

Abstract
A striking proportion of human cleavage-stage embryos exhibit chromosome instability (CIN). Notably, until now, no experimental model has been described to determine the origin and mechanisms of complex chromosomal rearrangements. Here, we examined mouse embryos deficient for the chromatin remodeling protein ATRX to determine the cellular mechanisms activated in response to CIN. We demonstrate that ATRX is required for silencing of major satellite transcripts in the maternal genome, where it confers epigenetic asymmetry to pericentric heterochromatin during the transition to the first mitosis. This stage is also characterized by a striking kinetochore size asymmetry established by differences in CENP-C protein between the parental genomes. Loss of ATRX results in increased centromeric mitotic recombination, a high frequency of sister chromatid exchanges and double strand DNA breaks, indicating the formation of mitotic recombination break points. ATRX-deficient embryos exhibit a twofold increase in transcripts for aurora kinase B, the centromeric cohesin ESCO2, DNMT1, the ubiquitin-ligase (DZIP3) and the histone methyl transferase (EHMT1). Thus, loss of ATRX activates a pathway that integrates epigenetic modifications and DNA repair in response to chromosome breaks. These results reveal the cellular response of the cleavage-stage embryo to CIN and uncover a mechanism by which centromeric fission induces the formation of large-scale chromosomal rearrangements. Our results have important implications to determine the epigenetic origins of CIN that lead to congenital birth defects and early pregnancy loss, as well as the mechanisms involved in the oocyte to embryo transition.
Keywords: Centromere, Heterochromatin, Kinetochore, Chromatin remodeling, CO-FISH, Death domain-associated protein (DAXX), Epigenetic modifications, Meiosis
HIGHLIGHTED ARTICLE: The chromatin remodelling protein ATRX is transmitted to the early zygote through the maternal germ line and is required to silence major satellite transcripts and control chromosome stability.
INTRODUCTION
Errors in chromosome segregation during mitotic and meiotic cell division lead to the formation of cells with an abnormal chromosome number. Aneuploidy is associated with cancer development and is also a leading cause of miscarriage and congenital birth defects (Hassold and Hunt, 2001; Kops et al., 2005). Human embryos are particularly susceptible to aneuploidy, which in the majority of cases is the result of abnormal meiosis in the female gamete (Hassold et al., 2007). However, post-zygotic chromosome instability (CIN) has recently emerged as a significant mechanism leading to chromosome imbalances and segmental chromosomal aberrations. For example, array-comparative genomic hybridization studies indicate that as many as 70-90% of in vitro fertilized human embryos are mosaic for structural or numerical chromosome abnormalities arising during the first cleavage divisions of the pre-implantation embryo (Vanneste et al., 2009; Robberecht et al., 2010; Voet et al., 2011; Chavez et al., 2012).
Analysis of segregation patterns for the mouse Y chromosome on the BALB/cWT inbred background provided the initial evidence to indicate that the first mitotic divisions in the embryo are prone to chromosomal non-disjunction (Bean et al., 2001). However, the underlying mechanisms predisposing to chromosome segregation errors during this critical developmental window as well as the cellular response of the early conceptus to CIN remain to be determined.
The transition to the first mitosis at the onset of pre-implantation embryo development is a unique and highly dynamic process, taking place in the absence of major transcriptional activity. The extensive chromatin remodeling of paternal and maternal genomes in the one cell zygote is quickly followed by active global DNA de-methylation of the paternal genome and results in the establishment of epigenetic asymmetry in which paternal and maternal pronuclei acquire a unique epigenetic landscape characterized by distinct DNA and histone methylation marks, and histone variants (Mayer et al., 2000b; Oswald et al., 2000; Santos et al., 2005; Santenard and Torres-Padilla, 2009; Banaszynski et al., 2010; Kota and Feil, 2010). Remarkably, epigenetic asymmetry is maintained during interphase in the two-cell embryo where distinct chromatin marks reveal the polar distribution of paternal and maternal genomes (Mayer et al., 2000a; Hayashi-Takanaka et al., 2011). The functional significance of the spatial separation of the parental genomes in the cleavage-stage embryo is not clear at present (Probst and Almouzni, 2011).
Heterochromatin formation is essential for centromere structure and function. As the site of kinetochore formation, the mammalian centromere is strictly required for accurate chromosome segregation and maintenance of a euploid chromosome complement. Functional centromeres are required to maintain sister chromatid cohesion until anaphase as well as to provide the assembly site for large protein complexes and crucial epigenetic marks required for kinetochore assembly and organization (Durand-Dubief and Ekwall, 2008; Verdaasdonk and Bloom, 2011). In the mammalian embryo, establishment and maintenance of pericentric heterochromatin (PCH) takes place during the first cleavage divisions and require extensive genome reprogramming. For example, deposition of the histone variant H3.3 and a burst of transcription of non-coding RNAs from major satellite sequences at the two-cell stage induce global remodeling and association of PCH domains into the nascent chromocenters of blastomere interphase nuclei (Probst et al., 2010; Santenard et al., 2010; Almouzni and Probst, 2011; Probst and Almouzni, 2011; Fadloun et al., 2013). Importantly, histone H3.3 is required at the early zygote stage to regulate ribosomal RNA transcription as well as chromatin condensation at the two-cell stage (Lin et al., 2013, 2014).
The α-thalassemia mental retardation X-linked protein ATRX is required for heterochromatin formation and chromosome segregation during mitosis and meiosis (Bérubé et al., 2000; De La Fuente et al., 2004; Ritchie et al., 2008; Baumann et al., 2010). Loss of ATRX function induces chromosome instability during early embryonic development, cell differentiation and malignant neoplastic transformation (Baumann et al., 2010; Berube, 2011; De La Fuente et al., 2011; Lovejoy et al., 2012). Spontaneous mutations result in ATRX syndrome, a neurodevelopmental condition in which individuals exhibit α-thalassemia and various degrees of gonadal dysgenesis (Gibbons and Higgs, 2000; Berube, 2011). Notably, exome sequencing studies revealed the presence of ATRX mutations in a high proportion of human pancreatic neuroendocrine tumors, malignant gliomas and neuroblastomas that exhibit high levels of chromosome instability (Elsasser et al., 2011; Jiao et al., 2011; Molenaar et al., 2012).
Transgenic expression of a hairpin double-stranded RNAi against the plant homeodomain (PHD) of ATRX under the control of a zona pellucida 3 promoter induces an efficient oocyte-specific functional ablation of ATRX during the final stages of oocyte growth (Baumann et al., 2010). Using this transgenic RNAi model, we have previously demonstrated that ATRX is required for pericentric heterochromatin formation, and its loss of function in pre-ovulatory oocytes induces centromere instability during the transition to the first mitosis in the early conceptus (Baumann et al., 2010). However, the role of ATRX in maintenance of genome integrity remained to be determined. Here, we present new evidence indicating that inheritance of ATRX through the female germ line is essential for centromere stability and transcriptional heterochromatin silencing in the maternal genome of the one-cell embryo. Notably, in addition to epigenetic asymmetry at PCH domains, the first mitotic cell division is characterized by a striking kinetochore size asymmetry, a process that might be crucial to maintain the spatial separation of the maternal and paternal genomes at the two-cell stage. Our results indicate that the specialized mechanisms required to accurately segregate epigenetically distinct parental centromeres during the first mitosis might underlie the higher susceptibility to chromosome segregation errors during this critical period of extensive genome reprogramming.
RESULTS
ATRX is required for silencing of major satellite transcripts in the maternal genome during the oocyte to embryo transition
We have previously demonstrated that ATRX is required to recruit DAXX to PCH in pre-ovulatory oocytes (Baumann et al., 2010). In somatic and embryonic stem cells, DAXX is essential for heterochromatin formation and deposition of histone variants (Drane et al., 2010; Goldberg et al., 2010; Lewis et al., 2010). However, the role of ATRX/DAXX interactions in heterochromatin formation during epigenetic reprogramming in the mammalian embryo is not known. Therefore, we used transgenic ATRX-deficient oocytes to determine the role of this chromatin-remodeling factor on DAXX localization during de novo heterochromatin formation in the mouse zygote. ATRX is transmitted to the one-cell embryo through the maternal genome where it remains colocalized with PCH domains of condensed chromosomes during anaphase II (Fig. 1A) and subsequently becomes enriched at the heterochromatin rim of peri-nucleolar like bodies (PNLBs) in the maternal pronucleus (Fig. 1B). Pericentric heterochromatin can be easily identified by the formation of a DAPI bright rim around the nucleolus initially in the maternal pronucleus at 6 h post-fertilization (6 hpf) and subsequently in the paternal pronucleus at 12 hpf (Fig. 1C). By contrast, ATRX is absent from decondensing sperm chromatin immediately after fertilization, and only appears as a diffuse nucleoplasmic signal upon male pronucleus formation (Fig. 1A,B). Notably, this helicase protein exhibits a dramatic redistribution to the heterochromatin rim of the paternal pronucleus by 10-12 hpf, where the levels of ATRX staining in both paternal and maternal heterochromatin are undistinguishable (Fig. 1C). This transition also coincides with recruitment of DAXX to the heterochromatin rim in both maternal and paternal pronuclei, where DAXX becomes enriched at the paternal heterochromatin rim by 12-15 hpf (Fig. 1C,D). Importantly, ATRX-deficient zygotes fail to recruit DAXX to both maternal and paternal heterochromatin (Fig. 1E,F). These results indicate that ATRX exhibits a dynamic localization pattern during male pronuclear formation where it is required to recruit DAXX to pericentric heterochromatin during the first embryonic cell cycle.
