

Abstract
Pharmacological preconditioning (PC) with tadalafil, a PDE5A inhibitor, enhances protein kinase G-1 (PKG-I) activity, resulting in stem cell survival. Protection by PC had two different phases, early (2 h) and late (24 h). However, the mechanism of protection during these phases remained grossly unknown. Mesenchymal stem cells (MSCs) from adult male Fischer-344 rats were cultured and pretreated with tadalafil (100 μM) for an hour and subjected to 2 h of hypoxia (1% O2), followed by reoxygenation (HR: in vitro model mimicking ischemia/reperfusion). We observed (i) increased MSC survival with reduced cell cytotoxicity as revealed by low lactate dehydrogenase release and trypan blue staining, respectively, in tadalafil-treated cells upon HR; (ii) decrease in TUNEL positivity as well as caspase activity; (iii) an increase in pAkt/Akt, iNOS, eNOS, and pGSK3β/GSK3β during the early protection phase of PC, and this protection seemed to be a spontaneous adaptive response of MSCs against HR and was independent of tadalafil, whereas an increase in Bcl2/Bax was tadalafil dependent; and (iv) during the late phase, we observed phosphorylation of STAT3 at serine727, leading to its entry inside the nucleus and binding onto the promoter of PKG-I by three-fold (P<0.05). In conclusion, an increase in Bcl2/Bax during the early phase and transcriptional upregulation of PKG-I by STAT3 during the late phase were responsible for stem cell protection by tadalafil against ischemic injury.
Introduction
Ischemia/reperfusion (IR) initiates various cellular and molecular changes often resulting in myocardial infarction and cardiac dysfunction [1]. Inhibition of enzymes that are involved in cell pathology could be potential therapeutic targets, especially phosphodiesterases (PDE) [2]. cGMP is known to be catabolized to GMP by specific members of the PDE superfamily [3], and the most widely studied cGMP esterase is PDE5A in cardiovascular disease [4]. PDE5A inhibitors already have clinical implication to treat erectile dysfunction and are FDA-approved drugs. Moreover, the PDE5A presence in the myocardium makes it a promising target [5] for tadalafil. Tadalafil, a PDE5A inhibitor, is a long-lasting drug with half-life (t1/2) of ∼17 h, more than other members of the family such as sildenafil and vardenafil, which have 4 to 8 h, respectively. In addition, it is a highly selective inhibitor of PDE5A and has a profound role in cardioprotection [6].
Protein kinase G-I (PKG-I) is an effector kinase of cGMP, which phosphorylates intracellular proteins and further downregulates physiological functions, such as control of vascular tone, cell differentiation, proliferation, and platelet aggregation [7]. It is a serine/threonine protein kinase having two isozymes found in eukaryotic cells, PKG-I and PKG-II. PKG-I, because of its deferential splicing of the N-terminus, produces two isoforms, α and β [8]. These isoforms also differ in their distribution; PKG-Iα is mainly found in the lung, heart, platelets, and cerebellum, whereas PKG-Iβ is highly expressed with PKG-Iα in smooth muscles of the uterus, vessels, intestine, and trachea. PKG-I is a widely studied cardioprotective protein; however, almost all of the previous work has focused on its activity alone with no clue about its expression and regulation.
We and other laboratories had previously established that preconditioning (PC) protects the heart from ischemic injury in two different phases, an early phase, which extends till 2–3 h, and the late phase or second window of protection (SWOP), which reappears after 12–24 h and extends till 3–4 days, that renders the heart relatively resistant to injury [9]. The late PC is mediated in part by iNOS [9,10], protein kinase C [11], Src protein tyrosine kinases [12], and NF-κB [13]. PDE inhibition protects the heart through signaling pathways involving activation of PKG-I, and endothelial and inducible nitric oxide synthases (eNOS/iNOS), along with opening of mitochondrial KATP (mitoKATP) channels [14–17].
Janus tyrosine kinase–signal transducers and activators of transcription (JAK-STAT) signaling result in transcriptional activation of target genes in the nucleus. The STAT family of proteins has seven isoforms (STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6). STATs are the substrate for JAKs, which phosphorylate the tyrosine residues in their Src homology 2 domain. Phosphorylated STAT proteins form dimmers, enter into the nucleus, and regulate the expression of STAT-responsive genes [18,19]. JAK-STAT3 has been reported to inhibit apoptosis in the infarcted heart [20]. The JAK-STAT pathway also has a role in the late phase of protection by PC, but its regulation of PKG-I is completely unknown.
