

Abstract
Determination of the patient-specific response to antiplatelet agents facilitates proper dosing for both acute and chronic prophylaxis. “Closed” systems (with or without flow) may fail to predict pharmacological potency in situations where platelets rapidly accumulate under flow conditions at a site of thrombosis (“Open” systems). Using an 8-channel microfluidic flow assay of human whole blood with corn trypsin inhibitor (± PPACK) perfused over focal zones of collagen, dose-response curves were measured for pharmacological agents at a wall shear rate of 210 s−1. The P2Y1 inhibitor MRS 2179 (IC50 = 0.233 ± 0.132 μM) and P2Y12 inhibitor 2-MeSAMP (IC50 = 2.558 ± 0.799 μM) were potent blockers of secondary platelet accumulation under flow, while the P2X1 inhibitor (NF 449) and apyrase failed to reduce platelet accumulation. MRS 2179 and 2-MeSAMP had undetectable effects on initial platelet adhesion to collagen. Numerical simulation of convective-diffusive transport and apyrase-mediated catalytic degradation of ADP indicated that ultra-high concentrations of apyrase (~2000 U mL−1) would be required to have the same effect under flow as much lower concentrations (1 U mL−1) currently used in closed systems (aggregometry or cone-and-plate viscometer). This is the first evaluation of IC50 values for P2Y12 and P2Y1 antagonists under controlled flow conditions. Evaluation of antiplatelet agents in open flow systems demonstrates that inhibition of either ADP by apyrase or antagonism of P2X1 signaling had no inhibitory effect on platelet accumulation. This technique provides a platform for rapidly investigating effects of antithrombotic therapies simultaneously in a model injury system.
Introduction
Antiplatelet therapies can prevent excessive clotting in patients with a wide range of pathologies.1,2 Targeting the thromboxane A2 pathway with aspirin3 or ADP feedback via the P2-family of platelet receptors4,5 reduces secondary platelet activation pathways6 and reduces pathologically excessive clot formation while not interfering with primary platelet adhesion and activation necessary for general hemostasis. However, bleeding risks and variability of patient response to antiplatelet therapies often mandate careful monitoring of patients. For example, a significant number of patients fail to respond to antiplatelet drugs, clopidogrel in particular.7
The most popular current target of antiplatelet therapy is the G protein-coupled receptor P2Y12, which is one of the three P2-family proteins found on the platelet surface.8 The receptor is activated by the binding of its ligand, ADP, released from activated platelets at the site of injury, which in turn causes activation of Gi2, inhibition of adenylyl cyclase and activation of PI3 kinase, a rise of cytosolic calcium, and finally granule release and shape change.9 P2Y1 is the second G protein-coupled receptor that has been targeted as an antiplatelet therapeutic target. Binding of ADP to this other receptor on the platelet surface, which is Gq coupled,10 leads to phospholipase β (PLCβ) activation and subsequent platelet activation. The third P2-family receptor, the P2X1 receptor, is an ATP ligand-gated ion channel,11 which directly leads to the influx of calcium from the extracellular milieu. The role of platelet P2X1 signaling is less well defined with respect to platelet activation and thrombus formation.12,13 Finally, the enzyme apyrase (ATP-diphosphohydrolase, EC 3.6.1.5) which actively hydrolyzes ATP to ADP and ADP to the hemostatically irrelevant AMP is used commonly in aggregometry, cone-and-plate viscometry, and other in vitro experimental techniques to remove the ligands of the P2 family receptors from the bulk solution. Endogenous ATP- and ADPases exist throughout the vaculature (CD39) to keep circulating concentrations below activating levels. The therapeutic potentials of recombinant apyrase14 or solubilized CD3915 are currently being explored as potential antiplatelet therapies.
Insight, innovation, integration.
Blood hemostasis and thrombosis are complex processes that occur in the presence of flow. The assembly of platelet aggregates and their pharmacological antagonism may be dependent on whether the assay system is open or closed and whether hemodynamic flow is present. Protein micropatterning and microfluidic devices provide the ideal setting to recreate focal thrombotic events and measure the potency of antiplatelet agents in the presence of flow. With such a device to image 8 simultaneous clotting events, platelet antagonists that targeted the P2Y1 or P2Y12 receptors resulted in dose-dependent inhibition of thrombosis, while apyrase (a potent inhibitor in closed systems) had no activity under flow. Numerical convection-diffusion- reaction simulations revealed the kinetic limitations where apyrase may increase thrombosis in some conditions.
Evaluation of platelet function and pharmacological agents can occur in closed systems of constant volume that lack flow (e.g. intracellular calcium measurement, automated calibrated thrombography) or include flow (e.g. aggregometry or cone- and- plate viscometry). However, these laboratory approaches fail to recreate the fact that intravascular thrombosis is an open system where blood is continually flowing over a thrombotic site. In open systems, the rapid accumulation of platelets at a surface leads to platelet concentrations greatly exceeding those found in whole blood and the delivery/removal of species by convection may impact the efficacy of pharmacological agents. Various in vitro flow systems allow precise control of hemodynamics, surface characteristics, and inlet blood pharmacology.16–22 If a pharmacological agent is potent in a closed system but lacks efficacy in an open system, then the therapeutic benefit of the agent may be limited with respect to reducing intravascular thrombosis under flow. This work explores the efficacy of one enzymatic and three pharmacological platelet activation inhibitors, showing that receptor antagonism is effective even in open flow systems, whereas enzymatic intervention can be ineffective or even detrimental to preventing platelet activation.
Experimental
Materials and methods
Blood collection and labeling
Blood was obtained from healthy individuals who had refrained from taking medications for at least ten days prior to donation. Informed consent was obtained for each volunteer and performed in accordance with IRB approval from the University of Pennsylvania’s Office of Regulatory Affairs and the Declaration of Helsinki.