Fig. 1.

ATRX is required for recruitment of the histone chaperone DAXX to pericentric heterochromatin in the paternal genome. (A,B) ATRX (red) is transmitted to the early zygote through the maternal germ line (arrows) where it becomes enriched at pericentric heterochromatin (PCH) upon pronuclear formation. ATRX becomes detectable in the paternal pronucleus (arrowheads) at 6 h post-fertilization (hpf). (C,D) Coincident with nucleolar formation, ATRX localizes to PCH in both maternal and paternal pronuclei at 12 hpf. DAXX (green) is recruited to PCH in both pronuclei but is enriched in the paternal pronucleus. (E,F) ATRX-deficient zygotes fail to recruit DAXX to PCH in both maternal and paternal pronuclei (inset). Scale bars: 10 µm.
To determine the functional consequences of ATRX depletion on PCH function, we compared the patterns of expression of major satellite transcripts in both maternal and paternal pronuclei in wild-type and ATRX-deficient zygotes using RNA-FISH. Consistent with previous studies (Puschendorf et al., 2008), major satellite transcripts can be detected associated with the heterochromatin rim of PNLBs in the paternal but not the maternal pronucleus in the majority of wild-type embryos (Fig. 2A; arrowheads). However, loss of ATRX function induced a significant increase (P<0.01) in the proportion (55.2%) of transgenic zygotes that exhibit major satellite transcript expression in both paternal and maternal pronuclei (Fig. 2A,B). These results reveal that ATRX is required for transcriptional repression of major satellite repeats in the maternal pronucleus. To determine the contribution of the maternal genome to major satellite transcription in the absence of ATRX function, we compared the levels of expression of major satellite transcripts between wild-type and ATRX-deficient parthenotes using real-time PCR. Consistent with our RNA-FISH results, quantitative transcriptional analysis revealed a twofold upregulation of major satellite transcripts in transgenic parthenogenetic embryos (Fig. 2C). These results are consistent with a role for ATRX in transcriptional repression of major satellite transcripts in the maternal pronucleus during the late zygote stage and demonstrate a crucial function for ATRX in maintenance of a transcriptionally repressive environment at PCH.
Fig. 2.
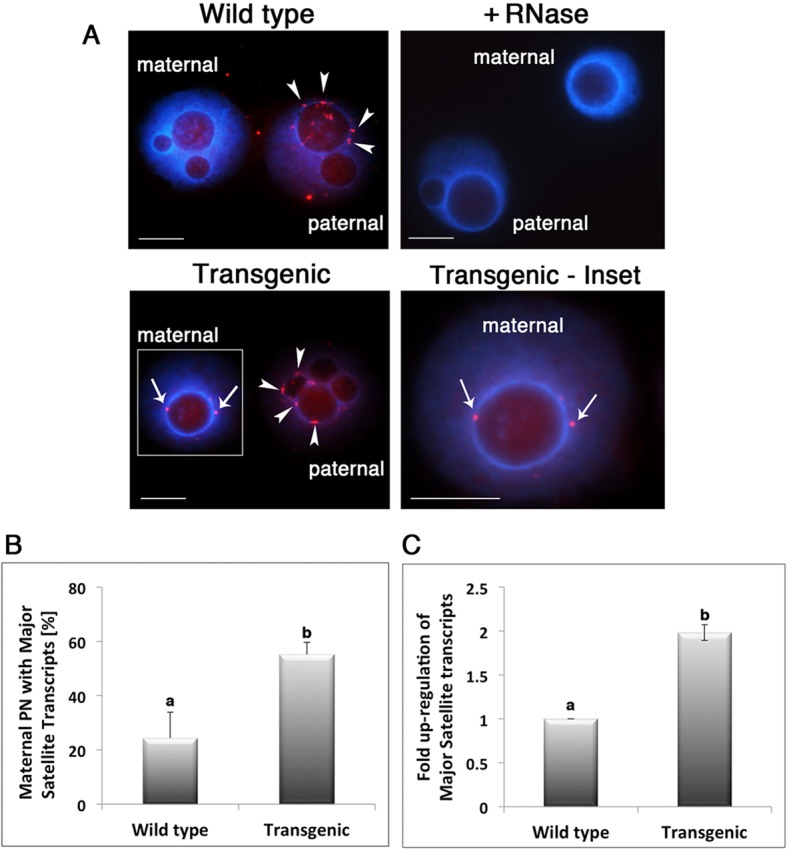
Loss of ATRX results in abnormal expression of major satellite transcripts in the maternal genome. (A) Detection of major satellite transcripts (red) in the paternal pronucleus with RNA-FISH (arrowheads). RNA tracks are present in the paternal pronucleus and absence of centromeric transcripts in the maternal pronucleus of wild-type zygotes. Treatment with RNAse (+) was used as a negative control for probe specificity. ATRX-deficient embryos exhibit major satellite transcripts (red) associated with PCH in both paternal (arrowheads) and maternal pronuclei (arrows). Expression of major satellite transcripts in the maternal pronucleus is indicated by the presence of two or more foci or RNA tracks in close apposition with the perinucleolar heterochromatin rim (see inset). (B) Loss of ATRX induces a significant (P<0.05) increase in the proportion of zygotes that exhibit centromeric transcripts in the maternal pronucleus. a and b indicate significant differences. (C) Quantitative analysis of major satellite transcripts using real-time PCR. ATRX-deficient parthenotes exhibit a twofold increase in the levels of centromeric transcripts at 12 hpf (P<0.05). All data are presented as the mean±s.d. of three replicates. Scale bars: 10 µm.
ATRX contributes to the epigenetic asymmetry of PCH in the maternal genome
Analysis of epigenetic marks at PCH during the transition to the first mitosis revealed that ATRX is absent from one set of condensed chromosomes during the first mitosis (Fig. 3A). Thus, transition to the first mitosis requires extensive heterochromatin remodeling that leads to the displacement of DAXX from condensing chromosomes and the acquisition of an asymmetric distribution of ATRX at PCH domains in the parental genomes (Fig. 3A). Next, to determine unequivocally whether ATRX associates with PCH of maternal or paternal chromosomes during the first mitosis, we used DNA fluorescence in situ hybridization (DNA-FISH) for the simultaneous detection of the sex chromosomes in one-cell embryos. ATRX was found highly enriched at PCH in the maternal chromosome complement bearing the X chromosome (green), while the paternal genome carrying the Y chromosome (red) in male zygotes exhibits negligible ATRX staining (Fig. 3A,B). Importantly, paternal chromosomes also exhibit reduced signals for the centromeric-specific proteins detected by the CREST antiserum (Fig. 3A,C). Similarly, both ATRX and CREST exhibit an asymmetric distribution to pericentric and kinetochore domains, respectively, in the maternal genome of female zygotes (Fig. 3C,D). We also confirmed that in both male and female embryos, large heterochromatin domains stained with ATRX colocalized with histone H3 tri-methylated at lysine 9 (H3K9me3; supplementary material Fig. S1), a histone mark that is known to specifically label maternal chromosomes in the zygote stage embryo (Puschendorf et al., 2008). By contrast, diploid parthenogenetic embryos carrying only a maternal chromosome complement exhibit no detectable differences in the levels of ATRX or CREST signals during the first mitosis (Fig. 3E,F).
Fig. 3.
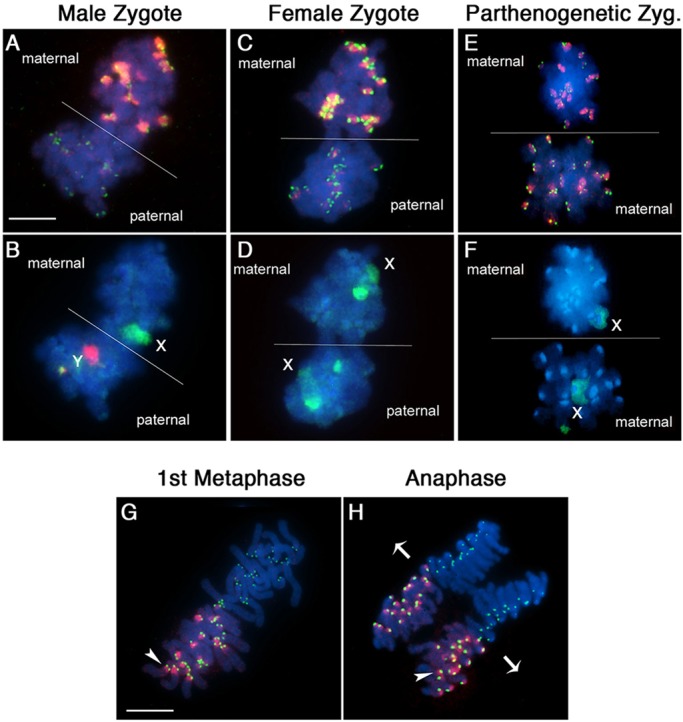
Maternal inheritance of ATRX confers post-meiotic centromere epigenetic asymmetry during the first mitosis. (A) Zygote stage embryo during the first mitosis. ATRX protein (red) is present in the pericentric heterochromatin in the maternal chromosome complement. Kinetochore domains are stained with CREST (green) and partially colocalize with centromere ATRX staining in the maternal but not the paternal chromosomes. (B) FISH analysis showing the localization of the X chromosome (green) on the maternal genome and the Y chromosome (red) on the paternal chromosomes of a male zygote. (C,D) ATRX decorates maternal centromeres, while paternal chromosomes show negligible or absent ATRX staining during the first mitosis in the female zygote. (E,F) No differences can be detected in ATRX staining in diploid parthenogenetic embryos in which only maternal chromosomes are present. (G) The asymmetric chromosomal localization of ATRX (red) clearly delineates the polar distribution of the maternal (arrowhead) and paternal genomes during the first mitosis. (H) The unique compartmentalization of parental genomes during the first mitosis ensures that only a single chromatid of each maternal (arrowhead) and paternal chromosome is precisely segregated to daughter cells during anaphase. Arrowheads indicate ATRX-labeled maternal centromeres. Arrows indicate pole-ward movement of single chromatids. Scale bars: 10 µm.
