

In this review, Durcan and Fon discuss how post-translational modifications are at the heart of how PARKIN and PINK1 function in mitochondrial quality control. They also ask how our current understanding of these proteins may impact the development of future therapies for Parkinson's disease.
Keywords: PARKIN, ubiquitin, deubiquitination, mitophagy, PINK1, phosphorylation
Abstract
Two Parkinson's disease (PD)-associated proteins, the mitochondrial kinase PINK1 and the E3-ubiquitin (Ub) ligase PARKIN, are central to mitochondrial quality control. In this pathway, PINK1 accumulates on defective mitochondria, eliciting the translocation of PARKIN from the cytosol to mediate the clearance of damaged mitochondria via autophagy (mitophagy). Throughout the different stages of mitophagy, post-translational modifications (PTMs) are critical for the regulation of PINK1 and PARKIN activity and function. Indeed, activation and recruitment of PARKIN onto damaged mitochondria involves PINK1-mediated phosphorylation of both PARKIN and Ub. Through a stepwise cascade, PARKIN is converted from an autoinhibited enzyme into an active phospho-Ub-dependent E3 ligase. Upon activation, PARKIN ubiquitinates itself in concert with many different mitochondrial substrates. The Ub conjugates attached to these substrates can in turn be phosphorylated by PINK1, which triggers further cycles of PARKIN recruitment and activation. This feed-forward amplification loop regulates both PARKIN activity and mitophagy. However, the precise steps and sequence of PTMs in this cascade are only now being uncovered. For instance, the Ub conjugates assembled by PARKIN consist predominantly of noncanonical K6-linked Ub chains. Moreover, these modifications are reversible and can be disassembled by deubiquitinating enzymes (DUBs), including Ub-specific protease 8 (USP8), USP15, and USP30. However, PINK1-mediated phosphorylation of Ub can impede the activity of these DUBs, adding a new layer of complexity to the regulation of PARKIN-mediated mitophagy by PTMs. It is therefore evident that further insight into how PTMs regulate the PINK1–PARKIN pathway will be critical for our understanding of mitochondrial quality control.
Phosphorylation and ubiquitination are two post-translational modifications (PTMs) that often occur together in the regulation of signaling cascades. Examples of pathways regulated by both include the epidermal growth factor (EGF) receptor and NFκB signaling pathways (Karin and Ben-Neriah 2000; Nguyen et al. 2013). Typically, the addition of a phosphate moiety onto a particular residue of a substrate protein regulates the ability of an E3-ubiquitin (Ub) ligase to attach Ub onto lysine residues within a substrate (Lin et al. 2002; Gallagher et al. 2006; Dou et al. 2012). Considering the prominent cross-talk between these PTMs, it is not surprising that they are also implicated in many disease-associated pathways, including cancer and neurodegenerative diseases such as Parkinson's disease (PD). PD is a progressive neurodegenerative disorder that in most cases occurs sporadically. However, the discovery of genes responsible for rare familial cases of PD has shed light on the pathogenesis of the disease. For instance, recessive loss-of-function mutations in the PARK2 and PARK6 genes encode the E3-Ub ligase PARKIN and the mitochondrially targeted kinase PINK1, respectively. These two proteins act together through an intricate interplay of phosphorylation and ubiquitination (Kitada et al. 1998; Valente et al. 2004a,b). In this review, we discuss how these PTMs are at the very heart of how PARKIN and PINK1 function in mitochondrial quality control. Moreover, we introduce the concept of PARKIN as a phospho-Ub-dependent E3 ligase. Finally, we ask how our current understanding of these proteins may impact the development of future therapies for PD.
PD and mitochondrial dysfunction
PD is the second most common neurodegenerative disorder, predominantly affecting individuals over the age of 65, although cases with a much earlier onset have been observed. Whereas many neuronal types are affected in PD, it is predominantly the dopamine (DA) neurons of the substantia nigra pars compacta (SNc) region that are progressively lost, causing a gradual depletion of the neurotransmitter DA. A striking feature of DA neurons is their high energetic demands in the form of ATP. Mitochondria serve as the primary producers of ATP within the neurons, with much of it generated via the electron transport chain (ETC) through a stepwise reaction that consumes oxygen. However, the production of high levels of ATP does come at a cost to the neuron—in the form of superoxide and reactive oxygen species (ROS)—that can be damaging to the mitochondria and toxic to the neuron (Guzman et al. 2010). By generating increased levels of ROS in a “vicious cycle” of oxidative damage, defective mitochondria can be deleterious to neighboring mitochondria, making the presence of surveillance mechanisms essential for promoting the removal of these damaged mitochondria. The inability to eliminate dysfunctional mitochondria can elicit a domino effect, with damage spreading to neighboring mitochondria, ultimately resulting in the death of the affected neuron.
Mitochondrial dysfunction is often cited as a contributing factor to the development of PD, with early evidence coming from observations with the synthetic opioid MPTP (Langston et al. 1983), the insecticide rotenone, and the herbicide paraquat (Day et al. 1999; Betarbet et al. 2000; Richardson et al. 2005), all of which are mitochondrial toxins that have also been associated with PD. In parallel with the identification of these agents, a number of studies have demonstrated a direct association between mitochondrial dysfunction and PD. First, reduced activity of complex I of the ETC has been demonstrated in the substantia nigra, skeletal muscle, and platelets of patients with PD (Mizuno et al. 1989; Parker et al. 1989; Schapira et al. 1989). Furthermore, complex I subunits derived from mitochondria from PD patients exhibited oxidative damage consistent with defects in the ETC (Keeney et al. 2006). In addition, mitochondrial DNA deletions were found in the DA neurons from the brains of PD patients (Bender et al. 2006; Kraytsberg et al. 2006). Thus, evidence from these studies strongly implicates mitochondrial dysfunction in the etiology of PD.
PD-associated proteins at the mitochondria
Considering how mitochondrial defects can lead to cellular dysfunction, it is not surprising that quality control pathways exist to protect against oxidative stress and ultimately remove damaged mitochondria whose presence might be deleterious to the cell. What was more surprising was the initial realization that PARKIN and PINK1 both play a key role in mitochondrial quality control. Indeed, soon after its discovery, PARKIN was shown to function in the Ub pathway (Shimura et al. 2000). However, whether or how it could impact mitochondria was unknown. The first clue that PARKIN was important for mitochondrial function came from Drosophila (Greene et al. 2003). PARKIN mutant flies exhibited locomotor deficits, indirect flight muscle degeneration, and male sterility associated with prominent abnormalities in mitochondrial morphology. Remarkably, PINK1-null flies exhibited a very similar phenotype that could be rescued by overexpressing human PARKIN. Conversely, overexpression of PINK1 failed to rescue the PARKIN mutant phenotype, suggesting that PINK1 acts upstream of PARKIN in a common genetic pathway that impacts mitochondrial function (Clark et al. 2006; Park et al. 2006; Yang et al. 2006).
