6273(01)00398-1 [DOI] [PubMed] [Google Scholar]
- Li A.-J., Suzuki S., Suzuki M., Mizukoshi E. and Imamura T. (2002). Fibroblast growth factor-2 increases functional excitatory synapses on hippocampal neurons. Eur. J. Neurosci. 16, 1313-1324. 10.1046/j.1460-9568.2002.02193.x [DOI] [PubMed] [Google Scholar]
- Lisman J. (2012). Excitation, inhibition, local oscillations, or large-scale loops: what causes the symptoms of schizophrenia? Curr. Opin. Neurobiol. 22, 537-544. 10.1016/j.conb.2011.10.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loilome W. Joshi A. D., ap Rhys C. M. J., Piccirillo S., Angelo V. L., Gallia G. L. and Riggins G. J. (2009). Glioblastoma cell growth is suppressed by disruption of fibroblast growth factor pathway signaling. J. Neurooncol. 94, 359-366. 10.1007/s11060-009-9885-5 [DOI] [PubMed] [Google Scholar]
- Mabb A. M. and Ehlers M. D. (2010). Ubiquitination in postsynaptic function and plasticity. Annu. Rev. Cell. Dev. Biol. 26, 179-210. 10.1146/annurev-cellbio-100109-104129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Peña A., Acebes A., Rodríguez J.-R., Sorribes A., de Polavieja G. G., Fernández-Fúnez P. and Ferrús A. (2006). Age-independent synaptogenesis by phosphoinositide 3 kinase. J. Neurosci. 26, 10199-10208. 10.1523/JNEUROSCI.1223-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason I. (2007). Initiation to end point: the multiple roles of fibroblast growth factors in neural development. Nat. Rev. Neurosci. 8, 583-596. 10.1038/nrn2189 [DOI] [PubMed] [Google Scholar]
- McAllister A. K. (2007). Dynamic aspects of CNS synapse formation. Annu. Rev. Neurosci. 30, 425-450. 10.1146/annurev.neuro.29.051605.112830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville H., Wang Y., Taub P. J. and Jabs E. W. (2010). Genetic basis of potential therapeutic strategies for craniosynostosis. Am. J. Med. Genet. A 152A, 3007-3015. 10.1002/ajmg.a.33703 [DOI] [PubMed] [Google Scholar]
- Moelling K., Schad K., Bosse M., Zimmermann S. and Schweneker M. (2002). Regulation of Raf-Akt cross-talk. J. Biol. Chem. 277, 31099-31106. 10.1074/jbc.M111974200 [DOI] [PubMed] [Google Scholar]
- Mohammadi M., Dionne C. A., Li W., Li N., Spivak T., Honegger A. M., Jaye M. and Schlessinger J. (1992). Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature 358, 681-684. 10.1038/358681a0 [DOI] [PubMed] [Google Scholar]
- Mohammadi M., Dikic I., Sorokin A., Burgess W. H., Jaye M. and Schlessinger J. (1996). Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol. Cell. Biol. 16, 977-989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata K., Abrams B., Grill B., Goncharov A., Huang X., Chisholm A. D. and Jin Y. (2005). Regulation of a DLK-1 and p38 MAP kinase pathway by the ubiquitin ligase RPM-1 is required for presynaptic development. Cell 120, 407-420. 10.1016/j.cell.2004.12.017 [DOI] [PubMed] [Google Scholar]
- O'Donovan M. C., Norton N., Williams H., Peirce T., Moskvina V., Nikolov I., Hamshere M., Carroll L., Georgieva L., Dwyer S. et al. (2009). Analysis of 10 independent samples provides evidence for association between schizophrenia and a SNP flanking fibroblast growth factor receptor 2. Mol. Psychiatry 14, 30-36. 10.1038/mp.2008.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver G., Sosa-Pineda B., Geisendorf S., Spana E. P., Doe C. Q. and Gruss P. (1993). Prox 1, a prospero-related homeobox gene expressed during mouse development. Mech. Dev. 44, 3-16. 10.1016/0925-4773(93)90012-M [DOI] [PubMed] [Google Scholar]
- Ong S. H., Hadari Y. R., Gotoh N., Guy G. R., Schlessinger J. and Lax I. (2001). Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc. Natl. Acad. Sci. USA 98, 6074-6079. 10.1073/pnas.111114298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M. and Itoh N. (2001). Fibroblast growth factors. Genome Biol. 2, reviews3005-reviews3005.12. 10.1186/gb-2001-2-3-reviews3005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornitz D. M., Xu J., Colvin J. S., McEwen D. G., MacArthur C. A., Coulier F., Gao G. and Goldfarb M. (1996). Receptor specificity of the fibroblast growth factor family. J. Biol. Chem. 271, 15292-15297. 10.1074/jbc.271.25.15292 [DOI] [PubMed] [Google Scholar]