Notably, the asymmetric distribution of ATRX at PCH is strictly maintained during the equatorial alignment of paternal and maternal chromosomes at the metaphase plate (Fig. 3G). This results in the precise segregation of one set of maternal chromatids exhibiting ATRX staining as a distinct unit and a set of paternal chromatids that exhibit only reduced CREST signals in a spatially separated and polar distribution during the anaphase-telophase transition (Fig. 3H). These results indicate that ATRX confers epigenetic asymmetry to PCH in the maternal genome and suggest that a unique mechanism might be set in place to detect different epigenetic modifications at the centromere in order to ensure the accurate segregation of maternal and paternal chromatids to distinct nuclear compartments in the two-cell embryo.
To determine the potential mechanisms regulating the unique chromosome segregation patterns of paternal and maternal chromosomes during the first mitosis, we used image analysis software to compare the size of kinetochore domains stained with CREST under high resolution using a 100× microscope lens followed by objective scoring and quantification of the size of each foci as a statistical descriptor, as described previously (Urani et al., 2013). Following ATRX staining (Fig. 3G), the maternal chromosome complement can be clearly distinguished in wild-type embryos (n=10). Strikingly, comparison of kinetochore domains between maternal and paternal chromosomes revealed the presence of kinetochore size asymmetry during the early stages of chromosome condensation leading to the first mitosis (Fig. 4A,B). Quantitative analysis of the average equivalent diameter (EqDiameter) of CREST signals using image analysis software (NIS-Elements) revealed that the mean diameter of maternal kinetochore domains (12.8±2.11 arbitrary units) was significantly larger (P<0.0001) than that of the paternal kinetochores (9.0065±1.02 arbitrary units) of mitotic chromosomes (Fig. 4A,B). Importantly, similar results were obtained after detection of the kinetochore-specific protein CENP-C (Fig. 4C). These results indicate that epigenetic asymmetry between parental genomes during the first mitosis extends to kinetochore domains at least until the early stages of anaphase. They also provide the first evidence indicating that post-meiotic kinetochore asymmetry might provide a unique mechanism for proper segregation of epigenetically distinct parental chromosome complements during the first mitosis. Notably, kinetochore size asymmetry persists in >90% of wild-type embryos at the second mitotic division (n=34) and is subsequently lost at the third cleavage division (n=12; supplementary material Fig. S2). These results indicate that there is a gradual conversion from meiotic to mitotic kinetochore domains during the oocyte to embryo transition. Importantly, kinetochore asymmetry was also detected in ATRX-deficient zygotes (supplementary material Fig. S3), suggesting that ATRX contributes to pericentric heterochromatin asymmetry but not to kinetochore size asymmetry.
Fig. 4.
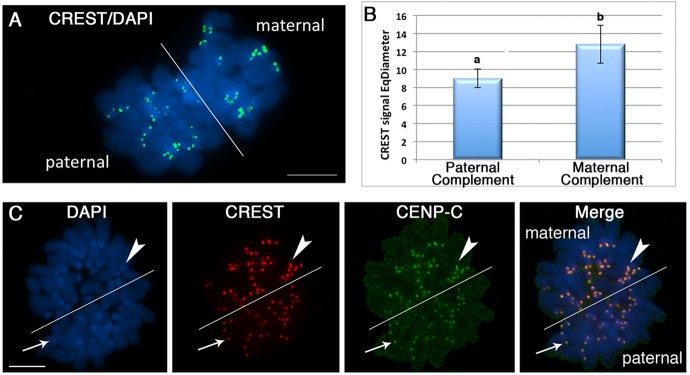
Post-meiotic kinetochore asymmetry is associated with enrichment of CENP-C in the maternal genome. (A) Zygote-stage embryo during the first mitosis. The maternal and paternal chromosome complements (DAPI; blue) exhibit significant differences (P<0.0001) in the levels of kinetochore-associated proteins stained with the CREST anti-serum (green). (B) Quantitative analysis of the average equivalent diameter of CREST signals revealed that the mean diameter of maternal CREST foci is ∼42% larger than those of the paternal chromosome complement. Data represent the mean±s.d. of four independent replicates. a and b indicate significant differences. (C) In addition to CREST signals (red), the kinetochore-specific protein CENP-C is preferentially enriched on the maternal chromosome complement (arrowhead), demonstrating the existence of a previously unrecognized asymmetry in the levels of several kinetochore-associated proteins during the first mitosis in the mammalian embryo. A diagonal line can be easily traced to distinguish the maternal (arrowheads) from the paternal (thin arrow) chromosome complement based on kinetochore size asymmetry. Scale bars: 10 µm.
Loss of ATRX results in abnormal centromeric mitotic recombination and double-strand DNA breaks
We have previously demonstrated that loss of pericentric heterochromatin structure in ATRX-deficient oocytes results in high levels of centromere instability, formation of micronuclei and severe interstitial chromosome damage arising following the transition to the first mitosis (Baumann et al., 2010). Therefore, we determined the chromosomal distribution of double-strand DNA breaks (DSBs) in ATRX-deficient embryos undergoing the first cleavage division. Comparison of control (n=13) and transgenic (n=13) zygotes revealed the presence of basal levels of diffuse γH2AX staining in the chromosomes of wild-type and transgenic embryos. Double-strand DNA breaks at interstitial chromosomal regions can be detected at low frequency (1.7±0.5 foci per metaphase) in 46.5% of wild-type embryos (Fig. 5C,D). However, γH2AX staining associated with DSBs can be clearly distinguished as bright irregular foci colocalized with chromosomal breaks in 91.7% of transgenic zygotes (Fig. 5B,D). In addition, ATRX-deficient embryos revealed a significant (P<0.05) increase in the frequency of DSBs per metaphase (7.9±3.4) that, although not exclusively, were found localized at proximal chromosomal segments exhibiting reduced or negligible CREST signals (Fig. 5B, insets). These results indicate that loss of ATRX function induces the formation of DSBs initially at PCH domains in the majority of transgenic zygotes. However, the presence of DSBs at interstitial chromosomal segments also revealed the induction of chromosome-wide DNA damage in transgenic embryos.
Fig. 5.

Lack of ATRX function results in the formation of double-strand DNA breaks (DSBs) at pericentric heterochromatin and interstitial chromosome segments. (A) Chromosomes from a wild-type zygote during the first mitosis exhibit sister chromatid cohesion (insets) and only negligible levels of γH2AX. Centromeres are stained with CREST (red). (B) Transgenic embryos exhibit a high incidence of DNA breaks (γH2AX; green). The presence of DSBs in pericentric heterochromatin is indicated by the localization of γH2AX signals in chromosomal segments that have lost the CREST signal due to centromeric breaks (arrowheads; insets). (C,D) Loss of ATRX function results in a significant increase (P<0.05) in the number of DSBs per metaphase, as well as the proportion of embryos that exhibit at least one DSB per metaphase. Data represent the mean±s.d. of three independent replicates. a and b indicate significant differences. Scale bars: 10 µm; 5 µm (insets).
Loss of maternal ATRX results in an increased rate of sister chromatid exchange at PCH in mammalian embryos (Baumann et al., 2010), and abnormal heterochromatin formation has been recently shown to induce illegitimate centromeric recombination in somatic cells (Jaco et al., 2008). Therefore, we determined whether centromeric instability in ATRX-deficient embryos is due to abnormal centromeric mitotic recombination events. Use of a strand-specific minor satellite DNA probe and chromosome orientation (CO-FISH) to compare the frequency of mitotic recombination in control and transgenic zygotes revealed that loss of ATRX induced a significant increase in the number of centromeric recombination events per chromosome (Fig. 6). As expected, chromosomes in wild-type embryos showed a single exclusively centromeric hybridization signal (green) corresponding to the leading strand of the minor satellite sequence on a single chromatid (Fig. 6A, inset). By contrast, ATRX-deficient embryos present a high frequency (P<0.05) of chromosomes exhibiting illegitimate centromeric recombination characterized by a dual CO-FISH signal (green) corresponding to the leading strand and a centromeric sister chromatid exchange within the minor satellite region, confirming the presence of mitotic centromeric recombination events (Fig. 6B, thick arrow and inset; Fig. 6C).
Fig. 6.