Clues to explain the cellular basis of this interaction came from the seminal studies from the laboratory of Richard Youle (Narendra et al. 2008), who found that in the presence of the uncoupler carbonyl cyanide m-chlorophenylhydrazone (CCCP), which dissipates the mitochondrial membrane potential (ΔΨm), PARKIN massively relocalized from the cytosol to the mitochondria. Moreover, once recruited to mitochondria, PARKIN promotes their clearance via the autophagy pathway (mitophagy). Remarkably, this process was strictly dependent on the presence of catalytically active PINK1, as kinase-dead PINK1 or PINK1 lacking an intact kinase domain is unable to promote PARKIN recruitment (Geisler et al. 2010; Matsuda et al. 2010; Narendra et al. 2010b). Considering the importance of PINK1 for the recruitment of PARKIN to mitochondria, these studies raise important questions about the molecular mechanisms involved. Mutations in the PINK1 gene are responsible for an autosomal recessive form of PD (Fig. 1A; Valente et al. 2004a,b). The PINK1 protein is a serine/threonine kinase, which under normal unstressed conditions is targeted to the mitochondria through its N-terminal mitochondrial targeting sequence (MTS), where it is imported via the translocase of outer membrane (TOM) and translocase of inner membrane (TIM) complexes (Lazarou et al. 2012). During its import through these channels, PINK1 undergoes a series of proteolytic cleavage steps, with the 64-kDa full-length form sequentially cleaved into 60- and 52-kDa fragments (Jin et al. 2010; Deas et al. 2011; Greene et al. 2012). The 52-kDa fragment is exported back into the cytosol, where it is degraded by the proteasome through the N-end rule pathway (Yamano and Youle 2013). As a result of this complex processing, PINK1 levels are maintained very low at steady state. PINK1 import requires an active proton gradient across the inner mitochondrial membrane to drive import. Indeed, depolarization of the mitochondria with either chemical or protein uncouplers prevents the import of full-length PINK1 via the TOM/TIM complex (Narendra et al. 2008; Grenier et al. 2014) and leads to the accumulation of full-length PINK1 on the outer mitochondrial surface. Overwhelming the import channels with excess PINK1 or preventing cleavage of PINK1 by knocking down specific mitochondrial proteases has the same effect, causing PINK1 to accumulate on the mitochondrial surface, where it can elicit PARKIN translocation from the cytosol (Narendra et al. 2010b; Greene et al. 2012). Moreover, if the N-terminal MTS is deleted or key residues in the MTS are mutated, PINK1 can accumulate on the mitochondrial surface and elicits PARKIN recruitment, suggesting the existence of an outer mitochondrial membrane localization domain between the N-terminal MTS and the transmembrane domain (Okatsu et al. 2015a). This domain might be involved in interactions with the TOM import channel, required for PINK1 accumulation at the outer mitochondrial membrane (Lazarou et al. 2012; Hasson et al. 2013; Okatsu et al. 2015a). Thus, accumulation of active PINK1 on the outer mitochondria surface is the first step in this quality control pathway.
Figure 1.
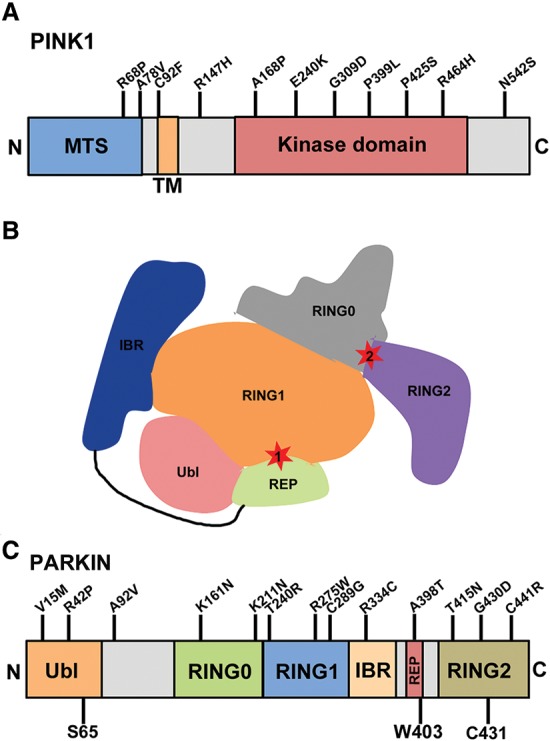
Schematic representation of the functional domains and PD-associated mutations in PINK1 and PARKIN. (A) Schematic of the domain structure and PD-associated mutations in PINK1. (B) Schematic highlighting sites of PARKIN autoinhibition. PARKIN activity depends on two functional sites: a binding site for the E2 on RING1 and a catalytic site at C431 in the RING2 domain. Both sites are occluded in the autoinhibited structure. (C) An outline of the domains in PARKIN and its PD-associated mutations. (Ubl) Ub-like domain; (RING) really interesting new gene; (IBR) in-between RINGs; (REP) repressor element of PARKIN domain. S65 in the Ubl is the site of PINK1-mediated phosphorylation. C431 is the PARKIN active site cysteine. W403 is a residue in the REP domain that, when mutated to an alanine (W403A), abolishes this autoinhibition and causes PARKIN to be hyperactive (Trempe et al. 2013). All indicated mutations in both PINK1 and PARKIN represent homozygous or compound heterozygous mutations associated with PD.
PARKIN: an autoinhibited E3-Ub ligase
While PINK1 initiates quality control, it is the E3-Ub ligase PARKIN that is responsible for directing the autophagic clearance of defective mitochondria (Narendra et al. 2008). PARKIN is also a PD-associated protein, and, to date, >120 loss-of-function mutations in the PARK2 gene have been linked to familial forms of PD (Kitada et al. 1998; Mata et al. 2004). As an E3-Ub ligase, PARKIN mediates the covalent attachment of the highly conserved 76-amino-acid protein Ub onto target proteins. The reaction involves the conjugation of the C-terminal glycine residue of Ub onto the ε-amino group of lysines within substrates. Ub itself contains seven lysines, any one of which can in turn be modified by another Ub molecule. This leads to the formation of polyUb chains with distinct architectures that have the potential to influence the function of a substrate in different ways depending on the linkages used to assemble the chains. The best-characterized linkage is Lys48 (K48), with K48-linked chains directing substrates for proteasomal degradation (Voges et al. 1999). However, Ub conjugates linked via one of the other six lysines or via the N-terminal α-amino group of Ub (head-to-tail linkage) can also regulate the function of the protein in a variety of distinct manners (Newton et al. 2008).
Soon after its discovery, PARKIN was demonstrated to function as an E3-Ub ligase, ubiquitinating a variety of substrates, including itself (Shimura et al. 2000; Zhang et al. 2000; Imai et al. 2001, 2002; Fallon et al. 2006; Trempe et al. 2009; Sarraf et al. 2013). However, PARKIN was found to exhibit rather poor E3 ligase activity at baseline in a variety of in vitro and cellular assays. Recent structural studies have shed light on the basis of this poor activity by revealing that PARKIN is natively autoinhibited (Riley et al. 2013; Spratt et al. 2013; Trempe et al. 2013; Wauer and Komander 2013). This autoinhibition is mediated by two distinct features of PARKIN (Fig. 1B): (1) A repressor element of the PARKIN (REP) domain occludes the RING1 (really interesting new gene 1) domain, preventing PARKIN from binding E2-Ub-conjugating enzymes, required for Ub transfer, and (2) an interaction between RING0–RING2 blocks access to the catalytic site, centered around Cys431 (C431) (Trempe et al. 2013). Indeed, when mutations are introduced that abolish these interactions (W403A, F463A, F146A, and RING0 deletion), PARKIN becomes constitutively active, leading to a dramatic increase in autoubiquitination (Trempe et al. 2013). In particular, the presence of a W403A mutation, which is predicted to dislodge the REP from RING1, enhances PARKIN-E2 binding, leading to a robust increase in PARKIN self-ubiquitination and accelerated recruitment onto depolarized mitochondria (Trempe et al. 2013). The structure also confirmed that PARKIN contains four zinc-binding “RING” domains along with an N-terminal Ub-like (Ubl) domain (Fig. 1C).
