

Abstract
Fluorescently tagged glycosides containing terminal α(1→3) and α(1→4)-linked thio-galactopyranosides have been prepared and tested for resistance to hydrolysis by α-galactosidases. Eight fluorescent glycosides containing either galactose or 5-thio-galactose as the terminal sugar were enzymatically synthesized using galactosyltransferases with lactosyl glycosides as acceptors and UDP-galactose or UDP-5′-thio-galactose, respectively, as donors. The glycosides were incubated with human α-galactosidase A (CAZy family GH27, a retaining glycosidase), Bacteroides fragilis α-1,3-galactosidase (GH110, an inverting glycosidase) or homogenates of MCF-7 human breast cancer cells and NG108-15 rat glioma cells. Substrate hydrolysis was monitored by capillary electrophoresis with fluorescence detection. All compounds containing terminal O-galactose were readily degraded. Their 5-thio-galactose counterparts were resistant to hydrolysis by human α-galactosidase A and the enzymes present in the cell extracts. B. fragilis α-1,3-galactosidase hydrolyzed both thio- and O-galactoside substrates, however, the thio-galactosides were hydrolyzed at only 1-3% of the rate of O-galactosides. The hydrolytic resistance of 5-thio-galactose was also confirmed by an in vivo study using cells in culture. The results suggest that 5-thio-galactosides maybe useful tools for the study of anabolic pathways in cell extracts or in single cells.
Keywords: thio-galactosides, glycosidases, hydrolytic stability
Introduction
Glycosphingolipids (GSLs) are ubiquitous, carbohydrate containing lipids that are especially abundant in cells of the nervous system. Within cells they undergo repetitive cycles of synthesis and degradation.[1] Synthesis takes place in the endoplasmic reticulum (ER) and Golgi apparatus, where glycosyltransferases add sugar residues to ceramide in a stepwise manner. Mature GSLs are then transported to the plasma membrane surface where they have roles in many cellular functions including in cell signaling events.[1] After a period of exposure on cell surfaces, GSLs are internalized to be partially or completely degraded by glycosidases and other hydrolases present in the endosomal/lysosomal system. Degradation products are then reused in synthesis. GSL turnover can be rather fast; half lives from two to five hours have been reported.[2]
The GSL composition of cells is characteristic for a given cell type and dramatic changes may occur during cell differentiation[3] and upon malignant transformation.[4] Changes in GSL composition are also seen in lysosomal storage diseases such as Tay-Sachs, Gaucher, Sandhoff and Fabry disease.[5, 6]
Analysis of GSL metabolism is required for understanding their roles in neuronal development and malignant transformation. Our method for studying GSL metabolism in cell extracts and in single cells utilizes fluorescently labeled GSLs that enter metabolic pathways together with endogenous GSLs.[7-9] Metabolism of fluorescently labeled GSLs after a given time of incubation can be monitored by disruption of the cells and analysis of the cellular contents by capillary electrophoresis with laser-induced fluorescence detection. The technique is sensitive enough to detect low zeptomole (10−21) levels of metabolites.[9] A drawback of the method has been that most of the metabolites observed are from GSL degradation in catabolic pathways.[7-9] One way to improve the study of anabolic pathways would be the use of GSL derivatives that are resistant to glycosidases but still function as substrates for endogenous glycosyltransferases.
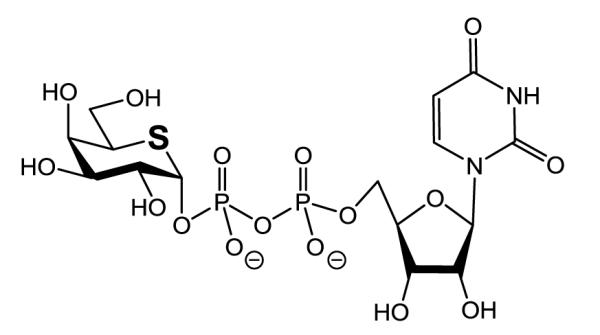
Promising candidates for such compounds are GSLs containing a terminal sugar in which the ring oxygen has been replaced by sulfur. Figure 1 shows the structure of UDP-5′-thio-galactose[10] used in this study. The sulfur atom is larger and more polarizable than oxygen. Moreover, the carbon-sulfur bond is longer, weaker and less polar than the carbon-oxygen bond and the C-S-C angle is more acute than the C-O-C angle, 95-100° compared to 110-113°. As a consequence, 5-thio-galactose shows differences in the anomeric effect, conformational behavior and chemical reactivity.[11-13] Also, molecular interactions of 5-thio-galactose and of other 5-thio-pyranoses with enzymes may differ from those of pyranoses containing oxygen. Some 5-thio-pyranoses are glycosidase inhibitors[14,15] and in vitro studies have shown that bacterial and plant glycosidases do not hydrolyze the sulfur derivatives[16-18] or do so only at a very low rate.[10,19] However, the hydrolytic stability of GSL-glycosides with terminal 5-thio-sugars has not been confirmed.
