

Abstract
Clostridium difficile is a prominent nosocomial pathogen, proliferating and causing enteric disease in individuals with a compromised gut microflora. We characterized the post-translational modification of flagellin in C. difficile 630. The structure of the modification was solved by nuclear magnetic resonance and shown to contain an N-acetylglucosamine substituted with a phosphorylated N-methyl-l-threonine. A reverse genetics approach investigated the function of the putative four-gene modification locus. All mutants were found to have truncated glycan structures by LC-MS/MS, taking into account bioinformatic analysis, we propose that the open reading frame CD0241 encodes a kinase involved in the transfer of the phosphate to the threonine, the CD0242 protein catalyses the addition of the phosphothreonine to the N-acetylglucosamine moiety and CD0243 transfers the methyl group to the threonine. Some mutations affected motility and caused cells to aggregate to each other and abiotic surfaces. Altering the structure of the flagellin modification impacted on colonization and disease recurrence in a murine model of infection, showing that alterations in the surface architecture of C. difficile vegetative cells can play a significant role in disease. We show that motility is not a requirement for colonization, but that colonization was compromised when the glycan structure was incomplete.
Introduction
The intestinal pathogen Clostridium difficile is a strictly anaerobic, Gram-positive bacterium. In recent years it has become a huge burden both economically and to human health due to its global spread throughout health care environments, where it causes disease ranging from mild diarrhoea to life threatening pseudomembranous colitis (Loo et al., 2005; Goorhuis et al., 2007; Kuijper et al., 2007). Its ability to differentiate into endospores, a metabolically dormant and highly robust cell form, is essential in its transmission as this allows it to survive external stresses such as desiccation, osmotic shock, and contact with chemicals such as disinfectants (Lawley et al., 2010; Deakin et al., 2012). Two large clostridial toxins, TcdA and TcdB are considered the main virulence factors associated with disease symptoms, their production leading to the disruption and eventual destruction of the intestinal epithelium (Voth and Ballard, 2005). The generation of at least one of these two potent toxins is essential for the pathogenesis of this organism (Lyras et al., 2009; Kuehne et al., 2010); however, other properties of C. difficile contribute to its success as a pathogen by promoting the colonization of the host, which may be influential in disease recurrence.
Bacterial surface-associated proteins are often involved in host colonization and immune evasion (Pizarro-Cerda and Cossart, 2006). C. difficile produces many such proteins, including peritrichous flagella, a paracrystalline S-layer, cell wall proteins and a putative sortase and sorted proteins (Hennequin et al., 2003; Eidhin et al., 2006; Kirby et al., 2009; Reynolds et al., 2011). Flagella are the organelles of bacterial propulsion and have been found to be important for the penetration of the mucous layer and adherence to the gut mucosa in enteric pathogens (Liu et al., 1988; Grant et al., 1993), although in C. difficile the flagella are not essential for virulence and colonization of the hamster or murine model (Dingle et al., 2011; Aubry et al., 2012; Baban et al., 2013).
Flagellin is the main protein component of the flagella; it forms the long whip-like filament which when rotated by the basal subunits of the structure causes motility. Some bacteria produce ‘simple’ flagella filaments which are composed of a single flagellin, while others produce ‘complex’ flagella filaments made up of multiple flagellins (Logan, 2006). In a number of Gram-positive and Gram-negative bacteria the flagellin is post-translationally modified with an O-linked oligosaccharide at serine and/or threonine residues on the highly variable, surface-exposed domains of the protein (Logan, 2006). Enzymes, often encoded alongside the flagella genes, transfer these post-translational modifications (PTMs) specifically to the flagellin. The nature of this specificity is yet to be determined as no consensus sequence is recognized by these enzymes. In many bacteria these flagellin PTMs are essential for motility and mutants of the PTM enzymes are not only unable to modify their flagellin but are also unable to secrete the flagellin from the cell, as has been described in Campylobacter jejuni, Helicobacter pylori, Caulobacter crescentus, Aeromonas caviae and Shewanella oneideinsis (Ewing et al., 2009; Asakura et al., 2010; Faulds-Pain et al., 2011; Parker et al., 2012; Sun et al., 2013). However, this is not the case for all bacteria, for example in Pseudomonas aeruginosa motility is unaffected when flagellin modification genes are mutated (Schirm et al., 2004; Verma et al., 2006).
C. difficile produces a simple flagella filament with a single flagellin which is post-translationally modified. As with most bacterial O-linked PTM systems, the genes encoding the modification enzymes are colocalized with those encoding the target protein. In C. difficile strains these enzymes are thought to be encoded among the flagella structural and regulatory genes, between the flagellin gene, fliC and the putative flagella basal body rod gene, flgB (Stabler et al., 2009).
It has been found that the C. difficile flagellin can be modified with at least two oligosaccharide structures, herein referred to as type A and type B, and that these structural variations are strain-specific and reflected in variations at the genome level (Fig. 1; Twine et al., 2009). The coding sequence (CDS) immediately downstream of fliC is conserved in the type A and type B strains and encodes a putative glycosyltransferase (GT) (Fig. 1A), which is essential for the modification of the flagellin and motility but not flagellin secretion or filament formation (Twine et al., 2009). The sugar associated with the flagellin protein in both type A and type B oligosaccharide modifications, has been identified by mass spectrometry to be an N-acetyl hexosamine (HexNAc) (Twine et al., 2009), suggesting that this first sugar is transferred to the flagellin by this common GT. Beyond this initial sugar neither the modification genes nor the structure of the PTMs are conserved in the type A and type B strains (Smith et al., 1981; Verma et al., 2006). The type A flagellin modification was previously proposed to be a methylated aspartic acid attached to the initial HexNAc via a phosphate bond in the non-epidemic strain 630 (Twine et al., 2009). The type B structure was identified among BI-I/NAP-I/ ribotype 027 epidemic strains. The oligosaccharide structure of these modifications is complex and consists of multiple monosaccharide residues which includes a HexNAc moiety through which the glycan is linked to the protein as well as two deoxyhexose residues and a heptose (Fig. 1B; Twine et al., 2009; Hitchen et al., 2010).
Fig 1.
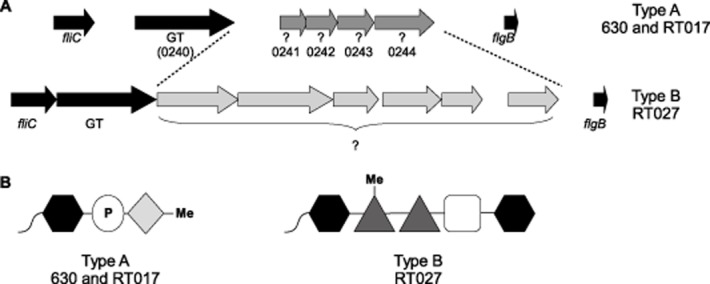
Schematic diagram illustrates the alternative flagellin modification gene loci type A and type B.A. The flagella structural genes fliC and flgB flank the genes predicted to be involved in flagellin modification in the ribotype 012 strain 630 and the ribotype 027 strains. The gene adjacent to fliC in both loci is a predicted glycosyltransferase (GT) and the ORFs in grey are the remaining putative modification genes, predicted to be involved in flagellin modification.B. The predicted modification structures of the type A 630/RT017 strains and the type B RT027 strains. The black hexagon represents a HexNAc, the white circle represents a phosphate group, Me denotes a methyl group, dark gray triangles represent a deoxyhexose and the white square a heptose, the light gray diamond represents an unidentified structure.
It has been hypothesized that as the type B PTM was identified in BI-I/NAP-I/ ribotype 027 outbreak strains, while the type A PTM was found in a non-epidemic strain, the type B PTMs could be contributing to the hypervirulence of these organisms (Stabler et al., 2009; Twine et al., 2009). In this study however we identified the presence of the type A loci in the toxin A negative epidemic ribotype 017 (RT017) strains of C. difficile, indicating that the relative importance of the type A and type B modifications might not be easily defined. We investigate the flagellin type A PTMs in both the 630 strain and an epidemic RT017 strain M68. A definitive structure for this modification was determined by NMR spectroscopy and the role of the putative modification genes were assessed. We also investigated the role of motility and flagellin modification in the colonization of C57BL/6 mice, finding that motility is not a requirement but that colonization was compromised when the glycan structure was incomplete.
Results
Orthologues of the type A modification gene cluster are found only in P. aeruginosa PA01 strains
Molecular typing and sequence analysis has revealed that there are at least five clonal lineages of C. difficile, designated clades one to five (He et al., 2010; Stabler et al., 2012), and that all have been involved in outbreaks of C. difficile-associated disease. Utilizing ACT comparisons; putative flagellin modification gene loci were compared among the five clonal lineages of C. difficile strains. Orthologues of the 630 type A modification genes were found in only one of these lineages which includes the epidemic RT017 strains.
To determine whether the type A modification genes are commonly found associated with flagella genes, BLASTP analysis was carried out with the sequences of strain 630 open reading frames (ORFs) CD0241, CD0242, CD0243 and CD0244. While orthologues of the ORFs CD0241 and CD0242 were occasionally identified together in other genomes (such as Helicobacter hepaticus) they were not associated with any known structural orthologues such as the flagella. In fact it was only in strain PA01 of P. aeruginosa where orthologues of CD0241, CD0242 and CD0243 were clustered together and like C. difficile they were found within the flagella genetic locus. It was noted previously that the flagellin modification of the PA01 flagellin was similar to that of C. difficile 630 (Twine et al., 2009), containing a methylated amino acid (in this case a tryptophan) attached via a phosphate bond to a deoxyhexose sugar on the surface of the flagellin (Verma et al., 2006). The genes PA1088, PA1089 and PA1090, located directly upstream of the flagellin GT and flagellin genes in P. aeruginosa PA01 strains, are orthologues of CD0243, CD0241 and CD0242 respectively and are involved in the modification of the PA01 flagellin. Their amino acid similarity, location and the similar structure of the modification indicate that these genes may have a similar function in C. difficile. No orthologue of the CD0244 ORF was found to be co-located with the other three ORFs in P. aeruginosa or any other organism and BLASTP searches only identified hypothetical proteins.
NMR analysis of the type A flagellin solves the structure of its post-translational modification
The structure of the C. difficile 630 flagellin glycan has been previously studied by LC-MS/MS and defined as a HexNAc sugar carrying a phosphate linked to a moiety which was suggested to be a methylated aspartic acid (Fig. 1B). This glycan was attached to the flagellin via the HexNAc at five sites (S141, S174, T183, S188 and S205 [S = serine and T = threonine]) (Twine et al., 2009). However, it has since been noted that at a mass of 115 Da the methylated amino acid is too small to be a methyl-aspartic acid (129 Da). In order to solve the structure of this modification, nuclear magnetic resonance (NMR) analysis of the flagellin glycan was carried out. Glycopeptides were released from the purified glycoprotein by Proteinase K treatment and isolated using size-exclusion and anion-exchange chromatography. Enzymatic hydrolysis did not cleave all amino acids and the product represented a mixture of glycosylated short peptides. NMR analysis using 2D spectra (gCOSY, TOCSY, ROESY, 1H-13C HSQC, 1H-13C HMBC, 1H-31P HMQC) indicated that the single HexNAc monosaccharide is a N-acetyl-β-glucosamine (GlcNAc) (all vicinal coupling constants 8–10 Hz), linked to a serine residue on the flagellin protein (Fig. 2). GlcNAc was phosphorylated at O-3, which caused a downfield shift of H/C-3 signals (Table 1). Additional signals were present, belonging to HOOC-CHX-CHX-CH3 fragment and a methyl group (Table 1). HMBC correlations between H/C of the methyl group and H/C-2 of this fragment indicated that methyl group was linked to the C-2 through a heteroatom. By the signal position of the C-2 at 70.5 ppm this heteroatom was most probably nitrogen. A downfield shift of the C-2 signal from its expected position of approximately 55 ppm was thus caused by N-methylation. C-3 carried a hydroxyl group which followed from the position of the C-3 signal at 73 ppm. The H-3 signal of the amino acid was shifted to 4.5 ppm due to phosphorylation. Overall this represents phosphorylated N-methylthreonine, in agreement with MS data indicating the mass of the glycosyl moiety at 398.1 amu (Fig. 2). Absolute l-configuration and identity of the N-methylthreonine was confirmed by GC of its acetylated ester with optically pure 2-octanol.