Interestingly, only RING1 adopts the fold of a canonical RING domain, whereas the configuration of the other three—RING0, IBR (in between RINGs), and RING2—adopt novel zinc-binding configurations. The three C-terminal zinc-binding domains (RING1, IBR, and RING2) form a distinct RBR superdomain, the hallmark of a family of 13 RBR-type E3s that include HHARI, Ariadne, HOIL, and HOIP (Spratt et al. 2014). Until recently, these RBR E3s were thought to function similarly to typical RING-type E3s. These act as scaffolds that merely promote the transfer of Ub from a Ub-charged E2 enzyme onto the substrate but do not form an intermediate with Ub in the conjugation reaction. However, studies with PARKIN and HHARI have disproven this model, demonstrating that PARKIN and other RBR-type E3s function as RING–HECT hybrid E3s (Wenzel et al. 2011). In this model, Ub is transferred from the E2 onto a catalytic cysteine in PARKIN, forming a short-lived PARKIN-Ub thioester intermediate before being transferred onto a lysine within the substrate (Lazarou et al. 2013; Riley et al. 2013). The crystal structure of PARKIN is largely consistent with this model, showing that C431 in RING2 is not bound to zinc and is therefore poised to function as the catalytic cysteine. Indeed, a PD mutation at this site (C431F) abolishes PARKIN E3-Ub ligase activity and its function in mitochondrial quality control (Lazarou et al. 2013). Moreover, mutating the catalytic cysteine to serine (C431S) can covalently trap Ub on PARKIN by way of an oxyester bond that is unable to release Ub onto substrates (Iguchi et al. 2013; Lazarou et al. 2013).
Activation of PARKIN by PINK1-mediated phosphorylation
In the context of mitochondrial quality control, these structural findings imply that PARKIN is predominantly inactive in the cytosol. Following the mitochondrial accumulation of PINK1, PARKIN is thought to undergo several activation steps that ultimately result in its translocation and clearance of damaged mitochondria. Central to this activation is PINK1-mediated phosphorylation. To elicit PARKIN recruitment, PINK1 exerts its effect in two ways: by phosphorylating PARKIN directly at S65 in the N-terminal Ubl domain (Kondapalli et al. 2012) and indirectly by phosphorylating Ub at the equivalent residue (Kane et al. 2014; Kazlauskaite et al. 2014; Koyano et al. 2014; Wauer et al. 2015). Evidence suggests that both PINK1-mediated phosphorylation of PARKIN and PARKIN binding to phospho-Ub activate PARKIN, triggering a feed-forward amplification loop to elicit further rounds of PARKIN-mediated ubiquitination and translocation onto the mitochondria. Moreover, PINK1 activity itself appears to be controlled by autophosphorylation, with the residues S228, T257, and S402 identified as sites of phosphorylation within PINK1 (Kondapalli et al. 2012; Okatsu et al. 2012; Aerts et al. 2015). These sites are critical for PINK1 function in the mitophagy pathway, as autophosphorylation at S228 and S402 enhances the ability of PINK1 to phosphorylate its substrates, PARKIN and Ub (Aerts et al. 2015). In line with these findings, mutation of S228 and S402 to alanine abolishes PINK1 activity, inhibiting the mitochondrial recruitment of PARKIN (Okatsu et al. 2012). However, the exact mechanism through which PINK1 phosphorylates itself and its substrates is just beginning to be elucidated.
We speculate that the addition of a phosphate onto S65 in both Ub and the PARKIN Ubl causes PARKIN to transition from a closed inhibited conformation to a more open active form via a stepwise series of events. Intriguingly, phosphorylated Ub can be incorporated into polyUb chains (Okatsu et al. 2015b; Wauer et al. 2015), and artificially targeting phospho-Ub or phosphomimetic chains to mitochondria and even lysosomes is sufficient to elicit recruitment of PARKIN in the absence of PINK1 (Shiba-Fukushima et al. 2014a; Okatsu et al. 2015b). Thus, in our model of PINK1-mediated activation and recruitment of PARKIN, we speculate that following its accumulation on depolarized mitochondria, PINK1 phosphorylates PARKIN and perhaps also pre-existing Ub conjugates on mitochondrial substrates that are located in close proximity. Indeed, it is likely that a small fraction of mitochondrial outer membrane proteins are conjugated with low levels of Ub at any given time. Upon accumulation of PINK1 at the surface, these Ub conjugates would become phosphorylated at S65, which increases their affinity for PARKIN. The combination of PINK1-mediated phosphorylation of PARKIN and phosphorylated Ub conjugates would effectively activate and recruit PARKIN to the mitochondria. Thus, a combination of interactions with phosphorylated Ub conjugates and direct phosphorylation of the Ubl activates PARKIN, leading to the polyubiquitination of multiple mitochondrial substrates. Such newly formed Ub chains would be in close proximity to PINK1 and thus ideally positioned for phosphorylation, which would elicit further PARKIN recruitment, activation, and substrate ubiquitination in a feed-forward mechanism (Fig. 2). Indeed, accumulation of phosphorylated Ub on depolarized mitochondria requires the presence of PARKIN, whereas PARKIN recruitment onto mitochondria also depends on Ub phosphorylation, and thus they are mutually interdependent (Okatsu et al. 2015b). Thus, we propose that PARKIN is a phospho-Ub-dependent E3 requiring a combination of interactions with phosphorylated Ub and direct phosphorylation of its own Ubl domain to be fully activated, ultimately leading to its mitochondrial recruitment and mitophagy. However, the precise sequence of phosphorylation and ubiquitination events still needs to be worked out. Moreover, it will be important to work out whether the same sequence of events is at play in vivo when parkin is activated in neurons in response to more physiological stimuli than chemical uncouplers.
Figure 2.
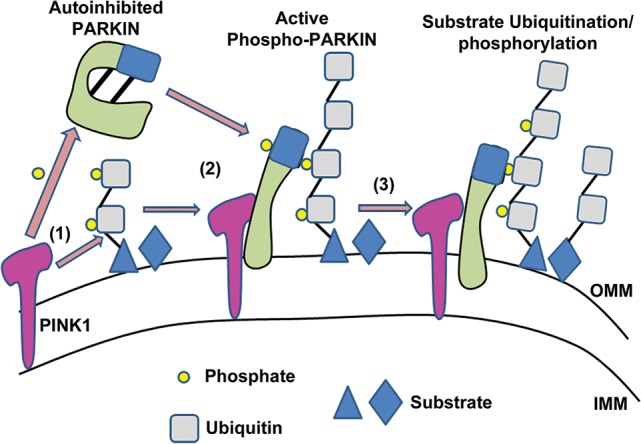
Model for PARKIN activation and recruitment onto damaged mitochondria. Following a loss in mitochondrial membrane potential, PINK1 accumulates on depolarized mitochondria. (1) When present on the OMM, PINK1 phosphorylates both S65 in the N-terminal Ubl of PARKIN and S65 within the Ub moieties in Ub conjugates that are attached to mitochondrial substrates on the outer mitochondrial membrane. (2) These phosphorylation events activate PARKIN and elicit its mitochondrial recruitment via direct interaction with the phospho-Ub conjugates on the mitochondria. (3) Active PARKIN promotes the ubiquitination of multiple mitochondrial substrates. PINK1 phosphorylates these Ub conjugates, which in turn acts as a feed-forward mechanism to further promote PARKIN activation and recruitment. (IMM) Inner mitochondrial membrane; (OMM) outer mitochondrial membrane.
