

Abstract
With the rise of high-throughput sequencing technology, traditional genotyping arrays are gradually being replaced by sequencing technology. Against this trend, Illumina has introduced an exome genotyping array that provides an alternative approach to sequencing, especially suited to large-scale genome-wide association studies (GWASs). the exome genotyping array targets the exome plus rare single-nucleotide polymorphisms (SNPs), a feature that makes it substantially more challenging to process than previous genotyping arrays that targeted common SNPs. Researchers have struggled to generate a reliable protocol for processing exome genotyping array data. The Vanderbilt epidemiology center, in cooperation with Vanderbilt Technologies for Advanced Genomics Analysis and Research Design (VANGARD), has developed a thorough exome chip–processing protocol. The protocol was developed during the processing of several large exome genotyping array-based studies, which included over 60,000 participants combined. The protocol described herein contains detailed clustering techniques and robust quality control procedures, and it can benefit future exome genotyping array–based GWASs.
Introduction
Background and application of the protocol
Exome sequencing refers to high-throughput sequencing technologies that specifically target the exome. Exome sequencing is an efficient way to screen for novel variation in the human exome; however, although the price of exome sequencing dropped substantially between 2009 and 2012, it has remained relatively stable since 2012. At around $750 per sample, it is still expensive to conduct large-scale GWASs using exome sequencing. Thus, the exome genotyping array was introduced as an affordable alternative to exome sequencing. Two major exome genotyping arrays are currently commercially available: the Illumina Infinium HumanExome BeadChip array and the Affymetrix Axiom exome array. Each approach offers slightly different marker selection variation and customizability. In the present protocol, we will focus on the Illumina Infinium HumanExome BeadChip array, hereafter referred to as exome chip.
At a cost of less than $50 per chip, exome chips have become cost-effective alternatives to exome sequencing for large GWASs. Furthermore, exome chip data require less hard drive space for storage and processing than exome sequencing data. For an exome chip genotyping project of 39,000 samples, ∼3.6 TB of storage is required to store and process the data. If the same size study were done by exome sequencing, ∼400 TB storage space could be required. However, exome sequencing enables researchers to identify novel SNPs across the whole exome region plus some intron and intergenic regions1,2, whereas the exome chip only enables researchers to test a predetermined set of variants, making it impossible to detect novel variants. Ideally, exome sequencing provides the highest probability for identifying novel disease-associated SNPs. However, at its current price, it is impractical to apply it to large-scale GWASs.
Several unique characteristics separate exome chips from other genotyping chips. As the name suggests, markers on exome chips are primarily exome SNPs. A detailed description of the Illumina exome chip can be found at its wiki site3. For example, in the exome chip 12v1 (where 12 denotes the number of samples that can be run on a single exome chip and v1 denotes version 1), over 92% of the SNPs are in the exome, compared with 5% in the Illumina Omni 1 chip (Supplementary Table 1), which is representative of a traditional GWAS chip.
The selection criterion for the exome chip design is such that the SNPs must be detected in at least several sequencing data sets. Furthermore, the exome chip is designed to concentrate on rare variants (rare SNPs) rather than on common ones, a design choice that affords exciting new opportunities, such as identifying low-frequency variants associated with various diseases and gene-focused CNS analysis4. Many GWASs conducted recently have taken advantage of the properties of an exome chip focused on rare variants5–9. The majority of the present protocol can be applied to other Illumina genotyping arrays, although the steps that are specific to rare SNP calling are most applicable to the Illumina exome chip because of the exome chip's focus on rare SNPs.
Previously, the CHARGE (Cohorts for Heart and Aging Research in Genomic Epidemiology)10 consortium released strategies for calling SNPs using exome chips11. The strategies reported therein are rather descriptive with no step-by-step instructions to follow. The protocol presented here provides additional and detailed practical stepwise instructions for SNP calling. Detailed steps for quality control (QC) and all scripts used in the protocol are also provided. The protocol presented here is designed for Illumina exome chips. Although some of the concept described here may apply to Affymetrix Axiom Exome arrays, this protocol has not been tested with the Affymetrix Axiom Exome array. A portion of this protocol involves manual reclustering using GenomeStudio, which is subject to human judgment. It is possible that two different persons might produce two slightly different results by following this protocol.
Development of the protocol
Although the use of the exome chip provides exciting new opportunities, it also introduces additional data-processing challenges. GenomeStudio, which is used to cluster all genotyping arrays from Illumina, is designed to identify common SNPs as opposed to rare SNPs. The algorithm that GenomeStudio uses for clustering, GenCall, was originally designed as part of BeadStudio for the Illumina 550K arrays many years before the introduction of exome chip in 2011. Multiple studies12,13 have shown that the GenCall algorithm is more suitable to the identification of common SNPs than to that of rare SNPs. The use of GenomeStudio to cluster exome chip genotyping data will result in many mis-clustered SNPs. Two types of such incorrect clusters exist: (i) genotypes that were incorrectly clustered and therefore assigned the wrong genotype; and (ii) genotypes that were set to ‘missing’ as the GenCall algorithm failed to interpret a genotype call. Novel strategies are required to correct these mis-clustered rare SNPs.
Assigning the strand orientation of a SNP has always been challenging for genetics researchers. Differences in strand orientation can cause confusion when comparing results across multiple studies and platforms. Four strand-naming conventions are commonly implemented: probe-target, plus-minus, forward-reverse and top-bottom14. The top-bottom convention was developed by Illumina, and it is used in all Illumina genotyping arrays. The idea behind the top-bottom strand orientation system is to enable Illumina systems to consistently designate the same SNP orientation and allele call, even if the SNP database (dbSNP) or the human reference changes. Although this idea has its merits, it is not widely understood. The rules for determining top and bottom strands are as follows: a SNP is in the top strand if the first allele is A and the second allele is either C or G; a SNP is in the bottom strand if the first allele is T and the second allele is C or G. However, as for A/T and C/G SNPs the definition of strands is still ambiguous, Illumina introduced a ‘sequence walking’ technique to designate strand and allele orientation for A/T and C/G SNPs15. The sequence walking technique works as follows: let the ambiguous SNP's position be n, and the positions before and after n can be denoted as n − 1, n − 2, …, and n + 1, n + 2…, respectively. Two walkers will move away from n to the left and to the right one nucleotide at a time. Thus, a new pair of nucleotides is observed each time (the first would be (n − 1/n + 1)). If an A or T is observed in the first unambiguous pair on the 5′ side of the SNP, then the SNP is defined to be in the top strand; if A or T is observed in the first unambiguous pair on the 3′ side of the SNP, then the SNP is defined to be in the bottom strand. However, even when Illumina put this new strand scheme into practice, numerous strand errors continued to be generated. When exporting SNP data from GenomeStudio, one option allows switching all SNPs' strand orientations to ‘forward’, defined by the forward strand recorded in the dbSNP. Some minor differences exist between the dbSNP ‘forward’ and HG19 ‘plus’ strand definitions. After converting to dbSNP-forward, 2,055 SNPs are not on the same strand as HG19-plus. The majority of these (except 35 SNPs; Supplementary Table 2) can be converted to the HG19-plus strand by using the method described in Steps 36 and 37 of the PROCEDURE.
GenomeStudio displays clusters in two formats: polar coordinates and Cartesian coordinates. The Cartesian coordinates display the cluster using the normalized intensity values A and B (denoting the two targeted alleles). Polar coordinates display the cluster using the normalized R and normalized θ values to denote the y and x axes, respectively. Normalized θ represents the angle of deviation from the pure A signal, where 0 denotes a pure A signal and 1.0 denotes a pure B signal. Normalized R represents the intensity of the B allele. Cartesian coordinates and polar coordinates represent the same clusters from different perspectives. For demonstration purposes, polar coordinates will be used throughout this manuscript. The color configuration for the clusters is defined as follows: red = AA, purple = AB and blue = BB.
Protocol overview
The protocol presented here was developed by the Vanderbilt Epidemiology Center in cooperation with VANGARD. This protocol has been used to process multiple large batches of exome chip data, and these data have been used in several genetic and cancer studies9,16.
This protocol can be organized into two major sections and 19 subsections.
GenomeStudio section
Loading the data into GenomeStudio
Performing automatic clustering
QC on SNPs located in a haploid genome
QC based on GenTrain score
QC based on cluster separation
QC based on Mendelian error and replication error
QC based on other criteria
Calling rare SNPs
Final fltering
Exporting from GenomeStudio
Post-GenomeStudio section
Converting all SNPs to the forward strand
Checking for gender mismatch
Checking for race mismatch
Checking for relatedness
Checking for Hardy-Weinberg equilibrium (HWE) outliers
Checking for heterozygosity outliers
Checking consistency between exome chip genotype and 1000 Genomes Project17 or HapMap18 genotype
Checking for minor allele frequency (MAF) consistency between exome chip and 1000 Genomes Project genotypes
Checking for batch effects
The first section of the protocol is carried out using GenomeStudio (Genotyping module v1.9.4), the software package that Illumina developed for various genomic analyses. GenomeStudio includes nearly a hundred QC parameters, with many holding crucial QC information. Because GenomeStudio is a commercial software package, however, many of its algorithms and parameter calculations are not transparent. To improve the cluster accuracy, a series of QC procedures can be carried out in sequential order. Screening through all of them would take months. On the basis of the recommendation of Illumina19 and our own experience, the protocol given here describes the most meaningful QC parameters to check in GenomeStudio and the proper order in which they should be checked. As to the post-GenomeStudio section, the order in which its subsections are implemented is not important. If desired, parallelization can be applied, except for the subsection in which SNPs are converted to forward strands, a task that should be performed first after exporting data from GenomeStudio.
It is important to remember that, unlike other fixed and exact protocols, the protocol described here involves a large amount of manual reclustering, the result of which is heavily affected by personal perception. Additional details of the steps and examples of cluster figures of all scenarios are provided in the ANTICIPATED RESULTS to minimize the variation introduced by differing perceptions of different observers.
Rare SNP clustering strategies, which can substantially increase the accuracy of rare SNP calls, will also be discussed. Once SNP data are exported into PLINK format, a series of additional QC procedures primarily using PLINK are described. Exact command lines used for conducting these QC steps are given. All R and Perl scripts used for these QC steps are also provided at https://github.com/slzhao/ExonChipProcessing/. Throughout the description of this protocol, some strategies that can potentially expedite exome chip data processing will also be presented. The final result will be a very clean data set that researchers can use for genetic studies with high confidence.
Experimental design
Throughout the PROCEDURE and in the ANTICIPATED RESULTS, specific examples are provided to illustrate potential problems that can arise by implementing this protocol and their relevant solutions. The examples presented here involve the use of data from over 39,000 participants from six study groups20. Genotyping of the study was performed by Vanderbilt Technologies for Advanced Genomics (VANTAGE) using the Illumina exome chip 12v1-1A. Rigorous QC measures were conducted during genotyping. Overall, 377 HapMap18 trios (Supplementary Table 3) and 179 replicated pairs have been used in this example for QC purposes. Including HapMap trio samples and duplicating them as controls in the genotyping plate has become common practice in GWASs. A few QC steps of our protocol are dependent on the presence of these control samples.
