

Abstract
In this review, we discuss T-cell activation, etiology, and the current therapies of autoimmune diseases (i.e., MS, T1D, and RA). T-cells are activated upon interaction with antigen-presenting cells (APC) followed by a “bull’s eye”-like formation of the immunological synapse (IS) at the T-cell–APC interface. Although the various disease-modifying therapies developed so far have been shown to modulate the IS and thus help in the management of these diseases, they are also known to present some undesirable side effects. In this study, we describe a novel and selective way to suppress autoimmunity by using a bifunctional peptide inhibitor (BPI). BPI uses an intercellular adhesion molecule-1 (ICAM-1)-binding peptide to target antigenic peptides (e.g., proteolipid peptide, glutamic acid decarboxylase, and type II collagen) to the APC and therefore modulate the immune response. The central hypothesis is that BPI blocks the IS formation by simultaneously binding to major histocompatibility complex-II and ICAM-1 on the APC and selectively alters the activation of T cells from TH1 to Treg and/or TH2 phenotypes, leading to tolerance.
Keywords: autoimmune diseases, immunological synapse, bifunctional peptide inhibitor (BPI), antigenic peptide
1. IMMUNOLOGICAL SYNAPSE (IS)
A. Mechanism of Formation of IS
T-cell activation is initiated upon interaction of antigen-presenting cells (APC) with T cells via major histocompatibility complex (MHC) and T-cell receptors (TCR) for generating adaptive immune response.1,2 Monks et al. were the first to report the formation of a three-dimensional cell–cell contact between a fixed single T cell and an antigen-presenting cell (APC).3 This cell–cell contact is an interaction of surface receptors and intracellular proteins in a well-organized and spatially distributed manner, leading to the formation of two concentric rings termed “supramolecular activation clusters” (SMAC). The inner ring is referred as the central TCR-SMAC (c-SMAC or Signal-1). It is composed of protein kinase C (PKC-θ) surrounded by an outer or peripheral SMAC (p-SMAC or Signal-2) enriched mainly with leukocyte function-associated antigen-1 (LFA-1) and talin. Initial contact between the T cell and APC involving TCR and MHC–peptide (MHC–p) and other costimulatory molecules is called the “immunological synapse” (IS).4 Grakoui et al. have shown that the formation of a nascent IS is initiated by Signal-2 (i.e., intercellular adhesion molecule-1 (ICAM-1) and LFA-1 interactions) at the central junction and by Signal-1 (TCR–MHC–p interactions) at the peripheral junction of the interface between APC and T cells.5 Signal-1 and Signal-2 then exchange places (translocate) via actin-based movement to form a stable Signal-1 cluster at the center and a Signal-2 cluster at the peripheral junction (Fig. 1).
Figure 1.

Mechanism of immunological synapse formation during T cell and APC interaction. (A) Initial contact between Signal-1 (TCR/MHC-II-peptide complex) and Signal-2 (LFA-1/ICAM-1complex). (B) Translocation of Signal-1and Signal-2 to form c-SMAC and p-SMAC of the immunological synapse. APC, antigen-presenting cell; SMAC, supramolecular activation clusters; ICAM-1, intercellular adhesion molecule-1; MHC, major histocompatibility complex; TCR, T-cell receptors; LFA, leukocyte function-associated antigen; p-SMAC, peripheral SMAC.
Many costimulatory molecules (Signal-2) have been discovered, including CD28/B7-1 (CD80) and CD28/B7-2 (CD86) as positive costimulatory signals and cytotoxic T lymphocyte antigen (CTLA-4)/B7-1 and CTLA-4/B7-2 as negative costimulatory signals (Fig. 2).6–8 The cytoskeletal protein talin and CD2-associated protein as well as intracellular signaling proteins, such as PKC-θ, LcK, ZAP, Fyn, and MEKK2, have also been identified.8,9 The roles of negative and positive costimulatory signals are to maintain the balance between the regulatory and effector functions of T cells in the immune system.
Figure 2.

Signaling molecules involved in the interface of T cell and APC interaction. The interaction between T cell and APC involves several pairs of receptors (Signal-1 and -2) and is associated with the release of cytokines (Signal-3). APC, antigen-presenting cell.