PDE5A inhibition with tadalafil promotes cGMP/PKG-I activity and provides longer and sustained protection of stem cells against H2O2-induced oxidative stress [21]. PDE inhibitors have a significant effect on stem cell proliferation and mobilization. Foresta et al. reported that vardendafil promoted significant mobilization of endothelial progenitor cells in patients [22]. Recently, Hare and colleagues reported that PC of cardiac stem cells with agonists of growth hormone-releasing hormone receptor promoted their proliferation and survival [23]. Besides the direct effect of PDE inhibitors on stem cell survival, overexpression of PKG-I, an important mediator of protection, through adenoviral transduction also increases stem cell survival upon oxygen glucose deprivation as well as cardioprotection against myocardial infarction [24]. The protective effect of PDE5A inhibition against oxidative stress is known; however, the mechanism involved in this early and late protection is grossly unknown. The overall objective of the present study was to critically examine the signaling pathways of pharmacological PC of mesenchymal stem cells (MSCs) with tadalafil during early and late phases of protection against hypoxia/reoxygenation (HR) injury. Our results demonstrated that pharmacological PC with tadalafil increased the survival of MSCs through an elevated Bcl2/Bax ratio during the early phase (2 h), whereas protection during the late phase of PC (24 h) was dependent on the STAT3/PKG-I pathway.
Materials and Methods
Isolation of MSCs from bone marrow
Animal experiments conformed to the Guidelines for Care and Use of Laboratory Animals published by US National Institutes of Health (NIH Pub. No. 85-23, Revised 1985), and the protocols were approved by the Institutional Animal Care and Use Committee at the University of Cincinnati. Rats were anesthetized by intraperitoneal injection of ketamine/xylazine (90–100 mg/kg/7–10 mg/kg). Bone marrow was harvested from 6- to 8-week-old male Fischer-344 rats and MSCs were isolated by flushing the cavities of femurs and tibias with basal DMEM cell culture medium. Bone marrow cells were seeded into 150-mm dishes and cultured in DMEM supplemented with 15% FBS and antibiotics. Nonadherent hematopoietic cells were removed by changing the fresh medium. The adherent spindle-shaped MSCs were expanded and cultured. MSCs were analyzed for their surface marker expression by flow cytometry (FACS Calibur, BD). Briefly, the cells were detached with cell dissociation solution (SIGMA) and washed with a buffer containing 1% bovine serum albumin (Sigma). After blocking for nonspecific binding with the buffer containing 10% FBS, the cells were incubated for 30 min at 4°C with the FITC-conjugated antibodies against rat CD34 (Santa Cruz), CD45, CD29 (BD Pharmingen), CD90 (Abcam), and CD117 (Chemicon). The labeled cells (1×104) were acquired and analyzed by flow cytometry using isotype-identical antibodies as controls.
HR protocol and experimental design
MSCs were treated with or without 100 μM tadalafil (dissolved in dimethyl sulfoxide; DMSO) for 1 h at 37°C and 5% CO2. Following pretreatment with tadalafil, they were then subjected to ischemia (1% hypoxia) for 2 h by replacing the medium with an ischemia buffer containing 118 mM NaCl, 24 mM NaHCO3, 1.0 mM NaH2PO4, 2.5 mM CaCl2-2H2O, 1.2 mM MgCl2, 20 mM sodium lactate, 16 mM KCl, and 10 mM 2-deoxyglucose (pH adjusted to 6.2) as described [25]. Reoxygenation was accomplished by replacing the ischemic buffer with DMEM medium kept under normoxic conditions (Fig. 1A).
FIG. 1.

PDE5A inhibition enhanced MSC survival against HR. (A) Hypoxia/reoxygenation protocol (HR; in vitro model): MSCs were pretreated with 100 μM of tadalafil (TAD) or DMSO (equal by volume) for 1 h and subjected to 2 h of hypoxia, followed by reoxygenation for 2 or 24 h. The arrow indicates the studies/assays being performed at 2 and 24 h. (B) Cell cytotoxicity (%) was calculated using LDH release from cells after 2 h of HR. Note that tadalafil-treated cells showed less damage concomitant with reduced LDH release. (C) Quantitative estimate of live and dead cells as determined by trypan blue staining. Values are the mean±SEM of three different experiments. *P<0.05 versus untreated cells (UT); #P<0.05 versus DMSO. DMSO, dimethyl sulfoxide; HR, hypoxia/reoxygenation; LDH, lactate dehydrogenase; MSC, mesenchymal stem cell. Color images available online at www.liebertpub.com/scd
Experimental groups
(i) UT: Untreated MSCs; (ii) DMSO+HR: MSCs were pretreated with DMSO (equivalent volume used for tadalafil) for 1 h at 37°C in 5% CO2 and subjected to 1% hypoxia for 2 h, followed by reoxygenation; (iii) TAD+HR: MSCs were pretreated with 100 μM tadalafil for 1 h at 37°C in 5% CO2 and subjected to 1% hypoxia for 2 h, followed by reoxygenation.
Cell viability and apoptotic assay
Cell viability was assessed by trypan blue exclusion assay and lactate dehydrogenase (LDH) release into the medium. Cells were treated with 0.4% trypan blue (Sigma-Aldrich) and counted at the end of 2 and 24 h of reoxygenation using the hemocytometer under a microscope. The cellular medium was collected, and LDH activity was monitored spectrophotometrically using a CytoTox-one assay kit (Promega) at the end of 2 and 24 h of reoxygenation (Excitation 560 nm/Emission 590 nm). Apoptosis was analyzed by Terminal dUTP nick end-labeling (TUNEL) staining, using a kit (Roche) that detects nuclear DNA fragmentation through a fluorescence assay (Excitation 540 nm/Emission 580 nm) as per the manufacturer's instructions. In brief, after 24 h of HR, the cells in the two slide chamber were fixed by 4% formaldehyde/phosphate-buffered saline at 4°C for 25 min and subjected to TUNEL assay according to the manufacturer's protocol. The slides were then counterstained with Vectashield mounting medium with 4′, 6-diamidino-2-phenylindole (a DNA intercalating dye for visualizing nuclei in fixed cells; catalogue number H-1200; Vector Laboratories). Cells were also visualized and photomicrographed with an inverted phase-contrast microscope (Olympus IX71) at 10×magnification.