Materials
Equipment
Δ CRITICAL Because Illumina claims that GenomeStudio was not designed to analyze data from studies with large sample sizes, and because projects of big magnitude are more vulnerable to corruption, we recommend using a high-powered Windows workstation similar to the one described below for large projects so as to lower the chance of file corruption and machine crash, because workstations provide more stability than a regular personal computer. A proper backup scheme should be implemented to avoid data loss. In our case, we configured our drives in Raid 5. Upon failure of a single drive, data could be recovered. GenomeStudio also provides a functionality called ‘Save Project Copy As…’ under the ‘File’ drop-down menu, which can be used to back up the project. We recommend regular backups of the project using this functionality. Δ CRITICAL Except for zCall, all packages reported under Software are required to complete this protocol. Different versions of the software reported may also work, but they have not been tested. Δ CRITICAL All software reported in this section should be installed by following the standard instructions provided by the software designer. No special requirements apply.
Files
Intensity files (.idat). These files are the raw data files from the exome chip. They should be provided by the facility that performed the genotyping
Sample sheets. The sample sheets are CSV files that contain sample information, such as plate ID, cell ID, gender and so on. The sample sheets should be provided by the genotyping facility
Hardware
Dell Precision T7600. Operating system: Windows 7 Professional (64 Bit), service pack 1. CPU: Intel Exon E5-2687 3.10 GHz, 16 cores, 2 processors. Memory: 192 GB, DDR3. Storage: 8 TB, 7,200 RPM, RAID 5. Network: Intel 82579LM Gigabit network connection Δ CRITICAL The Windows operating system is not optional because GenomeStudio is developed for Windows only. GenomeStudio's functionality and efficiency is highly dependent on memory availability particularly when the memory-based storage option is used. For best time efficiency, we recommend at least 128 GB memory for large projects (containing more than 10,000 samples). Extra CPU and memory allow multiple instances of GenomeStudio to be run simultaneously.
Dell PowerEdge R720xd (225-2110). Operating system: CentOS release 6.5. CPU: Intel Xeon E5-2670 2.60 GHz, 32 cores, 4 processors. Memory: 256 GB, RDIMM. Storage: 24 TB, 7,200 RPM, RAID 5. Network: Intel Corporation Ethernet controller 10-Gigabit X540-AT2 (rev 01) Δ CRITICAL A Linux machine is also needed, because one of the required software programs, EIGENSTRAT21, is Linux-specific, and two of the scripts are Linux shell scripts.
Software
GenomeStudio v2011.1 with Genotyping module v1.9.4. GenomeStudio is commercial software developed by Illumina, and it is the only commercial software required for this protocol. It contains many analysis modules and is the only genotyping module required to process Illumina exome chip data. GenomeStudio and the Genotyping module can be downloaded and purchased from http://www.illumina.com/informatics/sequencing-microarray-data-analysis/genomestudio.ilmn
PLINK22 v1.07. PLINK is a WGA analysis toolset with QC features that are useful for checking the integrity of the exome chip data. PLINK has been built for multiple operating systems; the Linux version is recommended. PLINK can be downloaded freely from http://pngu.mgh.harvard.edu/∼purcell/plink/
EIGENSTRAT (EIGENSOFT v4.2)21. EIGENSTRAT is a software package that can detect population stratification on the basis of SNP data using principal component analysis (PCA). It is used during the QC step of checking reported race accuracy. The EIGENSOFT package can be downloaded freely from http://www.hsph.harvard.edu/alkes-price/software/
R v3.02 64bit. R is a statistical programming language with excellent ability to create figures. R scripts have been provided to draw QC figures. R is built for multiple operating systems; Windows or Mac versions are recommended for better navigation. R can be downloaded freely from http://www.r-project.org/
Perl v5.10.1. Perl is a programming language best known for its ability to conduct text file processing. Perl scripts have been provided to conduct various QC checks. Perl is built for multiple operating systems; the Linux version is recommended because large data sets can more easily be processed in the Linux system than using Windows. Perl can be installed to Linux system directly by running the command ‘curl -L http://xrl.us/installperlnix | bash’ in a Linux shell; administrator privileges might be required
Python v2.7.4. Python is another scripting programming language. Python is built for multiple operating systems; the Linux version is recommended because large data sets can more easily be processed in the Linux system than using Windows. Python comes with all major Linux operating systems
zCall (ref. 23) v3.3. (Optional) zCall is a variant caller that can be used in postprocessing SNPs on the basis of the GenomeStudio report. zCall is optional, and the reason for this is explained in ANTICIPATED RESULTS. zCall can be downloaded freely at https://github.com/jigold/zCall
WinSCP 5.5.4. WinSCP is a secure file transfer tool. In this protocol, it can be used to transfer data between Window and Linux workstations. WinSCP 5.5.4 can be downloaded freely at http://winscp.net/eng/download.php
Other scripts and resources. 18 scripts and 8 resource files are necessary for the implementation of the present protocol. The detail of each script can be found in Supplementary Table 4, and the detail of each resource file can be found in Supplementary Table 5. The scripts are used to conduct QC and to generate figures, and the resource files are fixed input files used by these scripts. Because Illumina exome chip 12v1_A is used as the example chip in this protocol, all resource files provided are for version 12v1_A. For other versions of Illumina exome chip, the resource files can be downloaded from https://github.com/slzhao/ExonChipProcessing/. Implementation of the present protocol does not require any programming skill, as all scripts and their future updates can be downloaded from https://github.com/slzhao/ExonChipProcessing/. The protocol also does not assume knowledge of a majority of the listed software because all command lines are given. The protocol only assumes some basic knowledge of the proper use of GenomeStudio; thus, we will not describe the basic GenomeStudio protocol here. For complete GenomeStudio protocols, please refer to the GenomeStudio documentation and manual, which can be found at Illumina's support website at http://bioinformatics.illumina.com/informatics/sequencing-microarray-data-analysis/genomestudio.html
Procedure
Loading data into GenomeStudio • TIMING ∼8 h for 39,000 samples
1. Open GenomeStudio and load the intensity files (.idat) into GenomeStudio with the sample sheets. The .idat files are the raw data files from the exome chip. They should be provided by the facility that performed the genotyping. The sample sheets are .CSV files that contain sample information, such as plate ID, cell ID, gender and so on. The sample sheets should be provided by the genotyping facility. To decide how to perform the present task, please note that option A is suitable for smaller projects. For large projects, option A requires multiple manual loading steps because the .idat files and sample sheets are usually organized in batches. Option B is more suitable for large projects, where multiple batches are available. However, implementing option B requires breaking up the original .idat file storage structure. The original .idat file storage structure can be preserved if an extra copy of the .idat files storage directory is saved at the cost of extra hard drive storage space and time.
(A) Manual loading of data one sample sheet at a time
Follow the standard GenomeStudio protocol (see MATERIALS) to load the data one sample sheet at a time into GenomeStudio.
(B) Merging all sample sheets into one using supplement script ‘MergeSampleSheet.pl’
-
Run MergeSampleSheet.pl with the following operating system-independent command:
- > perl MergeSampleSheet.pl -d {sample.dir} -o {merged}.csv
- The parameter {sample.dir} is the directory in which all samples sheets are located, and {merged}.csv is the merged sample sheet. The use of merged sample sheets also requires all of the intensity files (.idat) to be in the same directory. The disadvantage of implementing the Step 1B is the loss of batch information. Batch information can be preserved by making an additional copy of the data at the cost of hard disk space or adding batch information into the sample sheet.
? TROUBLESHOOTING
Performing automatic clustering • TIMING ∼16 h
2. Follow the standard GenomeStudio protocol to perform automatic clustering. Automatic clustering will be prompted by GenomeStudio during the manual data loading step. Δ CRITICAL STEP To improve the overall performance of GenomeStudio, memory-based storage can be used. Under Tools → Options → Module → Genotyping, select ‘Use memory-based storage’. Using this option requires a large amount of memory. Approximately 40 GB of memory is used for an exome chip project that contains 39,000 samples.
3. In the ‘Samples Table’ of GenomeStudio (located by default in the lower left), select the ‘Call Rate’ column by clicking on the column header. Sort the Call Rate column in descending order by clicking on the ‘Sort column (Descending)’ button located at the top of the ‘Samples Table’.
4. Select all samples whose call rate is <95%, right-click to bring up the pop-up menu and Select ‘Exclude Selected Sample’ from the pop-up menu. After the mentioned samples are excluded, you will be prompted to update ‘sample’ heritability and reproducibility errors and to update ‘SNP statistics’. Select ‘Yes’ for both.
5. Remove the excluded SNPs from the cluster for clearer visualization: in the ‘SNP Graph’ (by default located at upper left), right-click to bring up the pop-up menu and unselect ‘Show Excluded Samples’. Δ CRITICAL STEP For studies including large numbers of samples, the clustering might look very messy owing to problematic samples. Typically, we estimate that 1–3% of samples might fail (call rate <98%) for various reasons during genotyping. Data from these samples appear as random dots on the cluster plot, which makes the real clusters difficult to identify by eye. This step minimizes the effect of these bad samples. Furthermore, in the toolbar just above the ‘SNP Graph’, the third button ‘Plot Normalized Values’ (denoted by a ‘1’) enables the user to switch the ‘SNP Graph’ between normalized values and un-normalized values. For some ambiguous SNP calls, we recommend checking both the normalized and un-normalized ‘SNP Graph’.
6. (Optional) To improve the overall clustering, first exclude all samples whose call rate is <99%, and then re-cluster with all samples with a call rate >99%. The resulting clusters should be cleaner, albeit at the cost of extra work time.
QC on SNPs located in a haploid genome • TIMING ∼4 h
7. In ‘Samples Table’, sort samples by ‘Gender’ and select all female samples.
8. Right-click on the selected samples and select ‘Configure Marks’; a ‘Configure Marks’ panel will pop up.
9. Click on ‘Add’ and a ‘Select Mark Name’ panel will pop up.
10. In the ‘Select Mark Name’ panel, type ‘female’ in the first text box, and select a color using the drop-down menu. Next, click ‘OK’ to close the ‘Select Mark Name’ panel and click ‘OK’ to close the ‘Configure Marks’ panel.
11. Repeat Steps 7–10 to identify male samples and select a different color for them.
12. Select ‘SNP Table’ by selecting the ‘SNP Table’ tab (located by default in the upper right).
13. In ‘SNP Table’ toolbar, click on ‘Import columns into the table’ (the fourth button); a pop-up menu named ‘Import’ will appear. By default, the ‘Column Import’ radio button should be selected. Click on ‘Browse…’ to open a file browsing window and select the ‘PAR_SNPs.txt’ file (provided as a resource file). Click the ‘Open’ button to close the file browse window. Click ‘OK’ to import and close the ‘Import’ pop-up menu. A new column with the header name ‘PAR’ (pseudoautosomal region) should be added to the ‘SNP Table’. The possible values for this column can be either 1, which denotes that the SNP is in a PAR, or empty, which denotes that the SNP is not in a PAR.
14. In ‘SNP Table’, click on the ‘Filter rows’ button at the toolbar on top of the SNP Table to bring up the ‘Filter Table Rows’ pop-up menu. In the ‘Filter Table Rows’ pop-up menu, select ‘Chr’ in the ‘Columns’ section. In the ‘Filter Table Rows’ pop-up menu, select the ‘=’ operator from the ‘Expression’ drop-down menu and type ‘X’ in the ‘Value’ text box. Next, click on the ‘⇒’ button to add this logical expression into the ‘Filter Tree View’.