The structure and function of IS are still not well understood because the formation of a mature IS occurs via a dynamic process. In other words, the formation of a mature IS is not merely the formation of protein clusters to sustain TCR signaling; it also requires the involvement of TCR-mediated tyrosine kinase signaling before IS maturation.10,11 This suggests that the process of IS formation could be preceded by T-cell activation10,11 and possibly the secretion of cytokines or cytotoxic agents by CD4+ and CD8+ T cells, respectively, upon interaction with APC.11–16 The IS enhances the interaction of CD28/B7-1/2 at the center.11 Lezzi et al.17 demonstrated that the activation and deletion of either naïve or effector T cells is dependent on the duration of antigenic stimulation. Prolonged antigenic stimulation is required for the activation of naïve T cells, but it causes apoptosis in effector T cells. Celli et al. found that more than 6 hr interaction between naïve CD4+ T cells and dendritic cells (DC) is necessary to produce T-cell clonal expansion and inhibition of TCR-MHC interactions that stops the T cell–DC interactions.18 Furthermore, TCR clustering enhances binding with MHC–p, and the cSMAC is an important site for strong TCR signaling.19,20 The internalization of TCR is required for downregulation of TCR signaling.19 The formation of IS may be required for gene activation to induce and secrete effector molecules and for intercellular transfer of MHC–p complex along with other receptors such as B7-1 from APC to T cell.8,21,22 This event may have biological repercussions, allowing T cells to behave as APC, altering the cellular functions and over all controlling the immune response itself.23
B. Signal-1
A complex of an immunogenic peptide and purified MHC-II (Ia) molecule is recognized by the TCR as a model for Signal-1,24,25 and the antigen presentations by MHC-I- or CD1 MHC-like molecules have been described previously.26–30 These findings were further confirmed by solving various crystal structures of peptide–MHC complexes.31–39 Later, the specific TCR recognition of MHC–p complex on the surface of APC as a result of antigen processing was termed as Signal-1.40,41 TCR consists of an α/β or γ/δ heterodimer connected via a disulfide bond with one variable and one constant region at the extracellular domain.42 The variable domain has the greatest diversity as a complementary-determining region 3 (CDR3).43 The sequence of CDR3 is primarily involved in recognizing the antigenic peptide presented by MHC molecules on APC.44 MHC class I and class II molecules on APC bind antigenic peptides derived from endogenous and exogenous proteins, respectively. Lipid antigens captured from exogenous antigens bind to CD1 MHC-like molecules.30,45
Upon activation, T cells can differentiate into TH1, TH2, and TH17 cells. TH1 cells produces cytokines such as IFN-γ and lymphotoxin.4,46 TH2 cells produce IL-4, IL-13, IL-5, and IL-6 cytokines,4,14,46 and TH17 cells generate IL-17.47 Naïve CD4+ T cells can also be differentiated into an independent lineage called regulatory T cells (Treg), which express fork-head transcription factor Foxp3 and CD25.47 CD4+ T cells execute many important effector functions, depending on the cytokines they secrete.46 Treg cells are also produced and primed with antigen in the thymus as part of mature cells for controlling immune response.48 CD8+ T cells are capable of differentiating into cytotoxic T lymphocytes (CTL);12 these cells recognize specific antigens presented by MHC class-I on their target cells and perform cell lyses.13,15
There are varieties of MHC-II (polymorphic) expressed on the surface of professional APC (pAPC) (i.e., B cells, DC, and macrophages). MHC-II molecules are generated as heterodimers (α- and β-chains) with α1–α2 and β1–β2 domains at the extracellular space.35 The α1- and β1-domains bind to the antigenic peptide for presentation to TCR.49 Except for the α1 domain of MHC-II, all other extracellular domains possess an intradomain disulfide bond. The transmembrane and cytoplasmic domains of MHC-II posses specific sequences required for efficient surface expression,50 signaling,51 antigen presentation,52,53 and lateral diffusion of MHC-II molecules on the surface of the APC.54
