

Abstract
Objective: Aberrant plasminogen activator inhibitor-1 (PAI-1) expression and activity have been implicated in bleeding disorders, multiorgan fibrosis, and wound healing anomalies. This study details the physiological consequences of targeted PAI-1 functional inhibition on cutaneous injury repair.
Approach: Dorsal skin wounds from FVB/NJ mice, created with a 4 mm biopsy punch, were treated topically with the small-molecule PAI-1 antagonist tiplaxtinin (or vehicle control) for 5 days and then analyzed for markers of wound repair.
Results: Compared to controls, tiplaxtinin-treated wounds displayed dramatic decreases in wound closure and re-epithelialization. PAI-1 immunoreactivity was evident at the migratory front in all injury sites indicating these effects were due to PAI-1 functional blockade and not PAI-1 expression changes. Stimulated HaCaT keratinocyte migration in response to recombinant PAI-1 in vitro was similarly attenuated by tiplaxtinin. While tiplaxtinin had no effect on keratinocyte proliferation, cell cycle progression, or apoptosis, it effectively reduced collagen deposition, the number of Ki-67+ fibroblasts, and incidence of differentiated myofibroblasts (i.e., smooth muscle α-actin immunoreactive cells), but not fibroblast apoptosis.
Innovation: The role for PAI-1 in hemostasis and fibrinolysis is established; involvement of PAI-1 in cutaneous wound healing, however, remains unclear. This study tests the effect of a small-molecule PAI-1 inhibitor in a murine model of skin wound repair.
Conclusion: Loss of PAI-1 activity significantly impaired wound closure. Re-epithelialization and fibroblast recruitment/differentiation were both reduced in tiplaxtinin-treated mice. Therapies directed at manipulation of PAI-1 expression and/or activity may have applicability as a treatment option for chronic wounds and scarring disorders.

Paul J. Higgins, PhD
Introduction
Plasminogen activator inhibitor-1 (PAI-1; SERPINE1), the major regulator of the pericellular proteolytic and fibrinolytic cascades, is a critical factor in the motile wound repair response in multiple cell lineages. PAI-1 is induced upon keratinocyte injury and restricted to the injury site where it is required for efficient keratinocyte migration in cell culture models of wound healing.1–3 These data are consistent with earlier findings that this SERPIN is among the most highly upregulated genes in the serum-activated wound-healing genomic program.4,5
Dysregulated PAI-1 expression is a causative factor in aberrant wound healing and fibrosis where this SERPIN may both dictate matrix turnover and the switch between the migratory and proliferative phenotypes.6 By acting as bait for urokinase and tissue-type plasminogen activators (uPA and tPA, respectively), PAI-1 limits the generation of plasmin and, subsequently, fibrin degradation.7 While the impact of PAI-1 in venous thrombosis and clotting disorders is well established, its role in the context of cutaneous injury is more paradoxical.8,9 PAI-1 and uPA are both upregulated in keratinocytes in healing wounds and highly expressed in cells adjacent to the denuded zone in cell culture models of epidermal injury.3,10 PAI-1 is deposited into keratinocyte migration trails; knockdown approaches, PAI-1 add-back rescue and use of neutralizing antibodies, and PAI-1−/− cells confirmed the requirement for PAI-1 in optimal monolayer wound repair.1,11 In recent years, factors (including PAI-1) that are involved in cell motility have been implicated in growth arrest in the “go or grow” proliferation/migration dichotomy.6,12 PAI-1 expressing keratinocytes at the wound margin are, in fact, less mitotically active than epithelial cohorts more distal from the injury site.1 While PAI-1−/− mice have accelerated wound closure,13 these genetically deficient mice are likely to have compensating mechanisms14 that could be avoided by targeted pharmacological approaches.
Several low-molecular-weight antagonists have been designed to assess the impact of PAI-1 functional inhibition in several in vitro and in vivo models of tissue injury. The well-studied small-molecule PAI-1 inhibitor, tiplaxtinin (PAI-039), which promotes a substrate-like conformation and PAI-1 cleavage, effectively reduces airway remodeling in an asthmatic mouse model and decreases the extent of hyperlipidemia, hyperglycemia, angiogenesis, and restenosis.15,16 To determine the requirement for the PAI-1 activity on cutaneous wound healing, tiplaxtinin was applied directly to the wound site immediately after and subsequent to injury initiation. Tiplaxtinin had no effect on keratinocyte proliferation or apoptosis, but significantly blocked wound closure and re-epithelialization by inhibiting keratinocyte migration and myofibroblast differentiation.
Clinical Problem Addressed
Aberrant wound healing, with its associated functional and cosmetic repercussions, affects the quality of life of millions of patients in the United States alone and is a major burden on the healthcare system. Dysregulated tissue levels of PAI-1 are common events in many repair anomalies, including persistent scarring and difficult-to-heal chronic wounds. Given the ubiquitous presence of PAI-1 in the injury field, small-molecule antagonists designed to attenuate the activity of this SERPIN have potential clinical utilization. Functional inhibition of PAI-1 with one such antagonist (tiplaxtinin) significantly inhibited wound closure, re-epithelialization, and myofibroblast differentiation. These findings suggest that targeting PAI-1 may be a therapeutic option in the context of pathogenic wound healing and fibrosis.
Materials and Methods
Ethics statement
All animal protocols were approved by the Institutional Animal Care and Use Committee of Albany Medical College (IACUC protocol #14-03003). Rodents were housed in the College Animal Resource Facility, which is licensed by the USDA and the NYS Department of Health.