15. In the ‘Filter Table Rows’ pop-up menu, select ‘PAR’ in the ‘Columns’ section, and select the ‘!=’ operator from the ‘Operation’ drop-down menu and type ‘1’ in the ‘Value’ text box. Confirm that under the ‘Sub—Statement’ section the ‘Action’ drop-down menu is set to ‘AND’. Click the ‘⇒’ button to add this logical expression into the ‘Filter Tree View’. Now, the logical expression under ‘Filter Tree View’ should be ‘(‘Chr’ = X) and (‘PAR’ != 1)’.
16. Click ‘OK’ to apply filters displayed in ‘Filter Tree View’ to ‘SNP Table’. The pop-up menu should disappear and the remaining SNPs should be only from the X chromosome and excluding SNPs in PAR.
17. In the ‘SNP Table’, select ‘AB Freq’ column by clicking on the column header. Sort this column in descending order by clicking on the ‘Sort column (Descending)’ button located at the top of the table.
18. Manually check the SNPs in descending order, and look for AB clusters with large numbers of male subjects. Because males should not appear in the AB cluster on the X chromosome, this manual check identifies mis-clustered X chromosome SNPs. Zero these SNPs, which is done by right-clicking on a SNP (a row) in the ‘SNP Table’, and by selecting ‘Zero Selected SNP’. The number of SNPs to be manually checked in this step is arbitrary, but we recommend checking at least the first 100 SNPs.
19. Sort the SNPs in the ‘SNP Table’ by call rate in descending order on the Y chromosome. The call rate for females on the Y chromosome should be at or near 0%. Zero the SNPs with large numbers of females called on the Y chromosome.
20. Sort the SNPs in ‘SNP Table’ by AB cluster frequency in descending order for mtDNA SNPs. mtDNA is diploid and therefore should show only AA and BB clusters, although the presence of some AB clusters may be due to heteroplasmy. Zero mtDNA SNPs with high AB cluster frequency.
21. Manually fix AB clusters for which the NormR position caused the cluster to carry some low-intensity samples into the cluster. To do this, move the AB cluster oval up on the y axis. Zero any SNPs that cannot be fixed in this way. Three cluster ovals are present in the polar coordinate ‘SNP Graph’, denoting the center areas of AA, AB and BB clusters, respectively. They can be moved and stretched.
22. Filter out SNPs that have already been manually examined: in the ‘SNP Table’. Use the filter rows functionality (described in Steps 14 and 15) to apply the following filters: Chr != X, Chr != XY, Chr != Y, Chr != MT, Call Freq = 0 and Edited = 0. Δ CRITICAL STEP To avoid redundant work, before advancing to the next step, always filter out previously checked and zeroed SNPs. For example, before performing Step 23, apply a filter to exclude SNPs on chromosomes X, XY, Y, as well as those in mtDNA, those with a call rate equal to zero and those that have been previously edited.
QC based on the Gentrain score • TIMING ∼4 h
23. Examine the GenTrain score in the ‘SNP Table’. In general, SNPs with GenTrain scores of >0.7 are clustered correctly.
24. For the clusters with low GenTrain scores (GenTrain score <0.7) that appear fixable, manually move the cluster ovals into more proper positions. A cluster oval might not be placed in the most ideal position by the GenCall algorithm; an incorrect placement of a cluster oval often results in a low GenTrain score. A user can spot these badly placed cluster ovals by examining the ‘SNP Graph’. If two or three clusters are clearly identifiable by eye, but are not identified by the GenCall algorithm, move the cluster oval to the center of these clusters, respectively (left: AA, middle: AB and right: BB).
25. In some cases, a low GenTrain score when there are three clusters visible results from a narrow angle of the AA or BB cluster. There is no way to call this SNP completely correctly. To resolve these cases partially, sacrifice the BB cluster by moving the BB cluster oval to the right. Repeat Step 22.
QC based on cluster separation • TIMING ∼4 h
26. Sort the SNPs in the ‘SNP Table’ by cluster separation scores in ascending order and examine clusters with low cluster separation scores. These low scores usually indicate overlapping clusters, very close clusters or SNPs with more than three possible clusters.
27. Manually move the cluster ovals to more appropriate positions to correct these SNPs with low cluster separation scores. Repeat Step 22.
QC based on Mendelian error and replication error • TIMING ∼4 h
28. Sort the parent-child (P-C) errors, parent-parent-child (P-P-C) errors and replicate (Rep) errors in descending order. Large error numbers (>10) indicate mis-clustering, a bad sample or bad genotyping.
29. Manually correct the positioning of the cluster ovals to resolve SNPs with large P-P-C and P-C errors.
30. Manually correct the positioning of the cluster ovals to resolve SNPs with large Rep errors. Repeat Step 22.
QC based on other criteria • TIMING ∼4 h
31. Sort by the additional parameters described in Table 1 and examine the SNPs at both ends of the spectrum of these parameters for errors. The number of SNPs to be examined is arbitrary. We recommend examining at least 100 SNPs from each end. Repeat Step 22 after examining each parameter.
Table 1.
GenomeStudio quality control parameters.
| Name | Type | Minimum | Maximum | Sort |
|---|---|---|---|---|
| GenTrain | Float | 0 | 1 | Ascending |
| Cluster separation | Float | 0 | 1 | Ascending |
| P-P-C Error | Integer | 0 | >100 | Descending |
| Rep Error | Integer | 1 | >100 | Descending |
| AB Freq | Float | 0 | 1 | Descending |
| Call Freq | Float | 0 | 1 | Ascending |
| AA T Mean | Float | 0 | 1 | Both |
| AA T Dev | Float | 0 | 1 | Descending |
| AB T Mean | Float | 0 | 1 | Both |
| AB T Dev | Float | 0 | 1 | Descending |
| BB T Mean | Float | 0 | 1 | Both |
| BB T Dev | Float | 0 | 1 | Descending |
| AA R Mean | Float | 0 | 1 | Both |
| AA R Dev | Float | 0 | 1 | Descending |
| AB R Dev | Float | 0 | 1 | Descending |
| BB R Mean | Float | 0 | 1 | Both |
| BB R Dev | Float | 0 | 1 | Descending |
Final data filtering • TIMING ∼20 min
32. Filter by sample call rate (98% for the Illumina exome chip).
33. Filter by SNP call frequency (95% for the Illumina exome chip).
Calling rare SNPs • TIMING ∼24 h
34. Call rare SNPs either with the variant caller zCall23 (option A) or manually (option B). Please consult the relevant section of the ANTICIPATED RESULTS to decide whether to implement option A or option B at this juncture of the PROCEDURE. Δ CRITICAL STEP This step is crucial for improving rare SNP calling accuracy. Owing to the design choice of the exome chip, which attempts to represent exome sequencing and thus concentrates on rare variants, GenomeStudio mis-clusters a large portion of the rare SNPs. As these rare SNPs are often a major reason for using an exome chip, it is important to recover the rare SNPs that can thus get lost.
(A) Calling rare SNPs in zcall
Create a GenomeStudio standard report by following the instructions provided by the zCall authors (https://github.com/jigold/zCall/blob/master/zCallTips.pptx)
- Run zCall using the following command in a Linux shell:
- > runZcall.py --zcall-dir {zcall_dir} --output-dir {dir_name} --use-weighted-lr --z-score 6 {GenomeStudioReport} > zCall.log
- The parameter ‘ --z-score 6’ was determined to be the most effective threshold for our example data set. Previous studies11,12,23 have also used 7 and 8 for --z-score threshold. The authors of zCall suggested that the optimal --z-score value can be determined by recalling common SNPs using various values and then computing the consistency with a reference, such as the 1000 Genomes Project data. The --z-score producing the highest consistency should be used. The ‘zCall.log’ is a log file on how zCall is run, which can be stored for future reference.
(B) Calling rare SNPs manually
In the ‘SNP Table’, select SNPs with MAF <0.01 and call frequency <0.9999 using the filter functionality. Filtering by MAF <0.01 ensures the selection of SNPs with very low MAF. The filter ‘Call Frequency <0.9999’ ensures the selection of SNPs with a small number of samples that are not called. When combining these two filters, the researcher obtains a list of SNPs with small or zero MAFs. This list contains the most likely rare SNPs mis-called by the GenCall algorithm.
Manually cluster the resulting SNPs (per our experience, about one out of every four SNPs of a total of ∼20,000–40,000).
Exporting the data • TIMING ∼4 h
35. Export the data from GenomeStudio in either the standard GenomeStudio format (option A) or the PLINK format (option B). GenomeStudio format is a text file format, and it can be used as input in other programs such as zCall. The PLINK format is also a text format and can be compressed into binary format, which substantially saves storage space. The PLINK format has become the standard accepted format for storing SNP data. Thus, we recommend exporting data into PLINK format, unless the user has a special circumstance that requires the GenomeStudio format. The remaining protocol Steps 36–57 are based on the PLINK format export.
(A) Exporting the data in the standard GenomeStudio format
Follow GenomeStudio's standard protocol to create a GenomeStudio standard report.
(B) Export the data in PLINK format
Download the PLINK format by exporting the plug-in from Illumina's website: http://support.illumina.com/array/array_software/genomestudio/downloads.html.
In GenomeStudio, go to Analysis → Reports → Report Wizard.
Select ‘Custom Report’, which by default should be PLINK.
In ‘Report Input Parameters’, select the parameter ‘UseForwardStrand’ as false. Click ‘Next’.
Choose ‘remove them from the report’. The other two options available at this point enable the user to export subset of samples. Click ‘Next’.
In the ‘Zeroed SNPs’ section, choose ‘Include zeroed SNPs in the report’. In the ‘Visible SNPs in the SNP Table’ section, choose ‘Include all SNPs in the report, regardless of whether or not they are visible’. The ‘Visible SNPs in the SNP Table’ section appears only when the current ‘SNP Table’ does not display all SNPs. Click ‘Next’.
Browse to the desired output directory in the ‘Output Path’ text box and input a report name in the ‘Report Name’ text box. Click ‘Finish’. A new folder named ‘PLINK_today's date’ will be created, which will contain .ped and .map files.
Transfer .ped, .map and phenotype files from the Windows workstation to Linux workstation using a secure FTP, such as WinSCP 5.5.4. When transferring text files from Windows to Linux systems using FTP, confirm that the files are transferred in ASCII mode.
- In the Linux operating system, convert .ped and .map files to .bed, .fam and .bim files to save hard disk space by using the following command:
- > plink --noweb --file {exome} --make-bed --out {exome}
- The parameter ‘ --noweb’ is required if the web-based version is not being used and the local version is installed.
Converting all SNPs to the HG19 plus strand • TIMING ∼1 h
36. Download the .bim file created by the Wellcome Trust Centre for Human Genetics to transfer all Illumina genotyping arrays to the forward strand: http://www.well.ox.ac.uk/∼wrayner/strand/.
37. Download update_build.sh and follow the instruction provided in http://www.well.ox.ac.uk/∼wrayner/strand/ to convert all SNPs to HG19 plus strand.