Figure 1.
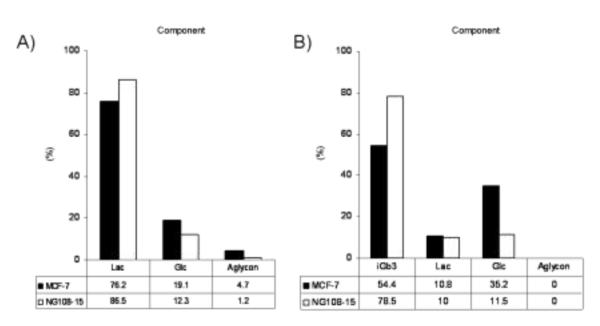
Composition of TMR-compounds after hydrolysis reaction using cell extracts. Substrates were A) Lac-O-TMR (3h reaction) and B) iGb3-O-TMR (24 h reaction). Each bar (black; MCF-7 and white; NG108-15) indicates the percentage of TMR-labeled compounds. The ratios on table were determined by capillary electrophoresis. Aglycon = O-TMR.
In the current investigation we synthesized eight fluorescently labeled glycosides containing either terminal O-galactose or the corresponding 5-thio-galactose residue. These were prepared using α(1→3)- and α(1→4)-galactosyltransferases, UDP-galactose (UDP-Gal) or UDP-5′-thio-galactose as the donors, and fluorescently tagged Lac-OR (2a and 2b, Figure 2) as acceptors. These were tested to see whether they are resistant to hydrolysis by two different types of galactosidase enzymes, human α-galactosidase A (CAZy GH27, a retaining glycosidase)[20] and Bacteroides fragilis α-1,3-galactosidase (FragB, CAZy GH110, an inverting glycosidase)[21] and enzymes present in mammalian cell extracts which are all retaining α-galactosidases. In addition to the in vitro assays, hydrolytic resistance of 5-thio-galactoside was explored in vivo.
Figure 2.
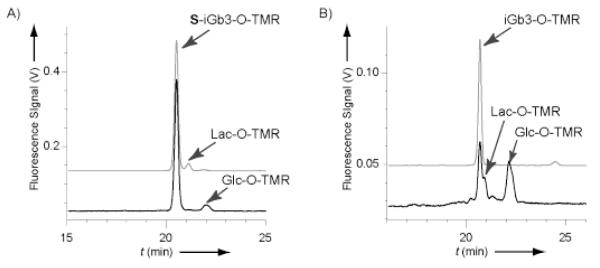
CE analyses of NG108-15 homogenates after incubation with (A) S-iGb3-O-TMR and (B) iGb3-O-TMR. Back and front lines represent before and after incubation, respectively. The S-iGb3-O-TMR in panel (A) contains Lac-O-TMR (7%) due to incomplete enzymatic synthesis.
Results and Discussion
Enzymatic synthesis of TMR labeled galactosides
Eight TMR labeled galactosidases (compounds 3 to 10, Figure 2) containing either galactose or 5-thio-galactose as the terminal sugar were synthesized with specific galactosyltransferases, that catalyze the transfer of galactose from UDP-Gal or UDP-5′-thio-Gal to Lac-O-TMR and Lac-Cer-TMR. Both α-1,3-galactosyltransferase and α-1,4-galactosyltransferase readily catalyzed the desired reactions to produce derivatives of iGb3 and Gb3 respectively.[22] The reactions with UDP-5′-thio-galactose were slower than those with the natural UDP-Gal donor. Reactions with UDP-5′-thio-galactose required a greater excess of donor due to enzymatic hydrolysis which is considerably faster than the enzymatic hydrolysis of UDP-Gal. Conversions above 90%, as estimated by TLC analysis, were seen for all reactions and products were isolated by preparative TLC. The various products differed with respect to aglycon structure, nature of the terminal glycosidic linkage, and the heteroatom in the ring of the terminal sugar. The structural details are provided in Figure 2.