Loss of ATRX results in enhanced centromeric mitotic recombination and large-scale chromosomal rearrangements. (A) Chromosome orientation fluorescence in situ hybridization (CO-FISH) using a strand-specific minor satellite DNA probe (green). Chromosomes were pseudo-colored in red. Wild-type zygotes exhibit a single centromeric signal (green) corresponding to the leading strand of the minor satellite sequence (see diagram). (B) A centromeric mitotic recombination event is detected by the presence of two minor satellite signals (thick arrows) in the same chromosome, resulting from an abnormal centromeric sister chromatid exchange (see diagram). Scale bars: 5 µm (left); 1 µm (right). (C) Loss of ATRX results in a significant increase (P<0.05) in the frequency of illegitimate mitotic recombination events per chromosome. Data represent the mean±s.d. of three replicates. a and b indicate significant differences. (D) Metaphase spread of a transgenic ATRX two-cell embryo showing a high frequency of interstitial sister chromatid exchanges (SCE; arrowheads). (E) Asynchronous DNA replication between sister chromatids in a normal chromosome showing BrdU incorporation (red) on a single chromatid. Arrow indicates the non-replicated chromatid, (F) Chromosome fragment (*) with partial BrdU staining (red) indicating that chromosome breaks frequently occur at the site of a SCE. (G) Sister chromatid exchange at a distal sub-telomeric region (arrowheads). Scale bars: 10 µm in D; 5 µm in E-G. (H) (Top) Spectral karyotyping of ATRX-deficient embryos at the four-cell stage. Scale bar: 10 µm. (Bottom) Sorted karyotype from a representative diploid male embryo showing 40 chromosomes, 40<2n> X-Y, that exhibit large-scale chromosomal rearrangements (insets). Scale bar: 5 µm. (I) SKY analysis reveals the presence of a partial deletion of chromosome 16 (Del16; left panel) and formation of a derivative chromosome 17 (Der 17; right panels and arrowheads) formed by the translocation of a fragment of chromosome 16 with the acrocentric region of chromosome 17: T (16; 17A1). Scale bar: 2.5 µm.
Importantly, by the two-cell stage, ATRX-deficient embryos showed a high frequency of interstitial sister chromatid exchanges (Fig. 6D), resulting in the formation of chromosomal breaks that frequently involved the sites of abnormal mitotic recombination events indicated by the presence of chromosomal fragments with asymmetrical regions of BrdU incorporation (Fig. 6F,G, red). Genome-wide analysis using spectral karyotyping revealed the formation of multiple large-scale chromosomal rearrangements in transgenic embryos by the four-cell stage, such as chromosome deletions and translocations. The formation of a derivative chromosome 17 showing centromeric fusion with a fragment of chromosome 16 is illustrated in Fig. 6H,I (inset and arrowheads). These results indicate that loss of ATRX induces abnormal centromeric mitotic recombination events that initially lead to centromeric breaks and chromosome fusions during subsequent cleavage division as a mechanism of large-scale chromosomal rearrangements in ATRX-deficient embryos.
Lack of ATRX function elicits chromosome instability with overexpression of centromeric cohesion and DNA repair factors in the cleavage-stage embryo
Analysis of histone H3 phosphorylated at serine 10 (H3S10ph), a maternal-specific epigenetic mark during the first mitosis (Almouzni and Probst, 2011; Hayashi-Takanaka et al., 2011), revealed the presence of abnormal segregation of maternal chromosomes in ATRX-deficient embryos, as determined by detection of lagging chromosomes of maternal origin stained with histone H3S10ph (Fig. 7B, arrowhead). Maternal chromosomes frequently remained excluded from the newly formed interphase nuclei of the early two-cell embryo in which the nuclear compartmentalization of paternal and maternal genomes appeared to be disrupted (Fig. 7B). Notably, quantitative transcriptional profiling of ATRX-deficient embryos using pathway-focused PCR arrays revealed a larger than twofold upregulation in transcript levels for aurora kinase B (P<0.05), DNA methyltransferase 1 (DNMT1; P<0.01), the ubiquitin-protein ligase DAZ-interacting zinc finger protein (DZIP3; P<0.01), the histone methyltransferase (EHMT1; P<0.05), as well as the centromeric cohesin acetyltransferase ESCO2 (P<0.05) compared with the transcript levels observed in control wild-type embryos (Fig. 7C,D). Consistent with our transcript analysis, ATRX-deficient embryos also exhibit overexpression of centromeric aurora kinase B protein at the four-cell stage (Fig. 7E, insets). These results provide the first evidence indicating that loss of ATRX function activates a crucial pathway to signal the presence of chromosome instability by increasing the expression of centromeric cohesion and DNA repair proteins.
Fig. 7.

ATRX functions to regulate a cellular response to chromosome instability in the cleavage-stage embryo. (A) Localization of histone H3 phosphorylation (H3S10ph; red) in the maternal genome (bold arrow) in wild-type zygotes. Epigenetic asymmetry results in a distinct nuclear compartmentalization of the maternal genome (thick arrow) during the formation of the interphase nucleus at the two-cell stage. H3S10ph staining is absent in the paternal genome (thin arrow). (B) Loss of ATRX results in the presence of lagging maternal chromosomes stained with H3S10ph (arrowhead). Scale bar: 10 µm. (C) Loss of ATRX results in increased expression of transcripts for centromere cohesion and DNA repair factors (**P<0.05, ***P<0.01). Data are from n=100 oocytes per replicate following three independent experiments. (D) Heat map of a panel of 84 chromatin-modifying enzymes analyzed at metaphase II stage. (E) ATRX-deficient embryos exhibit an overexpression of centromeric aurora kinase B protein (green) during mitosis at the four-cell stage (insets). Scale bars: 10 µm; 5 µm (inset).
DISCUSSION
A growing body of evidence obtained in both mouse and human cells indicates that ATRX is essential for maintenance of chromosome stability during mitosis and meiosis (De La Fuente et al., 2004; Ritchie et al., 2008; Baumann et al., 2010). A high frequency of human pancreatic neuroendocrine tumors and pediatric glioblastomas exhibit loss of ATRX function (Elsasser et al., 2011; Jiao et al., 2011; Molenaar et al., 2012) and mutations in the human ATRX gene have been recently detected in aggressive high-stage neuroblastomas that exhibit complex chromosomal rearrangements and chromothripsis, an extreme form of chromosome shattering (Jones and Jallepalli, 2012; Molenaar et al., 2012). However, the molecular mechanisms of ATRX function and its role in maintenance of chromosome stability are not known at present. Here, we provide new evidence indicating that maternal inheritance of ATRX is essential for centromere stability and transcriptional heterochromatin silencing at the onset of mammalian development.
Our studies indicate that ATRX is required for transcriptional repression of major satellite transcripts in the maternal genome and to prevent deleterious centromeric mitotic recombination events at minor satellite DNA sequences during the transition to the first mitosis. Moreover, we provide evidence indicating that loss of ATRX induces DNA double-strand breaks and large-scale chromosomal rearrangements by the four-cell stage. The enhanced post-zygotic centromeric recombination at minor satellites observed in transgenic embryos activates a pathway that increases the expression of centromeric cohesion and DNA repair proteins, suggesting a role for ATRX in regulating a potential functional crosstalk between pericentric heterochromatin and the epigenetic control of centromere function. Our studies demonstrate that ATRX participates in the establishment of epigenetic asymmetry of pericentric heterochromatin domains during the first mitosis and uncover a previously unrecognized kinetochore size asymmetry established by inheritance of CENP-C from the maternal genome. Collectively, these results provide novel insight into the cellular response of the early mammalian conceptus to chromosome instability and reveal that centromere fission is a major mechanism contributing to the formation of large-scale chromosomal rearrangements and aneuploidy in the cleavage-stage embryo.
Role of ATRX in heterochromatin formation and transcriptional silencing of major satellite transcripts
In somatic cells, the chromatin-remodeling complex formed by DAXX and ATRX is essential for deposition of histone variants to specialized chromatin domains (Drane et al., 2010; Lewis et al., 2010). Our previous studies indicate that ATRX is required for the recruitment of DAXX to centromeric heterochromatin in the mammalian oocyte (Baumann et al., 2010). However, the role of ATRX during de novo heterochromatin formation in the cleavage-stage embryo remained to be determined. Our current results reveal that, together with H3K9me3 and H4K20me3 (Santos et al., 2005; Puschendorf et al., 2008; Probst and Almouzni, 2011), ATRX functions as a transcriptionally repressive chromatin factor that confers epigenetic asymmetry to maternal centromeric domains. However, in contrast to other epigenetic marks that exhibit an exclusive association with the maternal genome, ATRX exhibits a transient and dynamic nuclear localization in the paternal genome, switching from a diffuse nucleoplasmic localization during the initial stages of pronuclear formation to an exclusive association with pericentric heterochromatin. Here, it is required to recruit DAXX to the perinucleolar heterochromatin rim, initially to the paternal pronucleus, and subsequently to the heterochromatin domains of both male and female pronuclei prior to syngamy. Thus, although a protein of maternal origin, ATRX actively participates in regulating the molecular composition of pericentric heterochromatin in the paternal genome.
