

Abstract
Foamy virus (FV) vectors are promising tools for gene therapy, but low titer is a major challenge for large-scale clinical trials. Here, we increased FV vector titer 50-fold by constructing novel vector plasmids and using polyethylenimine-mediated transfection. FV and lentiviral (LV) vectors were used separately to transduce human CD34+ cells at multiplicities of infection of 25, and those cells were transplanted into immunodeficient mice. FV vector transduction frequencies of repopulating human cells were 37.1 ± 1.9% in unstimulated cells and 36.9 ± 2.2% in prestimulated cells, and engraftment frequencies were 40.9 ± 4.9% in unstimulated cells and 47.1 ± 3.3% in prestimulated cells. Engraftment frequencies of FV vector-transduced cells were significantly higher than those of LV vector-transduced cells. Linear amplification-mediated PCR with Illumina paired-end runs showed that all human chromosomes contained FV provirus. FV had an integration preference near transcriptional start sites and CpG islands of RefSeq genes but not within genes. Repopulating lymphoid and myeloid cells contained common integration sites, suggesting that FV vector could transduce multilineage hematopoietic stem/progenitor populations. Our new FV vector backbone may be a suitable candidate for developing therapeutic FV vectors for use in clinical trials.
Introduction
Nonpathogenic foamy viruses (FVs) have several advantages over other retroviruses as gene therapy vectors. FV vectors have a large packaging capacity and broad host and cell-type tropism, and the cDNA of the viral genome is synthesized prior to infection, resulting in vector stability in nondividing cells.1 The integration preference of FV vectors is distinct from those of other retroviral vectors.2,3 FV DNA can enter the nucleus of G1/S-arrested cells, but no virus replication occurs.4 FV vectors can efficiently infect G0-arrested cells if they are capable of eventual cell division. The FV genome can persist in a stable form in growth-arrested cells and can integrate into the host genome when cells exit G0.1,5
Despite having good features as a gene therapy vector, the low titer of FV poses a major challenge for large-scale gene therapy.6 Therefore, improving titer is an important priority in FV vector research. For high-titer vector production, the packaging cells should be transfected at the highest possible level with vector plasmids, the gene transfer vector must produce large quantities of vector genome, and the packaging genes should produce optimal amounts of proteins to form the virus particles.
Hematopoietic stem cell (HSC) gene therapy is beneficial for patients with severe combined immunodeficiency (SCID).7,8 However, gene transfer of HSCs with Moloney murine leukemia virus (MLV) vectors have caused genotoxicity in some patients because of activation of proto-oncogenes, thereby limiting the clinical use of these vectors.9,10 Human immunodeficiency virus (HIV)–derived self-inactivating lentiviral vectors (LVs) pseudotyped with vesicular stomatitis virus (VSV)-G have proven effective in some clinical trials.1112
Several preclinical gene therapy studies using FV vectors in mice, dogs, and human cells have shown variable transduction and engraftment of cells.3,13,14 Because transduction of human-mobilized peripheral blood CD34+ cells with FV and LV vectors has not been compared in new NOD/SCID Gamma (NSG) mice, we investigated whether our vectors are more efficient in transducing and engrafting human HSCs.
Viral vectors may activate proto-oncogenes via transactivation by vector enhancers or read-through transcription from vector promoters or by integration within tumor suppressor genes that may inactivate their functions. Uncontrolled clonal growth of cells due to proto-oncogene activation or tumor suppressor gene inactivation by viral vectors has prompted efforts to decipher vector–host genome interaction.15,16 Analysis of vector integration sites by linear amplification–mediated (LAM)-PCR and high-throughput next-generation sequencing has greatly improved the retrieval of information regarding integration sites, with the goal of determining the clonal repertoire of analyzed samples.17–19 Paired-end reads can increase the accuracy of the sequence reads by precise alignment of the data generated by sequencing both ends, thereby improving the mapping of the integration site.
In this study, we successfully increased FV vector titer by constructing novel vector plasmids and using polyethylenimine (PEI)–mediated transfection.20 FV vectors were used to transduce human-mobilized peripheral blood CD34+ cells that were engrafted into immunodeficient mice. In addition, FV vector integration sites in SCID-repopulating cells were analyzed using LAM-PCR with Illumina paired-end runs.
Results
Modification of FV vector plasmids and transfection
FV vector was produced in 293T cells by transient transfection using the calcium phosphate method and a 4-plasmid system comprising genes encoding FV gag, pol, and env, and the gene transfer vector. The unconcentrated titer of the original FV-green fluorescent protein (GFP) vector was 2.0 ± 0.03 × 105 transducing units (TU)/ml in HT1080 cells (Figure 1a, left bar), similar to that previously reported.6 FV gene transfer vector plasmids were modified such that they would contain improved regulatory elements (see Supplementary Figure S1a, bottom), as used in our high-titer LV vector system.21 The transcriptional regulatory elements used in the original FV packaging gene plasmids seemed suboptimal for high-titer vector production (see Supplementary Figure S1b, top). Therefore, FV gag, pol, and env genes were cloned into either the pHDM (see Supplementary Figure S1b, middle) or pCAGGS plasmid backbones (see Supplementary Figure S1b, bottom). Studies have indicated that these plasmid systems are superior in expressing viral genes to achieve higher vector titers.21
Figure 1.