Mitophagy and PARKIN-mediated ubiquitination
Once PARKIN is in the active conformation, its E3-Ub ligase activity is essential for ubiquitinating and mediating the clearance of a wide range of mitochondrial proteins (Sarraf et al. 2013), which occurs in a stepwise manner during mitophagy. Among the earliest and most prominent targets of PARKIN-mediated ubiquitination are mitofusin 1 and mitofusin 2, two GTPases essential for mitochondrial fusion (Tanaka et al. 2010). By promoting proteasomal degradation of the mitofusins, mitochondria become increasingly fragmented, leading to their isolation from one another. This step appears to be critical for separating damaged mitochondrial fragments from the remaining healthy reticulum. Next, PARKIN ubiquitinates multiple substrate proteins, with a preference for outer mitochondrial membrane proteins, including TOM20, TOM70, and the VDACs (Sarraf et al. 2013; Bingol et al. 2014). The extremely large number of PARKIN substrates identified in recent proteomic studies (Sarraf et al. 2013; Bingol et al. 2014; Cunningham et al. 2015) combined with the new concept of Ub phosphorylation by PINK1 would suggest that PARKIN is a rather promiscuous E3 ligase, with its specificity conferred by the presence of phospho-Ub on substrates rather than the identity of the substrates per se. This unique mode of activity would define PARKIN as the first phospho-Ub-dependent E3. Interestingly, proteasome function appears to be required for the turnover of several of these ubiquitinated membrane proteins, perhaps acting via a VCP-dependent mechanism to extract them from the mitochondrial membrane (Tanaka et al. 2010; Chan et al. 2011; Kim et al. 2013). Concurrently with proteasomal degradation, ubiquitination of mitochondrial proteins promotes the recruitment of Ub-binding autophagy receptors such as p62, optineurin, and NBR1 (Geisler et al. 2010; Narendra et al. 2010a; Chan et al. 2011; Wong and Holzbaur 2014), which in turn elicits the targeting of the damaged mitochondria to LC3-positive phagophores for clearance in the lysosome (Narendra et al. 2010a; Okatsu et al. 2010). An area of recent interest has been the nature of the Ub chains formed on these substrates. Mass spectrometry-based studies of mitochondria purified from CCCP-treated cells reveals a predominance of K6, K11, K48, and K63 linkages in these Ub conjugates (Ordureau et al. 2014; Cunningham et al. 2015). The identification of K48 and K63 linkages was not surprising, as these have been shown previously (Geisler et al. 2010, 2014; Chan et al. 2011). K48-linked Ub chains are important for proteasomal targeting, whereas K63-linked chains have been proposed to recruit autophagy adaptors. However, the importance of K63-linked conjugates in PINK1- and Parkin-dependent mitophagy remains controversial, as the successful clearance of mitochondria can still occur in the absence of Ubc13, the main E2 enzyme responsible for K63-linked Ub chain formation (Shiba-Fukushima et al. 2014b). Moreover, although PARKIN was found previously to assemble K27-linked Ub chains on VDAC1 (Geisler et al. 2010), K27 was not detected in these mass spectrometry analyses (Ordureau et al. 2014; Cunningham et al. 2015), suggesting that either PARKIN does not assemble K27-linked chains, or their levels are extremely low.
Intriguingly, the noncanonical linkages K6 and K11 were also present to varying degrees in these conjugates on different mitochondrial substrates, with levels of both increasing following treatment with CCCP. When either K6 or K11 linkages were impeded from forming with the overexpression of K6R or K11R mutant Ub, mitochondrial clearance was impaired, arguing that these noncanonical linkages play an important role in the overall process of mitophagy (Cunningham et al. 2015). Besides PARKIN, the RING-type E3s BRCA1 and RING1b and the bacterial E3 NleL are the only other E3s to date to have been found to preferentially generate such K6-linked conjugates (Wu-Baer et al. 2003; Ben-Saadon et al. 2006; de Bie et al. 2010; Hospenthal et al. 2013). Consistent with these findings, when PARKIN is activated by PINK1-mediated phosphorylation or the presence of phosphorylated Ub, it forms K6-linked chains at a faster rate than any other except for K48 (Ordureau et al. 2014). In concert with PARKIN-mediated ubiquitination of multiple mitochondrial proteins (Sarraf et al. 2013), a robust increase in PARKIN self-ubiquitination can be observed following treatment with CCCP (Durcan et al. 2014). As with PARKIN substrates, these conjugates contain K6-, K11-, K48-, and K63-linked Ub, with K6 again being the most prominent (Durcan et al. 2014). The Ub conjugates on PARKIN are confined to Lys27, Lys48, and Lys76 in its N-terminal Ubl domain (Sarraf et al. 2013; Durcan et al. 2014). Ubiquitination at these sites may regulate the interaction of the Ubl with more C-terminal domains of PARKIN (Chaugule et al. 2011) as well as with other Ubl- and Ub-binding proteins (Fallon et al. 2006; Trempe et al. 2009; Aguileta et al. 2015). Thus, autoubiquitination of the PARKIN Ubl may act as a second PTM that, in concert with phosphorylation, acts as an added layer of regulation to control PARKIN activity and mitochondrial recruitment. However, the precise mechanisms remain to be elucidated. For instance, are chains assembled by PARKIN on a given site conjugated homogenously by a single type of lysine linkage (i.e., all K6-linked), or do they consist of mixed chains linked by varying combinations of K6, K11, K48, and K63 (Fig. 3)? What are the molecular determinants that influence how PARKIN assembles these chains, and how could these different chain topologies regulate mitophagy? What are the respective roles of PARKIN autoubiquitination versus mitochondrial substrate ubiquitination in mitochondrial quality control, and how are these two processes regulated?
Figure 3.
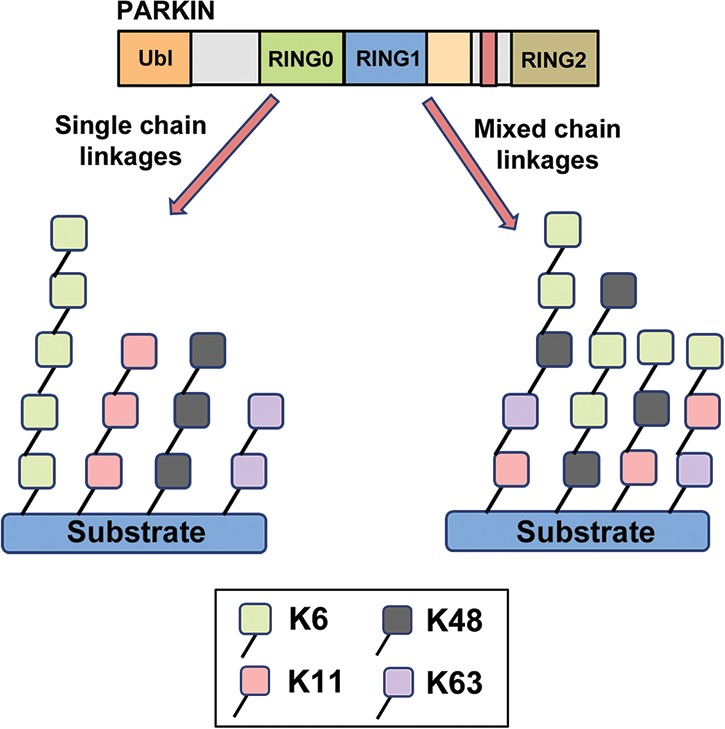
Model for the types of linkages in Ub conjugates formed by PARKIN. PARKIN forms Ub conjugates comprised of four distinct linkages: K6, K11, K48, and K63. (Left) In one potential scenario, PARKIN forms homotypic chains on its substrates comprised of one linkage only, albeit to varying degrees between chains. (Right) Alternatively, PARKIN can form chains that are mixed in nature, consisting of varying levels of each of the four linkages.