In the pre-implantation embryo, heterochromatin formation takes place during a period of extensive genome-wide chromatin remodeling, and histone H3.3 is essential for the establishment of heterochromatin domains (Puschendorf et al., 2008; Santenard et al., 2010; Probst and Almouzni, 2011; Lin et al., 2013, 2014). Previous studies using RNA-FISH and genetic polymorphisms have demonstrated a preferential expression of major satellite transcripts from the paternal genome in the zygote stage embryo (Puschendorf et al., 2008; Probst et al., 2010; Santenard et al., 2010; Casanova et al., 2013). However, the mechanisms responsible for maintaining transcriptional repression of major satellite transcripts in the maternal pronucleus during this crucial developmental transition remain to be determined. Here, we provide novel evidence indicating that loss of ATRX function results in the formation of a transcriptionally permissive chromatin environment and de-regulated transcription of major satellite transcripts in the maternal genome. Thus, ATRX is required for silencing of major satellite transcripts in the maternal pronucleus.
Studies in somatic cells reveal that major satellite transcripts are under a tight transcriptional regulation. Basal levels of expression of non-coding centromeric RNAs are essential for chromosome stability and the establishment of crucial epigenetic marks required for centromere function (Bouzinba-Segard et al., 2006; Ferri et al., 2009; Chan and Wong, 2012). However, the excessive accumulation of these transcripts induces changes in heterochromatin formation and results in abnormal epigenetic modifications at both PCH and kinetochore domains disrupting chromosome segregation (Bouzinba-Segard et al., 2006; Ferri et al., 2009; Chan and Wong, 2012). Consistent with these studies, abnormal major satellite transcription in ATRX-deficient embryos is associated with centromere instability through a mechanism involving enhanced centromeric mitotic recombination leading to the formation of sister chromatid exchanges. Our results indicate that major satellite transcripts might be involved in the epigenetic control of centromere function during this crucial period of genome reprogramming, as indicated by the presence of lagging chromosomes of maternal origin observed in ATRX-deficient embryos. These results also demonstrate that silencing of centromeric transcripts in the maternal pronucleus is required for the maintenance of chromosome stability during the transition to the first mitosis.
Post-meiotic epigenetic asymmetry in the mammalian centromere
The kinetics of ATRX nuclear localization in the one-cell embryo suggests a role in heterochromatin formation in both the male and female pronucleus. However, during the first mitosis of the zygote, the association of ATRX with pericentric heterochromatin in the paternal chromosome complement is drastically reduced. These results indicate that the chromosomal localization of ATRX is highly dynamic and that a developmentally programmed removal of ATRX from the paternal chromosomes, as well as the resulting centromere epigenetic asymmetry, might be of functional significance for de novo heterochromatin formation in the cleavage-stage embryo.
Importantly, our studies also revealed that the differences between the paternal and maternal chromosome complements extend not only to transcriptionally repressive heterochromatin marks (Santos et al., 2005; Puschendorf et al., 2008; Probst and Almouzni, 2011) but also to a striking kinetochore size asymmetry resulting from maternal inheritance and enrichment of the kinetochore-specific protein CENP-C on maternal chromosomes. CENP-C is essential for kinetochore assembly and accurate chromosome-microtubule interactions (Gopalakrishnan et al., 2009; Przewloka et al., 2011). Establishment and maintenance of kinetochore size in somatic cells is determined by deposition of CENP-C (Tomkiel et al., 1994) and studies using Xenopus oocyte extracts indicate that, in contrast to most kinetochore proteins, CENP-C is assembled at paternal kinetochores only shortly after fertilization (Milks et al., 2009).
Asymmetry in kinetochore components has been recently described in diploid yeast S. cerevisiae (Thorpe et al., 2009), where kinetochore proteins such as MTW1, NDC10 and CTF19 are twice as abundant in the kinetochore of mother cells, demonstrating that this is a feature of the whole kinetochore and not only a particular protein (Thorpe et al., 2009). In yeast, this unique form of post-meiotic kinetochore asymmetry was observed only in the first division immediately after meiosis, where it may be required to segregate chromosomes in a specific pattern (Thorpe et al., 2009). In the mammalian embryo, differences in maternal and paternal kinetochores persist until the second mitotic division, suggesting a requirement for progressive changes in kinetochore domains during the oocyte to embryo transition. The function of kinetochore asymmetry is not yet known but it might be involved in facilitating the segregation of sister chromatids to daughter cells during the first mitosis to allow for the conservation of epigenetic differences at maternal and paternal chromosomes. Importantly, this mechanism might contribute to maintaining the spatial separation of parental genomes at the two-cell stage by ensuring the co-segregation of paternal and or maternal chromosomes on the basis of distinctive epigenetic marks and parental origin. In the interphase nuclei of the two-cell embryo, distinct chromatin marks as well as major satellite transcripts reveal the polar distribution of paternal and maternal genomes (Mayer et al., 2000a; Santos et al., 2005; Puschendorf et al., 2008; Probst and Almouzni, 2011; Casanova et al., 2013). Maintaining this unique type of nuclear organization may be crucial to allow for parent-specific reprogramming and chromatin remodeling processes to take place during subsequent cell divisions until epigenetic equivalency is established in the pre-implantation embryo. Our results indicate that post-meiotic kinetochore asymmetry may be required to co-segregate the full complement of paternal and maternal chromatids as separate units to each daughter cell during the first cleavage division. Importantly, both epigenetic and kinetochore asymmetry during the first mitosis may impose unique requirements for the establishment of accurate chromosome-microtubule interactions that render the first cleavage division susceptible to chromosome segregation defects.
Centromere mitotic recombination and chromosome instability in the cleavage-stage embryo
We have previously demonstrated that loss of ATRX function induces centromeric breaks and micronuclei formation during the transition to the first mitosis in the mouse embryo (Baumann et al., 2010). However, the molecular mechanisms of centromere instability remained to be determined. Here, we provide evidence that centromeric breaks in ATRX-deficient embryos are due to enhanced centromeric mitotic recombination events at minor satellite sequences during the first cleavage division. Notably, unresolved double-strand DNA breaks lead to the formation of both centromeric and interstitial chromosome breaks, and micronuclei, resulting in large-scale chromosomal rearrangements in the form of deletions and translocations detectable by the four-cell stage. Elegant studies conducted in mouse embryonic stem cells have recently demonstrated that loss of the DNA methyltransferases DNMT3A/3B and DNA methylation at PCH results in an increased rate of centromeric mitotic recombination (Jaco et al., 2008). Our results indicate that abnormal pericentric heterochromatin formation due to loss of ATRX function induced increased mitotic recombination at minor satellite sequences and suggest a crosstalk between PCH and the epigenetic control of centromere function.
In somatic cells, DNA damage at PCH elicits a relocation of DSBs to the periphery of heterochromatin domains to facilitate repair of DNA lesions at this nuclear domain and mitigate illegitimate mitotic recombination at highly repetitive sequences (Jakob et al., 2011; Chiolo et al., 2011). However, persistent DNA damage has been found localized at both heterochromatin and euchromatin regions where the presence of unrepaired DNA lesions result in the generation of chromosomal breaks (Asaithamby et al., 2011). Similar mechanisms might be responsible for the spreading of chromosomal breaks to the interstitial segments of chromosomes at the four-cell stage. The resulting large-scale chromosomal rearrangements lead to the formation of non-recurrent translocations that are consistent with the presence of chromosome breakage-fusion-bridge cycles in ATRX-deficient embryos and inform the mechanisms inducing segmental chromosome aberrations in the pre-implantation embryo.
How the embryo responds to chromosomal breaks and DNA damage is unknown. However, our results reveal novel molecular pathways activated by chromosome instability during pre-implantation development. It is possible that the increase in ESCO2 transcript levels in ATRX-deficient zygotes is a consequence of DNA damage at heterochromatin domains. In addition to their well-characterized role in sister chromatid cohesion (Losada and Hirano, 2005), cohesin proteins are also required for post-replicative repair of DSBs (Unal et al., 2007; Caron et al., 2012). For example, the yeast cohesin acetyl transferase ECO1 is loaded around centromeres and chromosome arms, and is overexpressed in response to unresolved DSBs to activate cohesion in broken centromeres and to prevent large-scale chromosome rearrangements (Unal et al., 2007; Watrin and Peters, 2009). Consistent with this notion, overexpression of Dnmt1 and Dzip3 transcripts in ATRX-deficient zygotes may be part of a cellular response to DNA damage, as DNMT1 has been recently implicated in the recruitment of DNA damage repair factors independently of its methyltransferase activity, and both DNMT1 and DZIP3 are essential for mounting an early response to DNA damage (Zhou et al., 2009; Ha et al., 2011). The striking accumulation of aurora kinase B protein at centromeric domains might be part of a global response to improper chromosome-microtubule interactions. Previous studies indicate that aurora kinase B can be enriched up to threefold at the centromeres of misaligned chromosomes in human somatic cells (Knowlton et al., 2006). In addition, abnormal chromosome segregation induced by loss of Suv39h1-dependent H3K9 methylation results in increased aurora kinase B activity in somatic cells where centromeric enrichment of aurora B may function to increase the efficiency of error correction (Chu et al., 2012). Our results demonstrate that the aurora B-dependent mechanisms to sense misaligned chromosomes are present in the cleavage-stage embryo.