In vivo wounding
Adult FVB/NJ mice (6–10 weeks of age; Jackson Laboratories, Bar Harbor, ME) were anesthetized and shaved; four full-thickness wounds were made on the dorsal skin using a sterile 4 mm biopsy punch as described.17 Animals were separated into two groups, control (vehicle only) and tiplaxtinin treated. Immediately following wounding, each site received 10 μL of either tiplaxtinin (6 mg/kg; 1.25 μL of a stock 48 mg/mL tiplaxtinin solution solubilized in dimethyl sulfoxide [DMSO] and diluted in phosphate-buffered saline [PBS]) or vehicle (1.25 μL DMSO diluted in PBS). Each wound was then additionally treated for 4 consecutive days. On the fifth day, mice were euthanized, wounds surgically excised and bisected, fixed in 4% paraformaldehyde, embedded in paraffin, and sectioned (5 μm).
Quantitation of wound closure and re-epithelialization
Percent wound closure and re-epithelialization were quantified using the freehand line-drawing tool in ImageJ. Wound closure was calculated using the formula=(No. of pixels closed wound−No. of pixels open wound)/(No. of pixels closed wound)×100. The length (pixels) of the epithelial tongue was measured from the wound edge to the migratory front on either side of each wound margin and averaged for each wound.
Immunohistochemistry
Paraffin-embedded tissue sections (5 μm) were stained with hematoxylin and eosin (H&E). For immunohistochemistry, sections were incubated with antibodies to smooth muscle α-actin (1:6,400 dilution; Sigma-Aldrich, St. Louis, MO), Ki-67 (1:200 dilution; Abcam, Cambridge, United Kingdom), and PAI-1 (1:1,000 dilution; Affinity Bioreagents, Waltham, MA), washed, incubated with biotinylated anti-rabbit IgG and avidin D horseradish peroxidase (HRP; 1:1,000 dilution), and color reactions developed using an ABC kit before mounting coverslips in the Vectashield medium (Vector Labs, Burlingame, CA). For terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, tissue sections were incubated with biotinylated nucleotides and labeled using streptavidin HRP according to the manufacturer's instructions (Promega, Madison, WI). Isotype IgG controls were included. Sections were analyzed using a Nanozoomer 2.0RS digital microscope equipped with NDP 2.2.1 software (Hamamatsu Photonics, Hamamatsu, Japan).
Cell culture
Immortalized human keratinocytes (HaCaT cells) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin in a humidified atmosphere of 5% CO2 at 37°C.
Construction of a PAI-1 adenoviral vector
A full-length PAI-1 cDNA insert was excised from pcDNA3.1, using 5′-KpnI and 3′-NotI, and cloned into the 5′-KpnI and 3′-NotI sites of pAdTrack-CMV. PAI-1 cDNA was subcloned into the pAdTrack-CMV vector to produce recombinant adenovirus as described.18
Adenoviral infection
Asychronously growing HaCaT keratinocytes were infected with adenoviral particles encoding either PAI-1 or GFP for 6 h in serum-free and antibiotic-free media containing polybrene (Invitrogen Technologies, Carlsbad, CA). Reduced serum (5% FBS) media were replaced, cells were allowed to reach confluence, and then scratch wounded.
Scratch-wound migration assay
Confluent HaCaT cells were switched to serum-free media before scratch wounding and concomitant treatment with 1 nM recombinant PAI-1 1 (rPAI-1, 14-1b; Molecular Innovations, Novi, MI) or 10 μM tiplaxtinin for 24 h. Endotoxin levels of recombinant PAI-1 were determined using the Limulus Amebocyte Lysate kit QCL-1000 (Lonza, Basel, Switzerland) and found to be >0.14 EU/mL, the acceptable threshold.19 To assess the effect of PAI-1 on monolayer wounding, cells were fixed with 4% paraformaldehyde 24 h following scratch injury, stained with crystal violet, and wound closure quantitated using an ocular grid.
Flow cytometry
For all flow cytometric experiments, fluorescence intensity was measured using a FACS LSRII (BD Biosciences, San Jose, CA). Ten thousand events were counted for each sample and the data analyzed using FlowJo software (Tree Star, Inc., Ashland, OR).
Cell cycle analysis
HaCaT cells were trypsin harvested, collected by centrifugation, washed twice with ice-cold PBS, and fixed in 70% ethanol for 1 h. Fixed cells were washed twice with ice-cold PBS and incubated for 2 h in the dark in PBS containing 0.1% Triton-X 100, 20 μg/mL propidium iodide (PI; Sigma-Aldrich), and 10 μg/mL RNase A.
Cleaved caspase-3
HaCaT cells were harvested by trypsin release and centrifugation, washed with 0.2% bovine serum albumin in PBS, and fixed with 4% paraformaldehyde for 15 min. Cells were permeabilized in 90% cold methanol for 30 min, washed twice, and incubated with antibodies to cleaved caspase-3 (1:200 dilution, Asp175; Cell Signaling, Danvers, MA) followed by the Alexa fluor 488-tagged secondary antibody (1:1,000 dilution) for 60 min each. Cells were washed and fluorescence measured as described above.
Statistical analysis
Values are represented as mean±SEM. Student's t-test was used to determine statistical significance for experiments containing two experimental conditions. One-way ANOVAs with a Tukey post hoc analysis were used for statistical analysis for experiments containing three or more experimental conditions. p-Values<0.05 were considered statistically significant.