Deubiquitination and PARKIN-mediated mitophagy
Answers to some of these questions have come recently from work on deubiquitinating enzymes (DUBs). Ubiquitination is a reversible process, and DUBs oppose the activity of E3-Ub ligases by hydrolyzing the isopeptide bonds that link Ub chains. Deubiquitination is commonly used to regulate the activity of E3s, and many E3s have been shown to partner with one or more DUB. To date, close to 90 putative DUBs encoded by the human genome have been identified, belonging to five distinct subclasses (Nijman et al. 2005). The largest of these classes is the Ub-specific proteases (USPs). Similar to other E3s, the ability of PARKIN to ubiquitinate itself and its substrates is also regulated by deubiquitination. Ataxin-3, a member of the Josephin class of DUBs, was identified as the first DUB partner for PARKIN, binding it directly and opposing its ability to ubiquitinate itself (Durcan et al. 2011, 2012). However, ataxin-3 did not deubiquitinate PARKIN in a classical manner, as it was unable to hydrolyze the isopeptide linkage in PARKIN Ub conjugates after they had formed (Durcan et al. 2012). Rather, ataxin-3 was acting similarly to another unconventional DUB, OTUB1, that suppresses RNF168-mediated polyubiquitination by interacting with and inhibiting the Ubc13 E2-Ub-conjugating enzyme (Nakada et al. 2010). As with OTUB1, ataxin-3 binds the E2, in this instance UbcH7, directing the Ub from the E2-Ub thioester away from PARKIN and onto itself. This impedes the ability of PARKIN to ubiquitinate itself (Durcan et al. 2012). Although ataxin-3 regulates PARKIN self-ubiquitination, it does not appear to regulate the mitochondrial translocation of PARKIN or clearance of damaged mitochondria. However, as E3s are often regulated by multiple DUBs, it was likely that deubiquitination of PARKIN and its substrates by other DUBs was responsible for regulating its function in mitophagy.
One approach to identify such novel DUB partners for PARKIN involved using an siRNA screen to knock down all of the known DUBs to ascertain whether their silencing could affect the mitochondrial recruitment of PARKIN (Durcan et al. 2014). Knockdown of the endocytic DUB USP8 was found to delay PARKIN translocation onto depolarized mitochondria. Moreover, silencing of USP8 also delayed mitochondrial clearance at longer time points. In contrast to ataxin-3, USP8 deubiquitinated PARKIN directly, mediating the removal of Ub conjugates from PARKIN by specifically acting on the K6 linkages in these chains (Durcan et al. 2014). Thus, when levels of USP8 were reduced, an accumulation of K6-linked Ub conjugates on PARKIN delayed its overall function in the mitophagy pathway. Why an increase in K6-linked conjugates on PARKIN would impair PARKIN function is still unclear. Perhaps their presence interferes with PARKIN interactions with one or more protein required for clearance of damaged mitochondria. Indeed, if aberrant K6-linked Ub chains are present on the Ubl, the interaction of PARKIN with PINK1 and/or phosphorylated Ub may be impaired, thereby delaying the initial stages of PARKIN activation and recruitment. It also should be noted that K6-linked conjugates appear to persist longer on PARKIN following knockdown of USP8 (Durcan et al. 2014), and their presence may be impeding the interaction of PARKIN with autophagic adaptor proteins and other substrates. One possibility is that the ratio of linkages in the Ub conjugates on mitochondrial substrates may shift toward K6 relative to the other linkages. As a result, the altered Ub architecture on the mitochondria may impede interactions with the autophagic adaptors, thereby impairing the autophagic clearance of damaged mitochondria.
Deubiquitination of mitochondrial substrates
While many DUBs regulate their E3 partner by direct deubiquitination, others exert their effect indirectly by deubiquitinating substrates of their E3 partner. PARKIN is no exception, and separate studies have identified two DUBs that regulate PARKIN activity within the mitophagy pathway by deubiquitinating substrates of PARKIN (Bingol et al. 2014; Cornelissen et al. 2014; Dikic and Bremm 2014). In the first study, USP15 was identified as a binding partner of PARKIN by affinity purification (Cornelissen et al. 2014). Remarkably, when overexpressed, USP15 impaired PARKIN-mediated clearance of mitochondria, with no effect on the mitochondrial recruitment of PARKIN. Conversely, when USP15 was knocked down, clearance of mitochondria was enhanced, with an absence of USP15 rescuing the mitophagy defect observed in PARKIN mutant fibroblasts (Cornelissen et al. 2014).
In contrast to USP8, USP15 was unable to deubiquitinate PARKIN; rather, USP15 exerts its effect by deubiquitinating mitochondrial substrates of PARKIN. Thus, when absent, Ub conjugates normally hydrolyzed by USP15 are left intact, thereby accelerating the rate of mitophagy. When overexpressed, excessive USP15-mediated hydrolysis of these conjugates occurs, with diminished levels of Ub chains on mitochondrial substrates impairing mitophagy. Similar findings were reported for another DUB: USP30. USP30 overexpression impaired PARKIN-mediated mitophagy by deubiquitinating mitochondrial substrates that include TOM20, TOM70, and the VDACs (Bingol et al. 2014). It should be noted that USP30 was also overexpressed in the USP15 study, but no effect on mitophagy was observed (Cornelissen et al. 2014). Moreover, in addition to its role in regulating mitophagy, USP30 has recently been shown to deubiquitinate substrates of PARKIN in mitochondrial cell death pathways (Liang et al. 2015). Intriguingly, as with USP8, USP30 demonstrates a preference for chains with K6 linkages, hydrolyzing them at a faster rate compared with chains linked through one of the other lysines (Ordureau et al. 2014; Cunningham et al. 2015). Strikingly, USP30 overexpression and knockdown also had an effect on mitochondrial turnover in neurons. To monitor the rate of mitophagy in neurons, Bingol et al. (2014) used a mitochondrially targeted keima red fluorescent protein to measure turnover of the mitochondria in the lysosome. Using this system, knockdown of USP30 enhanced the rate of mitochondrial turnover, dependent on the presence of functional PARKIN and PINK1, even in the absence of CCCP. Conversely, when USP30 was overexpressed, the rate of turnover was decreased. Moreover, USP30 mutant Drosophila displayed increased resistance in models of oxidative stress and PD (Bingol et al. 2014). Thus, USP15 and USP30 both deubiquitinate substrates of PARKIN and oppose PARKIN function in mitophagy. Intriguingly, when USP30 was knocked down, the conjugates on mitochondrial substrates contained elevated levels of K6 linkages, which in turn appeared to accelerate mitochondrial turnover. When K6 linkages were prevented from forming with the overexpression of a K6R mutant Ub, mitophagy was impaired, highlighting a critical role for K6 linkages in the efficient clearance of mitochondria (Bingol et al. 2014; Cunningham et al. 2015). In contrast, when USP8 is knocked down, K6 conjugates that accumulate on PARKIN appear to impair its overall function (Durcan et al. 2014). Thus, K6 linkages appear to positively regulate mitophagy when present on mitochondrial substrates, while they appear to inhibit quality control when present at excessive levels on PARKIN. However, the reason for these opposing effects remains unclear and will require further study.