In conclusion, our results reveal the functional activation of an ATRX-dependent epigenetic response to cope with abnormal chromosome-microtubule interactions during the first mitosis and allow DNA repair mechanisms at heterochromatin domains. This process requires an exquisite coordination of dynamic epigenetic modifications and DNA repair in response to chromosome breaks at pericentric heterochromatin. The unique nature of chromosome microtubule interactions imposed by epigenetic and kinetochore asymmetry may impose additional requirements to chromosome segregation during the first mitosis and render this transition susceptible to CIN. Importantly, our studies provide new insight into the molecular mechanisms of centromere instability that lead to complex chromosomal rearrangements and provide direct evidence that centromere fission is an important cause of chromosome instability in the mammalian cleavage-stage embryo.
Note added in proof
While this manuscript was under revision, Noh et al. reported a role for ATRX in the silencing of satellite transcripts in mammalian primary neurons (Noh et al., 2014).
MATERIALS AND METHODS
Oocyte collection and culture
This study was approved by and performed in accordance to guidelines of The University of Georgia Institutional Animal Care and Use Committee (IACUC). Generation of a transgenic RNAi model with an oocyte conditional deletion of Atrx has been previously described (Baumann et al., 2010). Oocyte collection, in vitro fertilization and embryo culture were conducted as described previously (Baumann et al., 2010). Briefly, matured metaphase II oocytes were collected from super ovulated F1 hybrid (C57BL/6/DBA) wild-type and transgenic Atrx-RNAi females (B6SJL/CF1), 13-15 h after injection with 5 IU of human chorionic gonadotropin (HCG; Gibco). Oocytes were fertilized using epididymal sperm obtained from C57BL/6/DBA males in 500 µl drops of minimal essential medium MEM (Gibco) supplemented with 3 mg/ml BSA (Sigma) under mineral oil (Sigma). Alternatively, oocytes were parthenogenetically activated in Ca2+-free M16 medium containing 10 mM SrCl2 and 5 µg/ml cytochalasin B (Sigma) for 6 h.
Fluorescence in situ hybridization and spectral karyotyping
Antibodies used for chromosome immunofluorescence are described in methods in the supplementary material. Following image acquisition, slides were processed for fluorescence in situ hybridization analyses using mouse DNA probes specific for the X and Y chromosome (Cambio) as previously described (Baumann et al., 2010). Spectral karyotyping was conducted on surface spread chromosomes fixed with methanol:glacial acetic acid (3:1) using the SkyPaint DNA probes for mouse chromosomes [Applied Spectral Imaging (ASI)]. Image acquisition was performed with a COOL-1300 SpectraCube camera (ASI) mounted on an Olympus BX43 microscope using a spectral karyotyping (SKY) optical filter (ASI). Metaphase chromosomes were analyzed using HiSKY v6.0 software.
RNA-FISH
RNA-FISH (fluorescence in situ hybridization) experiments were conducted using a directly labeled Cy3 pan-centromeric probe (Cambio) (Kanellopoulou et al., 2005) with minor modifications. Zona-free pronuclear-stage zygotes (14 hpf) were melted onto glass slides in a solution of 1% PFA, 0.15% Triton X-100 in DEPC-H2O and fixed in 4% PFA/PBS for 20 min. Pronuclei were then permeabilized with a solution of 0.5% Triton X-100 in DEPC-PBS for 2 min at room temperature and washed three times in 2×SSC. Control slides were treated with 100 µg/ml RNase A (Roche) for 1 h at 37°C and subsequently washed extensively in 2× SSC. Major satellite transcripts were detected using a Cy3-conjugated Pan-centromeric probe (Cambio) at a dilution of 1:10 in manufacturer-supplied hybridization buffer following probe denaturation at 80°C for 8 min. Sample hybridization was carried out under non-denaturing conditions overnight at 37°C in a humidified chamber prior to two post-hybridization washes in 1× SSC in DEPC-H2O for 5 min at room temperature and mounting in Vectashield plus DAPI.
RNA isolation and quantitative RT-PCR
Total RNA was isolated from groups of 70 diploid wild type and Atrx-RNAi transgenic parthenotes (14 hpf) using the miRNeasy kit (Qiagen) and subsequently subjected to reverse transcription using random hexamer primers and the Superscript III first-strand synthesis system (Invitrogen). The resulting cDNA was used for quantitative expression analysis of major satellite transcripts (Probst et al., 2010) by real-time PCR using 2× Brilliant II real-time PCR reagents (Agilent) and Agilent MX3005p equipment. Real-time PCR results were normalized against β-actin transcript levels as a housekeeping control (primers: β-actin-Fwd, 5′-gat atc gct gcg ctg gtc gtc-3′; β-actin-Rev, 5′-acg cag ctc att gta gaa ggt gtg g-3′).
Chromatin-modifying enzyme PCR array
Pathway-focused PCR arrays (RT2 Profiler PCR Array Mouse Epigenetic Chromatin Modification, Qiagen) were conducted to compare mRNA expression profiles of a panel of 84 chromatin-modifying enzymes in metaphase II oocytes obtained from Atrx-RNAi transgenic and wild-type littermate females in three experimental replicates. Groups of 100 oocytes were used for mRNA extraction using the Micro-FastTrack 2.0 kit (Invitrogen), pre-amplified using RT2 PreAMP cDNA synthesis reagents and a pathway-specific primer mix (Qiagen). Expression profiles were established on a Roche LightCycler 480 system using RT2 SYBR Green qPCR Mastermix reagents (Qiagen). Raw threshold cycle data were compared using RT2 Profiler PCR Array Data Analysis software Version 3.5 (SA Biosciences) to perform all ΔΔCt-based fold-change calculations. Pairwise comparison (t-test) between groups of experimental replicates was conducted to define the fold up- or downregulation and statistical significant thresholds (P<0.05).
Chromosome orientation-FISH (CO-FISH) for minor satellite sequences
CO-FISH analyses were conducted using strand-specific FITC-labeled minor satellite PNA probes, as previously described (Jaco et al., 2008) with minor modifications. Briefly, zygotes were cultured in MEM/BSA medium containing 10 µM BrdU at 4 hpf to ensure BrdU incorporation during S phase. Metaphase chromosome complements were then surface spread as described above. Samples were stained with 500 µg/ml Hoechst 33258 for 15 min at room temperature and BrdU-incorporated DNA was subsequently UV-nicked for 30 min in 2× SSC and digested using 3 U/µl Exonuclease III (NEB Biolabs) for 30 min at 37°C before denaturation at 75°C for 15 min. Hybridization with the FITC-labeled leading strand probes was conducted at room temperature for 48 h. Stringency washes were conducted in 50% formamide in 2× SSC buffer for 5 min at room temperature, followed by incubation in 2× SSC for 5 min at room temperature before DNA counterstaining and mounting.
Image analysis and laser scanning confocal microscopy
Quantitative scoring of kinetochore foci was conducted following evaluation of statistical image descriptors (Urani et al., 2013). Immunochemical detection of kinetochores was conducted in 3% BSA in PBT buffer overnight at 4°C, using CREST antiserum (Europa Bioproducts; 1:100) and rabbit anti-CENP-C [1:1000 (Pluta and Earnshaw, 1996)]. Confocal z-stack images were recorded at 0.5 µm intervals over a range of 5 µm using identical image acquisition parameters for all zygote metaphase spreads before computing maximum projection images for quantitative analysis. To quantify kinetochore size, identical thresholds were established for all zygote metaphases (n=10) before transformation to binary layers and analysis using the NIS Elements 4.0 Automated Measurements Module (Advanced Research; Nikon Instruments) with multi-dimensional imaging software. The Equivalent Diameter (EqDiameter), a measure of the diameter of a circle with the same area as the measured object [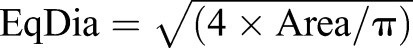 ], was recorded for each individual maternal (n=40) and paternal (n=40) CREST signal per metaphase spread, as distinguished by differential H3S10ph staining. Values were subjected to statistical analysis following calculation of the average EqDiameter per metaphase complement and corresponding standard deviations. In addition, average CREST signal intensities of the maternal and paternal complements were compared. Statistical significance testing was performed using Student's t-test with P<0.05.
], was recorded for each individual maternal (n=40) and paternal (n=40) CREST signal per metaphase spread, as distinguished by differential H3S10ph staining. Values were subjected to statistical analysis following calculation of the average EqDiameter per metaphase complement and corresponding standard deviations. In addition, average CREST signal intensities of the maternal and paternal complements were compared. Statistical significance testing was performed using Student's t-test with P<0.05.
Statistical analysis
Data presented as percentage values were analyzed by one-way analysis of variance (ANOVA) following arcsine transformation. Comparison of all pairs was conducted by the Tukey-Kramer HSD or Student's t-test using JMP Start Statistics (SAS Institute). Variation among individual replicates is indicated as the s.d. Differences were considered significant when P<0.05 and are indicated by different letters in the figures.