Results
PAI-1 activity is required for wound closure and re-epithelialization
To evaluate the contribution of PAI-1 to cutaneous wound healing, punch biopsy excisional wounds were treated topically with the low-molecular-weight PAI-1 functional antagonist, tiplaxtinin, daily, for 4 days after initial delivery immediately following injury. Compared to vehicle-treated controls (Fig. 1A), tiplaxtinin application resulted in a significant decrease in injury site closure and contraction (Fig. 1B, D); some tiplaxtinin-treated wounds exhibited a complete closure failure (Fig. 1C) and a substantially reduced re-epithelialization (Fig. 1E).
Figure 1.
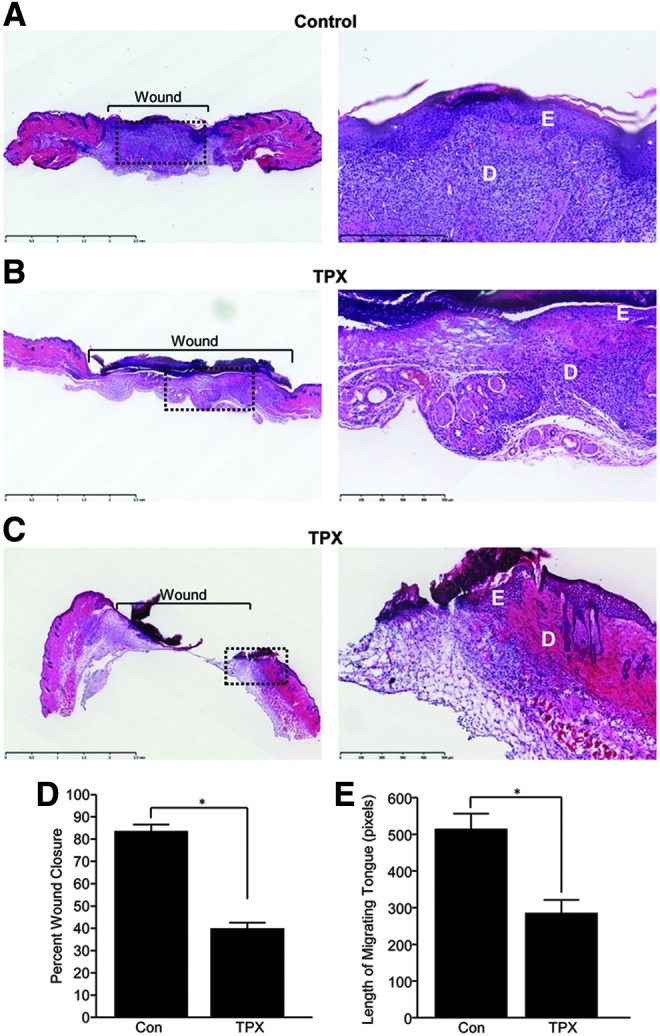
Inhibition of PAI-1 prevents wound closure and re-epithelialization. Paraffin sections (5 μm) of control (A) and tiplaxtinin-treated (B, C), 5 days postinjury, wounds were stained with H&E 5 days after wounding. Images were taken with a 10× (left; dashed box indicates location of magnified image) and 20× (right) objective (D, dermis; E, epidermis). (D) Quantitation of percent wound closure as described in the Materials and Methods section (data are presented as mean±SEM; n=5 in duplicate; *p<0.05). (E) Quantitation of re-epithelialization (data are presented as mean±SEM; n=5 in duplicate; *p<0.05). H&E, hematoxylin and eosin; PAI-1, plasminogen activator inhibitor-1; TPX, tiplaxtinin.
PAI-1 expression is evident in basal keratinocytes at the migratory front and stimulates wound closure in an in vitro model of injury repair
In both control and tiplaxtinin-treated wounds, PAI-1 expression is evident in both epithelial keratinocytes and dermal fibroblasts, particularly at the migratory front (Fig. 2A, B), consistent with previous findings10 and genomic profiling of the response to injury.20 PAI-1 expression is induced specifically at the migratory front in scrape-wounded cultured keratinocytes and is required for effective trauma site closure.3 Consistently, addition of a stable recombinant PAI-1 mutant (Fig. 2C) and adenoviral-mediated PAI-1 overexpression (Fig. 2D) stimulated injury resolution, while administration of tiplaxtinin, in contrast, significantly attenuated PAI-1-induced motility (Fig. 2E). Moreover, since PAI-1 is highly upregulated in response to the epidermal growth factor (EGF),21 it was important to evaluate the effect of PAI-1 functional blockade on EGF-stimulated motility. Tiplaxtinin inhibited migration stimulated by both recombinant PAI-1 (Fig. 2E) and EGF (Fig. 2F). These data suggest that the healing response in vivo (Fig. 1) is associated with increased PAI-1 expression and activity in the migratory keratinocyte cohort and these events can be recapitulated in an in vitro model of wound repair.
Figure 2.