Fig 2.
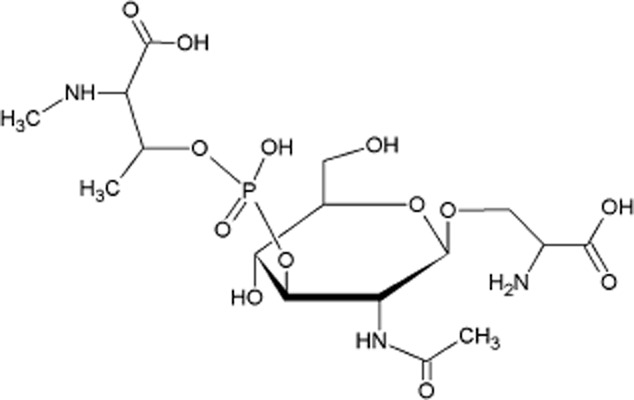
Schematic diagram illustrates the structure of the C. difficile 630 flagellar glycan shown in O-linkage to a serine residue, solved by NMR.
Table 1.
NMR data (δ, ppm; Varian INOVA 500 MHz, 25°C) for the C. difficile 630 glycopeptide
| H/C 1 | H/C 2 | H/C 3 | H/C 4 | H/C 5 | H/C 6 | Me | |
|---|---|---|---|---|---|---|---|
| GlcNAc | 4.65 | 3.85 | 4.17 | 3.63 | 3.51 | 3.79; 3.93 | |
| 100.1 | 55.6 | 79.9 | 70.5 | 76.6 | 61.6 | ||
| N-Me-l-Thr | 3.52 | 4.51 | 1.41 | 2.76 | |||
| 172.0 | 70.5 | 73.0 | 19.6 | 33.9 |
Exact mass of glycosyl cation 398.109035.
NAc signals C-1 176.1; H/C-2 2.08/23.8 ppm. 31P at −0.5 ppm.
The linkage between N-methylthreonine and GlcNAc was determined from the 1H-31P HMQC spectrum which showed correlations between H-3 of GlcNAc and H-3 of the N-methylthreonine and the same 31P signal at −0.5 ppm. To summarize, NMR revealed that the 630 glycan is formed of a single GlcNAc linked to a phosphorylated N-methyl-l-threonine at the oxygen at C3 of the sugar (Fig. 2).
The type A modification genes are essential for motility in strain 630
Flagellin modification is essential to motility in many organisms which modify their flagellins including C. difficile, in which mutation of the GT believed to transfer the first sugar to the flagellin protein causes a loss of motility in 630Δerm (Twine et al., 2009). To determine whether the other putative type A modification genes are required for C. difficile motility, mutations in the 630 ORFs CD0241, CD0242, CD0243 and CD0244 were generated by Clostron mutagenesis in 630Δerm (Table 2 for all strains and plasmids). These mutants were assessed for motility by different methods as have been previously described (Twine et al., 2009; Baban et al., 2013), the most reproducible and quantifiable results were achieved on C. difficile minimal media (CDMM) motility plates. In all assays the ORFs CD0241, CD0242 and CD0244 were found to be essential for the motility of C. difficile 630Δerm, with the normal swimming phenotype of the parental strain being completely abolished in these mutants, this was also observed in mutations of fliC and the initial GT CD0240 as was previously described (Fig. 3A; Twine et al., 2009). The non-motile phenotypes were all restored on complementation in trans (Fig. 3A), although the phenotypes of the fliC, CD0241 and CD0244 mutants were not restored to wild type levels. The mutant of CD0243 formed smaller swarms than the parental strain; however this difference was not statistically significant (Fig. 3A).
Table 2.
Bacterial strains and plasmids
| Strain/plasmid | Genotype or description | Reference(s) or source |
|---|---|---|
| Strains | ||
| E. coli | ||
| Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 nupG recA1 araD139 Δ(ara-leu)7697 galE15 galK16 rpsL(Strr) endA1 λ− | Invitrogen |
| CA434 | E. coli HB101 [F− mcrB mrr hsdS20(rB− mB−) recA13 leuB6 ara-14 proA2 lacY1 galK2 xyl-5 mtl-1 rpsL20(Smr) glnV44 λ−] containing plasmid R702 | Williams et al. (1990) |
| C. difficile | ||
| 630Δerm | Derivative of 630 strain, erythromycin sensitive | Hussain et al. (2005) |
| 630Δerm-fliC | fliC::CT insertion mutant derived from 630Δerm | Twine et al. (2009) |
| 630Δerm-0240 | 0240 mutant derived from 630Δerm by clostron insertion | Twine et al. (2009) |
| M68Δerm | M68Δerm: Derived from M68 by deletion of ermB | Faulds-Pain and Wren (2013) |
| M68ΔfliC | M68ΔfliC: Derived from M68 by deletion of fliC | Faulds-Pain and Wren (2013) |
| 630Δerm-0241::CT | 0241 mutant derived from 630Δerm by clostron insertion | This study |
| 630Δerm-0242::CT | 0242 mutant derived from 630Δerm by clostron insertion | This study |
| 630Δerm-0243::CT | 0243 mutant derived from 630Δerm by clostron insertion | This study |
| 630Δerm-0244::CT | 0244 mutant derived from 630Δerm by clostron insertion | This study |
| M68Δerm-0242::CT | 0242 mutant derived from M68Δerm by clostron insertion | This study |
| M68Δerm-0243::CT | 0243 mutant derived from M68Δerm by clostron insertion | This study |
| 630Δerm-0241 ΔfliC | Double mutant fliC deletion mutant derived from 630Δerm-0241::CT | This study |
| 630Δerm-0242 ΔfliC | Double mutant fliC deletion mutant derived from 630Δerm-0242::CT | This study |
| 630Δerm-0244 ΔfliC | Double mutant fliC deletion mutant derived from 630Δerm-0244::CT | This study |
| Plasmids | ||
| pMTL84151 | E. coli – C. difficile shuttle plasmid (pCD6;catP; ColE1+tra) | Heap et al. (2009) |
| pfliC_comp | pMTL84151, with C. difficile 630 fliC and its native promoter | This study |
| pMTL84153 | E. coli – C. difficile shuttle plasmid (pCD6;catP; ColE1+tra; fdx promoter) | Heap et al. (2009) |
| p0241_comp | pMTL84153 with CD0241 cloned behind the fdx promoter | This study |
| p0242_comp | pMTL84153 with CD0242 cloned behind the fdx promoter | This study |
| p0244_comp | pMTL84153 with CD0244 cloned behind the fdx promoter | This study |
| pMTL007C-E2-CD0241 | Clostron plasmid retargeted to CD0241 at 231/232s (DNA 2.0) | This study |
| pMTL007C-E2-CD0242 | Clostron plasmid retargeted to CD0242 at 156/157s (DNA 2.0) | This study |
| pMTL007C-E2-CD0243 | Clostron plasmid retargeted to CD0243 at 309/310a (DNA 2.0) | This study |
| pMTL007C-E2-CD0244 | Clostron plasmid retargeted to CD0244 at 556/557a (DNA 2.0) | This study |
| pMTLAFPfliC | pMTL82151 with + /− 630fliC 1200 bp | Faulds-Pain and Wren (2013) |
Fig 3.
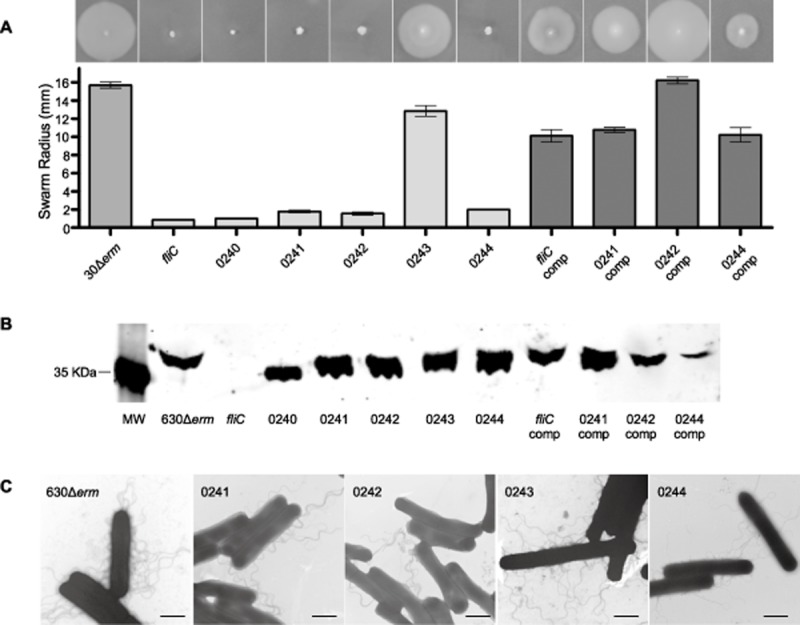
Motility and flagella production in 630 putative modification mutants.A. Motility assays of the 630Δerm parental strain compared to the fliC Clostron mutant and the mutations in the predicted modification genes were carried out in CDMM containing 0.3% Difco-bacto agar. The fliC mutant and the initial GT mutant were non-motile, as described previously, as were mutations in the ORFs CD0241, CD0242 and CD0244. These were all restored to motility by complementation.B. Flagellin from 630Δerm and the modification mutants were probed with an anti-FliC antibody by Western blot (MW = molecular weight, gene names and numbers below indicate mutation, comp = in trans complementation of mutant). All strains with the exception of the fliC mutant were found to produce flagellin, although the mass of the protein varied.C. The parental strain 630Δerm and the mutants of CD0241, CD0242, CD0243 and CD0244 were observed by TEM, the black scale bar in each image represents a length of 1 μm. All of the mutants appeared to have flagella associated with the surface of the cells.
It was previously observed by Twine and co-workers that despite being non-motile, the GT CD0240 mutant still produced polymerized flagella filaments, although they were truncated and fewer in number compared to the parental strain (Twine et al., 2009). To determine whether this was also true of the CD0241, CD0242, CD0243 and CD0244 putative flagellin modification mutants, we extracted the cell surface-associated proteins by glycine extraction and probed them with an anti-FliC antibody on a Western blot. All mutants were found to still produce flagellin, although its size varied (Fig. 3B). This is likely to be caused by alterations in the modifications leading to changes in the size of the protein.
Individual cells were visualized by negative stain transmission electron microscopy (TEM) by which the presence of flagella filaments associated with the surface of the cells in 630Δerm and the CD0241, CD0242, CD0243 and CD0244 (Fig. 3C). On further analysis of these electron micrographs it was observed that the flagella of the non-motile mutants of CD0241, CD0242 and CD0244 appeared to clump together, which was not the case for the motile parental strain, 630Δerm, or the motile mutant CD0243 whose flagella filaments were distributed evenly around the cell.