Supplementary Material
Acknowledgements
We thank Dr Maria Blasco and Dr W. Earnshaw for generously providing reagents, and we are grateful to Dr J. J. Eppig for insightful comments during manuscript preparation. We thank Julie M. Koeman for guidance with spectral karyotyping.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Author contributions
R.D.L.F., M.M.V. and C.B. conceived and designed the experiments; C.B. performed the experiments; C.B., M.M.V. and R.D.L.F. analyzed the data; M.M.V. and R.D.L.F. contributed reagents/materials/analysis tools; and M.M.V., C.B. and R.D.L.F. wrote the paper:
Funding
This research was supported by a grant from the National Institute of Child Health and Human Development (NICHD) National Institutes of Health (NIH) [2RO1-HD042740] and the Georgia Cancer Coalition to R.D.L.F. Research in M. M. Viveiros’ laboratory is supported by the NIH [HD071330]. Deposited in PMC for release after 12 months.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.118927/-/DC1
References
- Almouzni G. and Probst A. V. (2011). Heterochromatin maintenance and establishment: lessons from the mouse pericentromere. Nucleus 2, 332-338. 10.4161/nucl.2.5.17707 [DOI] [PubMed] [Google Scholar]
- Asaithamby A., Hu B. and Chen D. J. (2011). Unrepaired clustered DNA lesions induce chromosome breakage in human cells. Proc. Natl. Acad. Sci. USA 108, 8293-8298. 10.1073/pnas.1016045108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banaszynski L. A., Allis C. D. and Lewis P. W. (2010). Histone variants in metazoan development. Dev. Cell 19, 662-674. 10.1016/j.devcel.2010.10.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann C., Viveiros M. M. and De La Fuente R. (2010). Loss of maternal ATRX results in centromere instability and aneuploidy in the mammalian oocyte and pre-implantation embryo. PLoS Genet. 6, e1001137 10.1371/journal.pgen.1001137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean C. J., Hunt P. A., Millie E. A. and Hassold T. J. (2001). Analysis of a malsegregating mouse Y chromosome: evidence that the earliest cleavage divisions of the mammalian embryo are non-disjunction-prone. Hum. Mol. Genet. 10, 963-972. 10.1093/hmg/10.9.963 [DOI] [PubMed] [Google Scholar]
- Berube N. G. (2011). ATRX in chromatin assembly and genome architecture during development and disease. Biochem. Cell Biol. 89, 435-444. 10.1139/o11-038 [DOI] [PubMed] [Google Scholar]
- Bérubé N. G., Smeenk C. A. and Picketts D. J. (2000). Cell cycle-dependent phosphorylation of the ATRX protein correlates with changes in nuclear matrix and chromatin association. Hum. Mol. Genet. 9, 539-547. 10.1093/hmg/9.4.539 [DOI] [PubMed] [Google Scholar]
- Bouzinba-Segard H., Guais A. and Francastel C. (2006). Accumulation of small murine minor satellite transcripts leads to impaired centromeric architecture and function. Proc. Natl. Acad. Sci. USA 103, 8709-8714. 10.1073/pnas.0508006103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron P., Aymard F., Iacovoni J. S., Briois S., Canitrot Y., Bugler B., Massip L., Losada A. and Legube G. (2012). Cohesin protects genes against gamma H2AX induced by DNA double-strand breaks. PLoS Genet. 8, e1002460 10.1371/journal.pgen.1002460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casanova M., Pasternak M., El Marjou F., Le Baccon P., Probst A. V. and Almouzni G. (2013). Heterochromatin reorganization during early mouse development requires a single-stranded noncoding transcript. Cell Rep. 4, 1156-1167. 10.1016/j.celrep.2013.08.015 [DOI] [PubMed] [Google Scholar]
- Chan F. L. and Wong L. H. (2012). Transcription in the maintenance of centromere chromatin identity. Nucleic Acids Res. 40, 11178–11188. 10.1093/nar/gks921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez S. L., Loewke K. E., Han J., Moussavi F., Colls P., Munne S., Behr B. and Reijo Pera R. A. (2012). Dynamic blastomere behaviour reflects human embryo ploidy by the four-cell stage. Nat. Commun. 3, 1251 10.1038/ncomms2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiolo I., Minoda A., Colmenares S. U., Polyzos A., Costes S. V. and Karpen G. H. (2011). Double-strand breaks in heterochromatin move outside of a dynamic HP1a domain to complete recombinational repair. Cell 144, 732-744. 10.1016/j.cell.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu L., Zhu T., Liu X., Yu R., Bacanamwo M., Dou Z., Chu Y., Zou H., Gibbons G. H., Wang D. et al. (2012). SUV39H1 orchestrates temporal dynamics of centromeric methylation essential for faithful chromosome segregation in mitosis. J. Mol. Cell Biol. 4, 331-340. 10.1093/jmcb/mjs023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De La Fuente R., Viveiros M. M., Wigglesworth K. and Eppig J. J. (2004). ATRX, a member of the SNF2 family of helicase/ATPases, is required for chromosome alignment and meiotic spindle organization in metaphase II stage mouse oocytes. Dev. Biol. 272, 1-14. 10.1016/j.ydbio.2003.12.012 [DOI] [PubMed] [Google Scholar]
- De La Fuente R., Baumann C. and Viveiros M. M. (2011). Role of ATRX in chromatin structure and function: implications for chromosome instability and human disease. Reproduction 142, 221-234. 10.1530/REP-10-0380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drane P., Ouararhni K., Depaux A., Shuaib M. and Hamiche A. (2010). The death-associated protein DAXX is a novel histone chaperone involved in the replication-independent deposition of H3.3. Genes Dev. 24, 1253-1265. 10.1101/gad.566910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand-Dubief M. and Ekwall K. (2008). Heterochromatin tells CENP-A where to go. Bioessays 30, 526-529. 10.1002/bies.20763 [DOI] [PubMed] [Google Scholar]
- Elsasser S. J., Allis C. D. and Lewis P. W. (2011). New epigenetic drivers of cancers. Science 331, 1145-1146. 10.1126/science.1203280 [DOI] [PubMed] [Google Scholar]
- Fadloun A., Le Gras S., Jost B., Ziegler-Birling C., Takahashi H., Gorab E., Carninci P. and Torres-Padilla M.-E. (2013). Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat. Struct. Mol. Biol. 20, 332-338. 10.1038/nsmb.2495 [DOI] [PubMed] [Google Scholar]
- Ferri F., Bouzinba-Segard H., Velasco G., Hube F. and Francastel C. (2009). Non-coding murine centromeric transcripts associate with and potentiate Aurora B kinase. Nucleic Acids Res. 37, 5071-5080. 10.1093/nar/gkp529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons R. J. and Higgs D. R. (2000). Molecular-clinical spectrum of the ATR-X syndrome. Am. J. Med. Genet. 97, 204-212. 10.1002/1096-8628(200023)97:3<204::AID-AJMG1038>3.0.CO;2-X [DOI] [PubMed] [Google Scholar]
- Goldberg A. D., Banaszynski L. A., Noh K.-M., Lewis P. W., Elsaesser S. J., Stadler S., Dewell S., Law M., Guo X., Li X. et al. (2010). Distinct factors control histone variant H3.3 localization at specific genomic regions. Cell 140, 678-691. 10.1016/j.cell.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan S., Sullivan B. A., Trazzi S., Della Valle G. and Robertson K. D. (2009). DNMT3B interacts with constitutive centromere protein CENP-C to modulate DNA methylation and the histone code at centromeric regions. Hum. Mol. Genet. 18, 3178-3193. 10.1093/hmg/ddp256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K., Lee G. E., Palii S. S., Brown K. D., Takeda Y., Liu K., Bhalla K. N. and Robertson K. D. (2011). Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum. Mol. Genet. 20, 126-140. 10.1093/hmg/ddq451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T. and Hunt P. (2001). To err (meiotically) is human: the genesis of human aneuploidy. Nat. Rev. Genet. 2, 280-291. 10.1038/35066065 [DOI] [PubMed] [Google Scholar]
- Hassold T., Hall H. and Hunt P. (2007). The origin of human aneuploidy: where we have been, where we are going. Hum. Mol. Genet. 16, R203-R208. 10.1093/hmg/ddm243 [DOI] [PubMed] [Google Scholar]
- Hayashi-Takanaka Y., Yamagata K., Wakayama T., Stasevich T. J., Kainuma T., Tsurimoto T., Tachibana M., Shinkai Y., Kurumizaka H., Nozaki N. et al. (2011). Tracking epigenetic histone modifications in single cells using Fab-based live endogenous modification labeling. Nucleic Acids Res. 39, 6475-6488. 10.1093/nar/gkr343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaco I., Canela A., Vera E. and Blasco M. A. (2008). Centromere mitotic recombination in mammalian cells. J. Cell Biol. 181, 885-892. 10.1083/jcb.200803042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakob B., Splinter J., Conrad S., Voss K.-O., Zink D., Durante M., Lobrich M. and Taucher-Scholz G. (2011). DNA double-strand breaks in heterochromatin elicit fast repair protein recruitment, histone H2AX phosphorylation and relocation to euchromatin. Nucleic Acids Res. 39, 6489-6499. 10.1093/nar/gkr230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao Y., Shi C., Edil B. H., de Wilde R. F., Klimstra D. S., Maitra A., Schulick R. D., Tang L. H., Wolfgang C. L., Choti M. A. et al. (2011). DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 331, 1199-1203. 10.1126/science.1200609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M. J. K. and Jallepalli P. V. (2012). Chromothripsis: chromosomes in crisis. Dev. Cell 23, 908-917. 10.1016/j.devcel.2012.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C., Muljo S. A., Kung A. L., Ganesan S., Drapkin R., Jenuwein T., Livingston D. M. and Rajewsky K. (2005). Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 19, 489-501. 10.1101/gad.1248505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowlton A. L., Lan W. and Stukenberg P. T. (2006). Aurora B is enriched at merotelic attachment sites, where it regulates MCAK. Curr. Biol. 16, 1705-1710. 10.1016/j.cub.2006.07.057 [DOI] [PubMed] [Google Scholar]