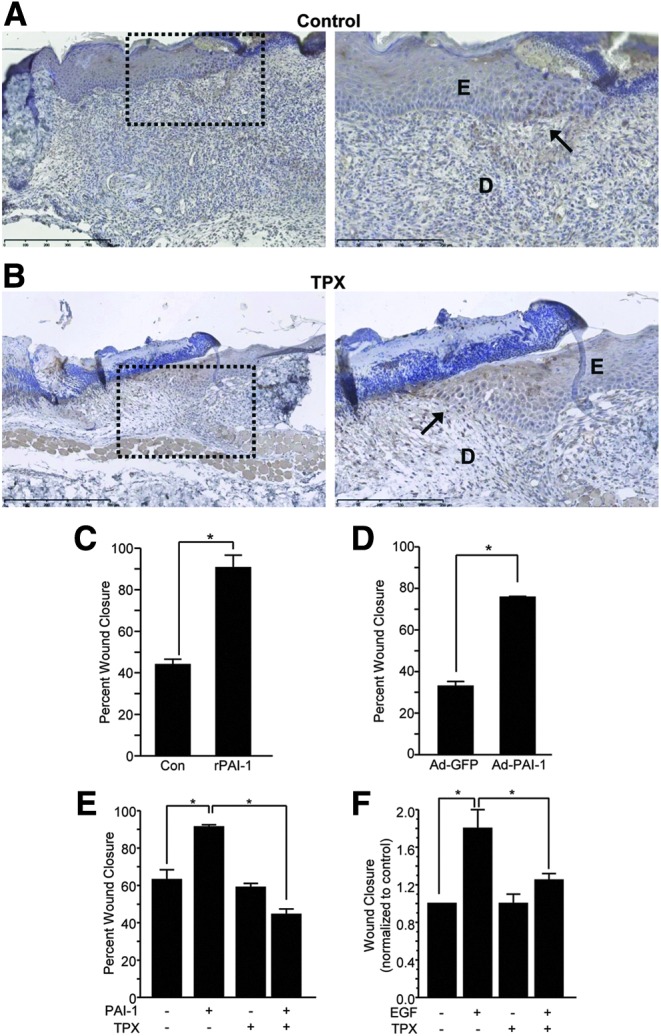
PAI-1 immunoreactivity is evident in basal keratinocytes at the migratory front. Paraffin sections (5 μm) of control (A) and tiplaxtinin-treated (B) wounds were probed with PAI-1 antibodies 5 days after wounding. Images were taken with a 10× (left; dashed box indicates location of magnified image) and 20× (right) objective (D, dermis; E, epidermis, wound edge is indicated by arrows). (C, E, F) PAI-1 (1 nM), EGF (10 ng/mL), and tiplaxtinin (10 μM) were added to HaCaT monolayers at the time of scratch injury in serum-free media. Cells were incubated for 24 h, fixed, stained with crystal violet, and wound closure quantitated using an ocular grid (data are presented as mean±SEM; *p<0.05). (D) Subconfluent cells were infected with adenoviral constructs encoding either GFP (control) or PAI-1. Cells were allowed to reach confluence, scratch wounded, incubated for 24 h, fixed, stained with crystal violet, and wound closure quantitated using an ocular grid (data are presented as mean±SEM; *p<0.05). EGF, epidermal growth factor.
PAI-1 inhibition has no effect on keratinocyte proliferation
Following injury, keratinocytes just distal to the wound edge undergo a proliferative burst, providing a cohort to support the migratory front.22 To determine if the small-molecule PAI-1 inhibitor altered keratinocyte growth, DNA content was analyzed in asynchronously growing HaCaT cells treated for 72 h with tiplaxtinin or vehicle alone. As compared to controls, cells incubated with tiplaxtinin did not exhibit changes in cell cycle progression (Fig. 3A). Control and tiplaxtinin-treated wounds were also stained with Ki-67. Consistent with normal healing, only the basal keratinocytes distal from the wound edge displayed a proliferative activity which was comparable between control and tiplaxtinin-treated mice (Fig. 3B, C). Moreover, there was no evidence of overt toxicity (necrosis) or cell death (i.e., sub-G1 keratinocytes), at least at the tiplaxtinin concentrations that effectively attenuated keratinocyte migration in vitro or tissue repair in vivo. Dermal fibroblast proliferation, in contrast, was significantly decreased as a function of tiplaxtinin treatment (Fig. 3B, D) as was the frequency of smooth muscle cell α-actin-positive cells (a differentiated myofibroblast marker) (Fig. 4A, B). Tiplaxtinin wounds had a less ordered collagen network compared to controls (Fig. 4C) and appeared less contracted (Fig. 1). Previous reports indicate impaired granulation tissue and deficient collagen deposition in diabetic wounds.23 These data suggest that pharmacologic inhibition of PAI-1 activity decreases fibroblast, but not keratinocyte, proliferation, decreases fibroblast-to-myofibroblast differentiation, and collagen deposition thereby modeling chronically impaired wound healing.
Figure 3.
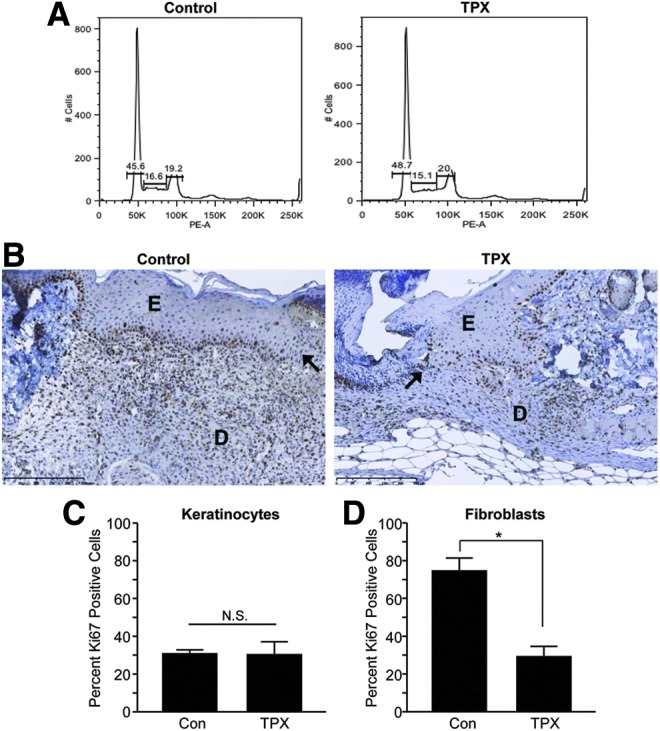
Tiplaxtinin has no effect on keratinocyte growth but inhibits fibroblast proliferation. (A) Subconfluent HaCaT cells were treated with 10 μM tiplaxtinin for 72 h. Cells were collected, stained with propidium iodide, and DNA content analyzed through FACS. (B) Paraffin sections (5 μm) of control and tiplaxtinin-treated wounds were stained with Ki-67 5 days after wounding. Images were taken with a 20× objective (D, dermis; E, epidermis, wound edge is indicated by arrows). (C, D) Quantitation of Ki-67-positive keratinocytes (C) versus fibroblasts (D) was calculated by direct cell counts in three different fields (data are presented as mean±SEM; *p<0.05).