Whole blood was drawn via venipuncture into corn trypsin inhibitor (CTI, Haematologic Technologies Inc., Essex Junction, VT, USA) to a final concentration of 50 μg mL−1 to inhibit Factor XIIa. Aliquots of 200 μL whole blood were labeled with 4 μL of AlexaFluor 647 conjugated anti-CD41 (αIIb) antibody (Clone PM6/248, AbD Serotec, Raleigh, NC, USA), which had no effect on platelet function (results not shown). In order to inhibit thrombin function in some experiments, H-D-Phe-Pro-Arg-chloromethylketone (PPACK, Calbiochem, San Diego, CA, USA) was added to a final concentration of 100 μM. CTI-treated whole blood was perfused through the fluidic chamber within 15 min of phlebotomy.
Microfluidic fabrication and protein patterning
Microfluidic devices were fabricated in poly(dimethylsiloxane) (PDMS, Sylgard 184, Ellsworth Adhesives, Germantown, WI, USA) using well established techniques.23 Detailed fabrication methods and patterning techniques were performed similarly to previously published methods,19 with device design and a sample device shown in Fig. 1. Briefly, glass slides (75 mm × 50 mm × 1 mm, Fisherbrand, Fisher Scientific, Pittsburgh, PA, USA) were cleaned and functionalized with Sigmacote (Sigma-Aldrich, St. Louis, MO, USA) to form a hydrophobic surface to aid collagen adsorption. Human Type-I collagen (Vitrocol 3.0 mg mL−1, Advanced BioMatrix, San Diego, CA, USA) was diluted to 300 μg mL−1 in HEPES buffered saline (HBS, 20 mM N-(2-hydroxyethyl)piperazine-N′-2-ethanesulfonic acid, 150 mM NaCl, pH 7.4) with 10 μL 0.1 M sodium hydroxide solution to neutralize pH to 7.0 to a final volume of 1 mL. Microfluidic devices were then filled with the solution and allowed to incubate at room temperature for 150 min to polymerize the collagen into a thin film on the surface of the slide. Excess solution was removed, immediately followed by placement of the flow device aligned on the formed collagen region. A solution of 0.5% bovine serum albumin (BSA) in HBS was then filled through the channels and allowed to sit at room temperature for at least 60 min in order to prevent non-specific platelet binding to both the PDMS and the un-reacted glass surface.
Fig. 1. Multiple-inlet single outlet flow device used for IC50-on-a-chip experiments.
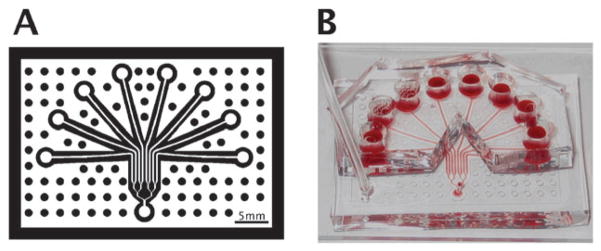
(A) Schematic of microfluidic flow device used to investigate the effect of eight different inhibitor concentrations on platelet aggregate formation simultaneously. (B) Photograph of actual device used. Physical device dimensions are 4.5 × 2 inches.
Flow cytometry and device characterization
Fluorescent particles (FTIC and PE-Cy5 Easy Calibration Kits, Spherotech, Lake Forest, IL, USA) were diluted 1 : 9 (v : v) in 0.5% BSA in HBS to a particle concentration of approximately 2 × 105 particles mL−1. Control mixtures of 10 μL of each type of fluorescent particle (FITC Peaks 2,3,4, and 5 and PE-Cy5 Peaks 2,3,4, and 5) were mixed in a microcentrifuge tube. Particles were assigned to input positions on the flow device using a random number generator and perfused through the device using a gastight syringe (250 μL, Hamilton Company, Reno, NV, USA) mounted on a syringe pump (PHD 2000, Harvard Apparatus, Holliston, MA, USA) at a volumetric flow rate of 16 μL min−1 for 5 min. The effluent was collected in Tygon tubing (0.020 inch ID, 0.060 inch OD, Fisher Scientific, Pittsburg, PA, USA) and subsequently infused into a microcentrifuge tube for assaying. Flow cytometry was performed on an Accuri A6 flow cytometer (Accuri Cytometers Inc., Ann Arbor, MI, USA) fitted with the CSampler automated plate sampler. Gates were applied in CFlow Plus software and kept constant across all samples analyzed.
Inhibitor preparation
2′-Deoxy-N6-methyl adenosine 3′,5′-diphosphate (MRS 2179), Grade VIII apyrase from potato (Solanum tuberosum), 2-methylthioadenosine 5-monophosphate (2-MeSAMP) (Sigma Aldrich, St. Louis, MO, USA), and 4,4′,4″,4‴-[carbonylbis(imino-5,1,3-benzenetriyl-bis(carbonylimino))] tetrakis-1,3-benzenedisulfonic acid (NF 449, Tocris Bioscience, Ellsiville, MO, USA) were dissolved in HBS to working concentrations. Dilutions of compounds were made to ten fold the desired final concentration in HBS on the day of the experiment, after which the inhibitor was added at a ratio of 1 : 9 to fluorescently labeled whole blood and allowed to incubate for 5 min at room temperature, with gentle mixing at 2 min, except for apyrase which was added immediately prior to perfusion. Final inhibitor concentrations used were: MRS 2179 (0–10 μM), 2-MeSAMP (0–200 μM), NF 449 (0–10 μM) and apyrase (0–100 U mL−1).
Platelet accumulation measurement
Blood samples incubated with the given inhibitors were introduced to the inlet of the flow device and immediately perfused through the device at a controlled volumetric flow rate of 15 μL min−1 for an average wall shear rate at the collagen patch of 210 s−1 (for rectangular channels of cross sectional dimensions of 60 μm × 250 μm). Assembled devices were mounted on an inverted microscope (IX81, Olympus America Inc., Center Valley, PA, USA) and imaged using a CCD camera (ORCA-ER, Hamamatsu, Bridgewater, NJ, USA) illuminated with a mercury lamp (100 W, 620 nm Ex/700 nm Em). Fields of view were captured using a 2× objective, integrating for 1000 ms, every 5 s for 5 min.