The antigens can be processed and presented by different APC in different ways. For example, the antigen presentation by MHC-II on DC is more effective for activating naïve T cells than that by B cells.55 Before the presentation of the peptide fragments of the antigen, the antigen is first internalized via phagocytosis, fluid-phase uptake by macropinocytosis, or receptor-mediated endocytosis via mannose and lectin-like receptors.56–58 Then, they are processed via early endosome and then transferred to late endosome before entering lysosomes.59–63 Because MHC-II compartments (MIIC) are rich in lysosomal enzymes, proteins are degraded by lysosomal proteases to generate peptide fragments; these peptides are loaded onto MHC-II molecules in the MIIC.64–66 The new MHC-II molecules are synthesized and assembled with invariant chain (Ii) in the endoplasmic reticulum to form MHC-II–Ii complexes, which are then transported to the Golgi apparatus and targeted to the endosomal/lysosomal compartments.67–70 The portion of Ii in MHC-II–Ii complex is then clipped off by proteases to produce MHC-II molecules with class II-associated invariant chain (CLIP). Then, the CLIP is exchanged for antigenic peptide to form the MHC-II–peptide (MHC-II–p) complex that is transported to the surface of the APC for presentation to a CD4+ T cell.28,66
C. Signal-2
Several roles of costimulatory signals (Signal-2) are to stabilize the physical interactions between TCR and MHC as well as modulating the TCR signal for T-cell activation and tolerance.71,72 The positive and negative costimulatory signals can be delivered by CD28/B7 and CTLA-4/B7 interactions (Fig. 2), respectively.73–75 Human CD4+ T cells and half of CD8+ T cells express CD28 with a possible increase in expression upon T-cell activation.76,77 A combination of TCR signaling and costimulation via CD2/B7 pathway can upregulate the production of IL-12 receptor,78–81 Bcl-χL,82 and cytokine transcription.83,84 Suppression of the positive costimulatory signal in the presence of TCR–MHC-II–antigen complex generates T-cell anergy, which can be reversed by delivering IL-12.85,86 Large and small molecules have been designed to block CD28/B7 signals for therapeutic agents to suppress autoimmune diseases. Therefore, it is necessary to balance the positive costimulatory signal with the negative costimulatory signal (CTLA-4/B7) to maintain tolerance. Normally, CTLA-4 is expressed in activated but not in resting T cells to dampen signals delivered via TCR. CTLA-4 expression is observed at 24–48 hr after priming,87 and the strength of expression correlates with stimulus by TCR88 upon recruiting Src homology 2 (SH2) domain-containing phosphatase-2 (SHP-2) during T-cell activation.89
APC normally express B7-1 (CD80) and B7-2 (CD86); these proteins have V- and C-like domains as well as cytoplasmic domains with phosphorylation sequences for PKC recognition.75,90–93 B7-1 and -2 can be upregulated upon activation,94 and they are recognized in lower avidity by CD28 than by CTLA-4.95,96 Both CD28 and CTLA-4 have a MYPPPY motif that is recognized as the extracellular V-like and C-type domains of B7-1 and -2.95,96
Inducible costimulator (ICOS) and programmed death-1 (PD-1) molecules are additional types of costimulatory molecules in the CD28 family. ICOS has a FDPPPF motif rather than a MYPPPY motif in the CD28 molecule; owing to the different in this recognition sequence, ICOS does not bind to B7-1 and B7-2.97,98 However, ICOS binds to B7-h molecule upon stimulation by TCR activation to increase T-cell proliferation97,99–101 as well as IL-4, IL-5, IFN-γ, TNF-α, and GM-CSF production.100 B7-h also regulates the production of IL-10 and plays a role in induction of tolerance.102,103 B cells and macrophages constitutively express B7-h, and its expression can be induced by inflammatory stimuli on fibroblasts as well as nonlymphoid, endothelial, and epithelial cells.