Phosphorylation and its effect on DUB activity
In addition to the differential effects of various types of Ub chains on mitophagy, a further layer of complexity is added by the potential for these chains to be phosphorylated by PINK1 (Shiba-Fukushima et al. 2014a; Okatsu et al. 2015b; Wauer et al. 2015). The functional consequences of PINK1-mediated Ub chain phosphorylation are currently unknown. Typically, the downstream effects of ubiquitination are mediated by proteins containing several families of Ub-binding domains (UBDs) (Husnjak and Dikic 2012). Do certain UBDs bind specifically or preferentially to chains containing phospho-S65 Ub? A recent study reporting the crystal structure of phospho-Ub suggested that this might be the case. Indeed, whereas phospho-Ub generally resembles unmodified Ub, it exhibited altered surface properties. Moreover, nuclear magnetic resonance (NMR) experiments revealed a minor species representing ∼30% of all phospho-Ub in solution that adopted a conformation in which β5-strand slippage retracts the C-terminal tail by two residues into the Ub core (Wauer et al. 2015). How this could impact Ub interactions and function is still not known. However, one possibility is that it would make the C terminus of Ub less accessible to enzymatic cleavage by DUBs. Indeed, the majority of DUBs tested was unable to hydrolyze the isopeptide linkages in phospho-Ub chains (Wauer et al. 2015). This included USP8, USP15, USP30, and ataxin-3, all DUBs known to regulate PARKIN activity. Even the relatively promiscuous DUB USP2, which deubiquitinates nearly every kind of Ub conjugate in vitro, was unable to cleave the isopeptide linkage in phospho-Ub chains (Wauer et al. 2015). Thus, phosphorylation of Ub appears to impede deubiquitination, suggesting that it may serve as a further mechanism to amplify the signal generated by PINK1 and PARKIN on mitochondria and perhaps also direct it toward a pathway involving a distinct set of phospho-Ub-binding proteins. Given their resistance to DUBs, how might the signal conferred by phospho-Ub chains be ultimately terminated? One potential mechanism could involve a phospho-Ub-specific phosphatase. Like the opposing cycles of ubiquitination/deubiquitination involving E3s and DUBs, dephosphorylation of PINK1-induced phospho-Ub and phospho-Ub chains is likely to be mediated by one or more phosphatases. By dephosphorylating phospho-Ub, this might enhance the accessibility of the Ub chains to DUBs and deubiquitination. One potential candidate is PGAM5, a phosphatase localized at the mitochondria that is released from the mitochondria following dissipation of the mitochondrial membrane potential (Sekine et al. 2012; Chen et al. 2014). Intriguingly, the presence of PGAM5 is required for PINK1 to accumulate during mitophagy, with a loss of PGAM5 reducing PINK1 levels and impairing mitophagy (Lu et al. 2014). Moreover, PGAM5 knockout mice exhibited a PD-like movement phenotype (Lu et al. 2014). Thus, these findings suggest that PGAM5 interacts with PINK1, but whether it is involved in dephosphorylating phospho-Ub is not yet clear. To further complicate matters, we also must consider the possibility that additional PTMs might regulate the formation of Ub conjugates during mitophagy. A recent mass spectrometry study demonstrated that Ub is acetylated at both K6 and K48, two critical residues in the formation of Ub conjugates in mitophagy (Ohtake et al. 2015). This implies that Ub chains generated by PARKIN might be phosphorylated and acetylated, with these PTMs regulating their overall structure and function. Taken together, PARKIN-mediated mitophagy is a complex pathway with an interwoven network of PTMs regulating the entire process.
Conclusions and perspectives
Mitochondria are under constant stress and need to cope with the high energetic demands of the neuron. Consequently, defects within the mitochondria can be catastrophic, causing the neuronal cell loss associated with PD and other neurodegenerative diseases. To cope with this stress, mitochondrial quality control pathways have evolved, with PINK1 and PARKIN central to these surveillance mechanisms. Regulation of PARKIN function in these pathways and in particular within the mitophagy pathway is complex, involving several overlapping PTMs. As an E3, the primary function of PARKIN is to attach Ub conjugates onto substrates, including itself, influencing the fate of the proteins. Intriguingly, PARKIN preferentially forms K6 and K11 chains in addition to K48 and K63, yet how the linkages in Ub conjugates influence the rate of mitophagy is unclear. As the field advances, it is critical that we work to understand the exact nature of these chains. Is every chain generated by PARKIN mixed in nature, or is it substrate-dependent? Moreover, what is the exact role of K6 linkages within these chains, and precisely how is it influencing the overall function of PARKIN and its substrates in this pathway.
PARKIN function is opposed either directly or indirectly by multiple DUBs. The endocytic DUB USP8 has recently been shown to function at the mitochondria and influence PARKIN function within the mitophagy pathway. Its absence causes K6-linked Ub conjugates to accumulate on PARKIN, delaying PARKIN recruitment and mitophagy (Durcan et al. 2014). Moreover, both USP15 and USP30 function indirectly, deubiquitinating substrates of PARKIN. Considering the importance of these DUBs in regulating PARKIN function, they represent important therapeutic targets that have the potential to be exploited as a means to enhance mitochondrial quality control. If one or more can be targeted to bolster PARKIN function within the mitophagy pathway, it might also protect neurons that would otherwise be overwhelmed by high levels of stress.
Cycles of ubiquitination and deubiquitination are central to the function and activity of PARKIN. However, these are not the only modifications that exert an effect. Phosphorylation is another PTM that is important for the regulation of PARKIN. Central to this process is PINK1-mediated phosphorylation of Ub and the Ubl of PARKIN at Ser65. However, the exact mechanism through which phosphorylated Ub chains are formed remains unknown and is likely the next important avenue to investigate. How they influence the overall process of mitophagy is unclear, and whether it is reversed by phosphatases also remains to be established. Further work is also required to determine whether other modifications, such as acetylation, might influence PARKIN activity.
From studies on PINK1- and PARKIN-mediated mitophagy, we have furthered our understanding of the PTMs that tightly regulate the pathway. However, other quality control mechanisms exist in which PTMs might be involved. Indeed, a second pathway has recently been demonstrated in which mitochondrial-derived vesicles (MDVs) bud from the mitochondria during conditions of oxidative stress that are targeted to lysosomes for degradation (Soubannier et al. 2012a,b; McLelland et al. 2014; Sugiura et al. 2014). Following incubation with anti-mycin A, which induces oxidative stress at complex III of the mitochondrial respiratory chain, PARKIN localizes to MDV bud sites and promotes their biogenesis and trafficking to the endosomal–lysosomal system for clearance. As with mitophagy, this process is both PINK1 and PARKIN dependent (McLelland et al. 2014). However, in contrast to mitophagy, little is still known about how MDV formation is regulated and whether PTMs are involved. As we move forward in our understanding of PINK1 and PARKIN and its regulation by PTMs in the mitophagy pathway, it will be imperative that we also extend our studies to better characterize their role in the MDV pathway. Indeed, it is becoming ever clearer that PINK1- and PARKIN-mediated mitochondrial quality control is a complex process with multiple layers of regulation, all intricately connected via PTMs. Moreover, each modification provides a target to be explored for potential therapeutic benefits. Considering the importance of a healthy mitochondrial reticulum for DA neuron function and survival, exploiting one or more of these regulatory pathways to enhance PINK1 or PARKIN function represents an important avenue to investigate in the development of new treatments for PD.
Acknowledgments
We acknowledge the Canadian Institutes of Health Research, Fonds de Recherche du Québec-Santé, and Parkinson's Society of Canada for their funding.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.262758.115.