Checking for gender mismatch • TIMING ∼2 h
38. To check for gender mismatch, the PLINK file must contain gender information. Gender information is automatically exported into the PLINK file, provided that it is available in the sample sheet at Step 1. In PLINK, type the following command:
> plink --noweb --bfile {exome} --maf 0.1 --check-sex --out {outfile}
The input exome is the GenomeStudio-outputted PLINK .bed file from Step 35B. The parameter ‘ --maf 0.1’ tells PLINK to only use SNPs with MAF >0.1, ‘ --check-sex’ tells PLINK to perform a gender check between reported and genotype-based gender and ‘ {outfile}’ specifies that the output file is named ‘ {outfile}.sexcheck’.
? TROUBLESHOOTING
39. Run the following command to perform gender check on the PLINK results:
> Rscript Gender.R {outfile}.sexcheck {gender}.jpeg
The input ‘ {outfile}.sexcheck’ is the output of the previous step, and ‘ {gender}.jpeg’ is the name of the figure generated.
Checking for race mismatch • TIMING ∼2 h
40. Extract ancestry-informative markers (AIMs) from the PLINK file exported from GenomeStudio with the following command:
> plink --noweb --bfile {exome} --extract AIMs.txt --out AIMs.txt
AIMs.txt is a text that contains all AIMs markers on this exome chip. It is provided as a resource file.
41. Run the following command to convert the PLINK file to EIGENSTRAT format:
> convertf -p Parameter.PED.EIGENSTRAT > eigen.log
- ‘ Parameter.PED.EIGENSTRAT’ is a parameter file that looks like the following:
- genotypename: {AIMs}.ped
- SNPname: {AIMs}.map
- indivname: {AIMs}.ped
- outputformat: EIGENSTRAT
- genotypeoutname: {filename}.geno
- SNPoutname: {filename}.SNP
- indivoutname: {filename}.ind
- familynames: NO
The parameters ‘ {AIMs}.ped’ and ‘ {AIMs}.map’ are output from Step 40. The ‘eigen.log’ is a log file recording how EIGENSTRAT is run; it can be stored for future reference.
? TROUBLESHOOTING
42. Use the three files produced in the previous step, ‘ {filename}.geno’, ‘ {filename}.SNP’ and ‘ {filename}.ind’, to run the command:
> perl smartpca.perl -i {filename}.geno -a {filename}.SNP -b {filename}.ind -o {out}.pca -p -m 0
The parameter ‘ -m 0’ turns off outlier removal, and ‘ {out}.pca’ is the output. Both convertf and smartpca. perl are scripts that come with the EIGENSTRAT package.
43. Draw a PCA plot in EIGENSTRAT using the following command:
> Rscript PCAPlot.R {out}.pca {racefile}
The input ‘ {out}.pca’ is the output of the script in Step 42, and the resulting figure is saved as ‘ {out.pca.jpeg}’. To produce a racefile, please see Step 55. The racefile needs to have the samples in the same order as they are in the ‘ {out}.pca’ file.
Checking for relatedness • TIMING ∼4 h
44. In PLINK, exclude SNPs not present in the autosome and independent SNPs with MAF >0.1 using the following command:
> plink --noweb --bfile {exome} --maf 0.1 --exclude chr23_26.txt --indep-pairwise 50 5 0.2 --out {indepSNP}
‘ chr23_26.txt’ is a text file that contains a list of SNPs from chromosomes X, Y, XY and from the mitochondria. The command ‘ --indep-pairwise’ is used for linkage disequilibrium-based SNP pruning. Its parameters ‘50 5 0.2’ stand for the window size, the number of SNPs to shift the window at each step and the multiple correlation coefficient for a SNP being regressed on all other SNPs simultaneously.
45. Check the relatedness of the SNPs not pruned in Step 44 using the following command:
> plink --noweb --bfile {exome} --extract {indepSNP.prune.in} --genome --out {outfile}
Checking for HWE outliers • TIMING ∼2 h
46. (Optional) If the PLINK files are not organized by race, split the PLINK files by race using the following command:
> plink --noweb --bfile {exome} --keep {Caucasian_list} --make- bed --out {Caucasian}
‘ Caucasian_list’ is a text file that contains two columns: the Family ID and Individual ID. This command will plotted using the followingproduce a new set of PLINK files for white participants. If other races are available in the data set, repeat this command using a race-specific list file such as ‘ Caucasian_list’ to get PLINK files for other races.
47. Perform the HWE test to identify SNPs that deviate from HWE using the following command:
> plink --noweb --bfile {Caucasian} --maf 0.05 --hardy --out {outfile}
This command generates a ‘ {outfile}.hwe’ file. The parameter ‘ --maf 0.05’ ensures that the HWE test is performed only on SNPs with MAF >0.05, because the HWE test is not appropriate for rare variants. As the assumptions of HWE (in particular random mating) may be violated in populations containing more than one race, if the data set contains multiple races repeat Step 46 using a race-specific list file, such as ‘ Caucasian_list’, to extract PLINK files for each race. Next, repeat the present step to compute the HWE test on that racial subset.
48. Examine the P values in the last column of ‘ {outfile}.hwe’. They can be plotted using the following command:
> Rscript PlotHWE.R {outfile}.hwe
This plot will draw histograms of HWE P values for all races under the same directory. Alternatively, SNPs that deviate severely from HWE can be identified by opening the .hwe file in excel and sorting the P value column (last column) from small to large.
Δ CRITICAL STEP Because PLINK does not perform multiple test correction, a large portion of the significant SNPs are false positives. Perform a conservative multiple correction test such as the Bonferroni correction24 or choose a much more conservative P value threshold25.
Checking for heterozygosity and inbreeding outliers • TIMING ∼2 h
49. Compute the inbreeding coefficient using the following command:
> plink --noweb --bfile {exome} --extract {indepSNP}.prune.in --het --out {outfile}
The file ‘ {indepSNP}.prune.in’ was produced during Step 44. This command produces an ‘ {outfile}.het’ file. The sixth column is the inbreeding coefficient F.
50. Compute heterozygosity and draw histograms of heterozygosity and inbreeding coefficient F using the following command:
> Rscript PlotHeterozygosity.R {outfile}.het
This command will produce two figures: ‘ {outfile}.heterozygosity.jpg’ and ‘{outfile}.F.jpg’.
Checking for genotype consistency • TIMING ∼2 h
51. Compute the genotype consistency between duplicated SNPs using supplement file ‘ ConsistencyDupSNP.sh’ and the following command:
> file= “{exome}” output=“{outfile}.result” sh ConsistencyDupSNP.sh
‘ {exome}’ is the PLINK format GenomeStudio output from Step 35B and the consistency results will be stored in ‘ {outfile}.result’.
52. If the study contains 1000 Genomes Project samples, check for consistency with the 1000 Genomes Project data using supplement file ‘Consistency1000G.sh’ and the following command:
> file=“{exome}” output=“{outfile}” sh Consistency1000G.sh
? TROUBLESHOOTING
Checking allele frequency consistency with 1000 Genomes project data by race • TIMING ∼2 h
53. Get allele frequency information by race by running the following shell script:
> G1kRace=“{race},.., {race}” sh AlleleFreq1000G.sh
The possible parameters for race are the population codes defined by the 1000 Genomes Project, and they are ‘CEU’, ‘ASW, ‘GBR’, ‘IBS’, ‘TSI’, ‘LWK’, ‘YRI’, ‘CLM’, ‘MXL’, ‘PUR’ and ‘FIN’. For detailed population code explanations, please see the 1000 Genomes Project population README file at http://ftp.1000genomes.ebi.ac.uk/vol1/ftp/README.populations. The results are stored in a file named ‘ {race}_af_1kg’.
54. Generate the input racefile. A racefile has three columns: Family ID, Individual ID and Race. Family ID and Individual ID are taken from PLINK file directly. The possible choices for Race are ‘W’, ‘B’ and ‘H’ for white, black and Hispanic, respectively.
55. Compute the allele frequency of exome chip data by running the following shell script:
> exome_dir=“{exomePath}” exomeRace=“{racefile}” sh AlleleFreqExome.sh
The input ‘ {exomePath}’ is the location of the exome chip binary PLINK files (e.g., .bim and .bed). There may be multiple PLINK files in this path to represent batches. The exome chip allele frequency will be stored in a file named ‘ final_{race}_g1k_exm’. If the racefile contains more than one race, more than one ‘ final_{race}_g1k_exm’ file will be produced.
56. Draw a figure to compare allele frequency using the following command:
> Rscript 1000GAlleleFreqPlot.R final_{race}_g1k_exm {out}.jpeg
Checking allele frequency consistency across batches • TIMING ∼2 h
57. Draw a figure using the supplement script ‘ BatchAlleleFreqMatrix.R’ and the following command:
> Rscript BatchAlleleFreqMatrix.R final_exm_${race} {out}.jpeg
The input ‘ final_exm_${race}’ is the output of Step 56.
? TROUBLESHOOTING
Troubleshooting advice can be found in Table 2.
Table 2.
Troubleshooting table.
| Step | Problem | Possible reason | Solution |
|---|---|---|---|
| 1 | GenomeStudio fails to locate intensity data | Incorrect directory is provided | Usually, the intensity files (.idat) are stored in folders with names consisting of just numbers that refer to the beadchip ID. Typically, intensity files for 12 samples are stored within one folder. The directory provided to GenomeStudio needs to be the folder that contains the subfolders that store the actual .idat files |
| GenomeStudio fails to load certain samples | Bad genotyping samples | These failures do not affect the loading of other samples, and there is no need to restart the loading process | |
| 38 | PLINK will not input the .bed file | The PLINK command for inputting a .bed file should not specify a file extension | Use the command ‘exome’ instead of ‘exome.bed’ |
| 41 | ‘convertf’ command will not run | The .fam file cannot have missing values of ‘−9’ for the sixth column | Use the following command in
Linux: ‘> awk ‘{$6=1;print}’ {original.ped} > modified.ped’ to change all ‘–9’ to ‘1’ columns of the .fam file to 1 |
| 52 | Two SNPs on the exome chip have the same genomic position | These SNPs represent different alleles at the same position | These SNPs are not duplicated SNPs and should be treated as independent of each other |
• Timing
The time it takes to implement this QC protocol is highly variable based on three different factors: sample size, computer power and user experience. The timing information provided here is based on a project involving 39,000 samples and the equipment listed in the Equipment section. The protocols were carried out by experienced individuals. For less experienced individuals, the overall time might be doubled.
Step 1, loading data into GenomeStudio: ∼8 h for 39,000 samples
Steps 2–6, performing automatic clustering: ∼16 h
Steps 7–22, QC on SNPs located in a haploid genome: ∼4 h
Steps 23–25, QC based on the GenTrain score: ∼4 h
Steps 26 and 27, QC based on cluster separation: ∼4 h
Steps 28–30, QC based on Mendelian error and replication error (scale with the number of trios and duplicated samples used in the study): ∼4 h
Step 31, QC based on other criteria: ∼4 h
Steps 32 and 33, final data filtering: ∼20 min
Step 34, calling rare SNPs: ∼24 h
Step 35, exporting the data, scale with sample size: ∼4 h
Steps 36 and 37, converting all SNPs to the HG19 plus strand (scale with sample size): ∼1 h
Steps 38 and 39, checking for gender mismatch (scale with sample size): ∼2 h
Steps 40–43, checking for race mismatch (scale with sample size): ∼2 h
Steps 44 and 45, checking for relatedness (scale with sample size): ∼4 h
Steps 46–48, checking for HWE outliers: ∼2 h
Steps 49 and 50, checking for heterozygosity and inbreeding outliers: ∼2 h
Steps 51 and 52, checking for genotype consistency: ∼2 h
Steps 53–56, checking allele frequency consistency with 1000 Genomes Project data by race: ∼2 h
Step 57, checking allele frequency consistency across batches (scale with the number of batches): ∼2 h
Anticipated Results
In this section, we discuss a few representative examples of the results to be expected by implementing this protocol using real data for a few of the steps.