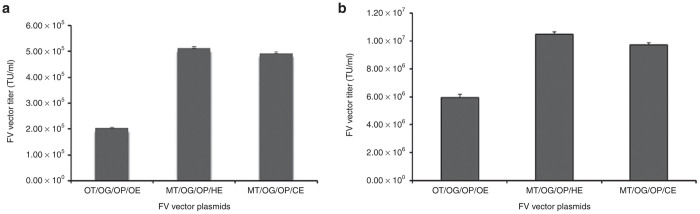
FV vector titers of 293T cells by using various combinations of vector plasmids and transfection methods. FV-GFP vector titers of (a) calcium phosphate transfection and (b) PEI transfection. HT1080 cells were transduced with FV vector at various concentrations. One week after transduction, cells were analyzed for GFP expression. Titers were determined as transducing units per milliliter (TU/mL). Data represent the mean and the standard error of mean (SEM) of three individual experiments. Left bars show original gene transfer (OT) vector, original gag (OG), original pol (OP), and original env (OE) vectors; middle bars show modified gene transfer vector (MT), original gag (OG), original pol (OP), and pHDM-env (HE) vectors; and right bars show modified gene transfer vector (MT), original gag (OG), original pol (OP), and pCAGGS-env (CE) vectors.
Thirty of 54 potential different combinations of FV original and modified gene transfer and packaging gene plasmids were used to optimize FV vector production (see Supplementary Figure S1c). Calcium phosphate–mediated transfection with our modified FV gene transfer and pHDM-env plasmids, coupled with the original gag and pol plasmids yielded the highest titer, 5.1 ± 0.06 × 105 TU/ml (2.5-fold increase; see Supplementary Figure S1a, middle bar). The pCAGGS-env plasmid with modified FV gene transfer and original gag and pol plasmids yielded the second highest titer, 4.9 ± 0.04 × 105 TU/ml (see Supplementary Figure S1a, right bar).
Polyethylenimine (PEI)-mediated transfection led to a 10-fold increase in vector production20,22 (J. Roy and J. Neil, personal communication). We used PEI to transfect 30 of 54 potential different combinations of FV original and modified gene transfer and packaging gene plasmids in 293T cells (see Supplementary Figure S1d). An unconcentrated FV-GFP vector titer of 1.1 ± 0.02 × 107 TU/ml (Figure 1b, middle bar), approximately 50-fold higher than that of the starting titer, was obtained using the modified FV-GFP gene transfer vector and pHDM-env coupled with original gag and pol packaging gene plasmids. When pCAGGS-env was used in place of the pHDM-env plasmid, a titer of 9.7 ± 0.1 × 106 TU/ml was obtained (Figure 1b, right bar).
Transduction of human CD34+ cells in vitro
Granulocyte colony-stimulating factor–mobilized CD34+ cells from peripheral blood of three human donors were studied in three individual experiments. Unstimulated or prestimulated cells were transduced with either a FV or LV vector containing GFP at a multiplicity of infection (MOI) of 25 (see Supplementary Figure S2). MOI of 25 represents FV vectors titrated on HT1080 cells and LV vectors titrated on 293T cells. FV vector-transduced cells expressed 49.9 ± 5.9% of GFP in unstimulated bulk culture (Figure 2a, FV-1st) and 53.3 ± 3.3% of GFP in prestimulated bulk culture (Figure 2a, FV-2nd) 1 week after transduction. LV vector-transduced cells expressed 89.0 ± 10.2% of GFP in unstimulated bulk culture (Figure 2a, LV-1st) and 89.5 ± 8.3% of GFP in prestimulated bulk culture (Figure 2a, LV-2nd). The percentages of GFP+ progenitors in MethoCult cultured colonies using FV vector were 57.0 ± 6.0% in unstimulated cells (Figure 2b, FV-1st) and 63.7 ± 2.0% in the prestimulated cells (Figure 2b, FV-2nd). The percentages of GFP+ LV vector-transduced colonies were 80.5 ± 8.5% in unstimulated cells (Figure 2b, LV-1st) and 84.3 ± 3.4% in prestimulated cells (Figure 2b, LV-2nd). The total number of colonies for untransduced and FV vector-transduced groups were similar, but there were significantly fewer in LV vector-transduced groups.
Figure 2.
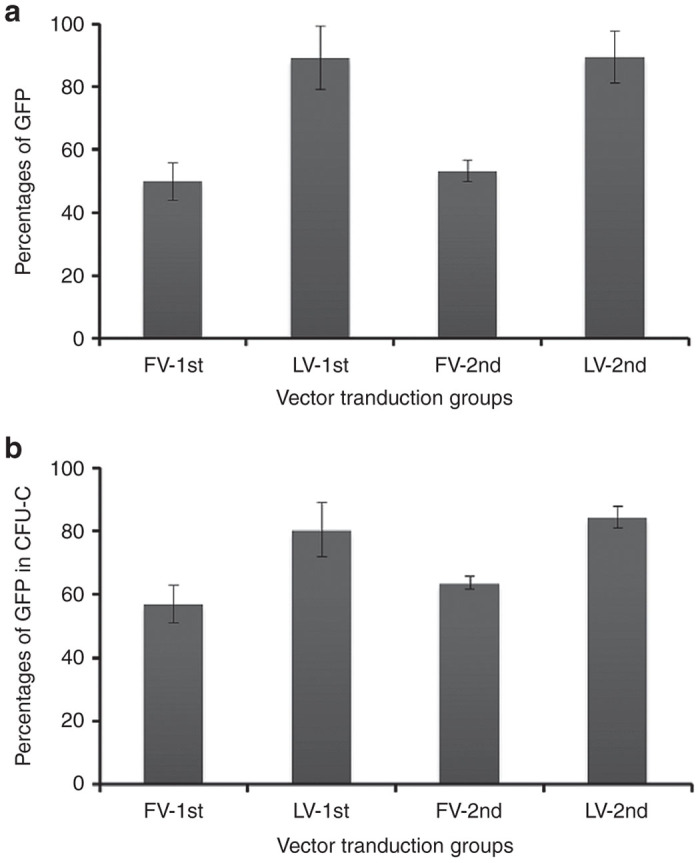
Vector transduction of bulk culture and clonogenic progenitors. (a) Percentages of GFP-expressing cells in bulk culture 1 week after vector transduction. (b) Percentages of GFP-expressing progenitor colonies on MethoCult medium 12 to 14 days after vector transduction. Unstimulated cells are designated FV-1st and LV-1st, and 24 h prestimulated cells are designated FV-2nd and LV-2nd. Both FV and LV vectors were transduced overnight on RetroNectin-coated plates at an MOI of 25. The experiments were done three times with mobilized human CD34+ cells from 3 donors. Data represent the mean and SEM.