Mutations in the type A modification genes cause alterations of the PTM
Disruption of the putative GT gene, CD0240, was previously reported to result in an inability to attach glycan to the sites of modification on the flagellin (Twine et al., 2009). In order to characterize the role of the remaining type A modification genes, the flagellin modifications of the CD0241, CD0242, CD0243 and CD0244 mutants were examined by Mass Spectrometry. Flagellin was isolated from wild type and mutant strains by shearing or glycine extraction and proteins were separated by SDS-PAGE. Gel bands corresponding to flagellin were excised, digested with trypsin and analysed by nLC-MS/MS. The corresponding MS/MS spectra were sequenced de novo, and the identified glycopeptides are listed in Table 3. Tandem mass spectrometry analyses of flagellin tryptic digests from the CD0241 mutant showed glycopeptides to be modified with a single HexNAc moiety. No peptides from the CD0241 mutant harboured the wild-type 398 Da glycan. Flagellin isolated from the CD0242 mutant also only harboured glycopeptides modified with a single HexNAc moiety. In contrast, flagellin isolated from the CD0243 mutant showed variable glycan modifications. A number of glycopeptides were modified with a single HexNAc moiety (Fig. 4A), and a number of peptides modified with a 384 Da glycan (Fig. 4B). The MS/MS spectrum of this structure showed typical peptide fragmentation and a glycan oxonium ion at m/z 385. Glycan-related fragment ions were also observed at m/z 284, 186 and 168, identical to the fragmentation pattern of the wild-type sugar (Twine et al., 2009). In the high m/z region of the MS/MS spectrum, neutral losses of 98 and 203 were observed, suggesting the 384 Da sugar is closely related to the wild type 398 Da sugar. The sugar structures appear to differ by 14 mass units, which indicates absence of the methyl group in the sugar of the CD0243 mutant strain. De novo sequencing of MS/MS spectra of flagellin from the CD0244 mutant showed a mixture of glycopeptides, modified with either a single HexNAc moiety, or the 398 Da wild-type sugar.
Table 3.
Summary of glycan modifications of C. difficile 630 flagellin glycosylation mutants
| Strain/plasmid | Detected glycopeptides (precursor m/z) | Glycan masses and predicted composition (Da) |
|---|---|---|
| 630Δerm | Various peptides | 398 |
| CD0241 mutant | LLDGTSSTIR (633.32+) | 203 |
| TMVSSLDAALK (669.82+) | 203 | |
| CD0241 mutant p0241_comp | AGGTTGTDAAK (577.32+) | 203 |
| TMVSLDAALK (697.42+) | 203 | |
| TMVSLDAALK (775.32+) | 398 (203-80-115) | |
| LLDGTSSTIR (633.32+) | 203 | |
| LLDGTSSTIR (730.82+) | 398(203-80-115) | |
| CD0242 mutant | TMVSSLDAALK+ox (677.82+) | 203 |
| CD0242 mutant p0242_comp | TMVSSLDAALK (679.32+) | 203 |
| 384(203-80-101) | ||
| 398(203-80-115) | ||
| CD0243 mutant | LLDGTSSTIR (633.32+) | 203 |
| LLDGTSSTIR (732.82+) | 384(203-80-101) | |
| TMVSSLDAALK (679.32+) | 203 | |
| TMVSSLDAALK (724.22+) | 384(203-80-101) | |
| CD0244 mutant | TMVSSLDAALK (679.32+) | 203 |
| TMVSSLDAALK (775.52+) | 398(203-80-115) | |
| LLDGTSSTIR (730.82+) | 398(203-80-115) | |
| CD0244 mutant p0244_comp | LLDGTSSTIR (730.82+) | 398(203-80-115) |
| TMVSSLDAALK (775.52+) | 398(203-80-115) |
Fig 4.
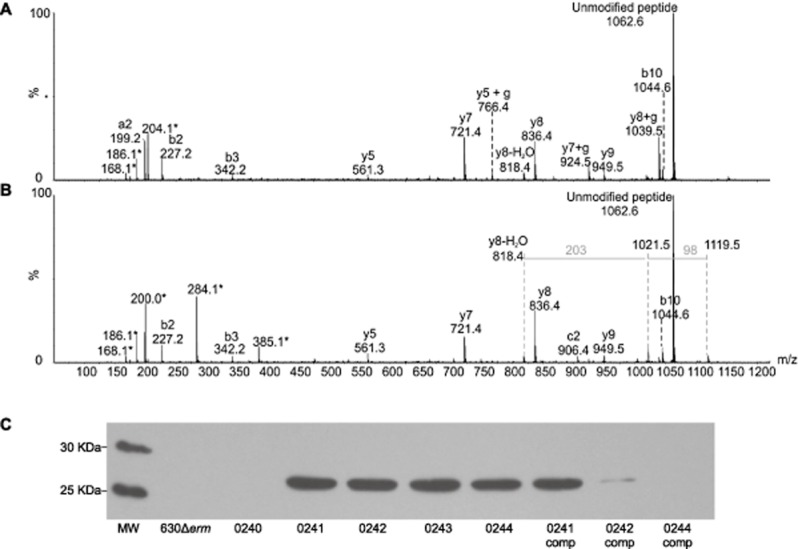
Mass spectrometry analyses of C. difficile flagellin tryptic glycopeptides from strain 630Δerm CD0243 mutant. Peptide MS/MS spectra were obtained from in gel digestion of protein bands from glycine extraction of whole cells.A. MS/MS spectrum of C. difficile flagellin glycopeptide LLDGTSSTIR. The unmodified peptide ion was observed at m/z 1062, with low intensity peptide type y and b ions confirming the peptide sequence. A moderately intense HexNAc oxonium ion was observed at m/z 204, with sequential dehydration giving rise to glycan related ions at m/z 186 and 168. Glycan related fragment ions are indicated with an asterisk.B. MS/MS spectrum of C. difficile flagellin glycopeptide LLDGTSSTIR, modified with a glycan of 384 Da. The MS/MS spectrum shows the typical peptide fragment ions and an additional, with a glycan oxonium ion at m/z 385. Glycan related ions were also observed at m/z 284, 186 and 168. In addition, in the higher m/z region of the spectrum neutral losses of 98 and 203 Da were observed. Together these data suggest the 384 Da glycan moiety is similar to the 398 Da wild type sugar, with the absence of a methyl group. C, Flagellin from 630Δerm and the modification mutants were analysed by Western blot, probing with an anti-β-O-GlcNAc antibody (MW = molecular weight, gene numbers below indicate mutation, comp = in trans complementation of mutant). With the exception of the CD0240 mutant, the flagellin of all mutants was found react with the anti-β-O-GlcNAc antibody, however the parental strain 630Δerm and the CD0244 complement did not.
In trans complementation of CD0241, CD0242 or CD0244 mutants either partially or fully restored modification of flagellin with the 398 Da wild type sugar (Table 3). For example, flagellin isolated from the complemented CD0241 mutant, showed glycopeptides modified with either HexNAc or the wild-type sugar. Flagellin isolated from the CD0242 complement harboured peptides modified with a 203 Da sugar, 384 Da sugar or the 398 Da wild-type sugar. Complementation of the CD0244 mutant appeared to fully restore wild type glycosylation.
Surface layer extracts of 630Δerm and the modification mutants were analysed by Western blot with an anti-β-O-GlcNAc antibody and confirmed the NMR structure that the initial sugar is a GlcNAc (Fig. 4C). Interestingly, there was no interaction of this antibody with the 630Δerm flagellin but it did bind to the flagellins of the CD0241, CD0242, CD0243 and CD0244 mutants, suggesting that the further modifications block the epitope for antibody binding to the GlcNAc residue in the parental strain glycan. As predicted no binding of the antibody to the flagellin of the CD0240 mutant, which cannot transfer glycan to the flagellin, was observed.
Alterations in the flagella PTM causes cell aggregation
When grown in liquid culture we observed that the CD0240, CD0241, CD0242 and CD0244 mutants all settle out of the suspension and formed sediment at the bottom of the growth vessel, while cultures of the parental strain 630Δerm and the CD0243 mutant remained in suspension (Fig. 5A). This phenotype was restored upon complementation in trans (Fig. 5A). As all mutants that formed sediment were also non-motile it was hypothesized that these two phenotypes could be related and that loss of motility led to sedimentation. However, when the non-motile aflagellate fliC mutant was grown in liquid it behaved as the motile parental strain and remained in suspension, suggesting that loss of motility alone cannot account for sedimentation of the modification mutants (Fig. 5A).
Fig 5.
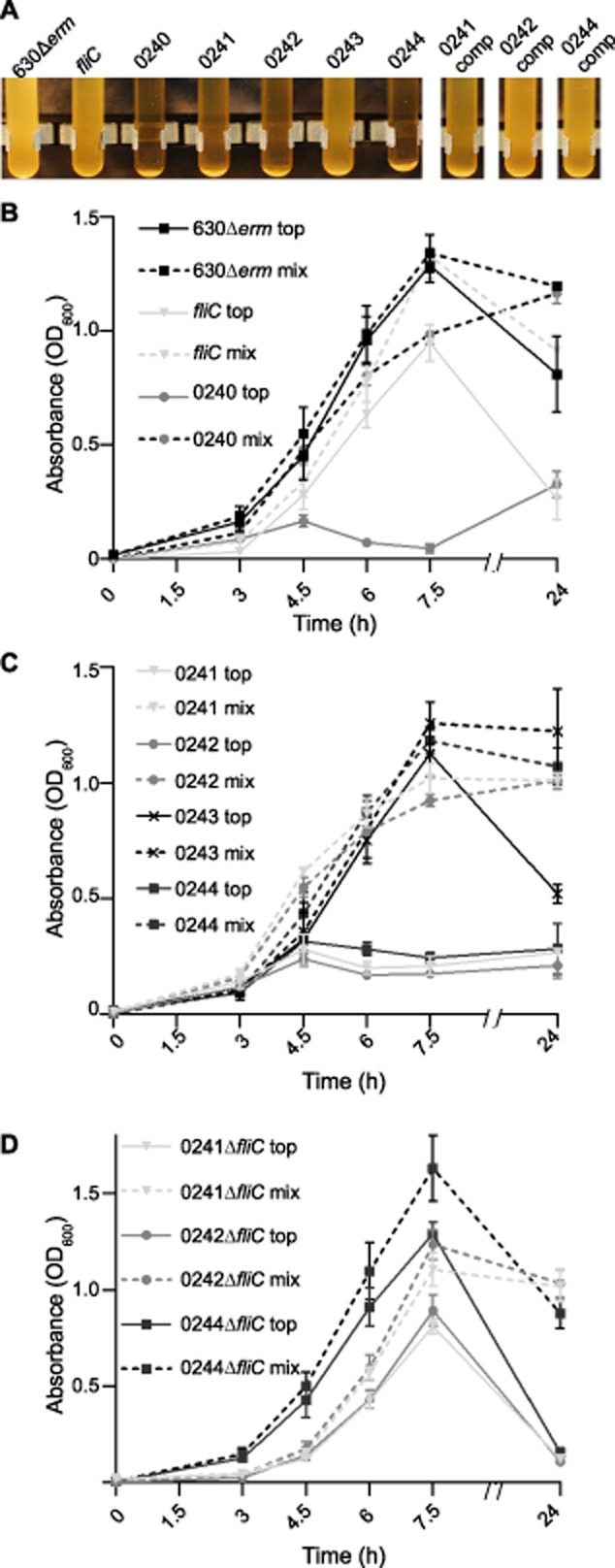
Sedimentation of the modification mutants.A. Mutations in the modification genes led cells to sediment in liquid culture with the exception of CD0243, this phenotype was restored by complementation in trans.B and C. This was quantified during growth by measuring the OD600 of the top 1 ml of culture (top – continuous lines) and a well mixed culture (mix – broken lines), demonstrating sedimentation in the CD0240, CD0241, CD0242 and CD0244 mutants, confirming the observations in A.D. The sedimentation in double mutants of CD0241 and fliC, CD0242 and fliC and CD0244 and fliC were also quantified. These double mutants had a growth profile similar to the fliC clostron mutant (B).