IC50 calculation
The far left and far right collagen regions were identified by hand, with intermediate and corresponding background regions interpolated using a custom MATLAB script (MathWorks, Natick, MA, USA). Background-corrected fluorescence values were fit using a four parameter logistical model (eqn (1)):
| (1) |
with ‘I’ representing the inhibitor concentration, ‘FI’ the background corrected fluorescence of the corresponding region, ‘A’ and ‘B’ the minimum and maximum intensities, respectively, and ‘C’ and ‘D’ the fit parameters. The data were fit using a least-squares non-linear curve fitting routine (MATLAB, Data Analysis Toolbox).
Computational model of apyrase activity in open and closed systems
In order to investigate the effect of convective mass transport on the hydrolysis of ADP and ATP by apyrase, a set of finite element method numerical simulations were performed. A commercial software package (COMSOL Multiphysics, Burlington, MA, USA) was used to solve both the steady-state flow profile and the coupled convection-diffusion-reaction system in a 2-dimensional domain approximating the 2 mm surrounding a 250 μm reactive zone with a height of 60 μm. The hydrolysis of ADP by apyrase was approximated by classical Michaelis–Menten kinetics using the published Km for apyrase with ADP and ATP.24 The influx of ADP and ATP at the reactive surface was set to an experimentally determined rate corresponding to full platelet activation25 which is comparable to previously estimated values,26,27 and a value calculated from the platelet flux to a collagen surface at the given shear rate28 and the approximate ADP concentration per platelet.29 Parameter values and boundary conditions are shown in Table 1. Grid independence was performed by evaluating the near-wall average concentration for a series of increasingly refined grids consisting of 13 840, 55 360, 105 429, and 221 440 triangular elements, each with less than 0.001% error from the estimate of the zero grid spacing solution using Richardson extrapolation.30 The intermediate grid (105 429 elements) was used in order to optimize solution time and accuracy. Steady-state momentum and mass balance calculations were performed using a direct solver (UMFPACK) on a workstation (dual 2.2 GHz processors, 12 GB RAM). The competition between ADP and ATP for hydrolysis by apyrase was modeled using classical Michaelis–Menten kinetics operating under the fast equilibrium assumption, shown by eqn (2), solved algebraically in eqn (3):
| (2) |
| (3) |
Table 1.
Physical parameters and boundary conditions used in numerical simulations
| Parameter | Value | Reference |
|---|---|---|
| Density of whole blood | 1.06 × 103 kg m−3 (25 °C) | 24 |
| Viscosity of whole blood | 4.7 × 10−3 Pa s (25 °C) | 24 |
| Diffusivity of ADP and ATP | 1.14 × 10−10 m2 s−1 | a |
| Flux of ADP (JADP) | 4.4 × 10−6 and 7.8 × 10−10 mol m×2 s−1 | 22, 33 |
| Flux of ATP (JATP) | 2/3 × JADP | 26 |
| vx,max | 4.18 × 10−3 m s−1 | b |
| Km, ADP | 0.25 mol m−3 | 21 |
| Km, ATP | 0.06 mol m−3 | 21 |
| kADP | 100 μmol min−1 mg−1 | 21 |
| kATP | 1210 μmol min−1 mg−1 | 21 |
Subdomain conditions: ADP: ∇·(DADP∇cADP) = kATPBATP − kADPBADP − vx(y)·∇cADP. ATP: ∇·(−DATP∇cATP) = −kATPBATP − vx(y)·∇cADP. Boundary conditions: ADP and ATP: Inlet: c = 0, Reactive surface: Flux = J, Outlet: Jout = vx(y)c (x = L,y), Walls and center line: ∇c = 0.
Calculated from the Wilkie-Chang correlation for molecular diffusivity.48
Determined by calculating the maximum velocity for a rectangular duct with cross sectional dimensions of 250 μm × 60 μm with a volumetric flow rate of 1.88 μL min−1.
In order to approximate the instantaneous ADP concentration in an aggregometry cuvette as a function of apyrase concentration, the same reactive system outlined in eqn (2) was used with a generation term to approximate ADP release from platelets in a well-mixed control volume shown in eqn (4):
| (4) |
which describes a normal distribution for ADP release to a cumulative total of ‘TADP’, with a time of maximal instantaneous release ‘τ’. The total ADP in the system was estimated by the typical volume and platelet count in an aggregometry cuvette (0.5 mL cuvette, 2 × 108 platelets mL−1) with the same approximate ADP concentration per platelet used in the open system,29 with the maximal release time estimated from the maximal rate of ADP release31,32 at 30 s, leading to a maximal ADP concentration of 6 μM in the absence of degradation.
Statistical analysis
Data were compared to appropriate controls using the Mann–Whitney U-test with p-values less than 0.01 considered statistically significant. All data are plotted as the mean ± standard deviation. All calculations were performed using the MATLAB Statistics Toolbox.
Results and discussion
Device characterization
To verify equal flow rates in each of the eight channels, each inlet reservoir was spiked with a unique bead tracer that was quantifiable by flow cytometry (Fig. 2A). By measuring each tracer bead concentration in the combined exit stream of the microfluidic device (total flow = 16 μL min−1 for 5 min; channel wall shear rate=222 s−1), the flow rates were verified to be equal across the chambers (Fig. 2B). This uniformity across the channels was validated in 15 different experiments using 5 different devices, thus confirming the designed intent that each reactive interaction zone is under statistically indistinguishable hemodynamics and transport conditions.
Fig. 2. Characterization of the multiple inlet single outlet device confirms equal flow through each channel.