Transduction analysis of SCID-repopulating human cells
Sixteen weeks post transplantation of human CD34+ cells, NSG mice were euthanized and bone marrow cells were collected. Analysis of human CD34+ cell-derived SCID-repopulating cells revealed considerable variation in the engraftment of human cells (CD45+) among mice. For the unstimulated group, engraftment frequencies of untransduced mock cells were similar to that of FV vector-transduced cells (37.6 ± 3.7% versus 40.9 ± 4.6%; P = 0.58; Figure 3a, Mock-1st and FV-1st), indicating that transduction of FV vectors at higher MOIs were nontoxic to human CD34+ cells. LV vector-transduced cells showed significantly lower engraftment than mock cells (37.6 ± 3.7% versus 24.4 ± 3.0%; P = 0.01; Figure 3a, Mock-1st and LV-1st). For the prestimulated group, engraftment frequencies were similar for mock cells and FV vector–transduced cells (43.0 ± 3.1% versus 47.1 ± 3.3; P = 0.36; Figure 3a, Mock-2nd and FV-2nd), whereas the engraftment frequencies of LV vector–transduced cells were lower than those of mock cells (43.0 ± 3.1% versus 22.3 ± 2.2%; P = 0.0001; Figure 3a, Mock-2nd and LV-2nd). For FV vector, unstimulated and prestimulated cells had similar levels of engraftment (Figure 3a, FV-1st and FV-2nd). For LV vector, both transduction conditions also had similar levels of engraftment (Figure 3a, LV-1st and LV-2nd).
Figure 3.

Transduction and engraftment of human cells in the bone marrow of NSG mice. (a) Percentages of engraftment of human CD45+ cells in the bone marrow of mice 16 weeks after transplantation. Each point represents the engrafted cells in an individual mouse. (b) Proportion of GFP-expressing cells in the engrafted human CD45+ cells. (c) Percentages of lymphoid (CD19+, black bars) and myeloid (CD33/15+, gray bars) cells in repopulating human CD45+ cells. (d) Proportion of GFP-expressing cells in lymphoid (CD19+, black bars) and myeloid (CD33/15+, gray bars) populations. Data represent the mean and SEM of three individual experiments.
Transduction frequencies of FV vectors were 37.1 ± 1.9% in unstimulated cells and 36.9 ± 2.2% in prestimulated cells (Figure 3b, FV-1st and FV-2nd). Transduction frequencies of LV vectors were 73.7 ± 1.8% in unstimulated cells and 72.9 ± 4.1% in prestimulated cells (Figure 3b, LV-1st and LV-2nd).
Engraftment frequencies of FV vector-transduced cells (Figure 3a, FV-1st and FV-2nd) were significantly higher than those of LV vector-transduced cells (Figure 3a, LV-1st and LV-2nd), whereas the transduction frequencies of FV vectors (Figure 3b, FV-1st and FV-2nd) were significantly lower than those of LV vectors (Figure 3b, LV-1st and LV-2nd) under both transduction conditions.
Lymphoid (CD19+) and myeloid (CD33/15+) populations within the human CD45+ cells from the NSG mouse bone marrow were analyzed.23,24 In unstimulated groups of cells, FV and LV vectors had similar levels of lymphoid populations (56.9–60.3%; Figure 3c, Mock-1st, FV-1st, and LV-1st, black bars) and myeloid populations (32.4–36.1%; Figure 3c, Mock-1st, FV-1st, and LV-1st, gray bars). In prestimulated groups of cells, FV and LV had similar levels of lymphoid populations (64.7–67.0%; Figure 3c, Mock-2nd, FV-2nd, and LV-2nd, black bars) and myeloid populations (27.0–30.8%; Figure 3c, Mock-2nd, FV-2nd, and LV-2nd, gray bars). The GFP marking in lymphoid and myeloid populations in both transduction conditions was 35.1–42.8% for the FV vectors (Figure 3d, FV-1st and FV-2nd), and 70.9–78.2% for the LV vectors (Figure 3d, LV-1st and LV-2nd).
CD34+ cells within human CD45+ population revealed similar percentages for both the FV and LV vector-transduced cells (10.7–13.1%; Figure 4a). GFP marking was consistent with the CD34+ population for both transduction conditions for FV and LV vectors (Figure 4b).
Figure 4.
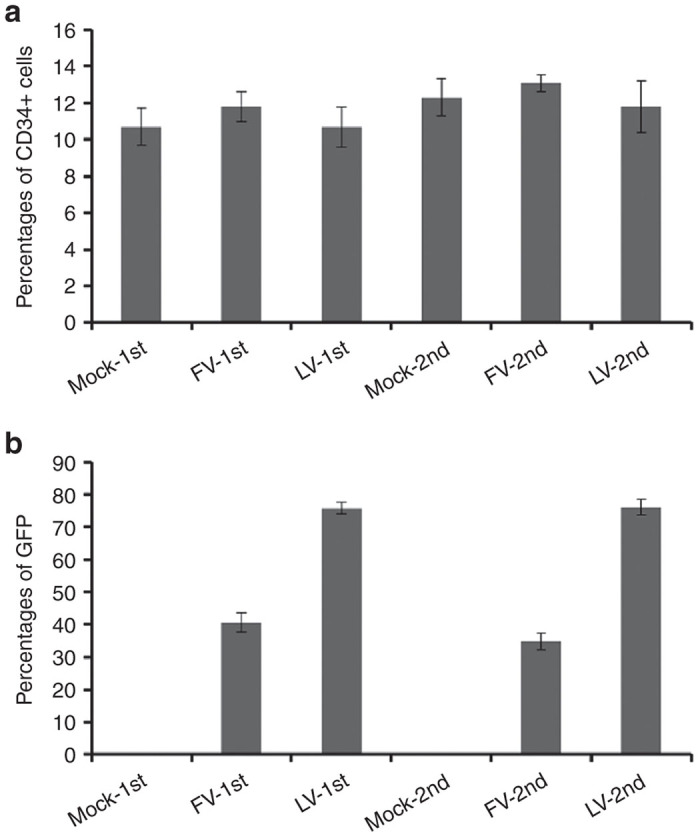
Human CD34+ cells in bone marrow of NSG mice. (a) Percentages of CD34+ cells in the engrafted human CD45+ cells. (b) Proportion of GFP+ cells in the CD34+ population. Data represent the mean and SEM of three individual experiments.