Step 1
Loading data into GenomeStudio can be a tedious process for large projects because many sample sheets can be generated, and GenomeStudio only allows one sample sheet to be loaded at a time. The sample sheet identifies each sample with a unique sample ID. The nonoptional fields in the sample sheet include Sample_Plate ID and Chip ID, which can be used to compute for batch effects. Batch effect in a genotyping study means a difference in probe intensity observed between sample batches. The batch can be defined by plate, chip, genotyping date, genotyping facility and so on. A Kruskal-Wallis26 test or Fligner-Killeen test of homogeneity of variances27 can be used to test for batch effects28. The optional phenotype variables include gender, pedigree information and replicate information. Many of these phenotype variables can be valuable parameters for QC at later processing steps.
For large projects, multiple sample sheets will usually be created, and for each sample sheet a folder that contains many subfolders is created to hold all of the intensity files (.idat). Each subfolder contains .idat files for 12 samples. The parent folder name is numerical and represents the BeadChip ID. The idat files are named according to the following convention: ‘{parent folder name}_{row number}_{column number}_{color}.idat’. For each sample, there are two .idat files by default, one for green and one for red.
Steps 2–6
GenomeStudio can perform automatic SNP clustering based on the loaded data, or it can fit loaded data on the basis of an existing cluster file. Performing automatic clustering will take substantially longer. For exome chip genotyping data from 39,000 samples, 24–48 h are usually required for automatic clustering, depending on computing power.
Steps 7–22
GenomeStudio has difficulty clustering on the X and Y chromosomes and mtDNA, because they are not diploid. Human males are heterogametic, having both X and Y chromosomes, and females are homogametic, having two X chromosomes. mtDNA is solely inherited maternally and thus is also a haploid genome. The cluster algorithm of GenomeStudio does not take haploid genetics into consideration, resulting in a large portion of mis-clustered SNPs in the X and Y chromosomes and mtDNA. There are many PAR variants on the exome chip, and they should be excluded owing to the high number of males within the AB cluster during examination of the X chromosome. Usually, the PAR variants are labeled as the XY chromosome. However, when using the Illumina-provided beadpool manifest, only two variants are present on the XY chromosome. Many PAR variants are annotated only to the X chromosome. Thus, these PAR variants need to be removed manually when examining the X chromosome.
Cases of mis-clustering are easily identified if distinct colors are used to denote data from male and female study participants. In Figure 1 are reported examples of misidentified clusters for haploid genomes. In Figure 1a is an example of a low-quality chromosome X cluster, in which data from male participants should not appear in the AB cluster. A PAR variant on the X chromosome might also look similar to the data reported in Figure 1a. Figure 1b is an example of a low-quality chromosome Y cluster, in which data from female participants should not be called. In Figure 1c is an example of heteroplasmy in the mitochondria, in which normally only AA and BB clusters should be called. In Figure 1d is an example of a low-quality cluster caused by the vertical position of the AB cluster oval. The AB cluster oval is too low, which caused some samples to be called as the AB genotype. This cluster can be fixed by manually moving the AB cluster oval slightly up on the y axis so that it can exclude the samples called as AB genotype.
Figure 1.
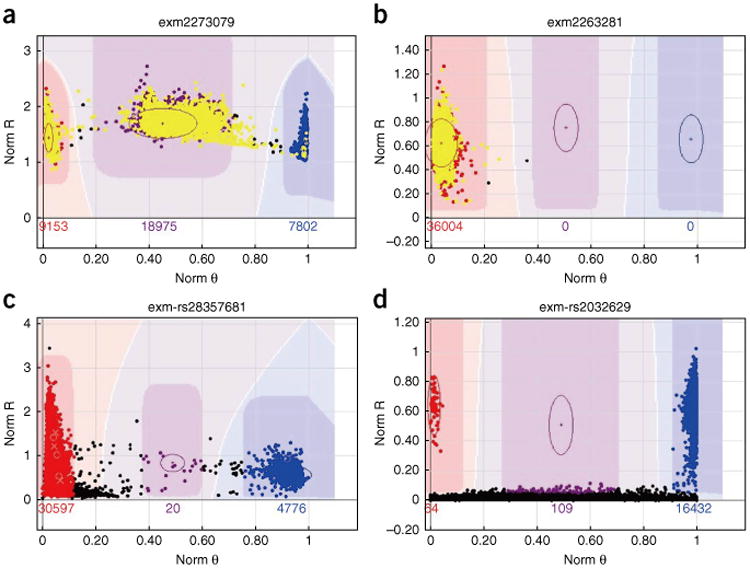
Examples of low-quality clusters for the haploid genomes described in Steps 7–22. The x axis denotes normalized θ, which represents the angle of deviation from the pure A signal, where 0 denotes a pure A signal and 1.0 denotes a pure B signal. The y axis denotes normalized R representing the intensity of the B allele. The color configuration for the clusters is defined as follows: red = AA, purple = AB, blue = BB and black = no call. The number reported above the x axis denotes the number of participants included in the corresponding cluster. Male SNPs are denoted in yellow; all non-yellow dots denote female SNPs. (a) An example of a low-quality chromosome X cluster, in which male SNPs should not appear in the AB cluster. (b) An example of a low-quality chromosome Y cluster, in which female SNPs should not be called. (c) An example of heteroplasmy in mitochondria, where normally only AA and BB clusters should be called. (d) An example of a low-quality cluster caused by the vertical position of the AB cluster oval. The AB cluster oval is too low, which caused some samples to be called as the AB genotype.
Steps 23–25
After dealing with chromosomes X, Y and mtDNA, the focus shifts to SNPs in the autosomal chromosomes. Several QC parameters can be used to identify problematic autosomal SNPs. One of the most important parameters is the GenTrain score. The GenTrain score is a number between 0 and 1, whereby higher values indicate better clustering. Even though it is designed to mimic evaluations made by a human expert's visual and cognitive systems, many of the SNPs with low GenTrain scores can be easily ‘fixed’ by manual clustering. Figure 2 reports example clusters with a low GenTrain score. Figure 2a is an example of a low GenTrain score caused by bad clustering by GenomeStudio, in which the AA and AB cluster ovals overlap. Figure 2b shows a SNP obtained after correction of the data reported in Figure 2a. This SNP is corrected by manually adjusting the cluster ovals’ positions. Figure 2c shows an example of low GenTrain score caused by the position of the AB and BB clusters being too close to each other. Data in Figure 2d show that by moving the BB cluster oval to the right, the AA and AB clusters are successfully called but the BB cluster is sacrificed. According to the criteria recommended by Illumina and the CHARGE consortium, both SNPs should be excluded from subsequent analysis, as they are very likely to fail further QC tests. In our example study, we flagged these two SNPs as to be treated with caution, so future association studies may exclude them at their discretion. Figure 3a reports the expected distribution of GenTrain scores after QC.
Figure 2.
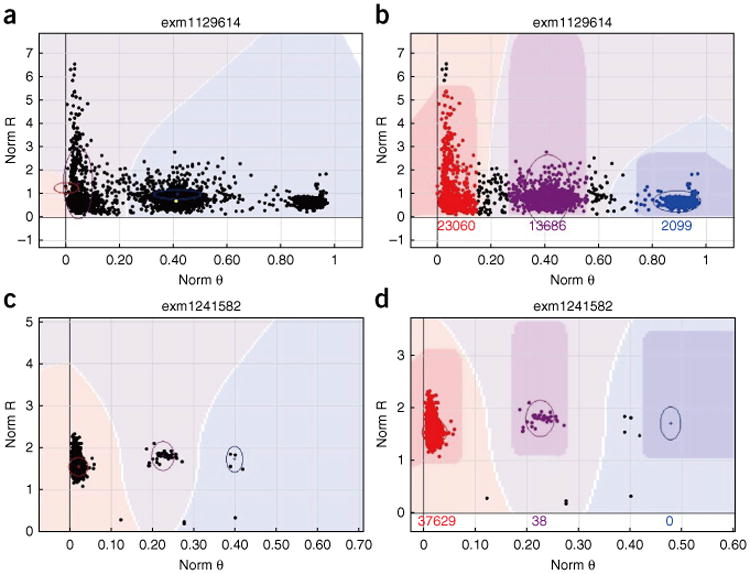
Example clusters relevant to Steps 23–25. The x and y axes are denoted as in Figure 1. (a) GenomeStudio cannot make a correct call on this SNP, as the AA and AB cluster oval overlap results in a very low GenTrain score. (b) This SNP can be correctly called by manually adjusting the cluster ovals’ positions. (c) The AB and BB clusters are too close to each other, causing this SNP to be a no call. (d) By moving the BB cluster oval to the right, the AA and AB clusters are successfully called, but the BB cluster is sacrificed.
Figure 3.
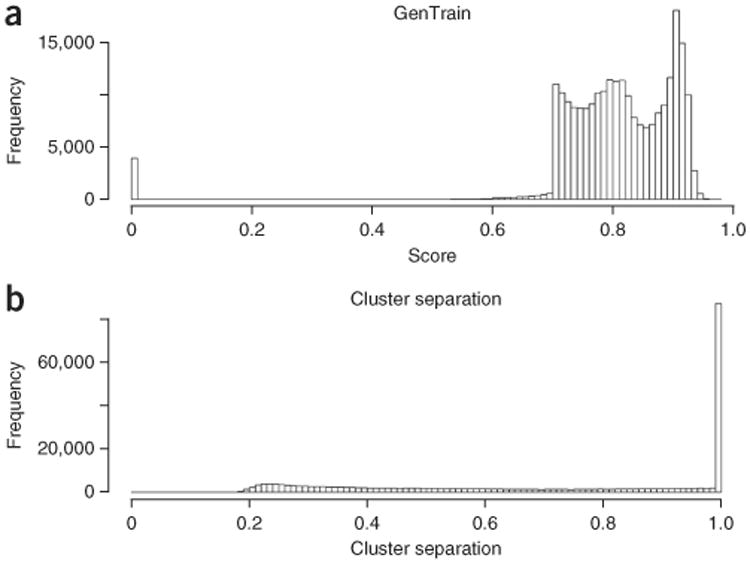
General distribution of the basic quality control (QC) parameters. (a) Distribution of GenTrain score after QC. (b) Distribution of cluster separation after QC.
Steps 26 and 27
Another useful parameter to identify problematic SNPs is the cluster separation score, which ranges between 0 and 1. The lower the score, the smaller the space between clusters. Figure 3b shows the expected distribution of cluster separation after QC. Sorting cluster separation scores in ascending order can help identify SNPs characterized by low cluster separation for further manual review and correction. In Figure 4a is reported an example of mis-clustered SNPs by GenomeStudio. The AA and AB cluster ovals are overlapping, which results in a very low cluster separation score. Figure 4b shows the SNP obtained after correction of the data reported in Figure 4a. Figure 4c shows an example of low cluster separation caused by two clusters in close proximity. In Figure 4d is an example of low cluster separation caused by having more than three clusters. The SNPs in Figure 4c,d are not recoverable.