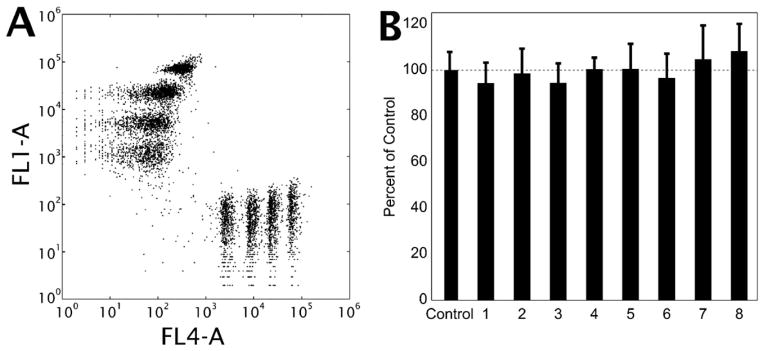
(A) Flow cytometry data indicating the clear identifications of eight distinct peaks in a single sample of eight volumetrically matched standards. Apparent scatter of each population is due to the variability in the background signal of the non-fluorescent channel. (B) Comparison of relative percent of particles in the efflux of the flow device from 15 experiments using 5 different devices (bars are standard deviation, p > 0.05 for all compared to control).
Since thrombus buildup reached heights of <20% of the chamber height for such short distances, the pressure drop across an individual ~250 μm long thrombus would be small compared to the pressure drop across the entire length of a channel. Thus, platelet accumulation at the collagen patch has a minimal effect on the flow distributions within the system.
IC50 for platelet GPCR inhibitors during thrombosis under flow
Three specific P2-family antagonists and apyrase were tested in a dose-response mode to calculate effective IC50 values for collagen-induced platelet aggregate formation inhibition, in the presence and absence of thrombin generation capability. In each experiment, a total of eight simultaneously forming thrombi were imaged in real-time (Fig. 3, and ESI† Fig. S1 and S4). Fluorescently labeled platelets accumulate only at the site of collagen exposure, with minimal non-specific upstream or downstream adhesion. The collagen displays substantial surface coverage due to platelet adhesion to collagen within 1 min and subsequent secondary platelet accumulation via platelet–platelet interactions over the 5 min experiment (ESI† Fig. S1). Determination of total surface fluorescence at 5 min allowed a determination of the IC50 for each pharmacological agent (Fig. 4, Table 2).
Fig. 3. Representative micrographs of fluorescently labeled platelet accumulation at the collagen patch.
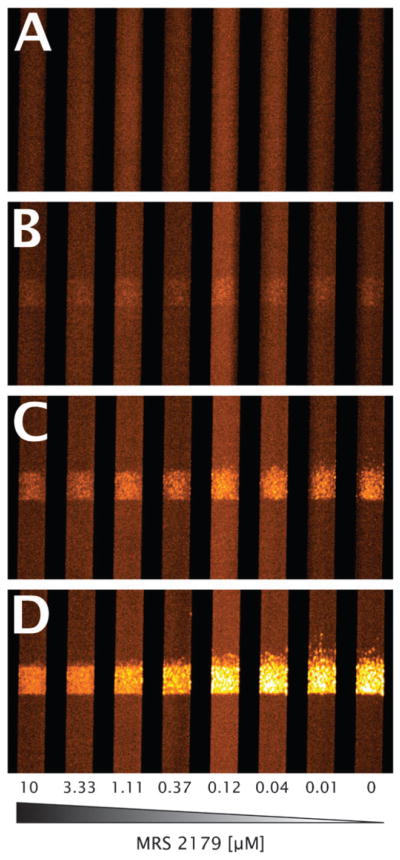
Images are shown at 0 (A), 1 (B), 2 (C), and 5 (D) min of perfusion. Flow is from bottom to top, with concentrations of MRS 2179 decreasing from left to right in human whole blood treated with CTI and PPACK. Channels are 250 μm in width, with reaction zones 250 μm in length. Images are pseudo-colored in order to increase visibility.
Fig. 4. IC50 curves for three P2-family receptors and apyrase under flow.

Representative IC50 curves for MRS 2179 (A, B), 2-MeSAMP (C, D), NF 449 (E, F), and apyrase (G, H), with (A, C, E, G), or without (B, D, F, H) thrombin inhibition. Measurements are shown as points, while the best fit curve is shown as a solid black line, with the calculated IC50. ‘IC50 = NA’ is shown when no clear dose dependence was displayed.
Table 2.
IC50 values for three P2-family receptor antagonists and apyrase in the presence and absence of thrombin generation capability (n=4 different donors, p>0.39 between CTI only and CTI PPACK)
| CTI only | CTI and PPACK | |
|---|---|---|
| P2Y1 (MRS 2179) [μM] | 0.171 ± 0.093 | 0.227 ± 0.098 |
| P2Y12 (2-MeSAMP) [μM] | 2.619 ± 0.994 | 2.497 ± 0.700 |
| P2X1 (NF 449) [μM] | NA | NA |
| Apyrase (U mL−1) | NA | NA |
The P2Y1 and P2Y12 inhibitors displayed remarkably robust dose-dependent reduction of platelets at a localized collagen region. This was in contrast to the P2X1 inhibitor which had no effect, as has been demonstrated previously at comparable shear rates in the lower physiological regime.33 The thrombin generation at 5 min in this system with CTI appears to be minimal, since there is no difference in the presence or absence of an irreversible thrombin inhibitor. While the presence and role of blood derived tissue factor exposed on or accumulating at collagen-induced platelet activation is still under intense investigation, the relatively short experimental time (5 min) without exogenous tissue factor results in minimal thrombin and fibrin generation in this model system as expected.34
In contrast to small molecule P2-receptor antagonists, apyrase operates by a different mechanism in preventing platelet activation by nucleotide substrates by enzymatic hydrolysis. In the microfluidic focal model, apyrase was unable to prevent collagen-initiated platelet accumulation, even at extremely high levels (100 U mL−1) of the enzyme. Incubating the blood sample with apyrase for up to 10 min prior to perfusion did not increase activity (results not shown). Enzyme activity was confirmed by reduction of ATP-mediated luciferase signal (ESI† Fig. S2). The slight enhancement of platelet deposition at >10 U mL−1 may be due to conversion of released ATP to ADP compared to the kinetically slower reaction and weaker binding of ADP to apyrase compared to ATP.