FV vector integration pattern in the human genome
Genomic DNA from FV vector-transduced repopulating human lymphoid and myeloid cells from 12 mice and untransduced human cells from 7 control mice were used to generate high-throughput FV vector integration sites. LAM-PCR products were used in Illumina paired-end runs, and vector integration sites were generated by analyzing the human genome-mapped sequence clusters. Only paired-end reads containing the 3′ long terminal repeat (LTR) and linker sequences (with intact sites for MluCI, MseI, or NlaIII) of at least 30 bp of human genomic sequences that uniquely aligned were used for analysis. The overlapping integration sites among samples were considered as a single site, and 83 integration sites (possibly endogenous FV-like sequences or cross-contamination of samples) identified in untransduced samples were excluded from FV vector–transduced samples. We identified 799 unique FV vector integration sites (see Supplementary Table S1), which were compared with three size-matched sets of 799 computer-generated random sites. FV vector proviruses were found throughout the human chromosomes (Figure 5a).
Figure 5.

FV vector integration sites in human chromosomes. (a) Percentages of FV vector integration sites and computer-generated random sites in each chromosome of repopulating human cells. (b) Percentages of FV vector integration sites near transcription start sites of the RefSeq genes. “−” denotes the upstream region of transcriptional start sites. (c) Percentages of FV vector integration sites near CpG islands. (d) Percentages of FV vector integration sites relative to the position of RefSeq genes. Genes were divided into 10 fragments, and the positions of FV integration sites were analyzed.
FV vector had a higher preference for integration near the transcription start sites than at random sites. The frequency curve of FV vector integration sites was bell-shaped relative to that of the transcription start sites. The percentages of FV vector integration sites were the highest 2 kb upstream of the transcription start sites (Figure 5b). CpG islands are found near promoter regions that regulate transcription.25 FV vector had a higher preference for integration near CpG islands than at random sites (Figure 5c), which is consistent with the frequency of FV vector integration near the transcription start sites. When RefSeq genes (NCBI Reference Sequence database) were divided into 10 segments and analyzed for FV vector integration sites, they showed a higher preference for targeting the first segment (5′ end) of the RefSeq genes than other segments (Figure 5d).
To decipher the pattern of integration into the genes, the percentages of FV vector integration sites within the RefSeq genes were analyzed. FV vector did not show a higher preference to integrate within genes (40.1 versus 45.4%; P = 0.034), in either exons (3.1 versus 2.8%; P = 0.77) or introns (36.9 versus 42.7%; P = 0.021), than at random sites (Table 1). The frequency of FV integration sites was higher in the upstream region (33.9% versus 20.4%; P = 1.53 × 10−9) but not in downstream region (24.8 versus 20.0%; P = 0.026) of genes than that of random sites (Table 1). These data suggest that FV vectors do not preferentially target genes during integration. However, we observed clustering of FV vector integration sites, which were quantified by measuring the distances between the neighboring integration sites (see Supplementary Figure S5a) and the number of hotspots, containing three integration sites in different-sized regions compared to random sites (see Supplementary Figure S5b). The list of regions in a window size of 50 kb with three closest integrants was shown (see Supplementary Table S2). These results are consistent with the previous study.2
Table 1. Distribution of FV vector integration sites in the human genome.
| Integration sites within | FV (n = 799) | Random (n = 799)a | P value |
|---|---|---|---|
| Gene (%) | 40.1 | 45.4 | 0.034 |
| Exon (%) | 3.1 | 2.8 | 0.77 |
| Intron (%) | 36.9 | 42.7 | 0.021 |
| Gene upstream (≤30 kb, %) | 33.9 | 20.4 | 1.53 × 10−9 |
| Gene downstream (≤30 kb, %) | 24.8 | 20.0 | 0.026 |
P value of ≤ 0.01 was considered statistically significant.
Average of three size-matched sets of 799 random sites.
We analyzed the common integration sites between lymphoid and myeloid populations. Approximately 10% of the integration sites were common between lymphoid and myeloid populations, suggesting that the FV vector can transduce HSC and progenitors of multi-lineage potential (Figure 6).
Figure 6.
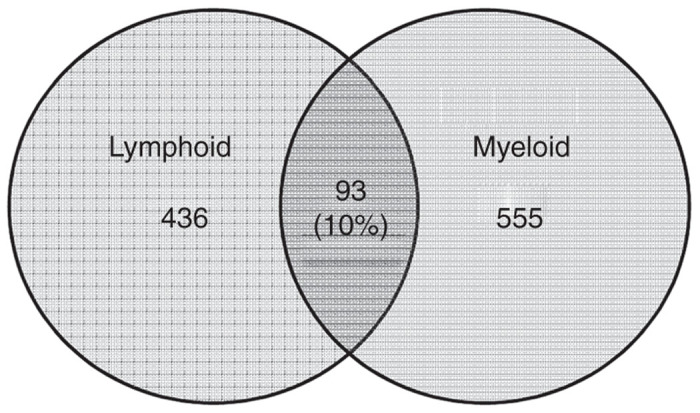
Common FV vector integration sites between lymphoid and myeloid populations.