Figure 4.
Example clusters relevant to Steps 26 and 27. The x and y axes are denoted as in Figure 1. (a) Three clusters are obviously identifiable; however, GenomeStudio is not able to make a correct call. AA and AB cluster overlap, which results in a very small cluster separation. (b) The situation in A can be easily fixed by manually re-positioning the cluster ovals. (c) Low cluster separation score caused by two very closely located clusters. (d) Low cluster separation score caused by four clusters being present rather than three.
Steps 28–30
The next QC parameters to be examined are the Rep and Mendelian errors. Rep error occurs when the genotypes of two replicate samples are different from each other. Mendelian error, which is described as P-P-C error in GenomeStudio, denotes apparent de novo mutations in the child. P-C error only records error between the child and one of the parents. These parameters provide additional QC checks beyond the GenTrain and the cluster separation scores. Although de novo mutations are of course possible, a high number of them is indicative of a problem in the data. The majority of SNPs should not contain Mendelian errors; however, occasionally, a particularly error-rich trio of samples (from child and the two parents) can contribute to high numbers of Mendelian errors. These low-quality samples can be filtered out by checking genotyping consistency with the 1000 Genomes Project. SNPs with excessive Mendelian errors (>10) are candidates for removal. Figure 5a shows an example SNP with good GenTrain and cluster separation scores. However, a few P-P-C errors are introduced owing to the lower BB cluster tail being called as AB. Figure 5b shows the manually corrected SNP from Figure 5a. In Figure 5c is reported another example of inaccurate clustering by GenomeStudio, which can be detected by checking for P-P-C error. Figure 5d reports the corrected cluster of the SNP from Figure 5c.
Figure 5.
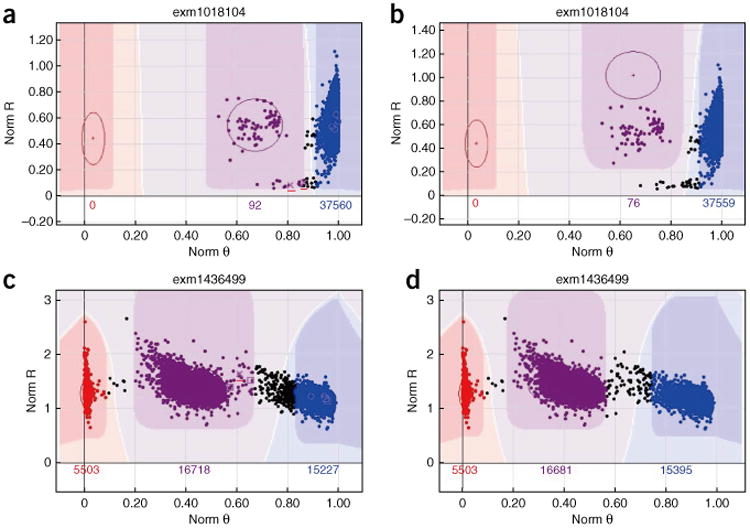
Example clusters relevant to Steps 28–30. The x and y axes are denoted as in Figure 1. (a) The small ‘x’ indicates P-P-C error (also marked with red line by us for clear viewing). This SNP has good GenTrain and cluster separation scores. However, a few P-P-C errors are introduced owing to the lower BB cluster tail being called as AB. (b) The problem highlighted in a can be fixed by moving the AB cluster oval up. (c) Another example of P-P-C errors introduced by inaccurate clustering of the AB cluster. (d) The P-P-C errors highlighted in c are fixed by narrowing the AB cluster oval.
Step 34
zCall can be used to correct mis-clustered rare SNPs. zCall is a variant caller that can be used to identify mis-called rare SNPs generated by GenomeStudio. The input of zCall is the standard report format of GenomeStudio. Unfortunately, even though zCall recovers a portion of the mis-called rare SNPs, it also introduces a large number of false positives. The most common type of false positive is inclusion of the wrong samples in the AB cluster, and some of the recovered rare SNPs by zCall are not complete (missing samples in the AB cluster). A recently published evaluation study29 also showed no significant improvement in rare variant clustering by using zCall. Furthermore, the zCall approach requires programming skills for interpretation and implementation; thus, it is an optional step and recommended for advanced users.
A better alternative approach to zCall is clustering by brute force, which in this case means manually clustering large numbers of SNPs. There are over 240,000 SNPs on the exome chip, and each manual cluster might take 10–30 s to perform. Manual clustering of 240,000 SNPs would take roughly 660–2,000 man-hours. The time required is strongly dependent on computer power and sample size. Thus, it is important to systematically select only the rare SNP candidates that might need manual re-clustering. The brute force method is still very time consuming, even after narrowing down the subset of SNPs; however, to achieve premium quality calling on rare SNPs, brute force manual clustering is the best approach.
Figure 6a reports an example of mis-clustering by zCall. zCall called two samples (on the very right of the AB cluster) as AB genotype. Figure 6b shows another example of mis-clustering by zCall. zCall is able to capture partially the AB cluster of this SNP, while still missing half the samples that should be in AB. The Cartesian plot of these two SNPs can be viewed in Supplementary Figure 1.
Figure 6.
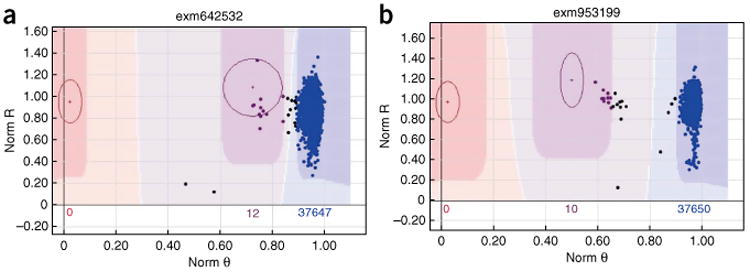
Example clusters relevant to Step 34A. The x and y axes are denoted as in Figure 1. (a) This SNP is recalled by zCall. zCall called two samples (on the very right of the AB cluster) as AB genotype. These two samples should be left as no call or BB cluster. (b) zCall is able to capture partially the AB cluster of this SNP, while still missing half the samples that should be in AB.
Step 35B
When exporting the SNP data to PLINK format, users should keep in mind that there are duplicated and tri-allelic SNPs on the exome chip. The duplicated SNPs are assigned different SNP ‘Names’. Each tri-allelic SNP is presented with two SNP ‘Names’ as well. Thus, for any given SNP ‘Name’, there can be only two alleles. When conducting association tests using exome SNP data, we recommend using the SNP with higher call frequency for duplicated SNPs. A user can choose to extract the tri-allelic SNPs to a separate PLINK file because different association tests can be applied to tri-allelic SNPs30.
Steps 38 and 39
Clinical information, such as gender and race, is subject to self-reporting and data input errors. Checking reported and genotyped gender consistency provides crucial information on data integrity. PLINK uses SNPs on chromosome X to estimate gender on the basis of the heterozygosity rate. For common SNP arrays, SNPs with MAF >0.2 are usually used. However, as the number of X chromosome SNPs present on the exome chip is limited, MAF >0.1 is used instead as the threshold value in the present protocol. The output of this command is a text file that contains six columns. Column 5 is ‘Status’, which can be PROBLEM or OK. PROBLEM indicates a problem with gender. Column 6 is the F value, which denotes the X chromosome inbreeding estimate. A PROBLEM is assigned if the self-reported and genotype-based genders do not match, or if the SNP data or pedigree data are ambiguous with regard to sex. A male call is made if F > 0.8; a female call is made if F < 0.2. The PLINK recommended threshold is rather strict for exome chip. For example, in a batch from our example study, which contained 6,000 samples, 141 samples had a gender ‘PROBLEM’ according to PLINK20. However, when drawing the inbreeding estimate distribution for males and females, the actual number of samples with gender problems was lower. For males, a majority of the samples had F values between 0.95 and 1, whereas only a few samples showed extremely low F values. Similarly, for females, the samples with outlier F values were clearly visible after drawing the distribution of inbreeding estimates. The actual number of samples with outlier F values was 25 compared with 141 identified by PLINK.
The possible causes of gender mismatch are data entry error, blood transfusions, sample mislabeling and cross-contamination. Except for cross-contamination, all of these scenarios can be checked if medical records are available. If any of the gender mismatch causes just described are recognized to be present, re-genotyping the samples with gender issues is recommended. Data in Figure 7a show the expected distribution of chromosome X inbreeding estimate for males. Inbreeding estimates should be close to 1 for males; some outliers are visible near 0. Data in Figure 7b show the expected distribution of chromosome X inbreeding estimate for females. In this case, inbreeding estimates should be in the range of −0.4 to 0.4; some outliers are visible near 1.
Figure 7.
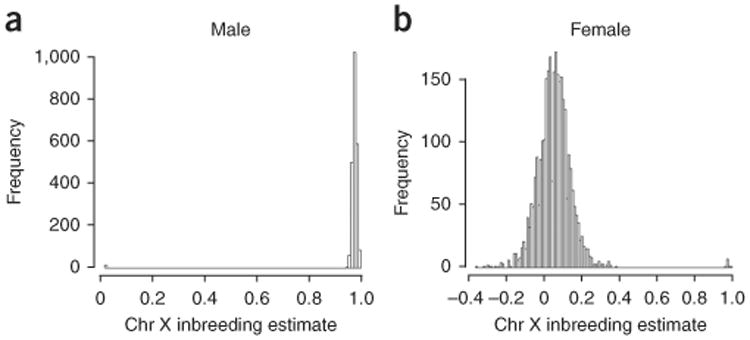
Example clusters relevant to Steps 38 and 39 of the PROCEDURE. Chr denotes chromosome. (a) Distribution of chromosome X inbreeding estimate for males. Inbreeding estimates should be close to 1 for males; some outliers are visible near 0 in a. (b) Distribution of chromosome X inbreeding estimate for females. Inbreeding estimates should be in the range of −0.4 to 0.4; some outliers are visible near 1.
Steps 40–43
The exome chip design contains over 3,000 AIMs distributed approximately one per megabase across the autosomes and chromosome X (Supplementary Table 6). These markers were selected because they have demonstrated strong differentiation power between African and European ancestry samples sequenced in the 1000 Genomes Project. By using EIGENSTRAT21, PCA on these AIM SNPs can be performed. EIGENSTRAT also has the functionality to draw PCA figures based on first and second principal components. However, a figure with reported race denoted would be more useful. An example of a PCA plot can be viewed in Figure 8. Figure 8a shows the first and second principal component plots from the 1000 Genomes Project. Figure 8b shows an example of a PCA plot from an exome chip study. Notice that in the example exome chip study a subset of interracial population between European Americans and African Americans is observable (Figure 8b). PCA can help estimate race distribution and check for discrepancies between estimated and reported ethnicities. For downstream association analysis, the PCA results from EIGENSTRAT can be used as covariates to control for population admixture within the models31.
Figure 8.