Caspase 3 activity assay
Active caspase was detected using the CaspaTag™ Pan-Caspase in situ assay kit (Chemicon) according to the manufacturer's instructions. In this assay, the cell-permeable noncytotoxic fluorochrome inhibitors of caspases bind covalently to a reactive cysteine residue on the large subunit of the active caspase heterodimer, thereby inhibiting further enzymatic activity. This kit uses a carboxyfluorescein-labeled fluoromethyl ketone peptide inhibitor of caspases-3 and -7 (SR-DEVD-FMK), which emits a green fluorescence. The green fluorescent signal is a direct measure of the amount of active caspase-3 in the cell at the time the reagent was added. The stained cells were immediately examined under a fluorescence microscope using a band pass filter (excitation 490 nm, emission 520 nm) to view the green fluorescence of active caspase-positive cells. Hoechst stain was detected by using a UV filter with excitation at 365 and emission at 480 nm.
Western blotting
Treated cells were lysed in RIPA buffer containing protease and phosphatase inhibitors. Equal amounts of protein lysate (as quantified by BCA and spot densitometry method) were electrophoresed in 10% SDS-PAGE and electroblotted on the PVDF membrane (Immun-Blot; BIO-RAD). The membrane was blocked with 5% fat-free milk in PBS with 0.05% tween 20 at RT for 1 h. Primary antibody binding was done at 4°C overnight in 5% FFM in PBST. The blot was then washed with 0.05% Tween-PBS thrice (10 min each). For secondary binding, the blot was incubated with HRP-conjugated respective antibody at 1:2000 dilution in 5% fat-free milk in PBS with 0.05% tween 20 for 1 h at room temperature. The blot was then washed again as before. Developing was done using ECL (GE Healthcare). Akt, pAkt, GSK3β, and pGSK3β antibodies were from Cell Signaling and iNOS, eNOS, Bcl2, Bax, PKG-I, pSTAT3, actin, and tubulin were from Santa Cruz.
PDE5A, PKG activity
cGMP PDE activity was assayed by a noncompetitive ELISA using a fluorometry method (Excitation 540 nm/Emission 580 nm; BioVision). PKG-I activity was assayed by colorimetric analysis (Absorbance at 450 nm; CycLex) and activities of both were checked using cellular lysates.
Cyclic GMP assay
Competitive enzyme-linked immunoassay was used to determine the cGMP level in cell lysate and absorbance was read at 450 nm (Cell Signaling).
Chromatin immunoprecipitation assay
As per the manufacturer's instructions (Pierce Agarose ChIP kit), briefly, proteins were cross-linked with genomic DNA using 1% formaldehyde and subjected to MNase (Micrococcal Nuclease) digestion. The digested chromatin was immunoprecipitated with STAT3 antibody and eluted out. DNA was recovered and real-time PCR was carried out using PKG-I primer.
cDNA synthesis and real-time PCR
Cells were transfected with siRNA using lipofectamine 2000 (Invitrogen) as per the manufacturer's instructions, and RNA was isolated from cells after treatment at appropriate time points by the RNA assay kit (Qiagen) as per the manufacturer's protocol and quantified using NanoDrop NanoVue(GE). Five hundred nanograms of RNA was converted to cDNA using the Omniscript Reverse Transcript Kit (Qiagen). Real-time PCR was done on a Biorad real-time PCR machine using Quantifect SYBR Green PCR Kits (Qiagen). The reference gene, namely β-actin, was used for normalization. The cycling condition was one cycle of initial activation at 95°C for 15 min, 40 cycles of denaturation at 94°C for 15 s, annealing at 55°C for 30 s, and extension at 72°C for 30 sc, and the expected product was confirmed by melt curve analysis. Relative quantitation was performed using the Relative Expression Software Tool (REST) available at www.gene-quantification.de/rest.html. Each real-time PCR was done in triplicates and repeated thrice. Sequences of real-time PCR primers were as follows:
Actin F: TCATGAAGTGTGACGTTGACATCCGT,
R: CCTAGAAGCATTTGCGGTGCACGATG and
PKG-I F: GCGGAGCCGCAGACCTACAG,
R: ACCAGTGACCCCACATCGCCT.
Statistics
Data are expressed as mean±standard error of mean (SEM). Differences between groups were calculated by analysis of variance, and a value of P<0.05 was considered as statistically significant.