Numerical simulation of apyrase-mediated ADP hydrolysis in open and closed systems
The closed system of an aggregometry cuvette was modeled using eqn (2) and (3), with the instantaneous ADP concentration at 2 min used to determine dose dependence, shown in Fig. 5A. In order to approximate the amelioration of the ‘active cloud’35 of ADP generated at the site of platelet activation by apyrase, average ADP concentrations in a region 5 μm above the length of the reactive zone were calculated using a finite element method coupled to a convective flow field. These calculations were made across a range of apyrase concentrations from 1 × 10−5 to 3 × 104 U mL−1, with representative samples shown in Fig. 5B. The steady-state concentrations in the reactive zone were plotted as a function of bulk apyrase concentration, calculating an effective IC50 using eqn (1), with the dose response shown in Fig. 5C. As shown in Fig. 5C, increasing the apyrase concentration effectively abolished any ADP in the closed system at 1 U mL−1, the concentration which is most commonly used in aggregometry.
Fig. 5. Results of numerical simulations of apyrase-mediated ADP flux hydrolysis.

The well-mixed aggregometer model ((A), eqn (2)–(4)) calculates the instantaneous ADP concentration for a set of apyrase concentrations in a closed system. (B) Steady-state solutions to the open system (finite element method convection-diffusion-reaction system with eqn (2) and (3)), with flow depicted from left to right in a 30 μm × 400 μm sub-region (scale bar is 50 μm). The 5 μm zone used to calculate the relevant ADP concentration is outlined in black. (C) Dose response curves of apyrase in convection-diffusion-reaction (red squares) and diffusion-reaction (blue circles) systems. The solid lines represent the fit from eqn (1) of the data used to calculate the IC50.
The slight but reproducible increase in platelet accumulation under high (10, 30 and 100 U mL−1) apyrase concentrations, shown in Fig. 4G and H (ESI† Fig. S4G and S4H), was hypothesized to be the result of preferential conversion of ATP to ADP by apyrase at the location of the growing thrombus. There are two prevalent isozymes of the apyrase enzyme, with relative ATPase : ADPase activity ratios of 12 : 1 and 1 : 1,36 with most apyrase reagents supplied as mixtures of the two. By running the numerical simulations with both pairs of binding and kinetic rate parameters, it was possible to show that in both the closed and open systems, an apyrase-mediated increase in ADP is generated due to preferential conversion of ATP released simultaneously during platelet degranulation as shown in Fig. 5C. The dose-dependent behaviors were not sensitive to the value of the surface flux used in the open simulation, which is important since literature estimates for the effective ADP flux from a growing thrombus vary over four orders of magnitude,25,28 indicating that the phenomena is independent of the actual ADP flux at the surface (ESI† Fig. S3).
The 8-channel microfluidic focal injury model allows the rapid determination of the effective concentration of antiplatelet and antithrombotic agents under flow conditions. By designing and fabricating a pair of microfluidic devices for patterning collagen and for perfusion with tight spatial control, this system is able to measure the accumulation of fluorescent platelets at 8 independent sites of collagen exposure simultaneously. By dosing small volumes (90 μL) of whole blood from a single donor with correspondingly small volumes (10 μL) of inhibitor for each flow channel, it is possible to generate a full dose response curve from a single experiment. The dose response behavior observed in this system for both 2-MeSAMP and MRS 2179 was similar to previously reported data using turkey erythrocyte inositol phosphate accumulation (MRS 2179 IC50 = 0.330 μM),37 human platelet ADP-induced calcium signaling depletion (MRS 2179 IC50 = 0.260 μM),38 and human platelet ADP-induced aggregation prevention (2-MeSAMP IC50~5 μM).39
The biochemical modes of action of the P2Y1 and P2Y12 inhibitors used in this work are different from the family of compounds currently used to inhibit the receptors as antiplatelet therapeutics (the thienopyridines). Such compounds require metabolism to turn the administered pro-drug into the active compound, which then binds its target receptor irreversibly and prevents ligand binding and subsequent G protein signaling. This mode of action is in contrast to the reversible binding, direct acting compounds used in this work (MRS 2179 and 2-MeSAMP) or next generation therapies under investigation (ARC69931MX). The reversibility of the compound interaction with the receptor may be a concern, as shown by a lack of efficacy leading to the early termination of the ARC69931MX trial (ClinicalTrials.gov identifier NCT00305162). However, the activity of MRS 2179 (IC50 ~200 nM) in this system was comparable to the published Ki of 138 nM.38 2-MeSAMP required a concentration approximately 10-fold higher than its calculated Kd (0.23 μM)40 for a half-maximal effect in this assay (IC50 ~2.5 μM).