Hydrolytic resistance assay of TMR-labeled galactosides to human α-galactosidase A (retaining glycosidase) and B. fragilis α-1,3-galactosidase (inverting glycosidase)
Compounds 1-10 were first tested for resistance to hydrolysis by human α-galactosidase A. Human α-galactosidase A is a lysosomal enzyme that cleaves terminal α-galactosyl residues from a variety of oligosaccharides with retention of configuration, with preferred substrates being trihexosylceramides such as Gb3 and iGb3 (Fig. 1). The presence of iGb3 in humans is currently a matter of debate.[23] Human α-galactosidase A is used as an enzyme replacement therapy for the treatment of Fabry disease, an X-linked inherited disorder caused by a defect in the α-galactosidase encoding gene. The enzyme used in this study is from and marketed by Shire. The hydrolysis rate and the products were determined by CE (Table 1).
Table 1.
Rate of enzyme-catalyzed hydrolysis of ten TMR-labeled compounds, using human α-galactosidase A (retaining enzyme) and B. fragilis α-1,3-galactosidase (inverting enzyme)
| Compound |
Human α-galactosidase A (pmol*min-1μg-1) |
B. fragilis α-1,3-galactosidase (pmol*min-1μg-1) |
|---|---|---|
| 1 (LacNAc-O-TMR) | n.h.[a] | n.h. |
| 2a (Lac-O-TMR) | n.h. | n.h. |
| 3 (iGb3-O-TMR) | 87 | 6400 |
| 4 (S-iGb3-O-TMR) | n.h. | 69 |
| 5 (Gb3-O-TMR) | 15 | n.h. |
| 6 (S-Gb3-O-TMR) | n.h. | n.h. |
| 7 (iGb3-Cer-TMR) | 17 | 1200 |
| 8 (S-iGb3-Cer-TMR) | n.h. | 36 |
| 9 (Gb-Cer-TMR) | 33 | n.h. |
| 10 (S-Gb3-Cer-TMR) | n.h. | n.h. |
n.h. = no hydrolysis, the reaction rate is less than 0.02 pmol*min−1μg−1.
Compounds containing natural α-galactose as the terminal sugar (3, 5, 7 and 9) were readily hydrolyzed to yield the corresponding lactosides. The reaction rate depended on the nature of the substrate. Generally O-TMR was preferred as the aglycon compared to the ceramide and the αGal(1→3)Gal linkage was hydrolyzed more rapidly than the αGal(1→4)Gal linkage. LacNAc-O-TMR (1) and Lac-O-TMR (2a), containing terminal β-galactose, were not hydrolyzed. On the other hand, galactosides containing terminal 5-thio-galactose (4, 6, 8 and 10) were resistant to hydrolysis by α-galactosidase A. Very slow degradation at a rate less than 0.02 pmol*min−1μg−1 enzyme was observed only after four days of incubation.
Guce et al., have reported X-ray crystal structures of human α-galactosidase in complex with substrates which showed that ring oxygen of galactoside has a van der Waals interaction with a cysteine residue on α-galactosidase.[20] The sulfur substitution for the oxygen atom might cause electron repulsion and destabilize the α-galactosidase-substrate complex.
The panel of substrates were next evaluated with B. fragilis α-1,3-galactosidase (FragB) which is specific for α-1,3 linkages including blood group B antigens.[21] This enzyme catalyzes hydrolysis with inversion of configuration. To date the FragB gene family has only been found in prokaryotes and no homologues or similar inverting enzymes have yet been found in eukaryotes.[21] As expected, α-1,3-galactosidase did not act on compounds lacking a α-1,3 galactoside linkage (1, 2a, 5, 6, 9 and 10) (Table 1, right). However, all iGb3 derivatives (3, 4, 7 and 8) including thio-galactoside compounds were hydrolyzed by the enzyme. Even though the hydrolysis rates of thio-galactosides 4 and 8 were very slow (1 or 3% of the O-galactosides), complete hydrolyses was observed in all iGb3 derivatives after long incubation times (data not shown).
Inverting glycosidase reactions are thought to occur via a single-displacement mechanism with an oxocarbenium ion-like transition state. Most retaining glycosidase reactions are believed to proceed via a double displacement mechanism (two transition states) with formation of a covalent enzyme-substrate intermediate.[24] Replacement of oxygen with a sulfur atom in the sugar ring might impair inverting galactosidases less than retaining enzymes if the transition state activation barrier is greater for thio-galactosides.
Hydrolytic resistance assay of TMR-labeled galactosides to enzymes from cell extracts
Since numerous α-galactosidases in cells can potentially hydrolyze Gb3 and iGb3, we therefore examined TMR-labeled compound (1-10) in extracts from MCF-7 and NG108-15 cells. The enzyme solution was prepared by lysis of the cell extracts and the protein concentrations were determined by the Bradford method; 3.8 mg/ml from MCF-7 and 5.0 mg/ml from NG108-15, respectively. By using the extracted enzyme solution, the hydrolytic stabilities of TMR-labeled galacotosides 1-10 were evaluated as was done for human α-galactosidase A and B. fragilis α-1,3-galactosidase.