Figure 4.
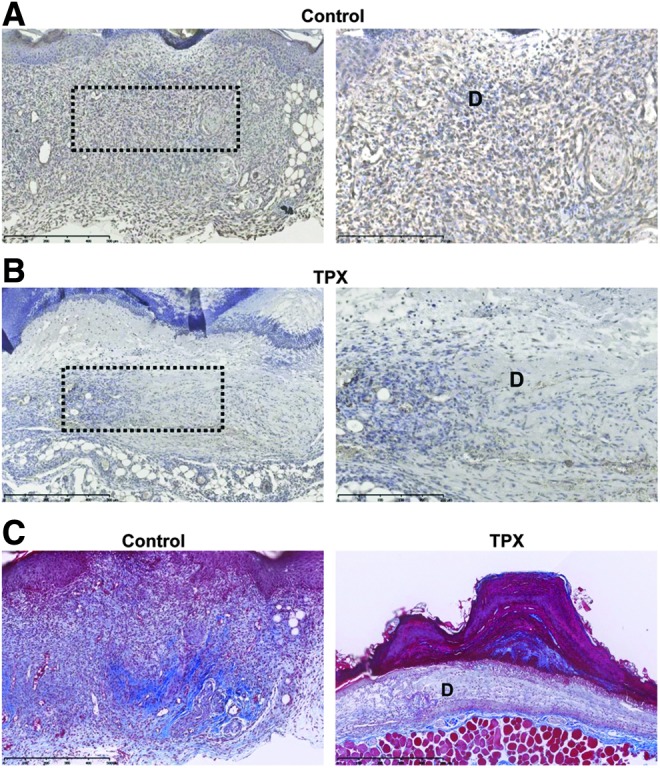
Tiplaxtinin reduces fibroblast differentiation. Paraffin sections (5 μm) of control (A) and tiplaxtinin-treated (B) wounds were stained with smooth muscle cell α-actin 5 days after wounding. Images were taken with a 10× (left; dashed box indicates location of magnified image) and 20× (right) objective (D, dermis). (C) Paraffin sections (5 μm) of control and tiplaxtinin-treated wounds were stained with trichrome reagent 5 days after wounding. Images were taken with a 10× objective (D, dermis).
Loss of PAI-1 activity does not alter wound bed cell apoptosis
One factor that might contribute to decreased re-epithelialization would be increased keratinocyte apoptosis as PAI-1 has been shown to be protective against cell death in various tissues and cell types.24–26 Both control and tiplaxtinin-treated wounds were subjected to TUNEL and were negative for apoptotic keratinocytes (compare Fig. 5A to B). Furthermore, tiplaxtinin treatment did not increase the apoptotic index of cultured keratinocytes as quantified by caspase-3 staining (Fig. 5E). Although a few apoptotic fibroblasts were evident, there was no discernible difference between control and tiplaxtinin-treated wounds, suggesting that pharmacologic PAI-1 inhibition has no effect on cutaneous cell survival.
Figure 5.