In addition to CTLA-4, PD-1 can deliver negative signals in CD8+ T cells to generate tolerance, and both CTLA-4 and PD-1 signals work in an additive fashion upon sustained connection with their respective ligands.104 The ligands for PD-1 are PD-L1 (B7-H1) (Fig. 2) and PD-L2 (B7-DC).104 PD-L1 is widely expressed on T cells, B cells, macrophages, monocytes, DCs, and nonlymphoid cells, including endothelial cells, syncytiotrophoblasts in the placenta, muscles, and pancreatic islets,105 whereas PD-L2 is only found on macrophages and DCs inducible by cytokines.106 The PD-1 molecule has roles in induction and/or maintenance of peripheral tolerance and acts as a negative regulator of T-cell responses. Mice with PD-1 deficiency developed a fatal dilated cardiomyopathy with early disease onset107 and lupus-like disease.104 PD-1 is expressed during thymic development primarily on CD4− CD28− and γ−δ− thymocytes and induced on peripheral CD4+ and CD8+ T cells, B cells, and monocytes upon TCR stimulation.105 Unlike CTLA-4, PD-1 inhibits the expression of the cell survival gene bcl-XL,105 and both CTLA-4 and PD-1 are known to limit glucose metabolism and Akt activation via different pathways. CTLA-4 inhibits Akt activation via protein phosphatase 2a, and PD-1 inhibits Akt activation via CD28-mediated activation of PI3K. Blocking CTLA-4 with antibody reduces CD8+ T-cell tolerance upon epitope presentation by resting DC; however, blocking CTLA-4 signal in the presence of PD-1 signal produces weak but constant T-cell priming.104
LFA-1 and ICAM-1 function as both adhesion and costimulatory molecules (Fig. 2) for T-cell activation.108 Functionally, LFA-1 has been shown to enhance IL-2 expression, leading to the induction of T-cell proliferation. LFA-1 is exclusively expressed on leukocytes and interacts with its ligands, such as ICAM-1, -2, and –3, to promote a variety of homotypic and heterotypic cell–cell adhesion necessary for normal and pathologic functions of the immune systems. The interaction of LFA-1 with its ligands allows for improved adhesion of leukocytes to vascular endothelium, an essential step in the recruitment and migration of leukocytes into inflamed tissue.109–111 Similarly, LFA-1-mediated adhesion is thought to facilitate antigen presentation to T cells, and numerous reports indicate that LFA-1 engagement decreases the minimal stimulatory dose of antigen by 10- to 100-fold.112–114
D. Signal-3
Signal-3, which is provided by the release of inflammatory cytokines by APC or T cells after initiation of Signal-1 and -2, is required for proliferation and differentiation of T cells.115,116 Signal-3 can polarize helper T cells to become type 1 helper T cells (TH1) or type 2 helper T cells (TH2).117 The “danger theory” proposed by Matzinger indicated that T cells must receive the “danger signal” from Signal-3 and costimulatory signals for their full activation to recognize the danger from pathogens.118,119 In contrast, in the absence of the “danger signal,” the immune system induces tolerance instead of an immunizing effect. To determine the effect of the “danger signal” on CD8+ and CD4+ T-cell proliferation, Mescher et al. utilized microspheres coated with immobilized MHC protein/peptide complexes (artificial APC) to provide Signal-1 and co-immobilized with B7-1 protein for Signal-2.115 IL-12 as Signal-3 was needed along with Signal-1 and -2 to stimulate and proliferate naïve CD8+ T cells and, in the absence of IL-12, the T cells did not differentiate into cytolytic effector cells. Similarly, IL-1 but not IL-12 was necessary to proliferate naïve CD4+ T cells. These results suggest that Signal-3 from inflammatory cytokines is necessary for proliferation of CD4+ and CD8+ T cells to prevent tolerance toward a specific antigen.
As a “polarizing” signal, Signal-3 is delivered to naïve helper T cells for their differentiation to either TH1 or TH2, and the differentiation depends on the type of cytokine delivered as the Signal-3. If the naïve helper T cells receive IL-12 signal, they differentiate into TH1. If they receive IL-4, the helper T cells differentiate into TH2.117 TH1 cells can produce interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) for directing T-cell immunity against intracellular pathogens. TH2 cells produce IL-4, IL-5, and IL-13 for optimal self-proliferation to control humoral immunity as well as fighting against extracellular parasites. The generation of Signal-1 and Signal-2 triggers signal transductions via several different pathways, including calcium–calcineurin, the RAS-mitogen-activated protein (MAP) kinase, and the nuclear factory-κB pathways (Fig. 2).116,120 IL-2 binds to IL-2 receptor (IL-2R) on T cells along with other cytokines to initiate T-cell proliferation through the rapamycin pathway.116 The mechanisms of action of cytokines in triggering the appropriate CD4+ and CD8+ T-cell responses are still not clear and need further investigation.115
2. ANTIGENIC PEPTIDES
Autoimmune diseases could involve a single organ (e.g., multiple sclerosis (MS), rheumatoid arthritis (RA), and diabetes) or no involvement of specific organs (e.g., systemic lupus erythematosus). T cells and B cells are engaged in inflammatory immune responses, leading to the onset and progression of autoimmune diseases. Activation of both T and B cells depends on antigen presentation. Although antigens are necessary for activation of these lymphocytes, several studies have shown that injections of antigenic peptides/proteins could also suppress autoimmune diseases. The mechanisms of suppression by antigens are thought to work by immune deviation and induction of Treg cells. In the following sections, etiologies and current treatments of various autoimmune diseases are discussed.