The corresponding compounds containing natural galactose (3, 5, 7 and 9) were readily degraded. The substrate preference observed was similar to that of α-galactosidase A, suggesting that this enzyme is also present in the extracts, despite the fact that a distinct band corresponding to human α-galactosidase A was not evident from SDS page analysis (data not shown). In this assay, LacNAc-O-TMR (1) and Lac-O-TMR (2a) were both hydrolyzed. The degradation pattern was different depending on whether the pure enzyme or an enzyme extract was used. While α-galactosidase cleaved only the terminal sugar, incubation with either cellular extract yielded further hydrolysis products as well, due to the action of β-galactosidases in the extracts.
Figure 3 presents the hydrolysis products from Lac-O-TMR (2a) and iGb3-O-TMR (3) after incubation with each cell extract. The β-linked galactoside in Lac-O-TMR was cleaved within 3 hours giving Glc-O-TMR and aglycon O-TMR as degradation products. Gb3-O-TMR was hydrolyzed more slowly than Lac-O-TMR, however, more than 20% hydrolysis occurred after 24 h incubation in both cell extracts. MCF-7 cellular extracts exhibited higher hydrolysis activity compared to NG 108-15 extracts.
Figure 3.
Structure of UDP-5′thio-galactose used in this study
Compounds (4, 6, 8 and 10) containing terminal 5-thio-galactose were resistant to degradation by enzymes present in the cell extracts. The thio-galactose therefore appears resistant to the action of cellular α- galactosidases.
In vivo stability of 5-thio-galactoside using NG108-15 cells
On the basis of the in vitro assay, we tested the hydrolytic resistance of 5-thio-galactosides in vivo with NG 108-15 cells. The cells were grown at 37 °C for 24 h with S-iGb3-O-TMR (4) added to the medium and then the intracellular contents in cell homogenate were analyzed by CE. Lac-O-TMR (2a) (7%) was present as a contaminant in this preparation of 4 due to incomplete enzymatic synthesis. If the two TMR-labeled compounds are taken up by the cells and show the hydrolysis properties observed in the in vitro assay (Table 2), only Lac-O-TMR should be digested by intracellular galactosidases. Gb3-O-TMR (3) was assayed in the same way as S-iGb3-O-TMR.
Table 2.
Rate in enzyme-catalyzed hydrolysis of ten TMR-labeled compounds, using cell extracts.
| Compound | MCF-7 (pmol*h−1) | NG108-15 (pmol*h−1) |
|---|---|---|
| 1 (LacNAc-O-TMR) | 82.5 | 18.5 |
| 2a (Lac-O-TMR) | 17.5 | 14.5 |
| 3 (iGb3-O-TMR) | 10.5 | 3.5 |
| 4 (S-iGb3-O-TMR) | n.h. | n.h. |
| 5 (Gb3-O-TMR) | 1.4 | 0.3 |
| 6 (S-Gb3-O-TMR) | n.h. | n.h. |
| 7 (iGb3-Cer-TMR) | 1.0 | 1.6 |
| 8 (S-iGb3-Cer-TMR) | n.h. | n.h. |
| 9 (Gb-Cer-TMR) | 0.5 | 0.5 |
| 10 (S-Gb3-Cer-TMR) | n.h. | n.h. |
n.h. = no hydrolysis, the reaction rate is less than 0.02 pmol*h−1.
Figure 4 shows the electropherogram of NG 108-15 cell contents after incubation with (a) S-iGb3-O-TMR (containing 7% Lac-O-TMR) or (b) iGb3-O-TMR. S-iGb3-O-TMR was not hydrolyzed in vivo; while the peak corresponding to Lac-O-TMR was greatly reduced with conversion to Glc-O-TMR. In contrast, iGb3-O-TMR was hydrolyzed to Lac-O-TMR and Glc-O-TMR. Anabolic glycosylated products were not generated from either iGb3 substrate. However, these results revealed that 5-thio-galactoside is stable towards hydrolysis in vivo as well as in vitro.
Figure 4.

TMR labeled compounds (1-10) and their enzymatic synthesis.
Conclusion
In summary, this report shows that glycosides containing terminal α-linked 5-thio-galactosyl residues are resistant to hydrolysis by mammalian α-galactosidases in vivo and in vitro. This investigation suggests that 5-thio-galactosides might be useful in the studies of anabolic pathways in cells.