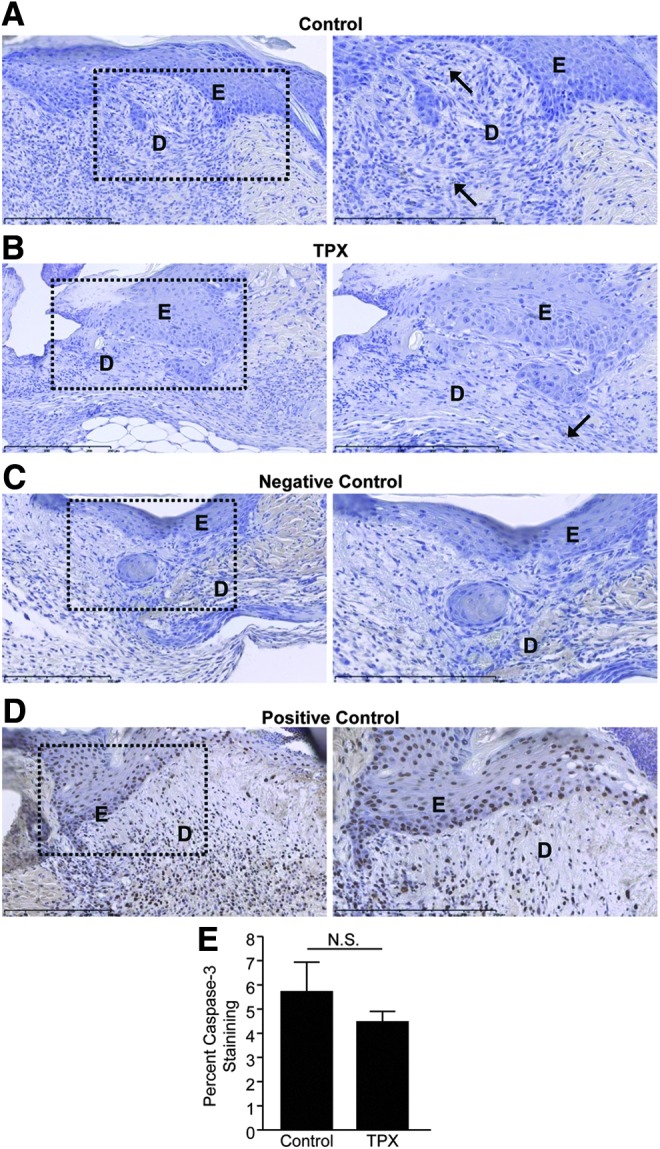
Tiplaxtinin does not alter wound site keratinocyte or fibroblast apoptotic index. Paraffin sections (5 μm) of control (A) and tiplaxtinin-treated (B) wounds were stained with TUNEL 5 days after wounding. In (A) and (B), arrows indicate TUNEL-positive apoptotic cells (C) No terminal deoxynucleotide transferase served as a negative control. (D) DNase treatment was used as a positive control. Images were taken with a 10× (left; dashed box indicates location of magnified image) and 20× (right) objective (D, dermis; E, epidermis). (E) Subconfluent HaCaT were pretreated with 10 μM tiplaxtinin or vehicle control for 24 h, as indicated. Cells were collected, stained with caspase-3, and analyzed by FACS. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Discussion
Wound resolution following cutaneous injury is a complex process that involves interactions, both spatially and temporally, between various cells and the repair site microenvironment. While most wounds heal rapidly and efficiently, many remain either chronic (e.g., diabetic lesions) or are pathologically remodeled (e.g., keloids, hypertrophic scars). The details as to the signaling components and cellular cues that drive repair remain incompletely understood.
A proliferative phase, characterized by keratinocyte proliferation and migration, serves to preliminarily re-epithelialize the injury site. Since PAI-1 has been implicated in mediating the go-or-grow migratory/proliferative switch,6 this study sought to determine the contribution of PAI-1 in cutaneous wound closure. Consistent with in vitro and in vivo models of wound healing,3,10 enhanced PAI-1 expression is evident at the migratory cohort of injured epidermal keratinocytes. The available data suggest that PAI-1 participates in the motile program by titering extracellular matrix proteolysis and altering the distribution of cellular adhesion molecules. In the early stages of wound healing, keratinocytes downregulate α3β1 and α6β4 integrins (to disrupt cell–cell contacts and dissociate hemidesmosomal adhesions) and upregulate vitronectin receptors (αvβ5, αvβ3).22,27 Vitronectin and other provisional matrix elements are deposited at the wound site; PAI-1, through vitronectin engagement, can displace uPAR and integrins, thereby stimulating keratinocyte detachment.28 A cryptic site on PAI-1 that binds the endocytic low-density lipoprotein receptor-related protein 1 (LRP1), moreover, is exposed upon complex formation with uPA.29 LRP1 endocytoses uPA/uPAR/PAI-1/integrin complexes; PAI-1 and uPA are lysosomally degraded, while uPAR and integrins are redistributed along the leading edge of migrating cells to promote cellular readhesion and focalize proteolytic activity.6,30 It is well appreciated that fibrin clot dissolution is required for effective keratinocyte migration.31 While tiplaxtinin reduces PAI-1-mediated uPA binding and protease inhibition, thereby potentially amplifying fibrinolysis, this small-molecule compound may also prevent cyclic deadhesion-/readhesion-driven keratinocyte motility. Together, these data suggest the effect of PAI-1 on cell migration and wound closure goes beyond its antiproteolytic role.
Concomitant with re-epithelialization, resident fibroblasts emerge from quiescence, begin to proliferate, and migrate into the wound bed within the provisional matrix.22 While wounds treated with tiplaxtinin did not exhibit changes in keratinocyte growth, fibroblasts appeared to have reduced proliferation. Once established at the injury site, fibroblasts differentiate into activated myofibroblasts, which stimulate wound contracture.32,33 Myofibroblast differentiation and persistence are governed by a feedback loop whereupon actin, bundling, and myosin-II light chain locomotion drives cellular contraction, concomitant with increases in tissue TGF-β1, and collagen deposition and remodeling.34 Elevated PAI-1, likely due to autocrine TGF-β1 production, is coincident with cutaneous and noncutaneous pathologically remodeled tissues (e.g., fibrosis, keloids and hypertrophic scars).14 Indeed, PAI-1−/− mice display reduced fibrosis and PAI-1 siRNA attenuates collagen levels in keloids.35,36 In addition to a decrease in smooth-muscle α-actin-positive myofibroblasts, tiplaxtinin-treated wounds appeared less contracted and have reduced organized collagen compared to control wounds. These data suggest that the PAI-1 activity promotes the myofibroblast contractile phenotype, is required for wound contracture, and reduced PAI-1 activity might lead to chronically impaired healing wounds as a function of deficient granulation tissue formation.