Results
PDE5A inhibition enhanced MSC survival and reduced necrosis/apoptosis
MSCs were pretreated with tadalafil (100 μM) for 1 h and subjected to 2 h of hypoxia (1% O2), followed by reoxygenation for defined time periods (2 & 24 h) as shown in Fig. 1A. Cell cytotoxicity, which was assessed by the release of LDH, was reduced in tadalafil-treated cells compared with DMSO (P<0.05) and untreated (P<0.05) at 2 h (Fig. 1B). Cell counting after trypan blue staining showed more live cells and limited cell death with tadalafil treatment at 24 h (P<0.05 vs. DMSO; Fig. 1C). Tadalafil treatment also reduced TUNEL positivity compared with controls (P<0.05; Fig. 2A, B) and was concomitant with limited caspase activity (P<0.05; Fig. 3A, B).
FIG. 2.

Tadalafil treatment reduced apoptosis upon ischemic stress. (A) MSCs were treated with tadalafil (TAD) 1 h before HR. Cells were stained with dUTP nick end-labeling (TUNEL) and counterstained with 4, 6-diamino-2-phenylindole (DAPI) after 24 h. (B) Quantitative estimation of cells using imageJ software. Values are the mean±SEM of three different experiments. *P<0.05 versus untreated cells (UT); #P<0.05 versus DMSO. Color images available online at www.liebertpub.com/scd
FIG. 3.

Caspase activity was reduced with tadalafil treatment. (A) MSCs were treated with tadalafil (TAD) 1 h before HR. Apoptotic cells detected by active caspase staining with fluorochrome inhibitors of caspases (FLICA) and counterstained with 4, 6-diamino-2-phenylindole (DAPI) after 24 h. (B) Quantitative estimation of cells using imageJ software. Values are the mean±SEM of three different experiments. *P<0.05 versus untreated cells (UT); #P<0.05 versus DMSO. Color images available online at www.liebertpub.com/scd
HR enhanced PDE5A activity
The activity of PDE5A was increased with HR, but was attenuated by tadalafil both at 2 and 24 h in comparison with DMSO and untreated controls (P<0.05; Fig. 4A). Concomitantly, concentration of cGMP also increased at both the time points in tadalafil-treated cells (P<0.05; Fig. 4B). The decrease in PDE5A activity associated with an increase in cGMP was not mirrored at the level of PKG-I and instead activity was increased both with DMSO and tadalafil-treated cells at 2 h (P<0.05 vs. UT; Fig. 4C), and no significant change was observed at 24 h.
FIG. 4.

Effect of HR on PDE5A activity. (A) MSCs were treated with tadalafil(TAD) 1 h before HR. Proteins were isolated after 2 h and 24 h. PDE5A activity expressed in the fluorescence unit (FU) per μg of protein in whole-cell lysate was performed and found to be decreased with tadalafil treatment both at 2 h and 24 h. (B) Concomitant increase in cGMP concentration per mg of protein in tadalafil-treated cells at 2 h and 24 h. (C) PKG-I activity per mg of protein was increased both in DMSO as well as with tadalafil samples at 2 h with no significant difference at 24 h. Values are mean±SEM of three different experiments. *P<0.05 versus untreated cells (UT); #P<0.05 versus DMSO; n.s., non significant. Color images available online at www.liebertpub.com/scd
Increased Bcl2/Bax during the early phase of protection
A significant increase in phosphorylation of Akt with a decrease in total Akt resulted in a net increase in pAkt/Akt both in tadalafil and DMSO-treated cells at 2 and 24 h (P<0.05 vs. UT; Fig. 5A, B). Expression of iNOS and eNOS was elevated during the early phase of protection both with tadalafil and DMSO, which gradually diminished during the late phase (P<0.05 vs. UT; Fig. 5A, C, D). Phosphorylation of GSK3β was increased in DMSO as well as in tadalafil-treated cells in the early phase with no significant change in the late phase (Fig. 5A). No change in the expression in total GSK3β was observed in both the phases of protection (Fig. 5A); therefore, an overall increase in pGSK3β/GSK3β was noted (P<0.05 vs. UT; Fig. 5E). Antiapoptotic protein, Bcl2, was significantly increased at 2 h specifically in tadalafil-treated cells, whereas proapoptotic Bax was slightly decreased both in tadalafil and DMSO groups compared with untreated cells (Fig. 5F). There was an increase in the Bcl2/Bax ratio at 2 h with no significant change at 24 h (P<0.05 vs. DMSO; Fig. 5G).
FIG. 5.