The inability of apyrase to prevent platelet accumulation at a collagen surface and the corresponding numerical simulations of both open and closed systems have several significant implications. The mode of action of an enzymatic degradation of receptor ligand in the bulk phase is fundamentally distinct from direct receptor antagonism. By preincubation of the small molecule inhibitors with the blood sample, ample time is given for diffusion to, recognition of, and equilibration with the antagonist’s given receptor, thereby disabling the ability of the receptor to bind its ligand and signal. However, enzymatic degradation of ligand must occur in the time between ligand exposure, in this case ADP release from dense granules upon primary platelet activation, and receptor binding. In closed, well mixed systems, such as aggregometry cuvettes, the ligand, enzyme, and receptor are all equally distributed throughout the control volume. Under flow conditions where platelets accumulate to levels that are 10 to 50-fold greater than in normal whole blood, apyrase is kinetically unable to decrease local ADP concentrations. The experiments and simulations were performed at relatively low (210 s−1) shear rates compared to the arterial vasculature, however convection is still the dominant mode of ADP transport compared to diffusion (Péclet number, , convection dominant). Additionally, a majority of the initial biochemical and platelet function experiments are performed in plasma systems or with washed platelets that may remove or reduce the activity of endogenous ectopyrase enzymes, artificially increasing the apparent effectiveness of exogenous apyrase activity.
Upon further investigation of the preferential hydrolysis of ATP to ADP for a specific isozyme of apyrase, it was determined that local ADP concentrations, in the presence of certain concentrations of apyrase, could actually be higher than in the absence of apyrase. As shown in Fig. 6, even an apyrase with equal kinetic parameters for both ATP and ADP would not be able to have an effect in an open system until very high concentrations. The system modeled here only takes into account the species released from platelets accumulating and activating at a collagen patch. It is well established that red blood cells are key factors in regulating vascular tone, using ATP as one of the main mediators.41 Red cells can release very significant amounts of ATP, especially in hypoxic conditions. Upwards of 2 × 107 molecules of ATP can be released per cell, leading to local ATP concentrations of over 160 μM in a normal hematocrit with approximately 4.8 × 1012 red cells per L. In this regard, red blood cells act as sources of ATP, which in the presence of circulating apyrase, could lead to increased ADP concentrations at growing thrombi, especially as surrounding tissues become oxygen starved during occlusive events such as heart attacks and ischemic strokes. This analysis implies that dosing high concentrations of adenosine phosphatases required to have an effect under flow conditions could actually increase platelet accumulation at the site of injury which was observed in this model system (at apyrase concentrations of 10, 30, and 100 U mL−1), putting individuals at risk for atherothrombosis at higher risk for pathological clot formation. Interestingly, apyrase can block shear-induced platelet activation (SIPA) detected at wall shear rates greater than 5000 s−1 in viscometers,42 but this result may not predict apyrase efficacy against SIPA in severe stenosis. In addition, the natural platelet inhibitors and nitric oxide production that endothelial cells provide in vivo are not present in this model system. These active processes maintain the quiescent state of platelets while circulating in the body by scavenging nucleotides released during normal homeostasis and raising intracellular platelet cyclic-AMP levels.
Fig. 6. Preferential ATP hydrolysis by apyrase increases local ADP concentration but does not alter effective dosing regime.

In order to investigate the effect of the estimated IC50 for apyrase mediated ADP depletion in both open (A) and closed (B) systems, numerical simulations were run using kinetic parameters for the two isozyme variants of apyrase with ATPase : ADPase activity ratio of 12 : 1 (closed symbols and solid fit line) and 1 : 1 (open symbols and dashed fit line). While the ADP concentrations increase over baseline for the ATP preferred isozyme, the concentration at which apyrase effectively decreases the ADP concentration is largely unaffected. The lines represent the fit of the data using eqn (1) to calculate the IC50 for each condition.
The open system described in this work is fundamentally different from classical aggregometry experiments. Rather than an isotropic release throughout the control volume, the focal nature of the injury necessitates that the ADP generation is also localized to the growing thrombus site. While laminar flow is characteristic of physiological conditions found in a large majority throughout the vasculature, it necessarily is accompanied by poor radial mixing, especially of non-ionic molecular species (MW>100 Da). The competing phenomena of reaction and convective transport is characterized by a dimensionless number, typically used to describe chemical reactor efficiency, called the Dämkohler number,43,44 defined by eqn (5).
| (5) |
with ‘U’, ‘L’, ‘V’, and ‘cA’ representing a characteristic reaction rate, length, velocity, and concentration, respectively. As the Dämkohler number approaches infinity, the reaction approaches completion as it would under no-flow conditions (i.e. V → 0) within the interaction zone. However, as the Dämkohler number approaches zero, convection dominates the system, meaning the reaction is minimal within the region of interest. In the experimental system and simulation setup, the Dämkohler number is 2 × 10−3, or close to zero. This is manifested in the fact that simulation results suggest that exceedingly high apyrase concentrations would be required to have an effect on the local ADP concentration for the given flow profile. It is worthwhile to reiterate that the experiments and simulations were performed at a wall shear rate of 210 s−1, a relatively low physiological shear rate. Higher shear rates, and correspondingly higher velocities, would only further decrease the Dämkohler number and decrease the effect of any bulk phase enzymatic-based agonist depletion.
The simple dimensional analysis provided by the Dämkohler number leads to several important clinical implications. This work indicates that enzyme-mediated ligand removal for localized surface-mediated events occurring under bulk convective transport will be limited, not due to enzyme specificity, but rather by the time required to react compared to convection away from the zone of interest. A number of nucleotide phosphohydrolase therapies currently undergoing testing for diminishing ADP-induced excessive platelet accumulation in thrombotic pathologies have shown early success in one of two major ways. The first is to use a model injury system that includes an element of stasis, either an ischemic stroke model45 or an ischemia/reperfusion thrombosis model.46 While these in vivo injury model systems are highly relevant, their intrinsic lack of blood flow at the site of injury necessarily provides the soluble enzyme the time required to act, in contrast to injury models in which blood flow is maintained. The second and newer approach is by which the enzymatic domain is covalently bound to a targeting moiety, hence allowing the local enzyme concentration to greatly increase compared to the bulk concentration at the site of injury.47 This surface-bound enzyme increases the residence time of the enzyme at the active site, providing more enzymatic activity at the site of ADP action.