References
- Aerts L, Craessaerts K, De Strooper B, Morais VA. 2015. PINK1 kinase catalytic activity is regulated by phosphorylation on serines 228 and 402. J Biol Chem 290: 2798–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguileta MA, Korac J, Durcan TM, Trempe JF, Haber M, Gehring K, Elsasser S, Waidmann O, Fon EA, Husnjak K. 2015. The E3 Ubiquitin ligase parkin is recruited to the 26S proteasome via the proteasomal ubiquitin receptor Rpn13. J Biol Chem 290: 7492–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, et al. 2006. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38: 515–517. [DOI] [PubMed] [Google Scholar]
- Ben-Saadon R, Zaaroor D, Ziv T, Ciechanover A. 2006. The polycomb protein Ring1B generates self atypical mixed ubiquitin chains required for its in vitro histone H2A ligase activity. Mol Cell 24: 701–711. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. 2000. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301–1306. [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. 2014. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510: 370–375. [DOI] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. 2011. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaugule VK, Burchell L, Barber KR, Sidhu A, Leslie SJ, Shaw GS, Walden H. 2011. Autoregulation of Parkin activity through its ubiquitin-like domain. EMBO J 30: 2853–2867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, et al. 2014. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54: 362–377. [DOI] [PubMed] [Google Scholar]
- Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. 2006. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166. [DOI] [PubMed] [Google Scholar]
- Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, et al. 2014. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23: 5227–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE. 2015. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol 17: 160–169. [DOI] [PubMed] [Google Scholar]
- Day BJ, Patel M, Calavetta L, Chang LY, Stamler JS. 1999. A mechanism of paraquat toxicity involving nitric oxide synthase. Proc Natl Acad Sci 96: 12760–12765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, Plun-Favreau H, Gandhi S, Desmond H, Kjaer S, Loh SH, Renton AE, Harvey RJ, Whitworth AJ, Martins LM, et al. 2011. PINK1 cleavage at position A103 by the mitochondrial protease PARL. Hum Mol Genet 20: 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bie P, Zaaroor-Regev D, Ciechanover A. 2010. Regulation of the Polycomb protein RING1B ubiquitination by USP7. Biochem Biophys Res Commun 400: 389–395. [DOI] [PubMed] [Google Scholar]
- Dikic I, Bremm A. 2014. DUBs counteract parkin for efficient mitophagy. EMBO J 33: 2442–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Buetow L, Hock A, Sibbet GJ, Vousden KH, Huang DT. 2012. Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat Struct Mol Biol 19: 184–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Thorarinsdottir T, Fallon L, Williams AJ, Djarmati A, Fantaneanu T, Paulson HL, Fon EA. 2011. The Machado-Joseph disease-associated mutant form of ataxin-3 regulates parkin ubiquitination and stability. Hum Mol Genet 20: 141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Kontogiannea M, Bedard N, Wing SS, Fon EA. 2012. Ataxin-3 deubiquitination is coupled to Parkin ubiquitination via E2 ubiquitin-conjugating enzyme. J Biol Chem 287: 531–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B, et al. 2014. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J 33: 2473–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon L, Belanger CM, Corera AT, Kontogiannea M, Regan-Klapisz E, Moreau F, Voortman J, Haber M, Rouleau G, Thorarinsdottir T, et al. 2006. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K–Akt signalling. Nat Cell Biol 8: 834–842. [DOI] [PubMed] [Google Scholar]
- Gallagher E, Gao M, Liu YC, Karin M. 2006. Activation of the E3 ubiquitin ligase Itch through a phosphorylation-induced conformational change. Proc Natl Acad Sci 103: 1717–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisler S, Holmstrom KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. 2010. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131. [DOI] [PubMed] [Google Scholar]
- Geisler S, Vollmer S, Golombek S, Kahle PJ. 2014. The ubiquitin-conjugating enzymes UBE2N, UBE2L3 and UBE2D2/3 are essential for Parkin-dependent mitophagy. J Cell Sci 127: 3280–3293. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. 2003. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci 100: 4078–4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greene AW, Grenier K, Aguileta MA, Muise S, Farazifard R, Haque ME, McBride HM, Park DS, Fon EA. 2012. Mitochondrial processing peptidase regulates PINK1 processing, import and Parkin recruitment. EMBO Rep 13: 378–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier K, Kontogiannea M, Fon EA. 2014. Short mitochondrial ARF triggers Parkin/PINK1-dependent mitophagy. J Biol Chem 289: 29519–29530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, Surmeier DJ. 2010. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 468: 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasson SA, Kane LA, Yamano K, Huang CH, Sliter DA, Buehler E, Wang C, Heman-Ackah SM, Hessa T, Guha R, et al. 2013. High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature 504: 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hospenthal MK, Freund SM, Komander D. 2013. Assembly, analysis and architecture of atypical ubiquitin chains. Nat Struct Mol Biol 20: 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husnjak K, Dikic I. 2012. Ubiquitin-binding proteins: decoders of ubiquitin-mediated cellular functions. Annu Rev Biochem 81: 291–322. [DOI] [PubMed] [Google Scholar]
- Iguchi M, Kujuro Y, Okatsu K, Koyano F, Kosako H, Kimura M, Suzuki N, Uchiyama S, Tanaka K, Matsuda N. 2013. Parkin-catalyzed ubiquitin-ester transfer is triggered by PINK1-dependent phosphorylation. J Biol Chem 288: 22019–22032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai Y, Soda M, Inoue H, Hattori N, Mizuno Y, Takahashi R. 2001. An unfolded putative transmembrane polypeptide, which can lead to endoplasmic reticulum stress, is a substrate of Parkin. Cell 105: 891–902. [DOI] [PubMed] [Google Scholar]
- Imai Y, Soda M, Hatakeyama S, Akagi T, Hashikawa T, Nakayama KI, Takahashi R. 2002. CHIP is associated with Parkin, a gene responsible for familial Parkinson's disease, and enhances its ubiquitin ligase activity. Mol Cell 10: 55–67. [DOI] [PubMed] [Google Scholar]
- Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. 2010. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. 2014. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu Rev Immunol 18: 621–663. [DOI] [PubMed] [Google Scholar]
- Kazlauskaite A, Kondapalli C, Gourlay R, Campbell DG, Ritorto MS, Hofmann K, Alessi DR, Knebel A, Trost M, Muqit MM. 2014. Parkin is activated by PINK1-dependent phosphorylation of ubiquitin at Ser65. Biochem J 460: 127–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeney PM, Xie J, Capaldi RA, Bennett JP Jr. 2006. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J Neurosci 26: 5256–5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, et al. 2013. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. Neuron 78: 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. 1998. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608. [DOI] [PubMed] [Google Scholar]
- Kondapalli C, Kazlauskaite A, Zhang N, Woodroof HI, Campbell DG, Gourlay R, Burchell L, Walden H, Macartney TJ, Deak M, et al. 2012. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating serine 65. Open Biol 2: 120080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, et al. 2014. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166. [DOI] [PubMed] [Google Scholar]
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. 2006. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet 38: 518–520. [DOI] [PubMed] [Google Scholar]
- Langston JW, Ballard P, Tetrud JW, Irwin I. 1983. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 219: 979–980. [DOI] [PubMed] [Google Scholar]