Experimental Section
Materials
UDP-Gal was from Merck, Darmstadt, Germany. UDP-5′-thio-galactose and tetramethylrhodamine (TMR) labeled substrates were prepared following literature procedures.[10] LacNAc-O-TMR (1), Lac-O-TMR (2a) and Lac-Cer-TMR (2b) were prepared as previously described.[25,26] Human α-galactosidase was provided by Shire, Lexington (MA). Bacteroides fragilis α-1,3-galactosidase, BfGal110B (FragB) which hydrolyzes blood group B and linear α1,3-Gal structures was expressed and purified as previously reported.[21] The production and purification of recombinant bovine α-1,3-galactosyltransferase[27] and α-1,4-galactosyltransferase from Neisseria meningitidis[28] have been described earlier. Sep-Pak C-18 cartridges were obtained from Waters. The rat glioma NG108-15 cells were from Dr. Ronald L. Schnaar, Johns Hopkins University, Baltimore. MCF-7 cells were provided by Dr. Mathilde Lerche, Albeda Research ApS, Copenhagen. Dulbecco’s modified Eagle’s medium (DMEM) (containing 4500 mg/L glucose, L-glutamine and sodium bicarbonate), fetal bovine serum (FBS), medium supplement containing hypoxanthine, aminopterin, and thymidine (HAT), penicillin-streptomycin solution and trypsin-EDTA solution were from Sigma. MALDI-TOF MS spectra were recorded on Bruker Microflex instruments using 2,5-Dihydroxybenzoic acid [10mg dissolved in 1 mL of 50/50 (v/v) acetonitrile/water with 0.1 % TFA] as matrix.
Enzymatic Synthesis of TMR labeled glycosides (3-10)
Eight glycosides containing galactose or 5-thio-galactose as the terminal sugar were enzymatically synthesized with regio-specific galactosyltransferases using UDP-Gal or UDP-5′-thio-galactose as donors, respectively. The enzyme reactions were carried out at ambient temperature and were monitored by TLC using aluminum backed silica gel 60 TLC plates and CHCl3:MeOH:H2O (65:35:5) as the eluent.
α-D-Galp-(1→3)-β-D-Galp-(1→4)Glcp-O-TMR (iso-globotriose-O-TMR, iGb3-O-TMR, 3) was synthesized from Lac-O-TMR (2a) and UDP-Gal using α-1,3-galactosyltransferase. The reaction mixture (100 μL) consisted of 20 nmol Lac-O-TMR, 40 nmol UDP-Gal and 0.14 mg α-1,3-galactosyltransferase in 23 mM MOPS buffer, pH 7.0, containing 150 mM NaCl, 3 mM MnCl2, 0.3 mM DTT and 0.01 % BSA. After 1.0 h another portion of donor (50 nmol) was added. The reaction was allowed to proceed for 1.5 h. MS (MALDI-TOF): m/z: calcd for C54H75N4O21 [M+H]+: 1115.49; found 1115.51.
The 5-thio-galactose analog of S-iGb3-O-TMR (4) was produced in the same way with the addition of another portion of UDP-5′-thio-galactose donor (25 nmol) after 1.5 h. After the second donor addition the reaction was allowed to proceed for another 0.5 h. MS (MALDI-TOF): m/z: calcd for C54H75N4O20S [M+H]+: 1131.47; found 1131.49.
α-D-Galp-(1→4)-β-D-Galp-(1→4)Glcp-O-TMR (Globotriose-O-TMR, Gb3-O-TMR, 5) was synthesized from Lac-O-TMR (2a) and UDP-Gal using α-1,4-galactosyltransferase. The reaction mixture (100 μL) consisted of 20 nmol Lac-O-TMR, 40 nmol UDP-Gal and 30 μg α-1,4-galactosyltransferase in 7 mM MOPS buffer, pH 7.0, containing 3 mM MnCl2, 0.01 % BSA and 0.6 mM DTT. The reaction was complete after 30 min at ambient temperature. MS (MALDI-TOF): m/z: calcd for C54H75N4O21 [M+H]+: 1115.48; found 1115.51.
S-Gb3-O-TMR (6) was produced the same way, except that two more portions of UDP-5′-thio-galactose donor (50 and 25 nmol) were added after 1.0 and 1.5 h , with a total reaction time of 1.75 h. MS (MALDI-TOF): m/z: calcd for C54H75N4O20S [M+H]+: 1131.47; found 1115.48.