Discussion
Although Patton et al.20 has initially reported the FV vector production by PEI, they did not clearly mention the FV vector titers. Large-scale clinical grade vector production is a major challenge in gene therapy clinical trials, necessitating the development of high-titer FV vectors.3 We increased FV vector titers 50-fold by systematically modifying and optimizing the FV vector plasmids and transfection methods.
PEI-mediated transfection of FV plasmids into 293T cells significantly increased the titers over those achieved with calcium phosphate. PEI has a high cationic charge density at physiological pH due to partial protonation of the amino groups in every third position. These amino groups form noncovalent complexes with negatively charged DNA, which leads to condensation and shielding of their negative charges, thereby allowing endocytosis into the cell.22 PEI has the ability to avoid trafficking to degradative lysosomes, and its buffering capacity leads to osmotic swelling and rupture of endosomes, resulting in the release of the vector particles into the cytoplasm.26
Adult HSCs are more difficult to culture ex vivo than cord blood HSCs. In addition, most cells are quiescent and are difficult to transduce with viral vectors.27–30 Interestingly, cells that are quiescent when exposed to FV vectors can become transduced when they divide.5 In our study, higher doses of FV vector transduced both unstimulated and prestimulated cells in similar efficiency (37.1 ± 1.9% and 36.6 ± 2.2%; P=0.86). A study using the transplantation of human CD34+ in a murine xenograft model showed that FV vector transduction frequencies were 26.1–42.2% in bone marrow, peripheral blood, and spleen cells.14 Two preclinical studies of dog models showed that FV vector transduction frequencies were 5–10% and 13–19% in peripheral blood cells.3,13 The variation of FV transduction may be due to the sources of CD34+ cells, vector doses, transduction conditions, or variation of viral receptor in cells.
When human CD34+ cells were transduced with our high-titer FV vectors, the engraftment frequencies were 40.9 ± 4.9% in unstimulated and 47.1 ± 3.3% in prestimulated cells. Several studies have reported variable frequencies of engraftment of FV vector-transduced cells.3,13,14 We have obtained less engraftment of LV vector-transduced cells than FV-transduced cells in NSG mice may be due to the toxicity of VSV-G envelope of LV vector to CD34+ cells at high doses.31 We used different cell lines for FV vector (HT1080) and LV vector (293T) titer estimation, which causes apparent variability of vector doses and subsequent transduction variation. In addition, FV and LV vectors may transduce different stem cell populations and may use different cellular receptors for transduction. These factors may contribute to the apparent transduction superiority of LV vectors over FV vectors in the current experiments.
LAM-PCR is dependent on the choice of the restriction enzymes, which is crucial for the performance of vector integration site analysis. However, the application of various enzymes with distinct restriction motifs is mandatory to reveal the majority of integration site repertoire.17 Even the combinatorial use of different enzymes that cleave at the highest frequency may not necessarily allow identification of the complete integration site pool present in a transduced sample, because some integration sites occur either too close to or too far from any specific restriction enzyme site, resulting in fragments that are too small to resolve or, alternatively, too long to be amplified, thus limiting the analysis to a subset of clones in a mixture.32
We identified 799 unique FV vector integration sites from 12 mice. The FV integration pattern appeared oligoclonal in human repopulating cells. The FV vector integrated preferentially near the CpG islands and transcription start sites but not within genes of human chromosomes. In a preclinical gene therapy study in dogs, FV vectors showed a lower integration preference for transcription start sites than did MLV vectors.3 MLV vectors have a strong affinity to integrate near transcription start sites.33 Although MLV and FV vectors have similar integration profile, the FV vector is not associated with clonal expansion and malignancies in dogs 4 years after transplantation.34 In contrast, transduction of cells with MLV-based vectors has caused genotoxicity through integration of the provirus near oncogenes in the host genome and their subsequent activation.9,10
FV vectors have a low potential for causing insertional inactivation of tumor suppressor genes because they do not preferentially target genes, which may reduce the risk of deleterious complications arising due to alteration or loss of protein function. The LV vectors show a strong affinity to integrate within actively transcribing genes, which may increase the risk of insertional inactivation of genes.12,35 In our in vivo study, 4.3% of FV vector integration sites were found within 50 kb of proto-oncogenes, which is consistent with findings of the dog study.3 In an in vitro study, 4.4% of FV vector integration sites were found around proto-oncogenes, compared with 6.7% for LV and 7% for MLV vectors.2,36 However, none of the integration sites were found near the oncogenes such as LMO2, EVI1, or HMGA2 (see Supplementary Table S3).
Our FV vector system consistently gives high titers, making it a valuable tool for large-scale gene therapy. In addition, our vector transduction and integration site analysis protocols will be useful for future gene therapy clinical trials.