Increased Bcl2/Bax during the early phase of protection against HR. (A) Effect of pharmacological preconditioning with tadalafil (TAD) on expression of survival and apoptotic proteins. MSCs were treated with tadalafil 1 h before HR. Proteins were isolated after 2 h and 24 h. Expression levels of pAkt, Akt, iNOS, eNOS, pGSK3β, and GSK3β. (B–E) Quantification of band intensities of western blots shown was performed using densitometry analysis software. Note that the increase in pAkt/Akt, iNOS, eNOS, and pGSK3β/GSK3β during the early phase of protection (2 h) both in tadalafil and DMSO-treated cells was due to adaptive response of stem cells against HR independent of tadalafil. (F) Expression levels of Bcl2 and Bax. (G) Quantification of band intensities of western blots shown was performed using densitometry analysis software. Increase in Bcl2 was tadalafil dependent and a plausible cause for protection during the early phase (2 h). Actin, a housekeeping gene used as loading control. Values are mean±SEM of three different experiments. *P<0.05 versus untreated cells (UT); #P<0.05 versus DMSO; n.s., non significant. Color images available online at www.liebertpub.com/scd
STAT3 regulates the expression of protein kinase G-I during the late phase of protection
It was noticed that the expression of proteins responsible for enhanced survival during the early phase was gradually diminished or the expression was not as robust at 24 h. The next logical question was as to how the cells were protected during the late phase. In this context, we found out that the phosphorylation of STAT3 was dramatically increased by three-fold (P<0.05 vs. DMSO) at 24 h in tadalafil-treated cells (P<0.05 vs. DMSO; Fig. 6A, B). This was accompanied with increased expression of PKG-I (P<0.05 vs. DMSO; Fig. 6A, C). It was interesting to note that phosphorylation of STAT3 was negligible at 2 h (data not shown).The high expression of PKG-I observed during the late phase with tadalafil could be either due to stabilization of PKG-I protein being formed previously and was not degraded upon ischemic stress or due to an increase in the overall transcriptional rate itself. To address the link between STAT3 and PKG-I, we checked the expression of PKG-I at the mRNA level, which was increased by three-fold in tadalafil-treated cells (P<0.05 vs. DMSO; Fig. 6D, E). Chromatin immunoprecipitation assay analysis was done with enzymatically digested chromatin against pSTAT3 antibody showing more than three-fold PKG-I promoter occupancy by pSTAT3 specifically in tadalafil-treated cells. IgG was used here as the isotypic control for the experiment (P<0.05 vs. DMSO and IgG; Fig. 6F, G). In addition, loss-of-function study was carried out by knocking down STAT3 using antisense against it before the treatment with tadalafil and HR as ischemic stress. The results showed more than two-fold decrease in PKG-I expression at the RNA level (P<0.05 vs. scramble; Fig. 6H). Furthermore, western blot analysis also confirmed almost total loss of PKG-I protein expression with STAT3 knockdown in cells (Fig. 6I, J).
FIG. 6.

STAT3 transcriptionally regulates PKG-I. (A) MSCs were treated with tadalafil (TAD) 1 h before HR and proteins were isolated after 2 h and 24 h time points. Western blotting was carried out for protein expression of phosphorylated STAT3, STAT3, and PKG-I using actin as internal control for normalization. (B, C) Quantification of western blots shown was performed using densitometry analysis software. (D, E) MSCs were treated with tadalafil 1 h before HR, and RNA was isolated after 24 h. Quantitative RT-PCR was performed and PKG-I expression at the mRNA level was determined and reaction mixture ran on agarose gel. (F, G) Chromatin immunoprecipitation (CHIP) assay was performed after 24 h of HR and 1 h pretreatment with tadalafil. Chromatin was digested and immunoprecipitated using pSTAT3β antibody and analyzed with real-time PCR with PKG-I primers showing PKG-I promoter occupancy by phosphorylated STAT3 antibody and also ran on agarose gel. IgG antibody here used as isotypic control. (H) STAT3 was knocked down using siRNA, 48 h later, the cells were treated with tadalafil or DMSO, then subjected to HR. RNA was isolated after 24 h of reoxygenation and real-time PCR done with PKG-I primer. (I) Western blot with STAT3 and PKG-I antibody after knocking down STAT3 using siRNA against it. (J) Quantification of band intensities of western blots shown was performed using densitometry analysis software. Actin used as internal control for normalization. Values are mean±SEM of three different experiments. *P<0.05 versus untreated (UT); #P<0.05 versus dimethyl sulfoxide (DMSO); £P<0.05 versus IgG; ΔP<0.05 versus DMSO treated and immunoprecipitated with pSTAT3; βP<0.05 versus scramble. Color images available online at www.liebertpub.com/scd
Discussion
The salient findings of our study are (i) pharmacological PC with tadalafil increased the survival of MSCs against HR by reducing apoptosis/necrosis, both during early and late phases; (ii) enhanced phosphorylation of Akt and GSK3β together with the induction of iNOS and eNOS during the early phase was a spontaneous adaptive response of MSCs against HR, but was independent of tadalafil treatment; (iii) increased Bcl2/Bax ratio was observed in tadalafil-treated cells and it was responsible for an early protection; (iv) PKG-I was upregulated and was responsible for the late protection. STAT3 was phosphorylated at ser727, leading to its entry inside the nucleus and binding onto the promoter of PKG-I, thereby transcriptionally activating PKG-I expression, which resulted in late phase protection against HR. Schematic representation of the signaling pathway involved is provided in Fig. 7.
FIG. 7.