In summary, this work has shown that a microfluidic focal injury model is capable of measuring the responsiveness of platelets to a set of inhibitors by measuring changes in collagen-induced accumulation under flow. While inhibitors of the P2Y1 and P2Y12 receptors displayed dose-dependent efficacy, the P2X1 antagonist did not have any effect in this model system. Similarly, apyrase was not able to reduce platelet accumulation by hydrolyzing ADP and preventing its action on subsequent activation of additional platelets. Numerical simulations have given us insight into why apyrase is able to function at moderate (1 U mL−1) concentrations in aggregometry and other closed systems, however significantly higher concentrations (2000 U mL−1) would be required for similar abolition of ADP-induced platelet activation under single-pass flow conditions. In addition, the preferential hydrolysis of ATP to ADP by a non-specific apyrase can increase the local ADP concentration to levels higher than in the absence of the enzyme, increasing platelet accumulation on a focal collagen region. This work represents a step towards what could be a functional assay used to rapidly estimate a patient’s response to antiplatelet therapy, prior to or during prophylactic treatment. Additionally, the results of this work describe the importance of considering method of action and transport issues relevant to specific therapeutic goals. Utilizing computational modeling to help interpret experimental results and exploring physically unrealizable situations to gain insight into observed results is shown as a powerful tandem in current biological systems research. By experimental data informing computational model inputs and model outputs guiding subsequent experiments, the complicated and rich interactions of biology can begin to be understood in ways that can rely upon the years of extensive research in the engineering disciplines.
Acknowledgments
This work is supported by grants from the United States National Institute of Health’s National Heart, Lung and Blood Institute grant numbers R33HL087317 (SLD, LFB), HL56621 (SLD), HL40387 (LFB), and T32HL007971 (SFM). Fabrication was performed at the Wolf Nanofabrication Laboratory at the University of Pennsylvania.
Footnotes
Electronic supplementary information (ESI) available: Supplementary figures, Fig. S1–S4. See DOI: 10.1039/b919728a
References
- 1.Brass LF, Zhu L, Stalker TJ. Arterioscler, Thromb, Vasc Biol. 2008;28:s43–50. doi: 10.1161/ATVBAHA.107.161026. [DOI] [PubMed] [Google Scholar]
- 2.Bhatt DL, Topol EJ. Nat Rev Drug Discovery. 2003;2:15–28. doi: 10.1038/nrd985. [DOI] [PubMed] [Google Scholar]
- 3.Wu KK. J Intern Med. 1996;239:17–34. doi: 10.1046/j.1365-2796.1996.331661000.x. [DOI] [PubMed] [Google Scholar]
- 4.Gachet C. Pharmacol Ther. 2005;108:180–192. doi: 10.1016/j.pharmthera.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 5.Hechler B, Cattaneo M, Gachet C. Semin Thromb Hemostasis. 2005;31:150–161. doi: 10.1055/s-2005-869520. [DOI] [PubMed] [Google Scholar]
- 6.Jackson SP, Schoenwaelder SM. Nat Rev Drug Discovery. 2003;2:775–789. doi: 10.1038/nrd1198. [DOI] [PubMed] [Google Scholar]
- 7.Wiviott SD, Antman EM. Circulation. 2004;109:3064–3067. doi: 10.1161/01.CIR.0000134701.40946.30. [DOI] [PubMed] [Google Scholar]
- 8.Kunapuli SP. Trends Pharmacol Sci. 1998;19:391–394. doi: 10.1016/s0165-6147(98)01248-6. [DOI] [PubMed] [Google Scholar]
- 9.Gachet C. Annu Rev Pharmacol Toxicol. 2006;46:277–300. doi: 10.1146/annurev.pharmtox.46.120604.141207. [DOI] [PubMed] [Google Scholar]
- 10.Offermanns S. Circ Res. 2006;99:1293–1304. doi: 10.1161/01.RES.0000251742.71301.16. [DOI] [PubMed] [Google Scholar]
- 11.Oury C, Kuijpers MJ, Toth-Zsamboki E, Bonnefoy A, Danloy S, Vreys I, Feijge MA, De Vos R, Vermylen J, Heemskerk JW, Hoylaerts MF. Blood. 2003;101:3969–3976. doi: 10.1182/blood-2002-10-3215. [DOI] [PubMed] [Google Scholar]
- 12.Nurden AT. J Thromb Haemostasis. 2007;5:907–909. doi: 10.1111/j.1538-7836.2007.02456.x. [DOI] [PubMed] [Google Scholar]
- 13.Mahaut-Smith MP, Tolhurst G, Evans RJ. Platelets. 2004;15:131–144. doi: 10.1080/09537100410001682788. [DOI] [PubMed] [Google Scholar]
- 14.Sun D, McNicol A, James AA, Peng Z. Platelets. 2006;17:178–184. doi: 10.1080/09537100500460234. [DOI] [PubMed] [Google Scholar]
- 15.Dwyer KM, Robson SC, Nandurkar HH, Campbell DJ, Gock H, Murray-Segal LJ, Fisicaro N, Mysore TB, Kaczmarek E, Cowan PJ, d’Apice AJ. J Clin Invest. 2004;113:1440–1446. doi: 10.1172/JCI19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Savage B, Ginsberg MH, Ruggeri ZM. Blood. 1999;94:2704–2715. [PubMed] [Google Scholar]
- 17.Diamond SL, Eskin SG, McIntire LV. Science. 1989;243:1483–1485. doi: 10.1126/science.2467379. [DOI] [PubMed] [Google Scholar]