Materials and Methods
Construction of FV gene transfer and packaging gene plasmids
Self-inactivating FV gene transfer vector plasmid, p∆Φ (delta phi)-GFP and packaging gene plasmids pCIGS∆ψ (gag), pCIPS (pol), and pCIES (env)20 were provided by Dr. David Russell (University of Washington, Seattle, WA). All plasmids were modified as described (see Supplementary Figure S1a,b). The original FV gene transfer plasmid p∆Φ-GFP (see Supplementary Figure S1a, top) contains a pBR322 origin of replication, which yielded low amounts of plasmid DNA from bacterial culture. To increase plasmid copy number and yield, the pUC origin of replication was introduced into the backbone of the plasmid (see Supplementary Figure S1a, bottom). To obtain a better yield of RNA genome of the FV gene transfer vector, a β-globin poly-adenylation [poly(A)] signal was introduced downstream of the 3′ LTR, which is relatively weak due to deletion of the U3 region (see Supplementary Figure S1a, bottom). In addition, an SV40 origin of replication was introduced into our modified FV gene transfer vector plasmids. SV40 T antigen of 293T cell interacts with SV40 origin of replication of vector plasmid and increases the copy number of plasmid in transfected cells. We also modified the original FV packaging gene plasmids (see Supplementary Figure S1b, top). The pHDM plasmid (a gift from Dr. Richard Mulligan, Harvard Medical School, Boston, MA) has a cytomegalovirus (CMV) enhancer-promoter with a 5′ untranslated region, a β-globin intron to transcribe the gene of interest, and a β-globin poly (A) signal to terminate the transcription (see Supplementary Figure S1b, middle). The pCAGGS plasmid has a CMV enhancer with a chicken β-actin promoter, a β-globin intron to transcribe the gene, and a β-globin poly(A) signal to terminate the transcription (see Supplementary Figure S1b, bottom). pCIGS∆ψ (gag), pCIPS (pol), and pCIES (env) were cleaved with NotI, and the corresponding gene was excised and cloned into the multiple cloning site of pHDM or pCAGGS plasmid. In addition, an SV40 origin of replication was introduced in all of the modified packaging gene plasmids.
FV vector production, concentration, and purification
The human embryonic kidney cell line 293T and the human fibrosarcoma cell line HT1080 were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, San Diego, CA) supplemented with 10% fetal bovine serum, 2 mmol/l L-glutamine, 1 mmol/l sodium pyruvate, 1 mmol/l nonessential amino acids, and 1% penicillin–streptomycin.
FV vectors were produced by calcium phosphate-mediated transient transfection37 with 12 µg of gene transfer vector, 12 µg of gag vector, 1.6 µg of pol vector, and 0.75 µg of env vector in a 10-cm cell culture dish. Before adding the transfection reaction mixture, cells were treated with 25 µmol/l chloroquine (Sigma-Aldrich, St Louis, MO), and 4–6 h after transfection, cells were treated overnight with 500 µmol/l sodium butyrate (Sigma-Aldrich). The next day, transfection medium was replaced with growth medium. The viral supernatant was harvested 3 days after transfection.6
FV vectors were also produced by PEI (Polysciences, Warrington, PA)-mediated transient transfection.20 Briefly, 12 µg of gene transfer vector, 12 µg of gag vector, 1.6 µg of pol vector, and 0.75 µg of env vector were dissolved in 400 µl of DMEM in an Eppendorf tube. To the plasmid DNA in DMEM, 60 µl of 1 mg/ml PEI was added dropwise while vortexing the tube. After 15 minutes, the reaction mixture was added dropwise to the cells in a 10-cm dish. The viral supernatant was harvested 3 days after transfection.38
FV vector supernatant was clarified by low-speed centrifugation, and fresh supernatant was directly used for transduction of the target cells or concentrated at 50,000 g in a Beckman SW28 rotor for 2 hours at room temperature. The vector supernatant was frozen in 5% dimethyl sulfoxide and stored at −80 °C. Vector was thawed, and dimethyl sulfoxide was removed by Viva Spin 20 (300k MWCO; Sartorius, Göttingen, Germany) before use.
Titers of the FV vector preparations were estimated after transduction of HT1080 cells and mentioned as transducing units per milliliter (TU/ml).6,38
HIV-based VSV-G pseudotyped LV production
HIV-based VSV-G pseudotyped LV vector production was described elsewhere. Titers of the LV vector preparations were estimated after transduction of 293T cells and mentioned as TU/ml.21
Transduction and transplantation of human CD34+ cells
The protocol for collecting CD34+ cells from human donors was approved by the St. Jude Children’s Research Hospital’s Institutional Review Board, and the studies were conducted in accordance with the Declaration of Helsinki. Mice studies were approved by the institutional animal care and use committee. A schematic diagram for transduction and transplantation of human CD34+ cells is shown (see Supplementary Figure S2). Granulocyte colony-stimulating factor–mobilized human CD34+ cells were harvested from peripheral blood mononuclear cells of healthy donors. Fresh CD34+ cells were cultured in X-Vivo 10 (Lonza, Allendale, NJ) or Stemline serum-free expansion medium (Sigma-Aldrich) containing 1% penicillin–streptomycin and L-glutamine. A cytokine mixture consisting of 100 ng/ml human stem cell factor, 100 ng/ml human thrombopoietin, and 100 ng/ml human FLT3 ligand (Cellgenix, Freiburg, Germany) was used for 106 cells/ml. Two transduction conditions were used in our studies. In the first condition (unstimulated), cells were not cultured for 24 hours. Cells were transduced in RetroNectin (Takara, Shiga, Japan)-coated plates (100 µg/ml) in growth medium overnight with concentrated FV-GFP or LV-GFP vectors at an MOI of 25. For LV transduction, 4 µg/ml protamine sulfate was added to the culture medium (Sigma-Aldrich). In the second condition (prestimulated), cells were cultured for 24 hours in cytokine-containing growth medium in the absence of RetroNectin. The next day, cells were transduced in RetroNectin-coated plates overnight with FV-GFP or LV-GFP vectors at an MOI of 25. Cells were collected, washed with phosphate-buffered saline, and resuspended in phosphate-buffered saline containing 2% fetal bovine serum. Cells (2 × 105) from each group were seeded in 6-well plates and analyzed for vector transduction for 3 weeks. From each group, 200 cells were seeded in a MethoCult (Stemcell Technologies, Vancouver, Canada) semisolid medium in duplicate in 35-mm plates for an in vitro colony assay. Ten 10-week-old female NOD/LtSz-scid IL2Rγc (NSG) mice (Jackson Laboratories, Bar Harbor, ME) in each group were given intraperitoneal injections of 35 mg/kg body weight of busulfan (Busulfex, PDL Biopharma, Redwood City, CA) 24 hours before transplantation. One million cells were injected into each mouse intravenously.