A. Multiple Sclerosis
MS is an autoimmune disease that affects the central nervous system; patients suffer from muscle weakness, changes in sensation and speech, vision problems, muscle spasms, and fatigue. In this disease, the autoreactive immune cells (i.e., T cells) attack and cause damage to the axonal via demyelination and inflammation.121 The myelin sheath damage hampers the communication between nerve cells due to the disruption of the electric impulses along the axons and affects the optic nerves, brainstem, spinal cord, cerebellum, and periventricular white matter. As the result of oligodendrocyte cell death and loss of axons, plaques and lesions can be observed in the brain white matter as well as in the spinal cord. This is a pathological marker of MS.
Nearly 80% of MS patients have clinical relapses referred to as relapsing–remitting MS (RRMS), which is characterized by inflammation of the brain and spinal cord along with white matter lesions.122 During the RRMS period, patients have unpredictable relapses followed by long remission with no signs and symptoms of the disease; about 65% of these patients will gradually progress toward a secondary progressive MS (SPMS) typified by an irreversible neurological defect.122 Some patients (20%) have a steady neurological decline without remission upon onset of the disease, which is called primary progressive MS (PPMS); however, PPMS is the least common type of MS. Axonal degeneration due to the loss of myelin sheath is commonly observed in PPMS and SPMS.
The cause of MS has not been fully elucidated; however, it is clear that MS patients have a leaky blood–brain barrier (BBB) with brain infiltrations of CD4+/CD8+ T cells, B cells, and monocytes.121,123,124 There is evidence to indicate that proteolipid protein (PLP), myelin basic protein (MBP), myelin oligodendrocyte glycoprotein (MOG), or some peptide epitopes from these proteins act as self-antigens for T-cell activation (Table I). In the CNS, naïve CD4+ T cells recognize the antigen presented by the local APC called microglia and they differentiate to TH1 effector cells, which produce IL-2, IFN-γ, and TNF-α.125 In addition, TH17 cells that secrete IL-17 cytokine are also activated by IL-6, IL-23, and TGF-β.126,127 Thus, MS patients have elevated levels of IL-17 in the blood and cerebrospinal fluid (CSF).128,129 It has been suggested that the BBB of MS patients is leaky and that IL-17 and -22 are responsible for the disruption of the BBB that aids the CNS infiltration of immune cells.122 Injection of anti-IL-17 monoclonal antibody (mAb) into SJL/J mice with autoimmune encephalomyelitis (EAE) suppresses the disease development, indicating that IL-17 has an important role in the pathogenesis of EAE and possibly MS.127,130 Contrary to this proposal, Haak et al. indicated that IL-17 may not be the major player in pathogenesis of EAE in mouse because IL-17F-deficient mice treated with anti-IL-17 mAb can still develop EAE.131 Their study indicated that IL-17 may not have a critical role in the pathogenic function of TH17 cells and TH1 cells are necessary to initiate CNS inflammation followed by CNS infiltration by TH17 cells.131 Haak et al. suggested that although eliminating IL-17 may not be a viable target for treating MS, it is still a good diagnostic marker for MS.131
Table I.
List of Candidate Autoantigens in MS, T1D, and RA
| Disease | Antigen | References |
|---|---|---|
| Multiple sclerosis | Proteolipid protein (PLP) Myelin basic protein (MBP) Myelin oligodendrocyte glycoprotein (MOG) Astrocyte-protein S100β |
309 |
| Type 1 diabetes | Glutamic acid decarboxylase (GAD) Insulin zinc transporter (ZNT8) Insulinoma antigen (IA-2) Insulin |
159–161 |
| Rheumatoid arthritis | Human cartilage gp-39 Cartilage proteins melanoma inhibitory activity (MIA) Type II collagen Citrullinated proteins (e.g., fibrin) |
310–316 |
Treatment of animal models of EAE with soluble myelin antigens can generate tolerance in diseased animals. For example, oral administration of MBP to Lewis rats before the EAE induction suppresses EAE as a result of clonal anergy or deletion of activated T cells with a reduced number of TH1 MBP-specific cells.132–135 Administration of bovine myelin antigens in a small group of MS patients provided a significant decrease in the number of T cells reactive toward MBP compared with the control group and a lower number of treated patients showing major attacks compared with the untreated control group.136 Unfortunately, a subsequent large clinical trial (Phase III) did not show the efficacy of a single oral dose of myelin antigens (MBP and PLP); the previous effects were considered to be a placebo effect. Thus, there are many unanswered questions about the effectiveness of this type of treatment.