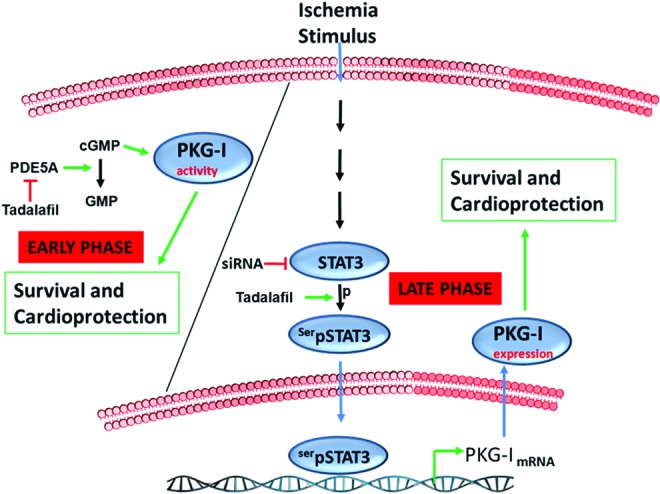
Signaling pathway of protection by tadalafil. Tadalafil, an inhibitor of phosphodiesterase, is highly specific for PDE5A and inhibits its activity. cGMP is increased upon inhibition of PDE5A. PKG-I is an effector kinase of cGMP and its activity is also increased during the early phase of protection. During the late phase, STAT3 was phosphorylated upon ischemic stimulus (HR) in the presence of tadalafil. It entered into the nucleus and bound to the promoter of PKG-I and thus transcriptionally enhanced its expression, which led to the late phase of protection. An increase in PKG-I expression by tadalafil, but not its activity, provided protection during the late phase. Color images available online at www.liebertpub.com/scd
Stem cells have the inherent property to proliferate and differentiate into varieties of cells. Thus, they have significant importance in cell-based therapy. However, a significant number of cells die once transplanted into the ischemic myocardium because of an unfavorable environment inside the heart [26]. PC evokes an endogenous protective mechanism against ischemic stress and it may therefore be a necessary step to enhance their survival and proliferation upon transplantation into the infarcted heart. PC with pharmacological drugs or short cycles of hypoxia/anoxia, followed by reoxygenation, increases the cell survival upon ischemic injury both in in vitro and in vivo models [12,21]. A previous report demonstrated the effect of tadalafil on stem cell survival in the ischemic environment [21]. However, its mechanism of action was not investigated. In our study, we did observe increased survival of stem cells against ischemia both at early and late phases of PC with tadalafil treatment, as observed with classical ischemic preconditioning.
Tadalafil, a PDE inhibitor, which prevents the breakdown of cGMP into GMP, is bound to its GAF domain [27]. PKG-I, a downstream effector kinase of cGMP, which binds to its regulatory domain, in turn activates it, especially in heart muscles. Our study has shown the enhanced PDE5A activity upon HR, but it was inhibited by tadalafil, consequently leading to an increase in cGMP. Since PKG-I is an effector kinase of cGMP, its activity, which was expected to increase, remained the same. The plausible explanation could be the activation of the PI3K-Akt pathway during the early phase of protection in both DMSO as well as tadalafil-treated cells. It is well known that through this pathway, phosphorylation and activation of nitric oxide synthase (NOS) could occur, leading to generation of nitric oxide (NO). NO in turn activates guanylate cyclase (GC), which consequently increases the cGMP generation, resulting in an increase in PKG-I activity [28]. Thus, here in this scenario, PKG-I activity could be regulated directly by PDE5A or indirectly through the NO pathway.
According to earlier studies, the PI3K/Akt signaling pathway plays a crucial role in PC of the heart against IR injury [29,30] through the activation of eNOS [31,32] and antiapoptotic pathways. The latter include inactivation of proapoptotic proteins, such as Bax and caspases [33,34]. Phosphorylation of GSK3β suppresses opening of the mitochondrial permeability transition pore by binding to adenine nucleotide translocase and reduces its affinity for cyclophilin D, which in turn increases cell survival [35]. Enhanced phosphorylation of Akt and GSK3β along with an induction of iNOS and eNOS during the early phase of pharmacological PC is supported by our data, but it was more likely a spontaneous adaptive stress response of MSCs against HR and independent of tadalafil treatment as suggested by DMSO controls. In addition, the survival effect of tadalafil was likely due to an increased ratio of Bcl2/Bax and reduced activity of caspase-3. Aforesaid proteins have been reported to have a role in protection and survival, as noted by tadalafil PC. Although these proteins upregulated as a result of PC have been expressed to a variable extent in the SWOP, their expression level was not robust, suggesting involvement of some other pathway in the late phase of protection.
The JAK-STAT pathway has previously been shown to provide protection from ischemic stress in the late phase of PC through iNOS upregulation [36,37]. However, we did not observe any change in iNOS or eNOS expression during the late phase. This discrepancy could arise due to different experimental protocols or types of cells used for PC. The data from our study suggest that transcriptional upregulation of PKG-I through phosphorylated STAT3 was responsible for the late phase protection. The latter entered inside the nucleus and bound to the promoter of PKG-I, resulting in upregulation of its expression, which initiated the late protection. It was of interest to note that phosphorylation of STAT3 was exhibited only at the later stage, that is, 24 h.