- Paek H., Gutin G. and Hébert J. M. (2009). FGF signaling is strictly required to maintain early telencephalic precursor cell survival. Development 136, 2457-2465. 10.1242/dev.032656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso B., Zucchini S. and Simonato M. (2013). Implication of fibroblast growth factors in epileptogenesis-associated circuit rearrangements. Front. Cell. Neurosci. 7, 152 10.3389/fncel.2013.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H. and Poo M.-M. (2013). Neurotrophin regulation of neural circuit development and function. Nat. Rev. Neurosci. 14, 7-23. 10.1038/nrn3379 [DOI] [PubMed] [Google Scholar]
- Partanen J., Schwartz L. and Rossant J. (1998). Opposite phenotypes of hypomorphic and Y766 phosphorylation site mutations reveal a function for Fgfr1 in anteroposterior patterning of mouse embryos. Genes Dev. 12, 2332-2344. 10.1101/gad.12.15.2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel V. N., Likar K. M., Zisman-Rozen S., Cowherd S. N., Lassiter K. S., Sher I., Yates E. A., Turnbull J. E., Ron D. and Hoffman M. P. (2008). Specific heparan sulfate structures modulate FGF10-mediated submandibular gland epithelial morphogenesis and differentiation. J. Biol. Chem. 283, 9308-9317. 10.1074/jbc.M709995200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers C. J., McLeskey S. W. and Wellstein A. (2000). Fibroblast growth factors, their receptors and signaling. Endocr. Relat. Cancer 7, 165-197. 10.1677/erc.0.0070165 [DOI] [PubMed] [Google Scholar]
- Sanes J. R. and Yamagata M. (2009). Many paths to synaptic specificity. Annu. Rev. Cell Dev. Biol. 25, 161-195. 10.1146/annurev.cellbio.24.110707.175402 [DOI] [PubMed] [Google Scholar]
- Shen K. and Cowan C. W. (2010). Guidance molecules in synapse formation and plasticity. Cold Spring Harb. Perspect. Biol. 2, a001842 10.1101/cshperspect.a001842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K. and Scheiffele P. (2010). Genetics and cell biology of building specific synaptic connectivity. Annu. Rev. Neurosci. 33, 473-507. 10.1146/annurev.neuro.051508.135302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Li Z., Xu Q., Wang T., Li T., Shen J., Zhang F., Chen J., Zhou G., Ji W. et al. (2011). Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat. Genet. 43, 1224-1227. 10.1038/ng.980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui T. J. and Craig A. M. (2011). Synaptic organizing complexes. Curr. Opin. Neurobiol. 21, 132-143. 10.1016/j.conb.2010.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Songyang Z., Shoelson S. E., Chaudhuri M., Gish G., Pawson T., Haser W. G., King F., Roberts T., Ratnofsky S., Lechleider R. J. et al. (1993). SH2 domains recognize specific phosphopeptide sequences. Cell 72, 767-778. 10.1016/0092-8674(93)90404-E [DOI] [PubMed] [Google Scholar]
- Southwell D. G., Nicholas C. R., Basbaum A. I., Stryker M. P., Kriegstein A. R., Rubenstein J. L. and Alvarez-Buylla A. (2014). Interneurons from embryonic development to cell-based therapy. Science 344, 1240622 10.1126/science.1240622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakou E. and Salinas P. C. (2014). Postsynaptic assembly: a role for Wnt signaling. Dev. Neurobiol. 74, 818-827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg Z., Myers C., Heim V. M., Lathrop C. A., Rebustini I. T., Stewart J. S., Larsen M. and Hoffman M. P. (2005). FGFR2b signaling regulates ex vivo submandibular gland epithelial cell proliferation and branching morphogenesis. Development 132, 1223-1234. 10.1242/dev.01690 [DOI] [PubMed] [Google Scholar]
- Stevens H. E., Smith K. M., Rash B. G. and Vaccarino F. M. (2010a). Neural stem cell regulation, fibroblast growth factors, and the developmental origins of neuropsychiatric disorders. Front. Neurosci. 4, 59 10.3389/fnins.2010.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens H. E., Smith K. M., Maragnoli M. E., Fagel D., Borok E., Shanabrough M., Horvath T. L. and Vaccarino F. M. (2010b). Fgfr2 is required for the development of the medial prefrontal cortex and its connections with limbic circuits. J. Neurosci. 30, 5590-5602. 10.1523/JNEUROSCI.5837-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A., Johnson-Venkatesh E. M., Toth A. B., Javed D., Sutton M. A. and Umemori H. (2010). Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465, 783-787. 