α-D-Galp-(1→3)-β-D-Galp-(1→4)Glcp-Ceramide-TMR (iGb3-Cer-TMR, 7) was synthesized from Lac-Cer-TMR (2b) and UDP-Gal using α-1,3-galactosyltransferase. The reaction mixture (65 μL) contained 70 nmol Lac-Cer-TMR, 70 nmol UDP-Gal and 0.1 mg α-1,3-galactosyltransferase in 22 mM MOPS buffer, pH 7.0, containing 150 mM NaCl, 3 mM MnCl2, 0.3 mM DTT and 0.01 % BSA. Two more portions of UDP-galactose (70 nmol each) were added after 1.1 and 3.3 h. The reaction was allowed to proceed for 6.4 h. MS (MALDI-TOF): m/z: calcd for C64H93N4O22 [M+H]+: 1269.63; found 1269.60.
S-iGb3-Cer-TMR (8) was produced the same way with UDP-5′-thio-galactose as the donor. MS (MALDI-TOF): m/z: calcd for C64H93N4O21S [M+H]+: 1285.61; found 1285.57.
α-D-Galp-(1→4)-β-D-Galp-(1→4)Glcp-Ceramide-TMR (Gb3-Cer-TMR, 9) was synthesized from Lac-Cer-TMR (2b) and UDP-Gal using α-1,4-galactosyltransferase. The reaction mixture (74 μL) contained 70 nmol Lac-Cer-TMR, 70 nmol UDP-Gal and 0.06 mg α-1,4-galactosyltransferase in 20 mM MOPS buffer, pH 7.0, containing 8 mM MnCl2, 0.04 % BSA and 1.7 mM DTT. Additional UDP-Gal (70 nmol) was added after 1.1 h. The reaction was terminated after 3.3 h. MS (MALDI-TOF): m/z: calcd for C64H93N4O22 [M+H]+: 1269.63; found 1269.47.
S-Gb3-Cer-TMR (10) was produced in a similar way, with the addition of two more portions of UDP-5′-thio-galactose donor (50 nmol each), after 3.3 and 4.4 h. Additional enzyme (0.02 mg) was added after 3.3 h. The reaction time was 5.1 h. MS (MALDI-TOF): m/z: calcd for C64H93N4O21S [M+H]+: 1285.61; found 1285.50.
Purification and quantification of TMR-Labeled Glycosides
The reaction products were purified by preparative TLC using aluminum backed silica gel 60 TLC plates and CHCl3:MeOH:H2O (65:35:5) as the eluent. The product bands, clearly visible due to the TMR label, were scraped from the plates. The TLC eluent (typically 10 mL) was used for product extraction from the silica scrapings. The solvent was removed in a rotary evaporator and the TMR labeled compounds were dissolved in water. The resulting aqueous solution was loaded onto a C-18 Sep-Pak cartridge, previously activated with methanol (10 mL) and then washed with MilliQ water (10 mL). The cartridges were washed with MilliQ water (50 mL) then the TMR-compounds were eluted with methanol (30 mL). The eluate was evaporated and dissolved in water. The concentration of the TMR labeled compounds was determined by comparison with TMR labeled compounds of known concentration using a Nanodrop UV-Vis spectrophotometer from Saveen & Werner (Limhamn, Sweden).
In vitro Hydrolysis Assay of TMR-labeled Glycosides with Human α-Galactosidase and B. fragilis α-1,3-galactosidase (FragB)
Human α-Galactosidase: The enzyme solution was prepared by diluting commercial human α-galactosidase solution (1mg/mL) with buffer, pH 4.6, containing 20 mM Na2HPO4, 30 mM citric acid and 150 mM NaCl. 100-fold diluted enzyme solutions were used for the O-compounds (1, 2, 3, 5, 7 and 9), and 10-fold diluted enzyme for thio-compounds (4, 6, 8 and 10). For the hydrolysis assay, 100 μM TMR-labeled glycoside solution (5 μL) and human α-galactosidase (5 μL) were mixed and incubated at 25 °C. In a time course (O-compound; 16 min and 31min and thio-compound; 1day and 5 days), reaction mixture (5 μL) was taken and added to 10% capillary electrophoresis (CE) buffer (50 μL) for analysis by CE. B. fragilis α-1,3-galactosidase; The enzyme solution (9.8 μg/mL) was diluted with 100 mM NaPO4 buffer, pH 6.8, containing 50 mM NaCl, 50-fold diluted enzyme for assaying compounds 3 and 10-fold diluted enzyme for compound 7. For the remaining compounds, undiluted enzyme solution was used. The assay was performed as human α-galactosidase with a time course of 16 and 31min at 25 °C.
Cell Culture
NG108-15 cells were cultured in DMEM supplemented with 5% (v/v) FBS and HAT. MCF-7 cells were maintained in DMEM supplemented with 10% (v/v) FBS and 2% penicillin/streptomycin. Both cell lines were incubated at 37 °C in a humidified atmosphere containing 5% CO2 and harvested with a 0.25% trypsin-EDTA solution.