In order to quantify this phenotype an assay was developed which allowed us to determine whether the cells settled out during growth or at stationary phase and whether the growth rate was affected by this phenotype. For this we measured both the optical density (OD) of a well mixed culture (referred to here as a mixed culture) in order to assess the overall growth rate of the strains and the OD600 from the top 1 ml of an undisturbed culture (referred to here as an unmixed culture), which when compared to the OD600 of the mixed culture would indicate whether the cells were coming out of suspension. As the process of measuring these OD600s involves mixing and altering the overall volume of the cultures, it meant that two separate cultures were required for the mixed and unmixed measurements and that new cultures would be required for each time point tested to avoid distorting the results.
Throughout exponential growth of the parental strain 630Δerm the mixed and unmixed cultures had very similar OD600s to one another, indicating that during growth of this strain little sedimentation occurs (Fig. 5B). At 24 h however the OD600s of the unmixed cultures were lower than the mixed (OD600 of approximately 0.8 and 1.2 respectively), suggesting that at stationary phase cells begin to sediment out of solution (Fig. 5B). No difference was observed in the overall growth of the fliC mutant compared to 630Δerm. In the unmixed culture of the fliC mutant the OD600 increases between 3 and 7.5 h, however these OD600s are lower than those of both the fliC mixed cultures and the 630Δerm unmixed cultures, indicating that some sedimentation occurs in this strain during growth (Fig. 5B). By 24 h the OD600 of the unmixed culture fell to an OD600 of approximately 0.2, while the mixed culture was around 1.0, this difference in OD600 at this time point is much greater than was observed in 630Δerm.
In the modification gene mutants CD0241, CD0242 and CD0244, overall growth was not significantly different to 630Δerm or the fliC mutant, however the sedimentation phenotype was much more pronounced and no increase in OD600 was observed in the unmixed culture beyond an OD600 of approximately 0.2 throughout the exponential growth phase (Fig. 5C). In line with other phenotypic observations, the knock-out mutant of CD0243, which encodes the putative methyltransferase, did not display a phenotype which was significantly different to the parental strain (Fig. 5A and C).
While it appears that the loss of the flagellin results in some sedimentation, mutations in the modification genes cause a more extreme phenotype which is continuous throughout growth. We therefore hypothesized that this phenotype is due to the presence of a flagella filament lacking the full repertoire of modifications. To address this hypothesis we took advantage of the recent progress in genetic tools for C. difficile (Faulds-Pain and Wren, 2013) and generated in-frame deletions of the fliC gene by allele exchange in the CD0241, CD0242 and CD0244 clostron mutant genetic backgrounds. Destructive growth curves were carried out as before and from the OD600 of the unmixed cultures it was found that sedimentation did occur during the growth of all three double mutants (Fig. 5D), however, unlike the single modification gene mutants, which remained at an OD600 of approximately 0.2 in the unmixed cultures, the double mutants did increase during growth phase, peaking at OD600 of between 0.8 and 1.2 at 7.5 h and had a comparable profile to the fliC single mutant (Fig. 5B). This supports the hypothesis that the sedimentation which occurs in these modification mutants is chiefly a consequence of an incompletely modified flagella filament.
To determine whether these cells are interacting with one another or with components of the growth medium, the mutants were tested for autoagglutination. Cells were first grown on solid BHIS agar then transferred to PBS, all strains were then normalized by absorbance to a starting OD600 of 10, as described previously (Howard et al., 2009; Reynolds et al., 2011). Following incubation overnight, sedimentation was observed in the CD0240, CD0241, CD0242 and CD0244 mutants (Fig. 6A). The results were quantified by calculating the difference between the absorbance of the top 1 ml of the solution and the absorbance of the whole volume, autoagglutination was calculated as the percentage of cells that had sedimented (Fig. 6B). This was found to be between 10 and 20% in the parental strain, the fliC mutant and the CD0243 mutant but 86% in the CD0240 mutant and between 65% and 72% in the CD0241, CD0242 and CD0244 mutants.
Fig 6.
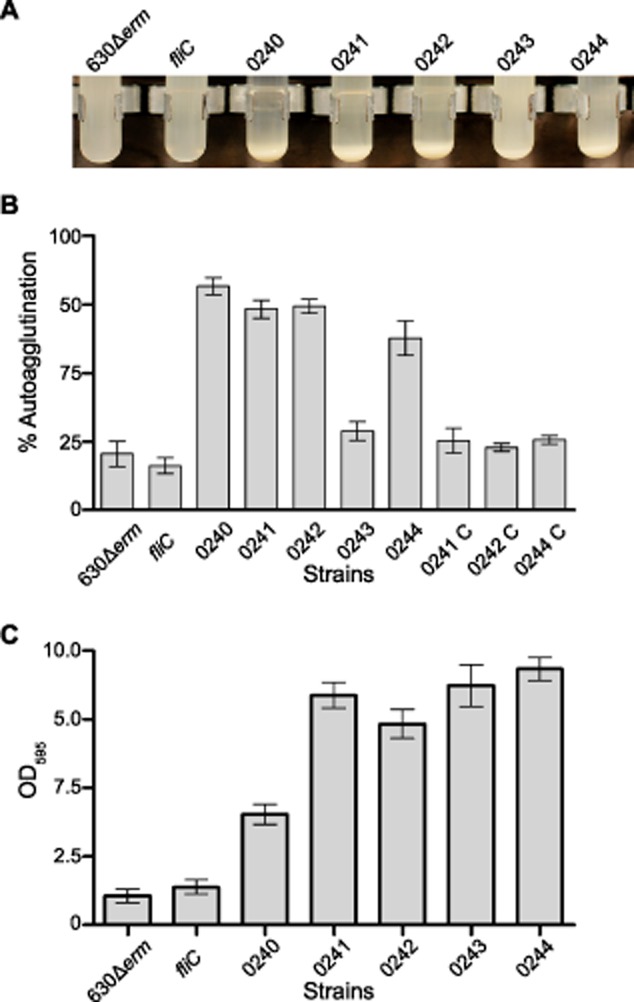
Agglutination of cells to one another and abiotic surfaces.A and B. The autoagglutination of the modification mutants was measured in the absence of media by growing them on solid media and transferring them to PBS. The total autoagglutination was calculated as the OD600 of the top 1 ml of cell solution compared to the total OD600 following 16 h incubation. Autoagglutination was found to be the highest in CD0240, CD0241, CD0242 and CD0244 mutants and could be restored on complementation, the fliC and CD0243 mutant were similar to the parental strain.C. The agglutination phenotype of the mutants was also investigated in their interaction with abiotic surfaces, strains were grown in 24-well plates for 5 days and the relative amount of cells bound was measured with crystal violet stain. All of the modification mutants showed a marked increase in binding to the surface of the wells compared to 630Δerm and the fliC mutant.
Alterations in the flagella PTMs leads to increased cell binding to abiotic surfaces
The autoagglutination observed in the CD0240, CD0241, CD0242 and CD0244 PTM mutants indicated that these cells had a ‘stickiness’ that caused them to group together and as a consequence settle out of solution. To determine whether this also made them more adherent to abiotic surfaces, C. difficile strains were grown in 24-well plates for 5 days and the comparative number of cells adhered to the surface of these plates were quantified by crystal violet staining (Fig. 6C; Dawson et al., 2012). All of the PTM mutations lead to an increased interaction with the surface of the plates; interestingly however this phenotype among the modification mutants was the least pronounced in the CD0240 mutant which had the strongest auto-aggregation phenotype. In contrast the CD0243 mutant, for which we previously observed no phenotypes, adhered to the abiotic surfaces as well as the CD0241, CD0242 and CD0244 mutants.
The role of PTM in the epidemic ribotype 017 strains
The configuration of the 630 type A modification locus was also identified in ribotype 017 strains, a distinct C. difficile lineage, members of which have caused a number of epidemics across Europe and Asia, but are toxin A negative (Drudy et al., 2007). To determine whether the properties associated with the PTM mutants in 630Δerm could be extrapolated to the orthologues in the ribotype 017 strains, mutants were made in the strain M68Δerm, a erythromycin-sensitive derivative of the ribotype 017 strain M68 isolated from an outbreak in Dublin (Drudy et al., 2007; Faulds-Pain and Wren, 2013). An in-frame deletion of the chromosomal ermB gene of M68, which we had previously constructed, makes this strain sensitive to macrolide, lincosamide, streptagramin (MLS) antibiotics which are typically required for mutagenesis by Clostron insertion (Faulds-Pain and Wren, 2013). The two genes M68-0242 and M68-0243 (orthologues of CD0242 and CD0243 respectively) were selected for mutagenesis as the former had a strong phenotype in 630Δerm and the latter a subtle phenotype. Motility of these mutants was assayed and compared to 630Δerm and M68Δerm and a fliC deletion mutant of M68, described previously (Faulds-Pain and Wren, 2013). As in the 630Δerm background, the M68-0242 mutant caused a complete loss of motility which was restored on complementation, while the M68-0243 mutant was slightly less motile than the parental strain (Fig. 7A and B).
Fig 7.
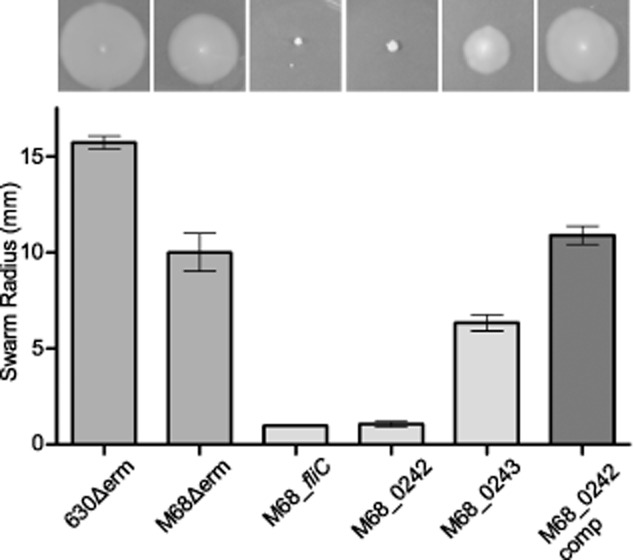
Motility in M68 putative modification mutants. Motility assays of both the 630Δerm and M68Δerm parental strains compared to the M68ΔfliC in-frame deletion mutant and the mutations in the predicted modification genes M68_CD0242 and M68_CD0243 were carried out in CDMM containing 0.3% Difco-bacto agar. M68Δerm was motile and the M68_fliC mutant non-motile, as described previously. A mutant of the M68_0242 gene was non-motile, this was restored upon complementation. The M68_0243 mutant was motile, with a slightly reduced motility compared to the parent.
Flagellin modification is essential for efficient colonization and recurrence in the murine model of infection
In a murine model, C. difficile can cause a self-limiting, chronic intestinal infection by which colonization and relapse can be studied (Chen et al., 2008; Lawley et al., 2009). We used this model to assess the role of the flagellin modifications in the establishment and recurrence of C. difficile infection. In order to clear the natural intestinal microflora and allow C. difficile to establish an infection, clindamycin was administered to 8 week-old C57BL/6 mice 5 days prior to challenge with C. difficile spores derived from either 630Δerm or the isogenic fliC, CD0241 or CD0243 mutants. Shedding of C. difficile in the faeces was used as an indirect measure of host colonization and was determined by plating emulsified faecal pellets and enumerating the colony forming units (cfu) per gram of faeces. In this model, colonization is transient and detection of bacteria in the faeces is difficult 14 days post infection. However, as low numbers persist, a second dose of clindamycin administered approximately 28 days post infection stimulates outgrowth and shedding, indicating relapse of infection, can be monitored.
Mice challenged with the parental strain 630Δerm shed the bacteria at high levels from day 1 until day 4 post challenge (approximately 5 × 106 cfu g−1 faeces), by day 7 the C. difficile load in the faeces had begun to fall (c. 1 × 104 cfu g−1 faeces) and by day 14 the load of C. difficile in the faeces was below the limit of detection (Fig. 8A). Recurrence of C. difficile shedding occurred 3 days after the administration of a second dose of clindamycin (experiment day 31). Shedding peaked after 4 days (day 32) (approximately 4.6 × 107 cfu g−1 faeces) and remained high at 7 days (day 35), after 14 days (day 42) levels had dropped almost to the level of detection of the experiment.