- 18.Okorie UM, Diamond SL. Biophys J. 2006;91:3474–3481. doi: 10.1529/biophysj.106.083287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neeves KB, Maloney SF, Fong KP, Schmaier AA, Kahn ML, Brass LF, Diamond SL. J Thromb Haemostasis. 2008;6:2193–2201. doi: 10.1111/j.1538-7836.2008.03188.x. [DOI] [PubMed] [Google Scholar]
- 20.Gutierrez E, Petrich BG, Shattil SJ, Ginsberg MH, Groisman A, Kasirer-Friede A. Lab Chip. 2008;8:1486–1495. doi: 10.1039/b804795b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karunarathne W, Ku CJ, Spence DM. Integr Biol. 2009;1:655–663. doi: 10.1039/b909873a. [DOI] [PubMed] [Google Scholar]
- 22.Ku CJ, Oblak TD, Spence DM. Anal Chem. 2008;80:7543–7548. doi: 10.1021/ac801114j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Duffy DC, McDonald JC, Schueller OJA, Whitesides GM. Anal Chem. 1998;70:4974–4984. doi: 10.1021/ac980656z. [DOI] [PubMed] [Google Scholar]
- 24.Del Campo G, Puente J, Valenzuela MA, Traverso-Cori A, Cori O. Biochem J. 1977;167:525–529. doi: 10.1042/bj1670525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neeves KB, Diamond SL. Lab Chip. 2008;8:701–709. doi: 10.1039/b717824g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grabowski EF, Franta JT, Didisheim P. Microvasc Res. 1978;16:183–195. doi: 10.1016/0026-2862(78)90054-7. [DOI] [PubMed] [Google Scholar]
- 27.Goldsmith HL, Turitto VT. Thromb Hemostasis. 1986;55:415–435. [PubMed] [Google Scholar]
- 28.Badimon L, Badimon JJ, Turitto VT, Vallabhajosula S, Fuster V. Circulation. 1988;78:1431–1442. doi: 10.1161/01.cir.78.6.1431. [DOI] [PubMed] [Google Scholar]
- 29.Holmsen H, Weiss HJ. Annu Rev Med. 1979;30:119–134. doi: 10.1146/annurev.me.30.020179.001003. [DOI] [PubMed] [Google Scholar]
- 30.Roache PJ. Verification and Validation in Computational Science and Engineering. Hermosapublishers; Albuquerque, NM: 1998. [Google Scholar]
- 31.Schraw TD, Rutledge TW, Crawford GL, Bernstein AM, Kalen AL, Pessin JE, Whiteheart SW. Blood. 2003;102:1716–1722. doi: 10.1182/blood-2003-01-0331. [DOI] [PubMed] [Google Scholar]
- 32.Detwiler TC, Feinman RD. Biochemistry. 1973;12:2462–2468. doi: 10.1021/bi00737a015. [DOI] [PubMed] [Google Scholar]
- 33.Hechler B, Lenain N, Marchese P, Vial C, Heim V, Freund M, Cazenave JP, Cattaneo M, Ruggeri ZM, Evans R, Gachet C. J Exp Med. 2003;198:661–667. doi: 10.1084/jem.20030144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okorie UM, Denney WS, Chatterjee MS, Neeves KB, Diamond SL. Blood. 2008;111:3507–3513. doi: 10.1182/blood-2007-08-106229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hubbell JA, McIntire LV. Biophys J. 1986;50:937–945. doi: 10.1016/S0006-3495(86)83535-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kettlun AM, Uribe L, Calvo V, Silve S, Rivera J, Mancilla M, Valenzuela MA, Traverso-Cori A. Phytochemistry. 1982;21:551–558. [Google Scholar]
- 37.Camaioni E, Boyer JL, Mohanram A, Harden TK, Jacobson KA. J Med Chem. 1998;41:183–190. doi: 10.1021/jm970433l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Baurand A, Raboisson P, Freund M, Leon C, Cazenave JP, Bourguignon JJ, Gachet C. Eur J Pharmacol. 2001;412:213–221. doi: 10.1016/s0014-2999(01)00733-6. [DOI] [PubMed] [Google Scholar]
- 39.Srinivasan S, Mir F, Huang JS, Khasawneh FT, Lam SC, Le Breton GC. J Biol Chem. 2009;284:16108–16117. doi: 10.1074/jbc.M809780200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jantzen HM, Gousset L, Bhaskar V, Vincent D, Tai A, Reynolds EE, Conley PB. Thromb Hemostasis. 1999;81:111–117. [PubMed] [Google Scholar]
- 41.Ellsworth ML, Forrester T, Ellis CG, Dietrich HH. Am J Physiol. 1995;269:H2155–2161. doi: 10.1152/ajpheart.1995.269.6.H2155. [DOI] [PubMed] [Google Scholar]
- 42.Oda A, Yokoyama K, Murata M, Tokuhira M, Nakamura K, Handa M, Watanabe K, Ikeda Y. Thromb Hemostasis. 1995;74:736–742. [PubMed] [Google Scholar]
- 43.Damkohler G. Der Chemie-Ingeniur. 1937;3:359–485. [Google Scholar]
- 44.Catchpole JP, Fulford G. Ind Eng Chem. 1966;58:46–60. [Google Scholar]
- 45.Pinsky DJ, Broekman MJ, Peschon JJ, Stocking KL, Fujita T, Ramasamy R, Connolly ES, Jr, Huang J, Kiss S, Zhang Y, Choudhri TF, McTaggart RA, Liao H, Drosopoulos JH, Price VL, Marcus AJ, Maliszewski CR. J Clin Invest. 2002;109:1031–1040. doi: 10.1172/JCI10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kohler D, Eckle T, Faigle M, Grenz A, Mittelbronn M, Laucher S, Hart ML, Robson SC, Muller CE, Eltzschig HK. Circulation. 2007;116:1784–1794. doi: 10.1161/CIRCULATIONAHA.107.690180. [DOI] [PubMed] [Google Scholar]
- 47.Dezfouli ZX, Crikis S, Dwyer K, Selan C, Robson S, D’Apice A, Cowan P, Nandurkar H. XXII Congress of the International Society on Thormbosis and Haemostasis; 2009; Session AS-TH-025. [Google Scholar]
- 48.Bird RB. Transport Phenomena. Wiley; New York: 1960. [Google Scholar]