Preparation of Enzyme Solution from Cell Extracts
10 million cells (NG-108-15 or MCF-7) were lysed by the addition of 1 mL of hypotonic buffer, pH 4.6, composed of 40 mM Na2HPO4, 22 mM citric acid, 0.1% TritonX-100 and protein inhibitors (Complete Mini, Roche). Lysis was carried out for 10 minutes on ice followed by centrifugation at 10,000 g for 15 minutes at 4 °C to remove cell debris. The supernatants were concentrated to 100 μL at 13,000 g for 20 minutes at 4 °C using a Vivaspin 500 column (10 kDa molecular weight cut-off membrane, Millipore). The protein contents were analyzed by SDS-PAGE and the protein concentration was determined by the Bradford method using a kit from Bio-Rad with bovine gamma globulin as a reference protein standard.
In vitro Assay of TMR-labeled Glycosides Using Enzyme Solutions from Cell Extracts
100 μM substrate solution (5 μL) and enzyme solution from cell extracts (5 μL) were incubated at 25 °C. Reaction mixture (3 μL) was taken in a time course (3, 24 and 48 h) and added to 10 % CE buffer (50 μL) for injection in CE.
In vivo Hydrolysis Assay of S-iGb3-O-TMR/iGb3-O-TMR in NG108-15 Cells
NG108-15 cells were seeded in 24-well plates (25,000 cells per well) and allowed to attach over night. The medium was then replaced with the complete medium (500 μL) containing 25 μM of iGb3-O-TMR (3) or 25 uM of S-iGb3-TMR (4) and 1.8 μM of Lac-O-TMR (2a) then cells were grown at 37 °C for 24 h. Cells were dissociated in trypsin-EDTA solution then washed 5 times with PBS. The cells were resuspended with ice-cold PBS (500 μL) and homogenized in a tissue grinder with 20 gentle strokes every 15 min over a 1.5 h period. The resulting homogenate was centrifuged and the supernatant was purified with C18 Sep-Pak cartridge as described above. The eluate was concentrated and resulting residue was dissolved with 10% CE buffer and analyzed by CE. As blank controls, cells were treated in the same way without TMR compound. All experiments were run in duplicate.
CE Analysis
CE analyses were performed on an automated PrinCE 560 CE system from PrinCE Technologies B.V., Emmen (The Netherlands). Separations were carried out in uncoated fused-silica capillary of 75 μm i.d. with a length of 60 cm using 50 mM borate, pH 9.3, and 150 mM sodium dodecyl sulfate (SDS) as running buffer. TMR-labelled compounds were detected and quantitated using an Argos 250B fluorescence detector (Flux Instruments, Switzerland) equipped with an excitation filter of 546.1/10 nm and an emission filter of 570 nm. All experiments were carried out at a normal polarity, that is inlet anodic. Data were processed by PrinCE 7.0 software. Most peaks were identified based on co-injection with authentic standards.
Acknowledgements
The authors wish to sincerely thank Dr. Boqian Wu (Carlsberg Laboratory, Copenhagen) and Dr. Mathilde Lerche (Albeda, Copenhagen) for their help and advice on cell culture experiments. This work was supported by grant R33DK70317 from the National Institutes of Health, USA, Danish Research Councils and the University of Copenhagen. The authors thank Micheline Wille MD from Shire HGT for having provided human α-galactosidase.