Fig 8.
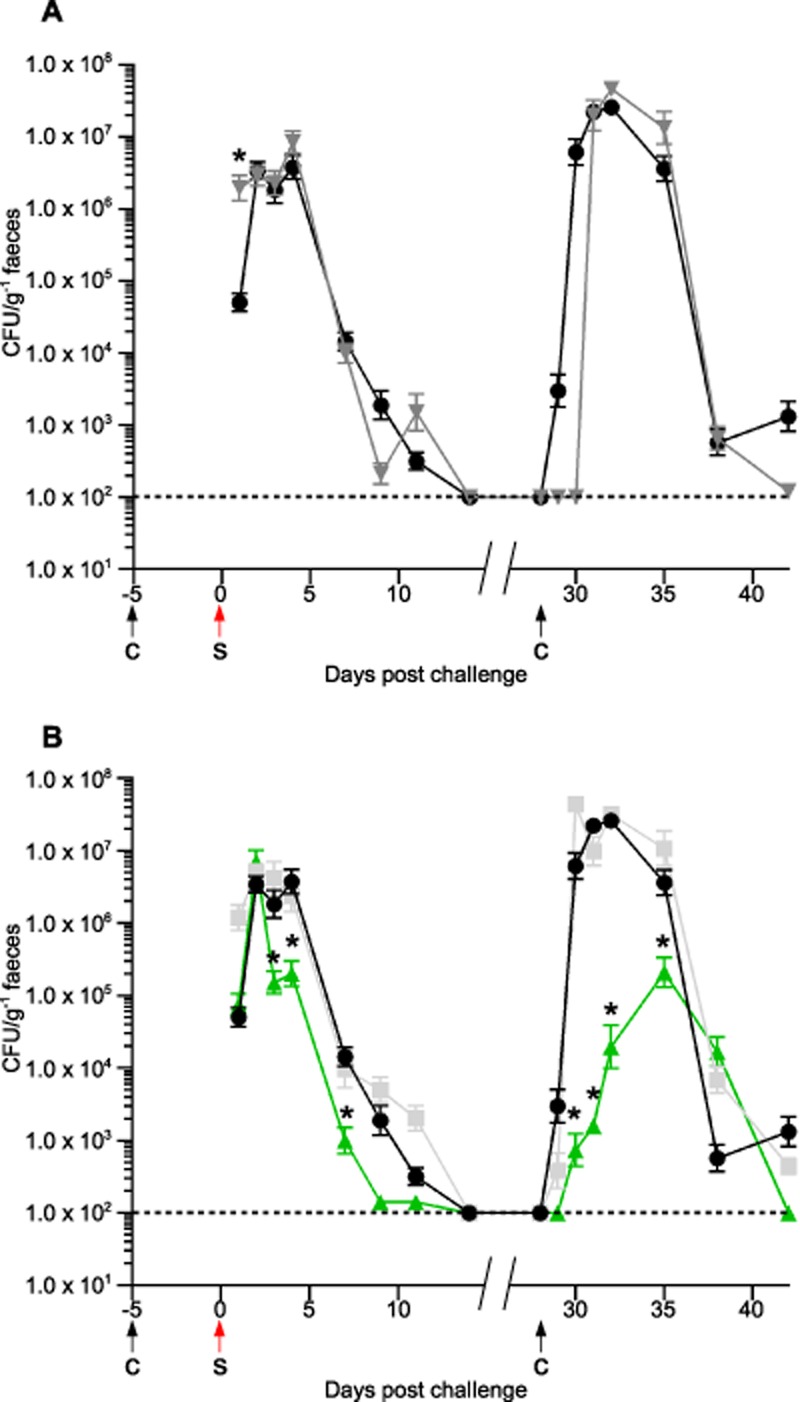
Flagellin modification is essential in the colonization and recurrence of C. difficile infection in C57BL/6 mice. Mice were challenged with C. difficile spores (S) 5 days following administration of clindamycin (C). Levels of C. difficile were measured by plating faecal samples and determining the C. difficile cfu g−1.A. Mice were challenged with the parental strain 630Δerm (inverted grey triangles) and the non-motile fliC Clostron mutant (black circles). Initial colonization is similar in these strains but relapse is delayed in 630Δerm by 2 days following the second dose of clindamycin.B. The initial colonization and relapse of the fliC mutant (black circles) were compared to the CD0241 and CD0243 modification mutants (green triangles and light grey squares respectively). While CD0243 has a similar initial colonization and recurrence rate to the fliC mutant, the CD0241 mutant is defective in both initial colonization and recurrence. Asterisks indicate a significant difference compared to the fliC mutant.
In these experiments mice challenged with the fliC mutant both the initial infection and the relapse of infection had a similar profile to the parental strain (Fig. 8A). The most significant difference between these two strains was that following the second dose of clindamycin C. difficile shedding in the faeces recurred after 1 day in the mutant (approximately 3 × 103 cfu g−1 faeces) compared to 3 days in the parental strain. After 4 days (experiment day 32) shedding of the fliC mutant peaked (approximately 2.6 × 107 cfu g−1 faeces) and remained high at 7 days (day 35), as was observed in the parental strain. At 14 days (day 42) C. difficile was still detectable in the faeces but in low numbers (approximately 1.2 × 103 cfu g−1 faeces). The shedding profile of mice challenged with the CD0243 mutant was not significantly different to strain the fliC mutant at any point post challenge or post second clindamycin treatment (Fig. 8B). The delay in re-colonization following the second dose of clindamycin observed in the parental strain did not occur in any of the mutants tested. As the Clostron mutants are marked with an ermB gene, which confers resistance to clindamycin [minimum inhibitory concentration (MIC) fliC = 16 μg ml−1, CD0241 = 8 μg ml−1 and CD0243 = 4 μg ml−1], while the parental strain, 630Δerm is sensitive to this class of antibiotics (MIC = 0.25 μg ml−1), it is likely that the clindamycin in the mice inhibits the recurrence of 630Δerm but not the mutants until the residual antibiotic is cleared.
Mice infected with spores of the CD0241 mutant reached a similar level of shedding as the parental strain by day 2 post challenge; however, while the parental strain and the fliC and CD0243 mutants continued to shed at high levels until day 4, shedding of the CD0241 mutant fell on day 3 and remained significantly lower than the fliC mutant at day 4 (P = 0.04; Fig. 8B). This trend continued at day 7 and at both days 9 and 11 the number of C. difficile cfu in the faeces was almost at the limit of detection. Following the second dose of clindamycin, recurrence of infection by the CD0241 mutant did occur, however the shedding of this mutant in the faeces was significantly less than the fliC mutant at all time points tested up to 10 days later (day 38) (P = 0.0011).
Weight change was monitored in the mice as another marker of infection. The animals were weighed before challenge and at 2 days post challenge with all four strains. Those infected with 630Δerm, the fliC mutant and the CD0243 mutant all lost weight in this time (between 2.3% and 6.3% of total body weight) compared to those challenged with the CD0241 mutant (gained 5% body weight). The difference in the change of weight in animals challenged with this strain was significantly different than 630Δerm (P = 0.002), the fliC mutant (P = 0.008) and the CD0243 (P = 0.01) mutant. It was also observed that the mice challenged with either 630Δerm, the fliC mutant and the CD0243 mutant produced sticky faeces in their cages, a sign of significant infection, this was not observed in those animals challenged with the CD0241 mutant.
In this experiment colonization of C57BL/6 mice and disease recurrence does not appear to be reliant on bacterial motility or the presence of the flagellin in C. difficile strain 630Δerm; however, disruption of the flagellin modification leads to attenuation in both colonization and relapse.
Discussion
Treatment of C. difficile disease is extremely difficult and relapse rates are high. Much work has been dedicated to the study of C. difficile toxins as they are important in disease and to sporulation and the spread of infection. However, little is known about the organisms' ability to interact with and colonize the intestinal epithelia of the host, a trait which may be vital in disease recurrence. In this study, we defined the structure of the type A flagellin post-translational modification by NMR and found that the resultant change altered the extent of colonization and outgrowth in the murine relapse model of infection.
Of the known flagellin modification structures only some P. aeruginosa strains produce a structure composed of a sugar plus a phosphate and a methylated amino acid, similar to the C. difficile type A flagellin modification (Verma et al., 2006). Interestingly, as with C. difficile, the P. aeruginosa flagellins are also subject to strain-specific variation of the PTMs, some being modified as the C. difficile type A strain such as strain PA01 and others, such as the strain PAK being modified with a larger glycan composed of a number of monosaccharide moieties (Verma et al., 2006). The precise role of this modification in P. aeruginosa has not been determined; however, flagella are known to be essential in the establishment of acute infections by P. aeruginosa (Cobb et al., 2004). The C. difficile type A flagellin modification was previously studied by LC-MS/MS and the initial structure was assigned as a HexNAc sugar with a methylated aspartic acid attached via a phosphate bond (Twine et al., 2009). On further analysis however it was realized that this amino acid was mis-identified as an aspartic acid, the mass of the moiety attached to the phosphate group being 115 Da is too small for an aspartic acid plus a methyl group. In this study, to definitively determine the structure of this modification a large-scale extraction of C. difficile 630 flagellin was completed. The flagellin protein was extensively digested with proteinase K and following purification the glycan structure was solved by NMR. The structural configuration of the HexNAc linking sugar was confirmed to be β-O linked GlcNAc and the remaining modification was linked via phosphate bond and determined to be an N-methylthreonine residue in the l-configuration.
Oligosaccharide flagellin modifications are commonly formed of a series of sugars, predominantly pseudaminic acid or its derivative legionaminic acid (Asakura et al., 2010; Parker et al., 2012; Sun et al., 2013). Specialist enzymes are required for the biosynthesis of these sugars as they are not synthesized by the cell for any other purpose (Parker et al., 2012; Sun et al., 2013). This is not the case for the C. difficile type A flagellin modification, as GlcNAc sugars are common precursors for many biosynthetic pathways, therefore no specialized biosynthesis genes are required. This is reflected in its modification genes (Fig. 1), which are predicted to be involved in the transfer of the modifications rather than the synthesis of precursors.
By generating mutations of the C. difficile type A modification genes and studying the resulting structures on the flagella filaments by LC-MS/MS, we were able to define more specifically the role each gene plays in the resulting modification. Flagellins of the CD0241 and CD0242 mutants are modified with the GlcNAc sugar only and both lack the phosphate group and the N-methylthreonine residue (Fig. 2). This indicates that the addition of the phosphate and the threonine to the GlcNAc is not sequential, but rather that they are added to the structure together. These genes encode proteins which have similarity with phosphoserine phosphatases and phosphocholine cytidylyltransferases respectively. Phosphoserine phosphatases catalyse the reversible reaction of serine and phosphate to phosphoserine and H2O, and it seems likely that CD0241 could encode a kinase which catalyses the addition of phosphate to threonine (Fig. 9). This is supported by a DXDX(T/V) motif encoded at the N-terminus of CD0241, this domain functions as an intermediate phosphoryl acceptor, indicative of a phosphatase (Osman et al., 2010). Phosphocholine cytidylyltransferases catalyse the reversible reactions of CTP plus choline phosphate to CDP-choline and diphosphate. As there is no evidence of phosphocholine in the flagellin modification, we hypothesize that the enzyme encoded by CD0242 catalyses the addition of the phosphothreonine to the GlcNAc residue (Fig. 9).
Fig 9.