By directly inhibiting PAI-1 activity through topical tiplaxtinin application, it is apparent that the PAI-1 activity is bifunctionally required for efficient wound closure. Attenuation of PAI-1 activity inhibits re-epithelialization as a consequence of reduced keratinocyte motility and decreased wound contracture as a result of decreased fibroblast proliferation and differentiation. Clinically, enhanced PAI-1 expression has been detected in pathological wounds, including keloids, hypertrophic scars, and fibrosis of several organ systems,14 while deficient PAI-1 activity might lead to the development of chronic wounds. Therapies directed at manipulation of PAI-1 expression and/or activity, therefore, may have applicability as a treatment option for chronic wounds and scarring disorders.
Innovation
In injury systems, PAI-1 can promote or attenuate wound responses depending on levels of plasminogen activators, proteases/protease inhibitors, and the growth factor repertoire in the injury site microenvironment. Given the diversity of PAI-1 targets and the complexities associated with deficient or overexpression strategies, including likely compensatory mechanisms in both genetic contexts, it would appear that functional inhibitor studies could shed new light on the impact of PAI-1 in cutaneous wound repair. The principal innovation of this study was the direct application of a small-molecule PAI-1 inhibitor to murine wounds to assess the requirement for PAI-1 activity on injury resolution.
KEY FINDINGS.
• Inhibition of PAI-1 activity with a small-molecule antagonist, tiplaxtinin, inhibits cutaneous wound closure and wound re-epithelialization in vivo.
• Tiplaxtinin reduces PAI-1-stimulated keratinocyte migration in vitro while having no effect on epithelial proliferation, cell cycle progression, or apoptosis.
• In a well-characterized in vivo mouse model of cutaneous injury, tiplaxtinin-treated wounds were less contracted, an effect likely due to reduced dermal fibroblast proliferation/recruitment and/or myofibroblast differentiation.
Abbreviations and Acronyms
- DMSO
dimethyl sulfoxide
- EGF
epidermal growth factor
- FBS
fetal bovine serum
- H&E
hematoxylin and eosin
- HaCaT
immortalized human keratinocytes
- HRP
horseradish peroxidase
- PAI-1
plasminogen activator inhibitor-1
- PBS
phosphate-buffered saline
- PI
propidium iodide
- SEM
standard error of the mean
- TGF-β1
transforming growth factor-β1
- tPA
tissue-type plasminogen activator
- TPX
tiplaxtinin
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
- uPA
urokinase plasminogen activator
Acknowledgments and Funding Sources
This work was supported by NIH GM057242 to P.J.H., an AHA Fellowship (14PRE18170012) to T.M. Simone, and an NCI Fellowship (F31CA174198) to W.M.L.
Author Disclosure and Ghostwriting
The authors do not have any commercial or financial conflicts of interest to declare. This article was written exclusively by the authors and no ghostwriters were involved.
About the Authors
Tessa M. Simone and Whitney M. Longmate are senior graduate students in the Center for Cell Biology and Cancer Research at the Albany Medical College in Albany, New York. Brian K. Law, PhD, is a faculty member in the Department of Pharmacology and Therapeutics at the University of Florida School of Medicine in Gainesville, Florida. Paul J. Higgins, PhD, is codirector of the Center for Cell Biology and Cancer Research at Albany Medical College. Dr. Higgins' research focuses on the molecular mechanisms underlying TGF-β1 target gene transcription and their involvement in wound healing, angiogenesis, and fibrosis.
References
- 1.Providence KM, Higgins PJ. PAI-1 expression is required for epithelial cell migration in two distinct phases of in vitro wound repair. J Cell Physiol 2004;200:297–308 [DOI] [PubMed] [Google Scholar]
- 2.Providence KM, Higgins SP, Mullen A, et al. SERPINE1 (PAI-1) is deposited into keratinocyte migration “trails” and required for optimal monolayer wound repair. Arch Dermatol Res 2008;300:303–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Providence KM, Kutz SM, Staiano-Coico L, Higgins PJ. PAI-1 gene expression is regionally induced in wounded epithelial cell monolayers and required for injury repair. J Cell Physiol 2000;182:269–280 [DOI] [PubMed] [Google Scholar]
- 4.Iyer VR, Eisen MB, Ross DT, et al. The transcriptional program in the response of human fibroblasts to serum. Science 1999;283:83–87 [DOI] [PubMed] [Google Scholar]
- 5.Qi L, Allen RR, Lu Q, et al. PAI-1 transcriptional regulation during the G0—>G1 transition in human epidermal keratinocytes. J Cell Biochem 2006;99:495–507 [DOI] [PubMed] [Google Scholar]
- 6.Simone TM, Higgins CE, Czekay RP, et al. SERPINE1: a molecular switch in the proliferation-migration dichotomy in wound-“activated” keratinocytes. Adv Wound Care (New Rochelle) 2014;3:281–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dellas C, Loskutoff DJ. Historical analysis of PAI-1 from its discovery to its potential role in cell motility and disease. Thromb Haemost 2005;93:631–640 [DOI] [PubMed] [Google Scholar]
- 8.Lijnen HR. Pleiotropic functions of plasminogen activator inhibitor-1. J Thromb Haemost 2005;3:35–45 [DOI] [PubMed] [Google Scholar]
- 9.Diebold I, Kraicun D, Bonello S, Gorlach A. The ‘PAI-1 paradox’ in vascular remodeling. Thromb Haemost 2008;100:984–991 [PubMed] [Google Scholar]