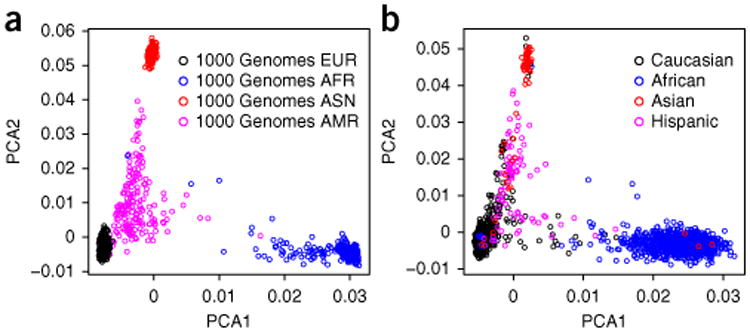
Example clusters relevant to Steps 40–43. The x axis denotes the first component of the PCA and the y axis denotes the second component of the PCA. (a) Scatter plot of first and second principal components for 1000 Genomes Project data. ASN, EUR, AFR and AMR denote East Asian, European, African and admixed American ancestry, respectively. (b) Example scatter plot of the first and second principal components using the example exome chip data batch 2.
Steps 44 and 45
A relatedness check can potentially help identify contaminated samples. The command ‘ --genome’ in PLINK computes identity by state (IBS) distance between pair-wise samples using estimates of pair-wise identity by descent (IBD). It outputs a file named ‘{outfile}.genome’ that contains several useful columns. One contains the parameter PI_HAT, which is computed as P(IBD = 2) + 0.5 × P(IBD = 1), where P denotes probablility. PI_HAT ranges between 0 and 1, where 0 indicates no relationship, and the higher the value the closer the relatedness. For two unrelated individuals, PI_HAT should be close to 0. Any value of >0.25 indicates relatedness or cross-contamination. In theory, for large a GWAS, there is always a chance of including related people in the study by chance, which can affect the results of association tests. If two samples with high PI_HAT value are observed, it is recommended to remove the one with the lower call rate, unless the study design is family-based. Because PI_HAT is computed between all possible pairs of samples, the number of test calculations performed is usually very large, so drawing a histogram will not help visualize the samples with high PI_HAT values. Rather, visualizing the data in Excel and using its filtering or sorting functionality to display the potential related samples is much more effective.
Steps 46–48
The HWE is a principle stating that the genetic variation in a population will remain constant from one generation to the next in the absence of disturbing factors. Departure from this equilibrium can be an indicator of potential genotyping errors, population stratification or even actual association with the trait under study25. Testing for deviation from HWE has been commonly used for QC purposes on large GWASs and is one of the few ways to identify systematic genotyping errors in unrelated individuals32,33. Because HWE is specific to a population, the HWE test should be applied to data from participants of each race separately. The last column of the output file contains the HWE test P value. For example, in one of the batches of the example exome chip study, 6,901 SNPs had P values of <0.05. Because PLINK does not perform multiple test correction, a large portion of the significant SNPs are actually false positives. One should perform a conservative multiple correction test such as the Bonferroni correction24, or choose a much more conservative P value threshold25. If the threshold used is P < 1 × 10−5, the number of SNPs out of HWE reduces to 842. Figure 9a shows the expected distribution of the HWE test P values.
Figure 9.
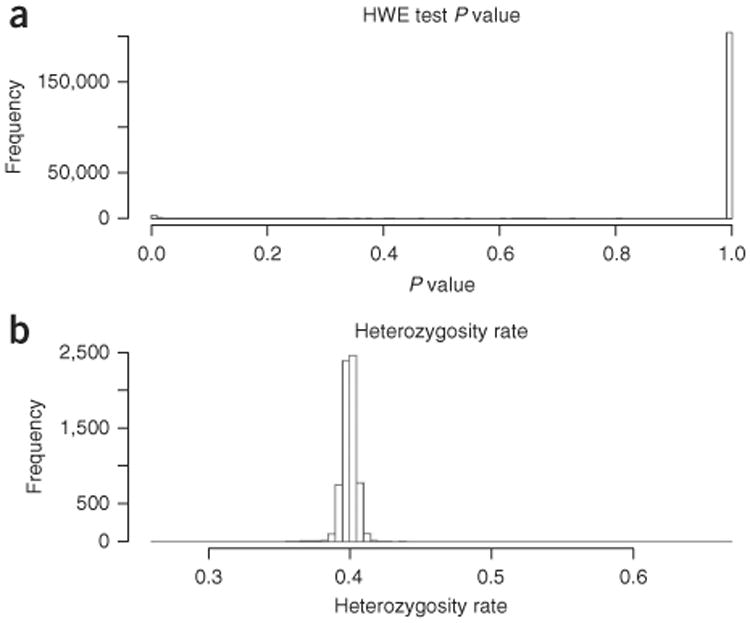
Distribution of HWE and heterozygosity rates relevant to Steps 46–50. (a) HWE test P value distribution. Note that the majority of the SNPs have P values near 1, and a minority of the SNPs have very low P values. There are P values spread out between 0 and 1, but they are not easily visible owing to their small numbers on the histogram. (b) Heterozygosity rate distribution of example data batch 2. The majority of the samples should be in the range of 0.35–0.45.
Steps 49 and 50
Given the large number of SNPs on the exome chip and a homogeneous sample, calculating the heterozygosity rate may also help identify problematic SNPs. Low heterozygosity may indicate inbreeding, which can lead to reduced fitness of a population with many homozygous genotypes, and high heterozygosity may indicate contamination. Similarly to the HWE test, the heterozygosity test should be performed separately on data from participants of each race. By using one of the batches in the example exome chip study20, a histogram of heterozygosity shows that the majority of the samples have a heterozygosity rate of 0.35–0.45. SNPs with heterozygosity that severely deviates from this range could indicate a problem. In Figure 9b is reported the expected distribution of heterozygosity rate.
Steps 51 and 52
Three types of genotyping consistencies exist that can be computed for the exome chip: (i) consistency with the 1000 Genomes Project, (ii) consistency between duplicated samples and (iii) consistency between duplicated SNPs. Consistency between duplicated samples can be checked in GenomeStudio. To check consistency with the 1000 Genomes Project–released data, several requirements must be met. First, the study must contain 1000 Genomes Project samples. The use of 1000 Genomes Project samples for QC has been a common practice for GWASs. However, only a select number of HapMap samples are included in the 1000 Genomes Project. In addition, only ∼60% of the SNPs on the HumanExome-12v1_A chip can be found within the 1000 Genomes Project, with the number varying slightly depending on the version of 1000 Genomes Project–released data used. Thus, the consistency computation is limited to the number of overlapped SNPs between the exome chip and the 1000 Genomes Project. There are 754 duplicated SNPs and 45 triallelic SNPs on the v1.1 exome chip (Supplementary Table 7); these SNPs provide additional opportunity for QC.
The 1000 Genomes Project Variant Call Format (VCF) files can be downloaded from http://www.1000genomes.org/data. However, the files are huge in size (∼1.2 TB), which makes the downloading problematic in some instances. We have made a smaller version that contains only the overlapping SNPs, and it is supplied as a supplementary file (G1000.vcf). This file can also be downloaded from https://github.com/slzhao/ExonChipProcessing/. The consistency computed by ‘Consistency1000G.sh’ is the heterozygous genotype consistency, and it is defined as the number of heterozygous genotypes consistent between the exome chip and the 1000 Genomes Project data, divided by the number of heterozygous genotypes identified by the exome chip. Computing heterozygous genotype consistency instead of the overall genotype consistency will yield more informative results, because exome chip targets rare SNPs, and the majority of the genotype of a sample genotyped on exome chip will be homozygous. Homozygous genotypes tend to be the same as the 1000 Genomes Project reference homozygous genotype, as all strands were already converted to the plus strand in Steps 36 and 37. Thus, the overall genotype consistency is inflated toward higher values2. Reasonable heterozygous consistency is usually >97%. Illumina exome chip genotyping also contains ∼800 duplicate markers. Computing genotype consistency between the duplicate markers provides additional insights into the integrity of the data. The expected consistency between the duplicated SNPs is 99%.
Steps 53–56
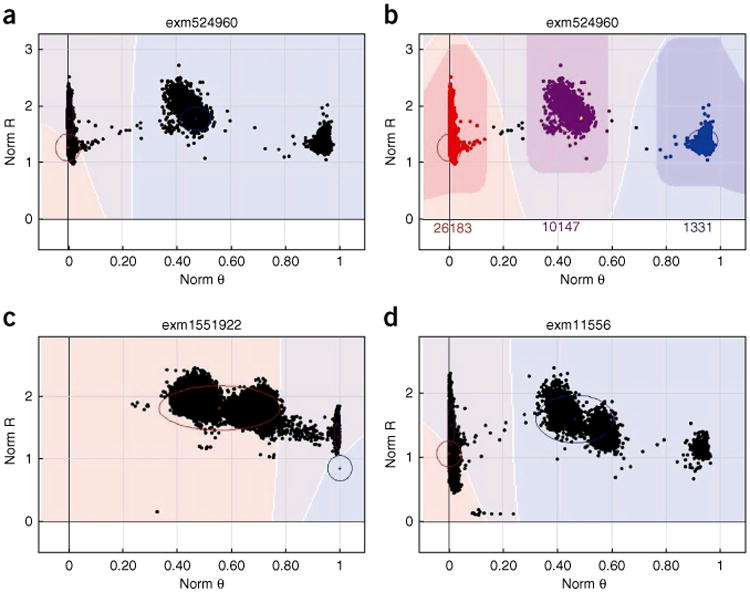
Another test for genotype quality can be performed by comparing the allele frequency generated by implementing the protocol with the allele frequency from a public database. Currently, two large public databases exist that are suitable for this purpose: the 1000 Genomes Project (n = 1,092) and the US National Heart, Lung and Blood Institute (NHLBI) Exome Sequencing Project (n = 6,500). Both projects include data on individuals from diverse racial backgrounds, and they are suitable for conducting allele frequency consistency analysis. An example allele frequency correlation plot can be seen in Figure 10. Notice that in this figure a few SNPs show no allele frequency in the example exome chip project but high allele frequency in the 1000 Genomes Project. The initial reaction may be that these SNPs must be mis-clustered by GenomeStudio and not caught in the QC process. However, after further investigation, we found that the clusters for these SNPs were rather clean (Y.G. and H.W., unpublished observations). For example, for SNP exm-rs8181166, the cluster can be seen in Figure 11a. Clearly, only the homozygous BB cluster was present. However, in the 1000 Genomes Project, the MAF for white participants for this SNP is 0.48. Further investigation was conducted into this SNP by examining the probe sequence provided by Illumina, and no evidence of error was found (Y.G., unpublished observations). It is possible that Illumina provided a corrected probe sequence in the documentation but used the wrong sequence on the actual chip. It is also possible that such phenomena are caused by the effect of neighboring SNPs, strand orientation problems or an error in the 1000 Genomes Project. Another example is SNP exm2216256, which is located in the mtDNA. In Figure 11b, clearly, the AA and BB clusters are presented and a few samples are in the AB cluster owing to heteroplasmy. In the 1000 Genomes Project, only one allele is reported for this SNP. Without clearly identifying the reason for such discordant SNPs between the exome chip project and the 1000 Genomes Project, SNPs with large MAF discrepancies should be excluded from further use. In addition, there are 339 SNPs with alleles not matching the alleles in the 1000 Genomes Projects (Supplementary Table 8); when dealing with these SNPs extra caution is required.
Figure 10.