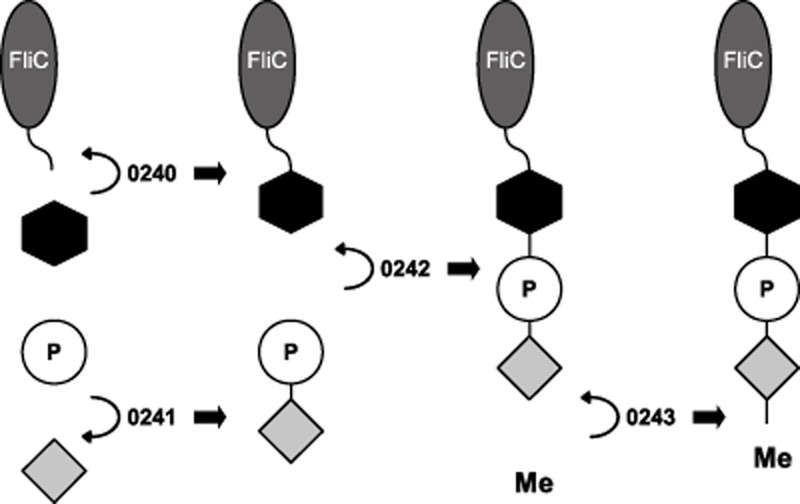
Schematic diagram illustrates the structure of the type A modification and the predicted contribution of the enzymes to the synthesis of the glycan. The FliC protein is modified at seven serine and threonine residues (grey oval represents a FliC monomer with the curved line representing either a serine or threonine amino acid within the FliC protein). To these residues a GlcNAc (black hexagon) is transferred by the GT encoded by CD0240. A phosphate group (white circle) is attached to a threonine (light grey diamond) by the enzyme encoded by CD0241 and this phosphorylated threonine is attached to the FliC-GlcNac by the enzyme encoded by CD0242. The methyltransferase encoded by CD0243 adds a methyl group to the threonine.
LC-MS/MS of the putative methyltransferase knockout (CD0243) identified a mixed population of glycan structures on flagellar peptides; either the GlcNAc residue alone or the extended structure where the threonine was no longer methylated. The former population indicates that the function of this enzyme is to transfer a methyl group to the threonine. This is represented as the final step in the formation of the glycan in Fig. 9, however from our data we cannot discern at what stage methylation occurs, whether it is when the threonine is in complex with the glycan, when it phosphorylated or when it is free threonine. The presence of some residues which are modified with GlcNAc alone is less easy to interpret; but it may indicate that when the methyl group is absent the modification is less stable. It is also unclear from our data what the function of the fourth gene in the locus, CD0244, might be. The LC-MS/MS analysis identifies a mixed population of both the wild-type structure and the GlcNAc alone; however the phenotypic characteristics of this mutant are the same as the CD0241 and CD0242 mutants. This predicted protein has some similarity with glycerophosphotransferase enzymes; consequently, we would predict that it is involved in the transfer of the phosphate group to either the threonine or the GlcNAc. This mixed population may indicate that this enzyme is partially redundant. Interestingly, the similar flagellin PTM cluster in P. aeruginosa PA01 strains contains only orthologues of CD0241, CD0242 and CD0243, with CD0244 absent, supporting this redundancy hypothesis. This protein may however have a function not identified in this current study.
Previously, the CD0240 mutant, predicted to encode the initial glycosyltransferase enzyme, was characterized and found to be non-motile (Twine et al., 2009). Unusually for strains with a mutation that prevents flagellin modification, this knock-out was still able to both secrete flagellin and form a filament. In this study we found that the CD0241, CD0242 and CD0244 mutants likewise produce flagella and are non-motile, but the mutant of CD0243 retained motility similar to the parental strain. Loss of motility in flagellin PTM mutants in most organisms can be explained by the absence of a flagella filament, this is not the case for the C. difficile PTM mutants suggesting that other properties of these unmodified or partially modified filaments are interfering with motility. We noted another property of these mutants was their tendency to aggregate to each other and to surfaces and consequently to sediment when grown in broth. This phenotype was independent of motility and was dependent on the presence of an unmodified or incompletely modified flagella filament. One possible explanation is that an alteration in the flagellin modifications affects the polarity of the cell surface, causing the aggregation of the cells which prevents them from swimming efficiently. An alternative hypothesis is that the alteration in the charge on the flagellins is causing deleterious changes in the regularity of filament pitch height and length and consequently affecting the organisms' ability to swim efficiently.
The type A modification gene locus identified in 630 has also been identified in the RT017 epidemic lineage of C. difficile. We found that the phenotypes associated with the type A modification mutants in 630Δerm were also associated with mutants of their orthologues in the RT017 strain M68Δerm.
Little is known about the advantages flagellin PTMs provide a bacterium, although they have been implicated in host colonization and virulence in a number of organisms (Balonova et al., 2012; Iwashkiw et al., 2012; Lithgow et al., 2014). Flagellin is a hugely abundant protein, prominently displayed on the surface of the cell and PTMs are localized to its externally exposed domains, indicating that their function may lie in the interaction with the environment. C57BL/6 mice have been shown to be a useful model for the study of C. difficile colonization and recurrence as they do not succumb to the overwhelming effects of the toxins during infection (Chen et al., 2008; Lawley et al., 2009). To investigate the role of flagellin, motility and PTMs in the infection and relapse, these mice were challenged with the parent strain, 630Δerm, and the fliC, CD0241 and CD0243 mutants. We found that the non-motile fliC mutant and the motile CD0243 mutant had the same profile of faecal C. difficile loads as 630Δerm during both the initial infection and relapse, although the relapse of clindamycin sensitive 630Δerm was delayed by 2 days compared to the mutants. The Clostron mutagenesis method used to generate our mutants requires an MLS antibiotic sensitive parent strain, in this case 630Δerm, and results in mutants marked with an ermB gene which confers MLS antibiotic resistance. As clindamycin is a MLS antibiotic and in this experiment it is used to trigger relapse, it is likely that the delay in 630Δerm recurrence is due to its sensitivity to clindamycin and the ability of the mutants to immediately re-grow is due to their resistance. During the initial infection the non-motile CD0241 mutant reached the same peak of bacterial load in the faeces as the other three strains however, this decreased much sooner and relapse of infection was slower, peaked later and reached a lower bacterial load in the faeces. These mice also avoided other symptoms of severe infection such as weight loss and sticky faeces.
We observed that neither flagella nor motility appear to be required for the efficient colonization of C. difficile in the mouse intestine, as had been described previously (Baban et al., 2013). The complete modification of the flagellin however does appear to be important in colonization and relapse. It is not known whether the CD0241 mutant aggregates in vivo as it does in vitro but it is possible that this phenotype is affecting the fitness of the bacteria or the hosts' ability to clear it from the intestine. Potentially the alteration in the modification structure could affect the interaction of this highly abundant protein with the hosts' immune system.
Experimental procedures
Bacterial strains and growth conditions
Escherichia coli Top10 (Invitrogen) and E. coli CA434 were grown at 37°C in Luria–Bertani (LB) growth media supplemented with chloramphenicol (12.5 μg ml−1) and CA434 with Kanamycin (50 μg ml−1) where appropriate. C. difficile was cultured at 37°C in an anaerobic work station (Don Whitley, Yorkshire, UK) and grown routinely in BHIS medium (Brain heart infusion medium supplemented with 5 mg ml−1 yeast extract and 0.1% w/v l-cysteine). C. difficile was activated from glycerol stocks on Braziers agar [CCEY (Bioconnections), 4% w/v egg yolk and 1% w/v defibrinated horse blood]. C. difficile was supplemented with d-cycloserine (250 μg ml−1), cefoxitin (8 μg ml−1), thiamphenicol (15 μg ml−1) and erythromycin (10 μg ml−1) where appropriate. Plasmids were transferred to C. difficile by conjugation from E. coli CA434 as described previously (Heap et al., 2007; Cartman and Minton, 2010).
C. difficile minimal media (CDMM) (Cartman and Minton, 2010) containing 0.3% bacto-agar (DIFCO) was used to assay motility of C. difficile strains, plates containing 35 ml media were inoculated with a single colony using a sterile toothpick and incubated for 3 days. Images were captured using a Canon 600D SLR camera. Minimum inhibitory concentration (MIC) assays were performed as described previously (Andrews, 2001).
General molecular biology techniques
Plasmids were extracted using a plasmid mini kit (Qiagen) and the genomic DNA with the DNeasy Blood and tissue kit (Qiagen) following treatment with lysozyme (10 mg ml−1 in phosphate-buffered saline at 37°C for 30 min), then SDS (10% w/v) at 65°C for 30 min (Cartman and Minton, 2010) or Chelex extraction [cell pellets vortexed in 5% chelex (Sigma) boiled for 10 min, pelleted and the supernatants removed and used] or by phenol-chloroform purification as described previously (Cartman and Minton, 2010). DNA was amplified for cloning using Phusion high fidelity polymerase (NEB), and for screening using Go-taq polymerase (Promega), both in accordance with the manufacturers' instructions. DNA was extracted from PCR reactions and agarose gels using the QIAquick PCR and gel extraction kits (Qiagen) respectively. Allele exchange and complementation plasmids were constructed by restriction/ligation cloning using restriction endonucleases, Antarctic phosphatase and T4 ligase (NEB). Clostron plasmids were designed at http://www.clostron.com and synthesized and cloned into pMTL007C-E2 by DNA2.0 (Heap et al., 2010).
Southern blot analysis was done using an intron specific probe. One microgram of genomic DNA was digested overnight with HindIII restriction enzyme (Promega). AlkPhosDirect™ Labelling and detection kit (GE Healthcare) and detection reagents, in accordance with the manufacturer's guidelines and visualized using CDP star (GE Healthcare).
Mutagenesis
C. difficile was mutated using the Clostron mutagenesis system as described previously (Heap et al., 2007; 2010,). Briefly, gene-specific re-targeted Clostron plasmids (Table 2) were transformed in to C. difficile by conjugation and transconjugants selected for with thiamphenicol, mutants were then positively selected with erythromycin and confirmed by PCR and Southern blot. Double mutants were made in C. difficile by allele exchange using the method described previously (Faulds-Pain and Wren, 2013). Briefly, the plasmid pAFP91 (Faulds-Pain and Wren, 2013) was transformed in to the C. difficile 630Δerm Clostron mutants of CD0241, CD0242 and CD0244 by conjugation and transconjugants selected for with thiamphenicol, a series of replica plating on thiamphenicol allowed single crossover integrants to be isolated, double crossovers were then isolated by replica plating without selection and identified by their inability to grow on thiamphenicol. Mutant construction was verified by PCR.
Construction of complementation vectors
The fliC complementation vector contained the genes own promoter and terminator, the primers amplified from the end of the TAA stop sequence of the gene immediately upstream of the fliC and 196 bp downstream of the fliC stop site. The PCR product was cloned into the C. difficile complementation vector pMTL84151 (Heap et al., 2009; Table S1 for primers and restriction sites). The CD0241 and CD0242 ORFs only were amplified and cloned into the complementation vector pMTL84153 (as the pMTL84151 vector but with an fdx promoter/RBS and terminator) at the NdeI site, immediately after the RBS in the vector and utilizes the ATG in its restriction sequence. The CD0244 ORF already contains an NdeI restriction site within its sequence therefore to utilize this restriction site as with the other two ORFs, the internal NdeI site was first removed by making a non-coding change in the sequence by SOE-PCR. All constructs were confirmed by restriction analysis and Sanger sequencing (SourceBioscience). In these three vectors expression of the complement genes are driven by the non-native constitutive promoter from the fdx gene of Clostridium pasteurianum.
Sedimentation growth curves
Sixty millilitres of BHIS broth was inoculated with a 1:100 dilution of an overnight culture, the inoculated broth was split into 5 ml aliquots which were each allowed to grow statically. At specific time points the top 1 ml of a 5 ml culture was carefully removed from one replicate, while the second replicate was mixed by vortexing and 1 ml of this mixed culture was removed, the OD600 of both were measured. These cultures were then disposed of and the subsequent OD600 reading taken from another pair of 5 ml cultures. Assays were repeated in triplicate. Statistical analysis was done in GraphPad Prism by two-way anova and groups compared using a Tukey's multiple-comparisons test.