- 10.Romer J, Lund LR, Eriksen J, et al. Differential expression of urokinase-type plasminogen activator and its type-1 inhibitor during healing of mouse skin wounds. J Invest Dermatol 1991;97:803–811 [DOI] [PubMed] [Google Scholar]
- 11.Providence KM, White LA, Tang J, et al. Epithelial monolayer wounding stimulates binding of USF-1 to an E-box motif in the plasminogen activator inhibitor type 1 gene. J Cell Sci 2002;115:3767–3777 [DOI] [PubMed] [Google Scholar]
- 12.Fedotov S Iomin A. Migration and proliferation dichotomy in tumor-cell invasion. Phys Rev Lett 2007;98:118101. [DOI] [PubMed] [Google Scholar]
- 13.Chan JC, Duszczyszyn DA, Castellino FJ, Ploplis VA. Accelerated skin wound healing in plasminogen activator inhibitor-1-deficient mice. Am J Pathol 2001;159:1681–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol 2012;227:493–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simone TM, Archambeault J, Higgins PJ. Small molecule targeting of PAI-1 function: a new therapeutic approach for treatment of vascular stenosis. J Mol Genet Med 2013;7:1000059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simone TM, Higgins PJ. Low molecular weight antagonists of plasminogen activator inhibitor-1: therapeutic potential in cardiovascular disease. Mol Med Ther 2012;1:101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longmate WM, Monichan R, Chu ML, et al. Reduced fibulin-2 contributes to loss of basement membrane integrity and skin blistering in mice lacking integrin alpha3beta1 in the epidermis. J Invest Dermatol 2014;134:1609–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Law ME, Corsino PE, Jahn SC, et al. Glucocorticoids and histone deacetylase inhibitors cooperate to block the invasiveness of basal-like breast cancer cells through novel mechanisms. Oncogene 2013;32:1316–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Degryse B, Neels JG, Czekay RP, et al. The low density lipoprotein receptor-related protein is a motogenic receptor for plasminogen activator inhibitor-1. J Biol Chem 2004;279:22595–22604 [DOI] [PubMed] [Google Scholar]
- 20.Fitsialos G, Chassot AA, Turchi L, et al. Transcriptional signature of epidermal keratinocytes subjected to in vitro scratch wounding reveals selective roles for ERK1/2, p38, and phosphatidylinositol 3-kinase signaling pathways. J Biol Chem 2007;282:15090–15102 [DOI] [PubMed] [Google Scholar]
- 21.Freytag J, Wilkins-Port CE, Higgins CE, et al. PAI-1 mediates the TGF-beta1+EGF-induced “scatter” response in transformed human keratinocytes. J Invest Dermatol 2010;130:2179–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martin P. Wound healing—aiming for perfect skin regeneration. Science 1997;276:75–81 [DOI] [PubMed] [Google Scholar]
- 23.Yue DK, Swanson B, McLennan S, et al. Abnormalities of granulation tissue and collagen formation in experimental diabetes, uraemia and malnutrition. Diabet Med 1986;3:221–225 [DOI] [PubMed] [Google Scholar]
- 24.Chen Y, Kelm RJ, Jr., Budd RC, Sobel BE, Schneider DJ. Inhibition of apoptosis and caspase-3 in vascular smooth muscle cells by plasminogen activator inhibitor type-1. J Cell Biochem 2004;92:178–188 [DOI] [PubMed] [Google Scholar]
- 25.Fang H, Placencio VR, Declerck YA. Protumorigenic activity of plasminogen activator inhibitor-1 through an antiapoptotic function. J Natl Cancer Inst 2012;104:1470–1484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bajou K, Peng H, Laug WE, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell 2008;14:324–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stefansson S, Lawrence DA. The serpin PAI-1 inhibits cell migration by blocking integrin alpha V beta 3 binding to vitronectin. Nature 1996;383:441–443 [DOI] [PubMed] [Google Scholar]
- 28.Czekay RP, Aertgeerts K, Curriden SA, Loskutoff DJ. Plasminogen activator inhibitor-1 detaches cells from extracellular matrices by inactivating integrins. J Cell Biol 2003;160:781–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stefansson S, Muhammad S, Cheng XF, et al. Plasminogen activator inhibitor-1 contains a cryptic high affinity binding site for the low density lipoprotein receptor-related protein. J Biol Chem 1998;273:6358–6366 [DOI] [PubMed] [Google Scholar]
- 30.Czekay RP, Kuemmel TA, Orlando RA, Farquhar MG. Direct binding of occupied urokinase receptor (uPAR) to LDL receptor-related protein is required for endocytosis of uPAR and regulation of cell surface urokinase activity. Mol Biol Cell 2001;12:1467–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Romer J, Bugge TH, Pyke C, et al. Impaired wound healing in mice with a disrupted plasminogen gene. Nat Med 1996;2:287–292 [DOI] [PubMed] [Google Scholar]
- 32.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol 2002;3:349–363 [DOI] [PubMed] [Google Scholar]
- 33.Hinz B, Phan SH, Thannickal VJ, et al. The myofibroblast: one function, multiple origins. Am J Pathol 2007;170:1807–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol 2007;179:1311–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu Y, Zhang Q, Ann DK, et al. Increased vascular endothelial growth factor may account for elevated level of plasminogen activator inhibitor-1 via activating ERK1/2 in keloid fibroblasts. Am J Physiol Cell Physiol 2004;286:C905–C912 [DOI] [PubMed] [Google Scholar]
- 36.Tuan TL, Wu H, Huang EY, et al. Increased plasminogen activator inhibitor-1 in keloid fibroblasts may account for their elevated collagen accumulation in fibrin gel cultures. Am J Pathol 2003;162:1579–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]