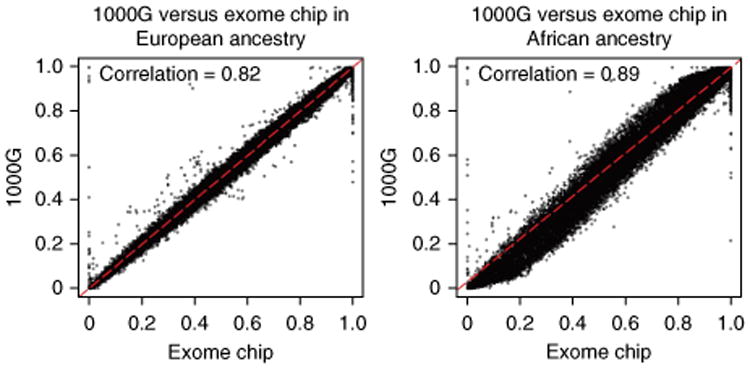
Example clusters relevant to the PLINK-related Steps 53–56. The x axis denotes the allele frequencies computed from example exome chip SNP data, and the y axis denotes the allele frequencies of the same alleles computed from the 1000 Genomes Project SNP data. (a) Scatter plot of allele frequency between the 1000 Genomes Project data and the example exome chip data for individuals of European ancestry. (b) Scatter plot of the allele frequency between the 1000 Genomes Project data and the example exome chip data for individuals of African ancestry. The outliers (defined as abs(x–y) >50%) should be checked.
Figure 11.
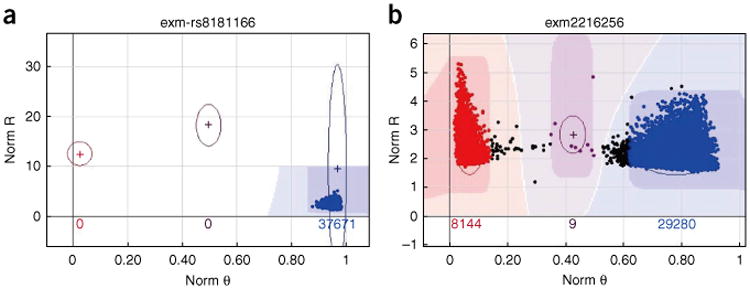
Example clusters relevant to the PLINK-related Steps 53–56. The x and y axes are denoted as in Figure 1. (a) This SNP showed zero MAF in the exome chip but the same SNP showed high MAF in the 1000 Genomes Project data. Although the exact reason for this discrepancy is not known, we recommend removing this SNP for cautionary purposes. (b) Mitochondrial SNP at position 3010, which is a known heteroplasmy site. Both AA and BB clusters should be presented. However, in the 1000 Genomes Project data, only one genotype is presented. In this case, it is more likely that the 1000 Genomes Project made an incorrect call.
Step 57
Batch effects can affect the genotype quality in GWASs34. For GWASs, batches can be defined as the 96 samples on a plate. For much larger studies, batches can be defined as each cohort of subjects. Many statistical tests can be performed to check for batch effects, such as the Kruskal-Wallis test26 and the Fligner-Killeen test of homogeneity of variances27. However, for a large study, these tests can be too sensitive, because large sample sizes enable the detection of minor differences. For a genotyping study containing thousands of samples, minor variation in probe intensity will result in statistically significant P values (<0.05). A better way to visually detect batch effects is to draw a scatter or box plot of allele frequency and call rate between cohorts and to compute the Pearson correlation between batches (Fig. 12). As in HWE and heterozygosity tests, owing to racial differences, allele frequencies should be computed separately by race. Figure 12a shows an example correlation matrix of allele frequency consistency between batches for subjects with European ancestry. Figure 12b shows an example correlation matrix of allele frequency consistency between batches for subjects with African ancestry. High correlation values indicate a low batch effect. If the correlation is <70%, we recommend checking the GenTrain score distribution by batches. If a batch has substantially lower GenTrain scores than the other batches in the same study, we recommend that the user re-perform QC on that batch by manually checking more SNPs. Afterward, if batch effects still exist, we recommend re-genotyping or removing this batch.
Figure 12.
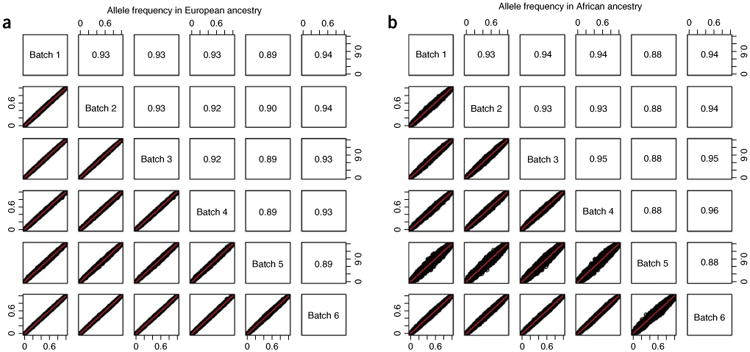
Example clusters relevant to the PLINK-related Step 57. (a) Correlation matrix of allele frequency consistency between batches for individuals with European ancestry. (b) Correlation matrix of allele frequency consistency between batches for individuals with African ancestry. A higher correlation indicates a lower batch effect.
Supplementary Material
{kind=link}
Acknowledgments
Development of this protocol is supported by Cancer Center Support Grant (CCSG) nos. (P30 CA068485) and R01CA158473. We thank M. Bjoring for editorial support.
Footnotes
Author Contributions Y.G. wrote the manuscript and designed the protocol with J.L.; S.Z., J.H., H.W., Q.S. and X.Z. contributed to script writing and generated the figures and tables; Y.S. and D.C.S. provided intellectual contributions to the overall design of the protocol.
Competing Financial Interests The authors declare no competing financial interests.
References
- 1.Samuels DC, et al. Finding the lost treasures in exome sequencing data. Trends Genet. 2013;29:593–599. doi: 10.1016/j.tig.2013.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo Y, et al. Exome sequencing generates high quality data in non-target regions. BMC Genomics. 2012;13:194. doi: 10.1186/1471-2164-13-194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Abecasis Lab. Exome Chip Design Wiki Site. http://genome.sph.umich.edu/wiki/Exome_Chip_Design.
- 4.Szatkiewicz JP, et al. Detecting large copy number variants using exome genotyping arrays in a large Swedish schizophrenia sample. Mol Psychiatry. 2013;18:1178–1184. doi: 10.1038/mp.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huyghe JR, et al. Exome array analysis identifes new loci and low-frequency variants influencing insulin processing and secretion. Nat Genet. 2013;45:197–201. doi: 10.1038/ng.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McElroy JJ, et al. Maternal coding variants in complement receptor 1 and spontaneous idiopathic preterm birth. Hum Genet. 2013;132:935–942. doi: 10.1007/s00439-013-1304-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moura R, et al. Exome analysis of HIV patients submitted to dendritic cells therapeutic vaccine reveals an association of CNOT1 gene with response to the treatment. J Int AIDS Soc. 2014;17:18938. doi: 10.7448/IAS.17.1.18938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seddon JM, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age-related macular degeneration. Nat Genet. 2013;45:1366–1370. doi: 10.1038/ng.2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosley JD, et al. Mechanistic phenotypes: an aggregative phenotyping strategy to identify disease mechanisms using GWAS data. PLoS ONE. 2013;8:e81503. doi: 10.1371/journal.pone.0081503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Psaty BM, et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ Cardiovasc Genet. 2009;2:73–80. doi: 10.1161/CIRCGENETICS.108.829747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grove ML, et al. Best practices and joint calling of the HumanExome BeadChip: the CHARGE Consortium. PLoS ONE. 2013;8:e68095. doi: 10.1371/journal.pone.0068095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perreault LP, et al. Comparison of genotype clustering tools with rare variants. BMC Bioinform. 2014;15:52. doi: 10.1186/1471-2105-15-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ritchie ME, Liu R, Carvalho BS, Irizarry RA. Comparing genotyping algorithms for Illumina's Infinium whole-genome SNP BeadChips. BMC Bioinform. 2011;12:68. doi: 10.1186/1471-2105-12-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nelson SC, Doheny KF, Laurie CC, Mirel DB. Is ‘forward’ the same as ‘plus’?…and other adventures in SNP allele nomenclature. Trends Genet. 2012;28:361–363. doi: 10.1016/j.tig.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Illumina. “TOP/BOT” Strand and “A/B” Allele. http://res.illumina.com/documents/products/technotes/technote_topbot.pdf.
- 16.Zhang Y, et al. Rare coding variants and breast cancer risk: evaluation of susceptibility loci identifed in genome-wide association studies. Cancer Epidemiol Biomarkers Prev. 2014;23:622–628. doi: 10.1158/1055-9965.EPI-13-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abecasis GR, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491:56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 19.Ilumina. Infnium Genotyping Data Analysis. http://res.illumina.com/documents/products/technotes/technote_infnium_genotyping_data_ analysis.pdf.
- 20.University Medical Center. BioVU. https://victr.vanderbilt.edu/pub/biovu/
- 21.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 22.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goldstein JI, et al. zCall: a rare variant caller for array-based genotyping: genetics and population analysis. Bioinformatics. 2012;28:2543–2545. doi: 10.1093/bioinformatics/bts479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc. 1955;50:1096–1121. [Google Scholar]
- 25.Wittke-Thompson JK, Pluzhnikov A, Cox NJ. Rational inferences about departures from Hardy-Weinberg equilibrium. Am J Hum Genet. 2005;76:967–986. doi: 10.1086/430507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kruskal WH, Wallis WA. Use of ranks in one-criterion variance analysis. J Am Stat Assoc. 1952;47:583–621. [Google Scholar]
- 27.Conover WJ, Johnson ME, Johnson MM. A comparative study of tests for homogeneity of variances, with applications to the outer continental shelf bidding data. Technometrics. 1981;23:351–361. [Google Scholar]
- 28.Guo Y, et al. Multi-perspective quality control of Illumina exome sequencing data using QC3. Genomics. 2014;103:323–328. doi: 10.1016/j.ygeno.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perreault LP, et al. Comparison of genotype clustering tools with rare variants. BMC Bioinform. 2014;15:52. doi: 10.1186/1471-2105-15-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guthridge JM, et al. Two functional lupus-associated BLK promoter variants control cell-type– and developmental-stage–specifc transcription. Am J Hum Genet. 2014;94:586–598. doi: 10.1016/j.ajhg.2014.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu C, DeWan A, Hoh J, Wang Z. A comparison of association methods correcting for population stratification in case-control studies. Ann Hum Genet. 2011;75:418–427. doi: 10.1111/j.1469-1809.2010.00639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gomes I, et al. Hardy-Weinberg quality control. Ann Hum Genet. 1999;63:535–538. doi: 10.1017/S0003480099007824. [DOI] [PubMed] [Google Scholar]
- 33.Hosking L, et al. Detection of genotyping errors by Hardy-Weinberg equilibrium testing. Eur J Hum Genet. 2004;12:395–399. doi: 10.1038/sj.ejhg.5201164. [DOI] [PubMed] [Google Scholar]
- 34.Hong H, et al. Assessing batch effects of genotype-calling algorithm BRLMM for the Affymetrix GeneChip Human Mapping 500 K array set using 270 HapMap samples. BMC Bioinform. 2008;9(suppl. 9):S17. doi: 10.1186/1471-2105-9-S9-S17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.