In this study, we attempted to investigate the survival effect of tadalafil upon IR and put forward the mechanism of early and late phase protection, which can be considered during MSC transplantation upon pharmacological PC to overcome massive cell death. This is the first report, which showed the novel mechanism of regulation of PKG-I by phosphorylated STAT3 during the late phase of protection. Moreover, tadalafil effect, which lasted over 24 h, protected the stem cells through activation of the STAT3 signaling pathway. Tadalafil appears to be an effective agent for PC of stem cells before transplantation to overcome massive cell death. The immediate effect of tadalafil is mediated by the Bcl2/Bax pathway, while the longer protection occurs through the STAT3/PKG-I signaling pathway.
Conclusions
The study reported a novel mechanism of stem cell protection over a prolonged period by an FDA-approved drug, which can be safely used in studies involving stem cells for therapeutic use against cardiovascular diseases. This work laid down the foundation for future studies for better understanding of the complete signaling pathway of crucial cardioprotective protein, PKG-I, in stem cell biology and its protection.
Acknowledgments
This study was supported by the National Institutes of Health (RO1-HL095375, HL 087246 to M. Ashraf). The authors thank Dr. Ibrahim Elmadbouh for culturing cells.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Fliss H. and Gattinger D. (1996). Apoptosis in ischemic and reperfused rat myocardium. Circ Res 79:949–956 [DOI] [PubMed] [Google Scholar]
- 2.Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y. and Kass DA. (2005). Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11:214–222 [DOI] [PubMed] [Google Scholar]
- 3.Rybalkin SD, Yan C, Bornfeldt KE. and Beavo JA. (2003). Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Cir Res 93:280–291 [DOI] [PubMed] [Google Scholar]
- 4.Reffelmann T. and Kloner RA. (2003).Therapeutic potential of phosphodiesterase 5 inhibition for cardiovascular disease. Circulation 108:239–244 [DOI] [PubMed] [Google Scholar]
- 5.Takimoto E, Champion HC, Belardi D, Moslehi J, Mongillo M, Mergia E, Montrose DC, Isoda T, Aufiero K, et al (2005). Cyclic GMP catabolism by PDE5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res 96:100–109 [DOI] [PubMed] [Google Scholar]
- 6.Sesti C, Florio V, Johnson EG. and Kloner RA. (2007). The phosphodiesterase-5 inhibitor tadalafil reduces myocardial infarct size. Int J Impot Res 19:55–61 [DOI] [PubMed] [Google Scholar]
- 7.Hofmann F. (2005). The biology of cyclic GMP-dependent protein kinases. J Biol Chem 280:1–4 [DOI] [PubMed] [Google Scholar]
- 8.Hofmann F, Ammendola A. and Schlossmann J. (2000). Rising behind NO: cGMP-dependent protein kinases. J Cell Sci 113:1671–1676 [DOI] [PubMed] [Google Scholar]
- 9.Bolli R. (2000). The late phase of preconditioning. Circ Res 87:972–983 [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Jones WK, Xuan YT, Tang XL, Bao W, Wu WJ, Han H, Laubach VE, Ping P, et al (1999). The late phase of ischemic preconditioning is abrogated by targeted disruption of the inducible NO synthase gene. Proc Natl Acad Sci U S A 96:11507–11512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ping P, Zhang J, Qiu Y, Tang XL, Manchikalapudi S, Cao X. and Bolli R. (1997). Ischemic preconditioning induces selective translocation of protein kinase C isoforms epsilon and eta in the heart of conscious rabbits without subcellular redistribution of total protein kinase C activity. Circ Res 81:404–414 [DOI] [PubMed] [Google Scholar]
- 12.Ping P, Zhang J, Zheng YT, Li RC, Dawn B, Tang XL, Takano H, Balafanova Z. and Bolli R. (1999). Demonstration of selective protein kinase C-dependent activation of Src and Lck tyrosine kinases during ischemic preconditioning in conscious rabbits. Circ Res 85:542–550 [DOI] [PubMed] [Google Scholar]
- 13.Xuan YT, Tang XL, Banerjee S, Takano H, Li RC, Han H, Qiu Y, Li JJ. and Bolli R. (1999). Nuclear factor-kappaB plays an essential role in the late phase of ischemic preconditioning in conscious rabbits. Circ Res 84:1095–1109 [DOI] [PubMed] [Google Scholar]
- 14.Das A, Smolenski A, Lohmann SM. and Kukreja RC. (2006). Cyclic GMP-dependent protein kinase Ialpha attenuates necrosis and apoptosis following ischemia/reoxygenation in adult cardiomyocyte. J Biol Chem 281:38644–38652 [DOI] [PubMed] [Google Scholar]
- 15.Das A, Ockaili R, Salloum F. and Kukreja RC. (2003). Protein kinase C plays an essential role in sildenafil-induced cardioprotection in rabbits. Am J Physiol Heart Circ Physiol 286:H1455–H1460 [DOI] [PubMed] [Google Scholar]