References
- [1].Wennekes T, van den Berg RJBH, Boot RG, van der Marel GA, Overkleeft HS, Aerts JMFG. Angew. Chem. Int. Ed. 2009;48:8848–8869. doi: 10.1002/anie.200902620. [DOI] [PubMed] [Google Scholar]
- [2].Tettamanti G. Glycoconjugate J. 2003;20:301–317. doi: 10.1023/B:GLYC.0000033627.02765.cc. [DOI] [PubMed] [Google Scholar]
- [3].Yu RK, Nakamati Y, Yanagisawa M. J. Lipid Res. 2009;50:S440–S445. doi: 10.1194/jlr.R800028-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Hakomori S. Cancer Res. 1996;56:5309–5318. [PubMed] [Google Scholar]
- [5].Xu Y-H, Barnes S, Sun Y, Grabowski GA. J. Lipid Res. 2010;51:1643–1675. doi: 10.1194/jlr.R003996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ohtsubo K, Marth JD, J.D. Cell. 2006;126:855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- [7].Cohen D, Dickerson JA, Whitmore CD, Turner EH, Palcic MM, Hindsgaul O, Dovichi NJ. Annu. Rev. Anal. Chem. 2008;1:165–190. doi: 10.1146/annurev.anchem.1.031207.113104. [DOI] [PubMed] [Google Scholar]
- [8].Whitmore CD, Hindsgaul O, Palcic MM, Schnaar RL, Dovichi NJ. Anal. Chem. 2007;79:5139–5142. doi: 10.1021/ac070716d. [DOI] [PubMed] [Google Scholar]
- [9].Whitmore CD, Olsson U, Larsson EA, Hindsgaul O, Palcic MM, Dovichi NJ. Electrophoresis. 2007;28:3100–3104. doi: 10.1002/elps.200700202. [DOI] [PubMed] [Google Scholar]
- [10].Yuasa H, Hindsgaul O, Palcic MM. J. Am. Chem. Soc. 1992;114:5891–5892. [Google Scholar]
- [11].Fernandez-Bolanos JG, Al-Masoudi NAL, Maya I. Adv. Carbohydr. Chem. Biochem. 2001;57:21–98. doi: 10.1016/s0065-2318(01)57015-8. [DOI] [PubMed] [Google Scholar]
- [12].Robina I, Vogel IP, Witczak ZJ. Curr. Org. Chem. 2001;5:1177–1214. [Google Scholar]
- [13].Aguirre-Valderrama A, Dobado JA. J. Carbohydr. Chem. 2006;25:557–594. [Google Scholar]
- [14].Izumi M, Tsuruta O, Kajihara Y, Yazawa S, Yuasa H, Hashimoto H. Chem. Eur. J. 2005;11:3032–3038. doi: 10.1002/chem.200400831. [DOI] [PubMed] [Google Scholar]
- [15].Yuasa H, Izumi M, Hashimoto H. Curr. Top. Med. Chem. 2009;9:76–86. doi: 10.2174/156802609787354270. [DOI] [PubMed] [Google Scholar]
- [16].Perlin AS. Pure Appl. Chem. 1978;50:1401–1408. [Google Scholar]
- [17].Hashimoto H, Kawanishi M, Yuasa H. Chem. Eur. J. 1996;2:556–560. doi: 10.1002/chem.19960020515. [DOI] [PubMed] [Google Scholar]
- [18].Morii Y, Matsuda H, Ohara K, Hashimoto M, Miyairi K, Okuno T. Bioorg. Med. Chem. 2005;13:5113–5144. doi: 10.1016/j.bmc.2005.05.028. [DOI] [PubMed] [Google Scholar]
- [19].Andrews JS, Weimar T, Frandsen TP, Svensson B, Pinto BM. J. Am. Chem. Soc. 1995;117:10799–10804. [Google Scholar]
- [20].Guce AI, Clark NE, Salgado EN, Ivanen DR, Kulminskaya AA, Brumer H, III, Garman SG. J. Biol. Chem. 2010;285:3625–3632. doi: 10.1074/jbc.M109.060145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu QP, Yuan H, Bennett EP, Levery SB, Nudelman E, Spence J, Pietz G, Saunders K, White T, Olsson ML, Henrissat B, Sulzenbacher G, Clausen H. J. Biol. Chem. 2008;283:8545–8554. doi: 10.1074/jbc.M709020200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Adlercreutz D, Weadge JT, Petersen BO, Duus JØ, Dovichi NJ, Palcic MM. Carbohydr. Res. 2010;345:1384–1388. doi: 10.1016/j.carres.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ahmad S. Nat. Rev. Immunol. 2007;7:325. [Google Scholar]
- [24].Rempel BP, Withers SG. Glycobiology. 2008;18:570–586. doi: 10.1093/glycob/cwn041. [DOI] [PubMed] [Google Scholar]
- [25].Zhang YN, Le XC, Dovichi NJ, Compston CA, Palcic MM, Diedrich P, Hindsgaul O. Anal. Biochem. 1995;27:368–376. doi: 10.1006/abio.1995.1293. [DOI] [PubMed] [Google Scholar]
- [26].Larsson AE, Olsson U, Whitmore CD, Martins R, Tettamanti G, Schnaar RL, Dovichi NJ, Palcic MM, Hindsgaul O. Carbohydr. Res. 2007;342:482–489. doi: 10.1016/j.carres.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Blanken WM, Van den Eijnden DH. J. Biol. Chem. 1985;260:12927–12934. [PubMed] [Google Scholar]
- [28].Wakarchuk WW, Cunningham A, Watson DC, Young NM. Protein Eng. 1998;11:295–302. doi: 10.1093/protein/11.4.295. [DOI] [PubMed] [Google Scholar]