Autoagglutination assay
Strains of C. difficile grown overnight on BHIS agar were resuspended in PBS and normalized to an OD600 of 10 in a volume of 5 ml. Following incubation for 16 h at 37°C in the anaerobic cabinet the top 1 ml of cells was removed and the OD600 measured and the OD600 of the whole solution measured. Autoagglutination was calculated as the difference between the OD600 of the whole solution and the OD600 of the top 1 ml as a percentage of the whole solution OD. Assays were repeated in triplicate.
Adherence to an abiotic surface
Overnight cultures of C. difficile were used to inoculate 2 ml of BHIS in high bind 24-well plates at a 1:100 dilution. The cultures were grown for 6 days after which the supernatant was carefully removed and the wells washed with PBS. 1% crystal violet was added to the wells and incubated at room temperature for 20 min, the wells were then washed twice with PBS and the remaining crystal violet was detached from the cells attached to the surface of the wells with methanol. The OD595 was measured in a Spectrophotometer (Biotek, UK). Assays were repeated in triplicate.
Transmission electron microscopy
Cultures of C. difficile were inoculated 1:100 from overnight culture and allowed to grow statically for the 6 h. An equal volume of fixative (2.5% paraformaldehide, 2.5% glutaraldehide, 0.1 M Na cacodylate at pH 7.4) was added to the cultures. Fixed samples were diluted in MilliQ water at a 1:6 dilution and 10 μl spotted on to a 400 mesh copper grid with a pioloform support film and incubated for 1 min. Excess liquid was then removed by pipetting and 10 μl of 0.3% phosphotungstic acid (PTA) added in order to stain the sample and incubated for 1 min. The excess PTA was drawn off with filter paper and the grid air-dried before examining on a Jeol JEM-1200EX II Transmission Electron Microscope fitted with a 2K side mounted AMT (Advanced Microscopy Techniques) CCD digital camera supplied by Debens UK Ltd (http://www.debens.co.uk), used to record the digital images.
Low-pH glycine extractions
Cells from 18 h overnight cultures of C. difficile strains were harvested, washed with phosphate buffered saline and resuspended in a 1:100 volume of low-pH glycine (0.2 M glycine-HCl, pH 2.2) and incubated at room temperature for 20 min with gentle shaking. The cells were removed by centrifugation at 4°C and the supernatant neutralized with the addition of 2M Tris to a pH of 7 to 8.
Purification of flagellar glycan for NMR analysis
Flagellar glycoprotein sample was digested with a large excess of proteinase K in 0.01 M TRIS-HCl buffer at pH 8 at 37°C for 48 h. The products of digestion or free oligosaccharides were separated on Bio-Gel P6 column (2.5 × 60 cm) and each fraction which eluted before the salt peak was dried and analysed by 1H NMR. Fractions containing sugars were separated by anion exchange chromatography on Hitrap Q column (5 ml size, Amersham) and the glycans eluted with a linear gradient of NaCl (0–1 M, 1 h). Desalting was performed on Sephadex G15 prior to analysis by NMR.
NMR spectroscopy analysis
NMR experiments were carried out on a Varian INOVA 500 MHz (1H) spectrometer with 3 mm gradient probe at 25°C with acetone internal reference (2.225 ppm for 1H and 31.45 ppm for 13C) using standard pulse sequences DQCOSY, TOCSY (mixing time 120 ms), ROESY (mixing time 500 ms), HSQC and HMBC (100 ms long range transfer delay). AQ time was kept at 0.8–1 s for H-H correlations and 0.25 s for HSQC, 256 increments was acquired for t1. For 1H-31P HMQC H-P coupling was set to 10 Hz.
Absolute configuration of N-methylthreonine
Glycopeptides were dephosphorylated with 48% HF (10 μl ml−1, 30 min, 30°C), dried by air stream, (R)-2-octanol (0.2 ml) and acetyl chloride (0.02 ml) were added at room temperature, heated at 100°C for 2 h, dried by air stream, acetylated (0.2 ml Ac2O–0.2 ml pyridine, 100°C, 30 min), dried, and analysed by GC-MS as described above. A standard was prepared from N-methyl-l-Thr (Sigma) by the same procedure without the dephosphorylation step with (R)- or (RS)-2-octanol. Analysis was performed on Agilent 6890 GC instrument with DB17 column at 160–280°C by 4°C min−1.
In-gel digestion of flagellin proteins for mass spectrum analysis
Flagellin gel bands were excised and destained using 25% ethanol: 10% acetic acid. The gel bands were subsequently dehydrated with the addition of acetonitrile. Fully dehydrated gel bands were air dried under a laminar flow hood and then digested over night with 20–50 μl of 20 ng μl−1 sequencing grade modified trypsin in 50 mM Ammonium bicarbonate. Digests were incubated at 37°C overnight. Peptides were extracted into fresh microfuge tubes and stored at −20°C.
Mass spectral identification and de novo sequencing of in-gel digested flagellins
Protein digests were analysed using a QTOF Ultima coupled to a nanoAcquity UPLC system. Briefly, 10 μl of each digested was injected onto a 180 μm ID × 20 mm, 5 μm symmetry C18 trap column in trapping mode, diverting flow through to waste. Peptides were eluted by reversed phase liquid chromatography (RPLC) to a 100 μm ID × 10 cm, 1.7 μm BEH130 C18 column (Waters) in analytical mode, using a linear gradient from 1% to 45% solvent B in 18 min, 45% to 85% solvent B for 2 min, 85% to 1% solvent B over 1 min, and hold for 8 min at 1% solvent B. Tandem mass spectrometry analyses were performed in data-dependent acquisition (DDA) mode. Peaklist files were generated and searched against the C. difficile 630 translated genome sequence, using MASCOT (Matrix Science) to identify unmodified peptides. The mass tolerance for precursor and fragment ions was 0.8 Da and an ion score of 30 or above indicated identity. In-house software ‘Glycan Hunter’ performed peak listing of MS/MS spectra of unmatched spectra that featured key glycan related ions, using information from previously published studies (Twine et al., 2009). Glycopeptides were identified by manual inspection of the MS/MS data, and de novo sequencing of peptide related ions.
Western blot
The flagellin was isolated by low-pH glycine extractions in all cases. For the anti-FliC Western blots the low-pH glycine extraction was carried out as described above. Samples were normalized according to the OD600 of the cultures and a total volume of 15 μl was run on a Nu-Page 12% Bis-Tris SDS-PAGE gel in MOPS running buffer (both Life Technologies). Proteins were transferred to an H + membrane in a semi-dry blotter in transfer buffer (0.24% Tris, 1.14% glycine and 20% Methanol). The membrane was blocked in PBS plus 2% milk (PBS-M) and probed with an anti-FliC antibody (a generous gift from the Armstrong lab) at a 1 in 10 000 dilution for 1 h at room temperature. Following standard membrane washing with PBS-T (PBS plus 0.1% Tween 20) a Li-cor Donkey anti-chicken IR Dye 680RD secondary antibody (Li-cor) was added at a 1: 5000 in PBS-M plus 0.1% Tween 20 and 0.01% SDS and incubated in the dark for 1 h. The membrane was washed as before and visualized using a Li-cor Odyssey.
For the anti-GlcNAc Western blots, low-pH glycine extraction was achieved by scraping overnight growth of C. difficile from plates and incubating the cells with low-pH glycine buffer. Samples were standardized according to the OD600 of the resuspended cells and protein separated on 12% Tris Glycine gels, and transferred to a PVDF membrane. The membrane was blocked with 5% milk-PBS-T then probed with 1 in 5000 dilution of anti-β-O-GlcNAc (Covance, Montreal, Canada) in PBS-T for 45 min at room temperature. After washing with PBS-T, reactivity was detected with anti-mouse IgM HRP conjugate (Sigma Aldrich, Oakville, Canada) secondary antibody at 1 in 10 000 dilution in PBS-T for 45 min at room temperature. Blots were imaged with ECL Prime Western blotting detection kit (GE Healthcare, Baie D'Urfe, Canada) according to manufacturer's guidelines, followed by exposure to X-ray film.
Ethics statement
All procedures were strictly conducted according to the requirements of the Animals (Scientific Procedures) Act 1986 approved by the Home Office, UK (project licence 60/4218).
Animal experiments
A total of 55 8-week-old C57BL/6 mice were housed in groups of 5/6 a week before dosing with 150 mg kg−1 clindamycin by oral gavage. Five days later animals were challenged with ∼ 1 × 105 spores/mouse C. difficile by oral gavage and mouse weight and body condition monitored throughout the study. Cages were changed weekly and the cabinet washed with perasafe after each group. To monitor C. difficile shedding faecal pellets from individuals were collected, serially diluted and plated on CCEY agar supplemented with cycloserine/cefoxitin, egg yolk emulsion and 15 μg ml−1 lincomycin (for mutants only). At 28 days post challenge, once C. difficile shedding levels were below the limit of detection, mice were given a second dose of clindamycin (150 mg kg−1) and shedding monitored for infection relapse. Results are expressed as mean ± SE of at least 10 animals from two independent biological replicates. All statistical analyses were performed using the GraphPad Instat 3.10 (GraphPad Instat Software). A Mann–Whitney analysis of variance analysis (anova) was used to determine significant difference in bacterial recoveries between all time points examined. P-values ≤ 0.05 were considered significant.
PCR was performed on colonies obtained from faecal shedding to determine the validity of the shedding strain. For the clostron mutants, gene-specific primers in combination with the universal EBS primer was used in PCR reactions, while for 630Δerm MLVA PCR analysis was used to confirm the identity of the shedding of this strain (Table S1). PCRs were done 1 day post challenge and weekly thereafter.
Acknowledgments
All authors have no conflict of interest to declare regarding commercial or other relationships. This work was supported by the Wellcome Trust and the Medical Research Council, UK. E.V. was supported by a Marie Curie Intra-European fellowship for career development (PIEF-GA-2009-252207-CDI). A.B. was supported by a Wellcome Trust grant 086418. We thank Kelly Fulton for preparation of flagellin digests for mass spectrometry analysis, Maria McCrossan at the LSHTM microscopy facility for the TEM work, Glen Armstrong for the provision of the FliC antibody and Peter Mullany for the provision of C. difficile 630Δerm.
Supporting information
Additional supporting information may be found in the online version of this article at the publisher's web-site.