10.1038/nature09041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terauchi A., Timmons K. M., Kikuma K., Pechmann Y., Kneussel M. and Umemori H. (2015). Selective synaptic targeting of the excitatory and inhibitory presynaptic organizers FGF22 and FGF7. J. Cell Sci. 128, 281-292. 10.1242/jcs.158337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thisse B. and Thisse C. (2005). Functions and regulations of fibroblast growth factor signaling during embryonic development. Dev. Biol. 287, 390-402. 10.1016/j.ydbio.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Tolias K. F., Duman J. G. and Um K. (2011). Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog. Neurobiol. 94, 133-148. 10.1016/j.pneurobio.2011.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N. and Grose R. (2010). Fibroblast growth factor signalling: from development to cancer. Nat. Rev. Cancer 10, 116-129. 10.1038/nrc2780 [DOI] [PubMed] [Google Scholar]
- Umemori H. (2009). Weaving the neuronal net with target-derived fibroblast growth factors. Dev. Growth Differ. 51, 263-270. 10.1111/j.1440-169X.2008.01079.x [DOI] [PubMed] [Google Scholar]
- Umemori H., Linhoff M. W., Ornitz D. M. and Sanes J. R. (2004). FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118, 257-270. 10.1016/j.cell.2004.06.025 [DOI] [PubMed] [Google Scholar]
- Wairkar Y. P., Toda H., Mochizuki H., Furukubo-Tokunaga K., Tomoda T. and DiAntonio A. (2009). Unc-51 controls active zone density and protein composition by downregulating ERK signaling. J. Neurosci. 29, 517-528. 10.1523/JNEUROSCI.3848-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Kan M., McKeehan K., Jang J.-H., Feng S. and McKeehan W. L. (1997). A homeo-interaction sequence in the ectodomain of the fibroblast growth factor receptor. J. Biol. Chem. 272, 23887-23895. 10.1074/jbc.272.38.23887 [DOI] [PubMed] [Google Scholar]
- Wang T., Zeng Z., Hu Z., Zheng L., Li T., Li Y., Liu J., Li J., Feng G., He L. et al. (2012). FGFR2 is associated with bipolar disorder: a large-scale case-control study of three psychiatric disorders in the Chinese Han population. World J. Biol. Psychiatry 13, 599-604. 10.3109/15622975.2011.650203 [DOI] [PubMed] [Google Scholar]
- Williams A. J. and Umemori H. (2014). The best-laid plans go oft awry: synaptogenic growth factor signaling in neuropsychiatric disease. Front. Synaptic Neurosci. 6, 4 10.3389/fnsyn.2014.00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M. E., de Wit J. and Ghosh A. (2010). Molecular mechanisms of synaptic specificity in developing neural circuits. Neuron 68, 9-18. 10.1016/j.neuron.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M. E., Wilke S. A., Daggett A., Davis E., Otto S., Ravi D., Ripley B., Bushong E. A., Ellisman M. H., Klein G. et al. (2011). Cadherin-9 regulates synapse-specific differentiation in the developing hippocampus. Neuron 71, 640-655. 10.1016/j.neuron.2011.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhams P. L., Webb M., Atkinson D. J. and Seeley P. J. (1989). A monoclonal antibody, Py, distinguishes different classes of hippocampal neurons. J. Neurosci. 9, 2170-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N.-J. and Henkemeyer M. (2012). Ephrin reverse signaling in axon guidance and synaptogenesis. Semin. Cell. Dev. Biol. 23, 58-64. 10.1016/j.semcdb.2011.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T., Uchida S. and Mishina M. (2009). Regulation of synaptic vesicle accumulation and axon terminal remodeling during synapse formation by distinct Ca 2+ signaling. J. Neurochem. 111, 160-170. 10.1111/j.1471-4159.2009.06309.x [DOI] [PubMed] [Google Scholar]
- Yu K., Xu J., Liu Z., Sosic D., Shao J., Olson E. N., Towler D. A. and Ornitz D. M. (2003). Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development 130, 3063-3074. 10.1242/dev.00491 [DOI] [PubMed] [Google Scholar]
- Zhang X., Ibrahimi O. A., Olsen S. K., Umemori H., Mohammadi M. and Ornitz D. M. (2006). Receptor specificity of the fibroblast growth factor family: the complete mammalian FGF family. J. Biol. Chem. 281, 15694-15700. 10.1074/jbc.M601252200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H., Yang T., Madakashira B. P., Thiels C. A., Bechtle C. A., Garcia C. M., Zhang H., Yu K., Ornitz D. M., Beebe D. C. et al. (2008). Fibroblast growth factor receptor signaling is essential for lens fiber cell differentiation. Dev. Biol. 318, 276-288. 10.1016/j.ydbio.2008.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.