Flow cytometry analysis of repopulating human cells
Sixteen weeks after transplantation, bone marrow cells were collected from experimental mice. Red blood cells were lysed. Cells were blocked with normal mouse IgG and stained with mouse antihuman CD45-APC, mouse antihuman CD15-PE, mouse antihuman CD19-PE-Cy7 (BD Biosciences, San Diego, CA), and mouse antihuman CD33-RPE (Dako, Carpinteria, CA) for 30 minutes. After washing, one portion of cells was used for flow cytometry analysis, and the other portion was sorted into myeloid (CD33/15) and lymphoid (CD19) populations. The sorted myeloid and lymphoid cells were 99% pure. Cells were also stained with mouse antihuman CD45-APC and mouse antihuman CD34-PE and were analyzed separately. The data were analyzed using FlowJo software (Treestar Ashland, OR). A schematic diagram of the analysis of repopulating human cells is shown in Supplementary Figure S3.
Linear amplification-mediated PCR of genomic DNA from repopulating human cells
Genomic DNA was purified individually from human myeloid and lymphoid cells engrafted in mice. LAM-PCR analysis was performed as described previously.17 Vector LTR junction sequences were amplified by linear PCR using 300 ng of genomic DNA, vector LTR–specific 5′ biotinylated primer, 5′-AGAACCTTGTGTCTCTCATCCC-3′, and PCR master mix. DNA was denatured at 94 °C for 5 minutes, followed by 50 cycles of amplification (94 °C for 45 seconds, 60 °C for 45 seconds, and 72 °C for 60 seconds), and a final extension at 72 °C for 10 minutes. Biotinylated PCR products were bound to streptavidin-conjugated magnetic beads overnight with mild shaking. After washing, double-stranded DNA was synthesized using a random hexamer (Invitrogen) and DNA polymerase I large fragment (NEB, Ipswich, MA). DNA was digested with MluCI, MseI, and NlaIII (NEB) and ligated to restriction enzyme-specific double-stranded linkers using the Quick Ligation Kit (NEB). DNA was denatured with 0.1 N NaOH and was PCR-amplified using LTR- and linker-specific primers (first nested PCR primer of LTR, 5′-ACCTCCTTCCCTGTAATACTC-3′, and primer of linker, 5′-GCACTCGTGCTCGACTGATAC-3′; second nested PCR primer of LTR, 5′-CCTGGTTTCTAGTGGCATTC-3′ and primer of linker, 5′-CCGTCGTATCGTAGCACAG-3′). A schematic diagram shows the LAM-PCR analysis (see Supplementary Figure S4).
Illumina high-throughput sequencing and FV vector integration site analysis in the human genome
PCR products were ligated to the index adapter oligonucleotide mix and resolved in a 2% agarose gel. DNA fragments of 300–500 bp were excised and amplified according to the protocol of the Illumina (San Diego, CA) sample preparation kit. The quality of the DNA samples was analyzed by using Agilent DNA 1000 gel electrophoresis and PCR analysis. Samples were denatured and loaded into an Illumina Miseq personal sequencer by using DNA chips. The adapter sequences were trimmed from the reads. The paired-end reads were aligned with the FV LTR sequence and the linker sequence specific for MluCI, MseI, or NlaIII. Paired-end reads with both LTR and linker sequences containing at least 30 nucleotides of human genomic DNA sequence were uniquely aligned to a human reference genome (UCSC assembly hg19, GRCh37, February 2009) using CLC Genomics Workbench v5.1 (CLC Bio, Denmark). The viral integration sites were established on mapped clusters with at least 10× reads coverage plus 1% of total sample reads and confirmation of the restriction enzyme site on the opposite end of paired-end read from the integration sites. As a control, three size-matched sets of 799 random sites were created by computer simulation using the following criteria: (i) the DNA sequences must be in a range of 30–100 nucleotides in length containing one of the three restriction enzyme sites, outside of the sequences, in a window of 300 nucleotides upstream or downstream of the sequence, (ii) the sequences must be uniquely aligned to the human genome, and (iii) the random sites were selected at one end of the sequences that is farther away from the nearest restriction enzyme site.
Statistical analysis
Statistical analysis was done using the Student’s two-tailed t-test to determine statistically significant differences between mean values of data sets using Microsoft Excel (Microsoft Corporation, Redmond, WA) and GraphPad Prism (GraphPad, San Diego, CA), and a P value of ≤ 0.05 was considered statistically significant. A two-tailed Fisher’s exact test was used to compare differences between FV vector integration sites and random sites, and a P value of ≤ 0.01 was considered statistically significant.
Acknowledgments
We thank David W Russell, University of Washington, Seattle, WA, for generously providing FV vector plasmids. This work was supported by the National Heart, Lung, and Blood Institute PO1HL053749, Basic and Translational Research Program Sickle Cell Disease Grant U54HL070590, the Cooley’s Anemia Foundation, and the American Lebanese Syrian Associated Charities.
The authors declare no conflict of interest.