- 16.Salloum F, Yin C, Xi L. and Kukreja RC. (2003). Sildenafil induces delayed preconditioning through inducible nitric oxide synthase-dependent pathway in mouse heart. Circ Res 92:595–597 [DOI] [PubMed] [Google Scholar]
- 17.Das A, Xi L. and Kukreja RC. (2008). Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem 283:29572–29585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Darnell JE., Jr. (1999). STATs and gene regulation. Science 277:1630–1635 [DOI] [PubMed] [Google Scholar]
- 19.Heim MH. (1999). The Jak-STAT pathway: cytokine signalling from the receptor to the nucleus. J Recept Signal Transduct Res 19:75–120 [DOI] [PubMed] [Google Scholar]
- 20.Negoro S, Kunisada K, Tone E, Funamoto M, Oh H, Kishimoto T. and Yamauchi-Takihara K. (2000). Activation of JAK/STAT pathway transduces cytoprotective signal in rat acute myocardial infarction. Cardiovasc Res 47:797–805 [DOI] [PubMed] [Google Scholar]
- 21.Haider HKh, Lee YJ, Jiang S, Ahmed RP, Ryon M. and Ashraf M. (2010). Phosphodiesterase inhibition with tadalafil provides longer and sustained protection of stem cells. Am J Physiol Heart Circ Physiol 299:H1395–H1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Foresta C, Lana A, Cabrelle A, Ferigo M, Caretta N, Garolla A, Palù G. and Ferlin A. (2005). PDE-5 inhibitor, Vardenafil, increases circulating progenitor cells in humans. Int J Impot Res 17:377–380 [DOI] [PubMed] [Google Scholar]
- 23.Florea V, Majid SS, Kanashiro-Takeuchi RM, Cai RZ, Block NL, Schally AV, Hare JM. and Rodrigues CO. (2014). Agonists of growth hormone-releasing hormone stimulate self-renewal of cardiac stem cells and promote their survival. Proc Natl Acad Sci U S A. 111:17260–17265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang L, Pasha Z, Wang S, Li N, Feng Y, Lu G, Millard RW. and Ashraf M. (2013). Protein kinase G1 α overexpression increases stem cell survival and cardiac function after myocardial infarction. PLoS One 8:e60087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rakhit RD, Edwards RJ, Mockridge JW, Baydoun AR, Wyatt AW, Mann GE. and Marber MS. (2000). Nitric oxide-induced cardioprotection in cultured rat ventricular myocytes. Am J Physiol Heart Circ Physiol 278:H1211–H1217 [DOI] [PubMed] [Google Scholar]
- 26.Hodgetts SI, Beilharz MW, Scalzo AA. and Grounds MD. (2000). Why do cultured transplanted myoblasts die in vivo?. DNA quantification shows enhanced survival of donor male myoblasts in host mice depleted of CD4+ and CD8+ cells or Nk1.1+ cells. Cell Transplant 9:489–502 [DOI] [PubMed] [Google Scholar]
- 27.Rybalkin SD, Rybalkina IG, Shimizu-Albergine M, Tang XB. and Beavo JA. (2003). PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J 22:469–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garlid KD, Costa AD, Quinlan CL, Pierre SV. and Dos Santos P. (2009). Cardioprotective signaling to mitochondria. J Mol Cell Cardiol 46:858–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tong H, Chen W, Steenbergen C. and Murphy E. (1999). Ischemic preconditioning activates phosphatidylinositol-3-kinase upstream of protein kinase C. Circ Res 87:309–315 [DOI] [PubMed] [Google Scholar]
- 30.Datta SR, Brunet A. and Greenberg ME. (1999). Cellular survival: a play in three Akts. Genes Dev 13:2905–2927 [DOI] [PubMed] [Google Scholar]
- 31.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R. and Zeiher AM. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399:601–605 [DOI] [PubMed] [Google Scholar]
- 32.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ. and Rosenzweig A. (2001). Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation 104:330–335 [DOI] [PubMed] [Google Scholar]
- 33.Weston CR, Balmanno K, Chalmers C, Hadfield K, Molton SA, Ley R, Wagner EF. and Cook SJ. (2003). Activation of ERK1/2 by deltaRaf-1:ER represses Bim expression independently of the JNK or PI3K pathways. Oncogene 22:1281–1293 [DOI] [PubMed] [Google Scholar]
- 34.Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S. and Reed JC. (1998). Regulation of cell death protease caspase-9 by phosphorylation. Science 282:1318–1321 [DOI] [PubMed] [Google Scholar]
- 35.Nishihara M, Miura T, Miki T, Tanno M, Yano T, Naitoh K, Ohori K, Hotta H, Terashima Y. and Shimamoto K. (2007). Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J Mol Cell Cardiol 43:564–570 [DOI] [PubMed] [Google Scholar]
- 36.Xuan YT, Yiru G, Hui H, Yanqing Z. and Bolli R. (2001). An essential role of the JAK-STAT pathway in ischemic preconditioning. Proc Natl Acad Sci U S A 98:9050–9055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xie Q, Kashiwabara Y. and Nathan C. (1994). Role of transcription factor NF-ΠB/Rel in induction of nitric oxide synthase. J Biol Chem 269:4705–4708 [PubMed] [Google Scholar]