- Kops G. J. P. L., Weaver B. A. A. and Cleveland D. W. (2005). On the road to cancer: aneuploidy and the mitotic checkpoint. Nat. Rev. Cancer 5, 773-785. 10.1038/nrc1714 [DOI] [PubMed] [Google Scholar]
- Kota S. K. and Feil R. (2010). Epigenetic transitions in germ cell development and meiosis. Dev. Cell 19, 675-686. 10.1016/j.devcel.2010.10.009 [DOI] [PubMed] [Google Scholar]
- Lewis P. W., Elsaesser S. J., Noh K.-M., Stadler S. C. and Allis C. D. (2010). Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc. Natl. Acad. Sci. USA 107, 14075-14080. 10.1073/pnas.1008850107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-J., Conti M. and Ramalho-Santos M. (2013). Histone variant H3.3 maintains a decondensed chromatin state essential for mouse preimplantation development. Development 140, 3624-3634. 10.1242/dev.095513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-J., Koh F. M., Wong P., Conti M. and Ramalho-Santos M. (2014). Hira-mediated H3.3 incorporation is required for DNA replication and ribosomal RNA transcription in the mouse zygote. Dev. Cell 30, 268-279. 10.1016/j.devcel.2014.06.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losada A. and Hirano T. (2005). Dynamic molecular linkers of the genome: the first decade of SMC proteins. Genes Dev. 19, 1269-1287. 10.1101/gad.1320505 [DOI] [PubMed] [Google Scholar]
- Lovejoy C. A., Li W., Reisenweber S., Thongthip S., Bruno J., de Lange T., De S., Petrini J. H. J., Sung P. A., Jasin M. et al. (2012). Loss of ATRX, genome instability, and an altered DNA damage response are hallmarks of the alternative lengthening of telomeres pathway. PLoS Genet. 8, e1002772 10.1371/journal.pgen.1002772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer W., Smith A., Fundele R. and Haaf T. (2000a). Spatial separation of parental genomes in preimplantation mouse embryos. J. Cell Biol. 148, 629-634. 10.1083/jcb.148.4.629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer W., Niveleau A., Walter J., Fundele R. and Haaf T. (2000b). Embryogenesis: demethylation of the zygotic paternal genome. Nature 403, 501-502. 10.1038/35000656 [DOI] [PubMed] [Google Scholar]
- Milks K. J., Moree B. and Straight A. F. (2009). Dissection of CENP-C–directed centromere and kinetochore assembly. Mol. Biol. Cell 20, 4246-4255. 10.1091/mbc.E09-05-0378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molenaar J. J., Koster J., Zwijnenburg D. A., van Sluis P., Valentijn L. J., van der Ploeg I., Hamdi M., van Nes J., Westerman B. A., van Arkel J. et al. (2012). Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 483, 589-593. 10.1038/nature10910 [DOI] [PubMed] [Google Scholar]
- Noh K.-M., Maze I., Zhao D., Xiang B., Wenderski W., Lewis P. W., Shen L., Li H. and Allis C. D. (2014). ATRX tolerates activity-dependent histone H3 methyl/phos switching to maintain repetitive element silencing in neurons. Proc. Natl. Acad. Sci. USA. 10.1073/pnas.1411258112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald J., Engemann S., Lane N., Mayer W., Olek A., Fundele R., Dean W., Reik W. and Walter J. (2000). Active demethylation of the paternal genome in the mouse zygote. Curr. Biol. 10, 475-478. 10.1016/S0960-9822(00)00448-6 [DOI] [PubMed] [Google Scholar]
- Pluta A. F., Earnshaw W. C. (1996). Specific interaction between human kinetochore protein CENP-C and a nucleolar transcriptional regulator. J. Biol. Chem. 271, 18767-18774. 10.1074/jbc.271.31.18767 [DOI] [PubMed] [Google Scholar]
- Probst A. V. and Almouzni G. (2011). Heterochromatin establishment in the context of genome-wide epigenetic reprogramming. Trends Genet. 27, 177-185. 10.1016/j.tig.2011.02.002 [DOI] [PubMed] [Google Scholar]
- Probst A. V., Okamoto I., Casanova M., El Marjou F., Le Baccon P. and Almouzni G. (2010). A strand-specific burst in transcription of pericentric satellites is required for chromocenter formation and early mouse development. Dev. Cell 19, 625-638. 10.1016/j.devcel.2010.09.002 [DOI] [PubMed] [Google Scholar]
- Przewloka M. R., Venkei Z., Bolanos-Garcia V. M., Debski J., Dadlez M. and Glover D. M. (2011). CENP-C is a structural platform for kinetochore assembly. Curr. Biol. 21, 399-405. 10.1016/j.cub.2011.02.005 [DOI] [PubMed] [Google Scholar]
- Puschendorf M., Terranova R., Boutsma E., Mao X., Isono K.-i., Brykczynska U., Kolb C., Otte A. P., Koseki H., Orkin S. H. et al. (2008). PRC1 and Suv39h specify parental asymmetry at constitutive heterochromatin in early mouse embryos. Nat. Genet. 40, 411-420. 10.1038/ng.99 [DOI] [PubMed] [Google Scholar]
- Ritchie K., Seah C., Moulin J., Isaac C., Dick F. and Berube N. G. (2008). Loss of ATRX leads to chromosome cohesion and congression defects. J. Cell Biol. 180, 315-324. 10.1083/jcb.200706083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robberecht C., Vanneste E., Pexsters A., D'Hooghe T., Voet T. and Vermeesch J. R. (2010). Somatic genomic variations in early human prenatal development. Curr. Genomics 11, 397-401. 10.2174/138920210793175967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santenard A. and Torres-Padilla M.-E. (2009). Epigenetic reprogramming in mammalian reproduction: contribution from histone variants. Epigenetics 4, 80-84. 10.4161/epi.4.2.7838 [DOI] [PubMed] [Google Scholar]
- Santenard A., Ziegler-Birling C., Koch M., Tora L., Bannister A. J. and Torres-Padilla M.-E. (2010). Heterochromatin formation in the mouse embryo requires critical residues of the histone variant H3.3. Nat. Cell Biol. 12, 853-862. 10.1038/ncb2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos F., Peters A. H., Otte A. P., Reik W. and Dean W. (2005). Dynamic chromatin modifications characterise the first cell cycle in mouse embryos. Dev. Biol. 280, 225-236. 10.1016/j.ydbio.2005.01.025 [DOI] [PubMed] [Google Scholar]
- Thorpe P. H., Bruno J. and Rothstein R. (2009). Kinetochore asymmetry defines a single yeast lineage. Proc. Natl. Acad. Sci. USA 106, 6673-6678. 10.1073/pnas.0811248106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomkiel J., Cooke C. A., Saitoh H., Bernat R. L. and Earnshaw W. C. (1994). CENP-C is required for maintaining proper kinetochore size and for a timely transition to anaphase. J. Cell Biol. 125, 531-545. 10.1083/jcb.125.3.531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unal E., Heidinger-Pauli J. M. and Koshland D. (2007). DNA double-strand breaks trigger genome-wide sister-chromatid cohesion through Eco1 (Ctf7). Science 317, 245-248. 10.1126/science.1140637 [DOI] [PubMed] [Google Scholar]
- Urani C., Corvi R., Callegaro G. and Stefanini F. M. (2013). Objective scoring of transformed foci in BALB/c 3T3 cell transformation assay by statistical image descriptors. Toxicol. In Vitro 27, 1905-1912. 10.1016/j.tiv.2013.06.011 [DOI] [PubMed] [Google Scholar]
- Vanneste E., Voet T., Le Caignec C., Ampe M., Konings P., Melotte C., Debrock S., Amyere M., Vikkula M., Schuit F. et al. (2009). Chromosome instability is common in human cleavage-stage embryos. Nat. Med. 15, 577-583. 10.1038/nm.1924 [DOI] [PubMed] [Google Scholar]
- Verdaasdonk J. S. and Bloom K. (2011). Centromeres: unique chromatin structures that drive chromosome segregation. Nat. Rev. Mol. Cell Biol. 12, 320-332. 10.1038/nrm3107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voet T., Vanneste E., Van der Aa N., Melotte C., Jackmaert S., Vandendael T., Declercq M., Debrock S., Fryns J.-P., Moreau Y. et al. (2011). Breakage–fusion–bridge cycles leading to inv dup del occur in human cleavage stage embryos. Hum. Mutat. 32, 783-793. 10.1002/humu.21502 [DOI] [PubMed] [Google Scholar]
- Watrin E. and Peters J.-M. (2009). The cohesin complex is required for the DNA damage-induced G2/M checkpoint in mammalian cells. EMBO J. 28, 2625-2635. 10.1038/emboj.2009.202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W., Wang X. and Rosenfeld M. G. (2009). Histone H2A ubiquitination in transcriptional regulation and DNA damage repair. Int. J. Biochem. Cell Biol. 41, 12-15. 10.1016/j.biocel.2008.09.016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.