Supporting information
References
- Andrews JM. Determination of minimum inhibitory concentrations. J Antimicrob Chemother. 2001;48(Suppl. 1):5–16. doi: 10.1093/jac/48.suppl_1.5. [DOI] [PubMed] [Google Scholar]
- Asakura H, Churin Y, Bauer B, Boettcher JP, Bartfeld S, Hashii N, et al. Helicobacter pylori HP0518 affects flagellin glycosylation to alter bacterial motility. Mol Microbiol. 2010;78:1130–1144. doi: 10.1111/j.1365-2958.2010.07393.x. [DOI] [PubMed] [Google Scholar]
- Aubry A, Hussack G, Chen W, KuoLee R, Twine SM, Fulton KM, et al. Modulation of toxin production by the flagellar regulon in Clostridium difficile. Infect Immun. 2012;80:3521–3532. doi: 10.1128/IAI.00224-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baban ST, Kuehne SA, Barketi-Klai A, Cartman ST, Kelly ML, Hardie KR, et al. The role of flagella in Clostridium difficile pathogenesis: comparison between a non-epidemic and an epidemic strain. PLoS ONE. 2013;8:e73026. doi: 10.1371/journal.pone.0073026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balonova L, Mann BF, Cerveny L, Alley WR, Jr, Chovancova E, Forslund AL, et al. Characterization of protein glycosylation in Francisella tularensis subsp. holarctica: identification of a novel glycosylated lipoprotein required for virulence. Mol Cell Proteomics. 2012;11(M111):015016. doi: 10.1074/mcp.M111.015016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartman ST. Minton NP. A mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Appl Environ Microbiol. 2010;76:1103–1109. doi: 10.1128/AEM.02525-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Katchar K, Goldsmith JD, Nanthakumar N, Cheknis A, Gerding DN. Kelly CP. A mouse model of Clostridium difficile-associated disease. Gastroenterology. 2008;135:1984–1992. doi: 10.1053/j.gastro.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Cobb LM, Mychaleckyj JC, Wozniak DJ. Lopez-Boado YS. Pseudomonas aeruginosa flagellin and alginate elicit very distinct gene expression patterns in airway epithelial cells: implications for cystic fibrosis disease. J Immunol. 2004;173:5659–5670. doi: 10.4049/jimmunol.173.9.5659. [DOI] [PubMed] [Google Scholar]
- Dawson LF, Valiente E, Faulds-Pain A, Donahue EH. Wren BW. Characterisation of Clostridium difficile biofilm formation, a role for Spo0A. PLoS ONE. 2012;7:e50527. doi: 10.1371/journal.pone.0050527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deakin LJ, Clare S, Fagan RP, Dawson LF, Pickard DJ, West MR, et al. The Clostridium difficile spo0A gene is a persistence and transmission factor. Infect Immun. 2012;80:2704–2711. doi: 10.1128/IAI.00147-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dingle TC, Mulvey GL. Armstrong GD. Mutagenic analysis of the Clostridium difficile flagellar proteins, FliC and FliD, and their contribution to virulence in hamsters. Infect Immun. 2011;79:4061–4067. doi: 10.1128/IAI.05305-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drudy D, Fanning S. Kyne L. Toxin A-negative, toxin B-positive Clostridium difficile. Int J Infect Dis. 2007;11:5–10. doi: 10.1016/j.ijid.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Eidhin DN, Ryan AW, Doyle RM, Walsh JB. Kelleher D. Sequence and phylogenetic analysis of the gene for surface layer protein, slpA, from 14 PCR ribotypes of Clostridium difficile. J Med Microbiol. 2006;55:69–83. doi: 10.1099/jmm.0.46204-0. [DOI] [PubMed] [Google Scholar]
- Ewing CP, Andreishcheva E. Guerry P. Functional characterization of flagellin glycosylation in Campylobacter jejuni 81–176. J Bacteriol. 2009;191:7086–7093. doi: 10.1128/JB.00378-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulds-Pain A. Wren BW. Improved bacterial mutagenesis by high-frequency allele exchange, demonstrated in Clostridium difficile and Streptococcus suis. Appl Environ Microbiol. 2013;79:4768–4771. doi: 10.1128/AEM.01195-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulds-Pain A, Birchall C, Aldridge C, Smith WD, Grimaldi G, Nakamura S, et al. Flagellin redundancy in Caulobacter crescentus and its implications for flagellar filament assembly. J Bacteriol. 2011;193:2695–2707. doi: 10.1128/JB.01172-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goorhuis A, Van der Kooi T, Vaessen N, Dekker FW, Van den Berg R, Harmanus C, et al. Spread and epidemiology of Clostridium difficile polymerase chain reaction ribotype 027/toxinotype III in The Netherlands. Clin Infect Dis. 2007;45:695–703. doi: 10.1086/520984. [DOI] [PubMed] [Google Scholar]
- Grant CC, Konkel ME, Cieplak W., Jr Tompkins LS. Role of flagella in adherence, internalization, and translocation of Campylobacter jejuni in nonpolarized and polarized epithelial cell cultures. Infect Immun. 1993;61:1764–1771. doi: 10.1128/iai.61.5.1764-1771.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He M, Sebaihia M, Lawley TD, Stabler RA, Dawson LF, Martin MJ, et al. Evolutionary dynamics of Clostridium difficile over short and long time scales. Proc Natl Acad Sci USA. 2010;107:7527–7532. doi: 10.1073/pnas.0914322107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST, Carter GP. Minton NP. The ClosTron: a universal gene knock-out system for the genus Clostridium. J Microbiol Methods. 2007;70:452–464. doi: 10.1016/j.mimet.2007.05.021. [DOI] [PubMed] [Google Scholar]
- Heap JT, Pennington OJ, Cartman ST. Minton NP. A modular system for Clostridium shuttle plasmids. J Microbiol Methods. 2009;78:79–85. doi: 10.1016/j.mimet.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Heap JT, Kuehne SA, Ehsaan M, Cartman ST, Cooksley CM, Scott JC. Minton NP. The ClosTron: mutagenesis in Clostridium refined and streamlined. J Microbiol Methods. 2010;80:49–55. doi: 10.1016/j.mimet.2009.10.018. [DOI] [PubMed] [Google Scholar]
- Hennequin C, Janoir C, Barc MC, Collignon A. Karjalainen T. Identification and characterization of a fibronectin-binding protein from Clostridium difficile. Microbiology. 2003;149:2779–2787. doi: 10.1099/mic.0.26145-0. [DOI] [PubMed] [Google Scholar]
- Hitchen PG, Twigger K, Valiente E, Langdon RH, Wren BW. Dell A. Glycoproteomics: a powerful tool for characterizing the diverse glycoforms of bacterial pilins and flagellins. Biochem Soc Trans. 2010;38:1307–1313. doi: 10.1042/BST0381307. [DOI] [PubMed] [Google Scholar]
- Howard SL, Jagannathan A, Soo EC, Hui JP, Aubry AJ, Ahmed I, et al. Campylobacter jejuni glycosylation island important in cell charge, legionaminic acid biosynthesis, and colonization of chickens. Infect Immun. 2009;77:2544–2556. doi: 10.1128/IAI.01425-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain HA, Roberts AP. Mullany P. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J Med Microbiol. 2005;54:137–141. doi: 10.1099/jmm.0.45790-0. [DOI] [PubMed] [Google Scholar]
- Iwashkiw JA, Seper A, Weber BS, Scott NE, Vinogradov E, Stratilo C, et al. Identification of a general O-linked protein glycosylation system in Acinetobacter baumannii and its role in virulence and biofilm formation. PLoS Pathog. 2012;8:e1002758. doi: 10.1371/journal.ppat.1002758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby JM, Ahern H, Roberts AK, Kumar V, Freeman Z, Acharya KR. Shone CC. Cwp84, a surface-associated cysteine protease, plays a role in the maturation of the surface layer of Clostridium difficile. J Biol Chem. 2009;284:34666–34673. doi: 10.1074/jbc.M109.051177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A. Minton NP. The role of toxin A and toxin B in Clostridium difficile infection. Nature. 2010;467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- Kuijper EJ, van Dissel JT. Wilcox MH. Clostridium difficile: changing epidemiology and new treatment options. Curr Opin Infect Dis. 2007;20:376–383. doi: 10.1097/QCO.0b013e32818be71d. [DOI] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Walker AW, Goulding D, Stabler RA, Croucher N, et al. Antibiotic treatment of Clostridium difficile carrier mice triggers a supershedder state, spore-mediated transmission, and severe disease in immunocompromised hosts. Infect Immun. 2009;77:3661–3669. doi: 10.1128/IAI.00558-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawley TD, Clare S, Deakin LJ, Goulding D, Yen JL, Raisen C, et al. Use of purified Clostridium difficile spores to facilitate evaluation of health care disinfection regimens. Appl Environ Microbiol. 2010;76:6895–6900. doi: 10.1128/AEM.00718-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lithgow KV, Scott NE, Iwashkiw JA, Thomson EL, Foster LJ, Feldman MF. Dennis JJ. A general protein O-glycosylation system within the Burkholderia cepacia complex is involved in motility and virulence. Mol Microbiol. 2014;92:116–137. doi: 10.1111/mmi.12540. [DOI] [PubMed] [Google Scholar]
- Liu SL, Ezaki T, Miura H, Matsui K. Yabuuchi E. Intact motility as a Salmonella typhi invasion-related factor. Infect Immun. 1988;56:1967–1973. doi: 10.1128/iai.56.8.1967-1973.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan SM. Flagellar glycosylation – a new component of the motility repertoire? Microbiology. 2006;152:1249–1262. doi: 10.1099/mic.0.28735-0. [DOI] [PubMed] [Google Scholar]
- Loo VG, Poirier L, Miller MA, Oughton M, Libman MD, Michaud S, et al. A predominantly clonal multi-institutional outbreak of Clostridium difficile-associated diarrhea with high morbidity and mortality. N Engl J Med. 2005;353:2442–2449. doi: 10.1056/NEJMoa051639. [DOI] [PubMed] [Google Scholar]
- Lyras D, O'Connor JR, Howarth PM, Sambol SP, Carter GP, Phumoonna T, et al. Toxin B is essential for virulence of Clostridium difficile. Nature. 2009;458:1176–1179. doi: 10.1038/nature07822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osman C, Haag M, Wieland FT, Brugger B. Langer T. A mitochondrial phosphatase required for cardiolipin biosynthesis: the PGP phosphatase Gep4. EMBO J. 2010;29:1976–1987. doi: 10.1038/emboj.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker JL, Day-Williams MJ, Tomas JM, Stafford GP. Shaw JG. Identification of a putative glycosyltransferase responsible for the transfer of pseudaminic acid onto the polar flagellin of Aeromonas caviae Sch3N. MicrobiologyOpen. 2012;1:149–160. doi: 10.1002/mbo3.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerda J. Cossart P. Bacterial adhesion and entry into host cells. Cell. 2006;124:715–727. doi: 10.1016/j.cell.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Reynolds CB, Emerson JE, de la Riva L, Fagan RP. Fairweather NF. The Clostridium difficile cell wall protein CwpV is antigenically variable between strains, but exhibits conserved aggregation-promoting function. PLoS Pathog. 2011;7:e1002024. doi: 10.1371/journal.ppat.1002024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirm M, Arora SK, Verma A, Vinogradov E, Thibault P, Ramphal R. Logan SM. Structural and genetic characterization of glycosylation of type a flagellin in Pseudomonas aeruginosa. J Bacteriol. 2004;186:2523–2531. doi: 10.1128/JB.186.9.2523-2531.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Markowitz SM. Macrina FL. Transferable tetracycline resistance in Clostridium difficile. Antimicrob Agents Chemother. 1981;19:997–1003. doi: 10.1128/aac.19.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009;10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabler RA, Dawson LF, Valiente E, Cairns MD, Martin MJ, Donahue EH, et al. Macro and micro diversity of Clostridium difficile isolates from diverse sources and geographical locations. PLoS ONE. 2012;7:e31559. doi: 10.1371/journal.pone.0031559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Jin M, Ding W, Yuan J, Kelly J. Gao H. Posttranslational modification of flagellin FlaB in Shewanella oneidensis. J Bacteriol. 2013;195:2550–2561. doi: 10.1128/JB.00015-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twine SM, Reid CW, Aubry A, McMullin DR, Fulton KM, Austin J. Logan SM. Motility and flagellar glycosylation in Clostridium difficile. J Bacteriol. 2009;191:7050–7062. doi: 10.1128/JB.00861-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma A, Schirm M, Arora SK, Thibault P, Logan SM. Ramphal R. Glycosylation of b-Type flagellin of Pseudomonas aeruginosa: structural and genetic basis. J Bacteriol. 2006;188:4395–4403. doi: 10.1128/JB.01642-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth DE. Ballard JD. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev. 2005;18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DR, Young DI. Young M. Conjugative plasmid transfer from Escherichia coli to Clostridium acetobutylicum. J Gen Microbiol. 1990;136:819–826. doi: 10.1099/00221287-136-5-819. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information