- Lazarou M, Jin SM, Kane LA, Youle RJ. 2012. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazarou M, Narendra DP, Jin SM, Tekle E, Banerjee S, Youle RJ. 2013. PINK1 drives Parkin self-association and HECT-like E3 activity upstream of mitochondrial binding. J Cell Biol 200: 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JR, Martinez A, Lane JD, Mayor U, Clague MJ, Urbe S. 2015. USP30 deubiquitylates mitochondrial Parkin substrates and restricts apoptotic cell death. EMBO Rep 10.15252/embr.201439820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. 2002. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J 21: 4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Karuppagounder SS, Springer DA, Allen MD, Zheng L, Chao B, Zhang Y, Dawson VL, Dawson TM, Lenardo M. 2014. Genetic deficiency of the mitochondrial protein PGAM5 causes a Parkinson's-like movement disorder. Nat Commun 5: 4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Lockhart PJ, Farrer MJ. 2004. Parkin genetics: one model for Parkinson's disease. Hum Mol Genet 13: R127–R133. [DOI] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, et al. 2010. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. 2014. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33: 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, Oya H, Ozawa T, Kagawa Y. 1989. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Commun 163: 1450–1455. [DOI] [PubMed] [Google Scholar]
- Nakada S, Tai I, Panier S, Al-Hakim A, Iemura S, Juang YC, O'Donnell L, Kumakubo A, Munro M, Sicheri F, et al. 2010. Non-canonical inhibition of DNA damage-dependent ubiquitination by OTUB1. Nature 466: 941–946. [DOI] [PubMed] [Google Scholar]
- Narendra D, Tanaka A, Suen DF, Youle RJ. 2008. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. 2010a. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. 2010b. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, et al. 2008. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134: 668–678. [DOI] [PubMed] [Google Scholar]
- Nguyen LK, Kolch W, Kholodenko BN. 2013. When ubiquitination meets phosphorylation: a systems biology perspective of EGFR/MAPK signalling. Cell Commun Signal 11: 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R. 2005. A genomic and functional inventory of deubiquitinating enzymes. Cell 123: 773–786. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Sakamoto K, Ohtake K, Nishikawa H, Tsuchiya H, Ohta T, Tanaka K, Kanno J. 2015. Ubiquitin acetylation inhibits polyubiquitin chain elongation. EMBO Rep 16: 192–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, et al. 2010. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15: 887–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Oka T, Iguchi M, Imamura K, Kosako H, Tani N, Kimura M, Go E, Koyano F, Funayama M, et al. 2012. PINK1 autophosphorylation upon membrane potential dissipation is essential for Parkin recruitment to damaged mitochondria. Nat Commun 3: 1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Kimura M, Oka T, Tanaka K, Matsuda N. 2015a. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J Cell Sci 128: 964–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okatsu K, Koyano F, Kimura M, Kosako H, Saeki Y, Tanaka K, Matsuda N. 2015b. Phosphorylated ubiquitin chain is the genuine Parkin receptor. J Cell Biol 209: 111–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, et al. 2014. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. Mol Cell 56: 360–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, et al. 2006. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161. [DOI] [PubMed] [Google Scholar]
- Parker WD Jr, Boyson SJ, Parks JK. 1989. Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 26: 719–723. [DOI] [PubMed] [Google Scholar]
- Richardson JR, Quan Y, Sherer TB, Greenamyre JT, Miller GW. 2005. Paraquat neurotoxicity is distinct from that of MPTP and rotenone. Toxicol Sci 88: 193–201. [DOI] [PubMed] [Google Scholar]
- Riley BE, Lougheed JC, Callaway K, Velasquez M, Brecht E, Nguyen L, Shaler T, Walker D, Yang Y, Regnstrom K, et al. 2013. Structure and function of Parkin E3 ubiquitin ligase reveals aspects of RING and HECT ligases. Nat Commun 4: 1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. 2013. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. 1989. Mitochondrial complex I deficiency in Parkinson's disease. Lancet 1: 1269. [DOI] [PubMed] [Google Scholar]
- Sekine S, Kanamaru Y, Koike M, Nishihara A, Okada M, Kinoshita H, Kamiyama M, Maruyama J, Uchiyama Y, Ishihara N, et al. 2012. Rhomboid protease PARL mediates the mitochondrial membrane potential loss-induced cleavage of PGAM5. J Biol Chem 287: 34635–34645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Arano T, Matsumoto G, Inoshita T, Yoshida S, Ishihama Y, Ryu KY, Nukina N, Hattori N, Imai Y. 2014a. Phosphorylation of mitochondrial polyubiquitin by PINK1 promotes Parkin mitochondrial tethering. PLoS Genet 10: e1004861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiba-Fukushima K, Inoshita T, Hattori N, Imai Y. 2014b. Lysine 63-linked polyubiquitination is dispensable for Parkin-mediated mitophagy. J Biol Chem 289: 33131–33136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, et al. 2000. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302–305. [DOI] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. 2012a. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22: 135–141. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM. 2012b. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7: e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt DE, Julio Martinez-Torres R, Noh YJ, Mercier P, Manczyk N, Barber KR, Aguirre JD, Burchell L, Purkiss A, Walden H, et al. 2013. A molecular explanation for the recessive nature of parkin-linked Parkinson's disease. Nat Commun 4: 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt DE, Walden H, Shaw GS. 2014. RBR E3 ubiquitin ligases: new structures, new insights, new questions. Biochem J 458: 421–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM. 2014. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33: 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. 2010. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trempe JF, Chen CX, Grenier K, Camacho EM, Kozlov G, McPherson PS, Gehring K, Fon EA. 2009. SH3 domains from a subset of BAR proteins define a Ubl-binding domain and implicate parkin in synaptic ubiquitination. Mol Cell 36: 1034–1047. [DOI] [PubMed] [Google Scholar]
- Trempe JF, Sauve V, Grenier K, Seirafi M, Tang MY, Menade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, et al. 2013. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, et al. 2004a. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304: 1158–1160. [DOI] [PubMed] [Google Scholar]
- Valente EM, Salvi S, Ialongo T, Marongiu R, Elia AE, Caputo V, Romito L, Albanese A, Dallapiccola B, Bentivoglio AR. 2004b. PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 56: 336–341. [DOI] [PubMed] [Google Scholar]
- Voges D, Zwickl P, Baumeister W. 1999. The 26S proteasome: a molecular machine designed for controlled proteolysis. Annu Rev Biochem 68: 1015–1068. [DOI] [PubMed] [Google Scholar]
- Wauer T, Komander D. 2013. Structure of the human Parkin ligase domain in an autoinhibited state. EMBO J 32: 2099–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wauer T, Swatek KN, Wagstaff JL, Gladkova C, Pruneda JN, Michel MA, Gersch M, Johnson CM, Freund SM, Komander D. 2015. Ubiquitin Ser65 phosphorylation affects ubiquitin structure, chain assembly and hydrolysis. EMBO J 34: 307–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel DM, Lissounov A, Brzovic PS, Klevit RE. 2011. UBCH7 reactivity profile reveals parkin and HHARI to be RING/HECT hybrids. Nature 474: 105–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC, Holzbaur EL. 2014. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci 111: E4439–E4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu-Baer F, Lagrazon K, Yuan W, Baer R. 2003. The BRCA1/BARD1 heterodimer assembles polyubiquitin chains through an unconventional linkage involving lysine residue K6 of ubiquitin. J Biol Chem 278: 34743–34746. [DOI] [PubMed] [Google Scholar]
- Yamano K, Youle RJ. 2013. PINK1 is degraded through the N-end rule pathway. Autophagy 9: 1758–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, Yang L, Beal MF, Vogel H, Lu B. 2006. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci 103: 10793–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. 2000. Parkin functions as an E2-dependent ubiquitin-protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci 97: 13354–13359. [DOI] [PMC free article] [PubMed] [Google Scholar]