References
- Linial ML.2007. Foamy viruses. In: FieldsBNKHowleyDMPeterM. (eds.). Fields Virology, vol. 1 Lippincott Williams & Wilkins; Philadelphia. pp. 2245–2262. [Google Scholar]
- Trobridge GD, Miller DG, Jacobs MA, Allen JM, Kiem HP, Kaul R. Foamy virus vector integration sites in normal human cells. Proc Natl Acad Sci USA. 2006;103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer TR, Jr, Allen JM, Hai M, Tuschong LM, Khan IF, Olson EM. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat Med. 2008;14:93–97. doi: 10.1038/nm1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saïb A, Puvion-Dutilleul F, Schmid M, Périès J, de Thé H. Nuclear targeting of incoming human foamy virus Gag proteins involves a centriolar step. J Virol. 1997;71:1155–1161. doi: 10.1128/jvi.71.2.1155-1161.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge G, Russell DW. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. J Virol. 2004;78:2327–2335. doi: 10.1128/JVI.78.5.2327-2335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trobridge G, Josephson N, Vassilopoulos G, Mac J, Russell DW. Improved foamy virus vectors with minimal viral sequences. Mol Ther. 2002;6:321–328. doi: 10.1006/mthe.2002.0672. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, Gross F, Yvon E, Nusbaum P. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- Aiuti A, Slavin S, Aker M, Ficara F, Deola S, Mortellaro A. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296:2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, von Kalle C, Schmidt M, Le Deist F, Wulffraat N, McIntyre E. A serious adverse event after successful gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2003;348:255–256. doi: 10.1056/NEJM200301163480314. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiem HP, Allen J, Trobridge G, Olson E, Keyser K, Peterson L. Foamy-virus-mediated gene transfer to canine repopulating cells. Blood. 2007;109:65–70. doi: 10.1182/blood-2006-04-016741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson NC, Trobridge G, Russell DW. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Hum Gene Ther. 2004;15:87–92. doi: 10.1089/10430340460732481. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M, Schwarzwaelder K, Bartholomae C, Zaoui K, Ball C, Pilz I. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- Deichmann A, Hacein-Bey-Abina S, Schmidt M, Garrigue A, Brugman MH, Hu J. Vector integration is nonrandom and clustered and influences the fate of lymphopoiesis in SCID-X1 gene therapy. J Clin Invest. 2007;117:2225–2232. doi: 10.1172/JCI31659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarzwaelder K, Howe SJ, Schmidt M, Brugman MH, Deichmann A, Glimm H. Gammaretrovirus-mediated correction of SCID-X1 is associated with skewed vector integration site distribution in vivo. J Clin Invest. 2007;117:2241–2249. doi: 10.1172/JCI31661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton GS, Erlwein O, McClure MO. Cell-cycle dependence of foamy virus vectors. J Gen Virol. 2004;85 Pt 10:2925–2930. doi: 10.1099/vir.0.80210-0. [DOI] [PubMed] [Google Scholar]
- Hanawa H, Kelly PF, Nathwani AC, Persons DA, Vandergriff JA, Hargrove P. Comparison of various envelope proteins for their ability to pseudotype lentiviral vectors and transduce primitive hematopoietic cells from human blood. Mol Ther. 2002;5:242–251. doi: 10.1006/mthe.2002.0549. [DOI] [PubMed] [Google Scholar]
- Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josephson NC, Vassilopoulos G, Trobridge GD, Priestley GV, Wood BL, Papayannopoulou T. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proc Natl Acad Sci USA. 2002;99:8295–8300. doi: 10.1073/pnas.122131099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larochelle A, Vormoor J, Hanenberg H, Wang JC, Bhatia M, Lapidot T. Identification of primitive human hematopoietic cells capable of repopulating NOD/SCID mouse bone marrow: implications for gene therapy. Nat Med. 1996;2:1329–1337. doi: 10.1038/nm1296-1329. [DOI] [PubMed] [Google Scholar]
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol. 1987;196:261–282. doi: 10.1016/0022-2836(87)90689-9. [DOI] [PubMed] [Google Scholar]
- Akinc A, Thomas M, Klibanov AM, Langer R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J Gene Med. 2005;7:657–663. doi: 10.1002/jgm.696. [DOI] [PubMed] [Google Scholar]
- Williams DA.2000. Stem cell model of hematopoiesis. In: HoffmanRBenzEJShattilSJ (ed). Hematology: Basic Principles and PracticeChurchill Livingstone: London, UK; 126–138. [Google Scholar]
- Miller DG, Adam MA, Miller AD. Gene transfer by retrovirus vectors occurs only in cells that are actively replicating at the time of infection. Mol Cell Biol. 1990;10:4239–4242. doi: 10.1128/mcb.10.8.4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takatoku M, Sellers S, Agricola BA, Metzger ME, Kato I, Donahue RE. Avoidance of stimulation improves engraftment of cultured and retrovirally transduced hematopoietic cells in primates. J Clin Invest. 2001;108:447–455. doi: 10.1172/JCI12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glimm H, Oh IH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0) Blood. 2000;96:4185–4193. [PubMed] [Google Scholar]
- Burns JC, Friedmann T, Driever W, Burrascano M, Yee JK. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc Natl Acad Sci USA. 1993;90:8033–8037. doi: 10.1073/pnas.90.17.8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Jares A, Winkler T, Xie J, Metais JY, Dunbar CE. High efficiency restriction enzyme-free linear amplification-mediated polymerase chain reaction approach for tracking lentiviral integration sites does not abrogate retrieval bias. Hum Gene Ther. 2013;24:38–47. doi: 10.1089/hum.2012.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- Ohmine K, Li Y, Bauer TR, Jr, Hickstein DD, Russell DW. Tracking of specific integrant clones in dogs treated with foamy virus vectors. Hum Gene Ther. 2011;22:217–224. doi: 10.1089/hum.2010.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- Uchiyama T, Adriani M, Jagadeesh GJ, Paine A, Candotti F. Foamy virus vector-mediated gene correction of a mouse model of Wiskott-Aldrich syndrome. Mol Ther. 2012;20:1270–1279. doi: 10.1038/mt.2011.282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston RE, Chen CA, Rose JK. Calcium phosphate transfection. Curr Protoc Mol Biol. 2003;Chapter 9:Unit 9.1. doi: 10.1002/0471142727.mb0901s63. [DOI] [PubMed] [Google Scholar]
- Nasimuzzaman M, Persons DA. Cell Membrane-associated heparan sulfate is a receptor for prototype foamy virus in human, monkey, and rodent cells. Mol Ther. 2012;20:1158–1166. doi: 10.1038/mt.2012.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.