Alternatively, intrathecal, intravenous (i.v.), subcutaneous (s.c.), or nasal administration of antigenic peptides has been investigated to induce tolerance in patients. Two phase I trials conducted by Warren et al. demonstrated that administration of MBP peptides, MBP75–95 or MBP85–96, in chronic progressive MS patients did not produce any adverse effects.137,138 Induction of tolerance was monitored by detecting the reduction of anti-MBP antibodies in CSF. Intrathecal administration resulted in transient neutralization, whereas i.v. administration resulted in long-lasting tolerance or reduction in anti-MBP antibodies for up to 1 year after a second i.v. dose of antigen. However, s.c. administration of the antigenic peptides did not affect the reduction of anti-MBP antibodies in CSF, clearly demonstrating that s.c. injection failed to induce tolerance in these patients.138 Unfortunately, these studies failed to relate the clinical significance of the reduction in antibodies against MBP and its effect on the prognosis of the disease in MS patients.
Antigenic peptides derived from MBP were successfully delivered via the nasal route and were effective in inhibiting EAE disease in a murine model.139–142 It has been suggested that proliferation of antigen-specific Treg cells in EAE can be triggered by antigenic-peptide presentation by plasmacytoid dendritic cells (pDC) as part of the professional APC.143 The mechanism of tolerance in this model is due to MBP-reactive specific Treg cells that produce IL-10. In addition, these cells were found to express higher levels of IL-4 and TGF-β mRNA.141,144 Similarly, altered peptide ligands (APL) are mutated antigenic peptides with increased affinity for MHC-II and/or TCR, and they have been shown to control RA and EAE in animal models. A suggested mechanism of action for APL in inhibiting antigen-specific T-cell activation is increasing the expression of indoleamine 2, 3-dioxygenase (IDO) enzyme in activated T cells. Presumably, the increased amount of IDO converts tryptophan to its metabolites (i.e., 3-hydroxykynurenic acid (3-HKA), picolinic acid (PA), and quinolinic acid (QA)) and these metabolites suppress the EAE relapse by reducing the production of inflammatory cytokines and stimulating the production of IL-4 and IL-10 that are related to TH2 and Treg cellular differentiation.145
Another promising approach developed for the treatment of MS involves the use of recombinant T-cell ligands (RTL). A human recombinant T-cell receptor ligand (RTL1000) consisting of DR2 α1+β1 domains of MHC-II molecules linked covalently to MOG35–55 peptide was shown to reverse clinical and histological signs of EAE146 and was evaluated for safety in a phase 1 randomized, placebo-controlled, escalating dose study in 34 patients with MS.147 Preclinical studies of RTL showed that these molecules were effective in ameliorating both active and passive MOG-induced EAE in C57BL/6 mice146 and chronic EAE in SJL/J mice.148 One of the constructs, RTL401 (RTL with bound PLP139–151), was able to suppress EAE in SJL/J mice immunized with PLP139–151 but could not treat MOG35–55-induced EAE.148 Furthermore, RTL401 could not treat EAE in mice immunized singly with PLP178–191 or MBP84–104 peptides, and the same was also true with other RTL constructs linked to different peptides.148 This suggests antigen-specific suppression of EAE by RTL modulation of the cognate T-cell specificity. Interestingly, RTL401 was able to ameliorate EAE induced with syngenic whole spinal cord homogenate, indicating that treatment with a single RTL can suppress EAE induced with multiple T-cell specificities provided that one of the immunizing peptides is present in the RTL being used for treatment. This demonstrates that treatment with a single RTL can deviate autoimmune responses of cognate T-cell specificities while inducing bystander suppression that reverses EAE in mice immunized with multiple encephalitogenic peptides.148 It is thought that the mechanism of action of RTL involves direct reduction of proinflammatory cytokines, such as IL-17 and TNF-α, while concomitantly inducing the production of IL-10, IL-13, and TGF-β and inhibiting the migration of inflammatory cells into the CNS via a reduction in the expression of chemokines/receptors, VCAM-1, and ICAM-1.146,148
Current strategies that are approved for the treatment of MS include two classes of drugs: anti-inflammatory and disease-modifying therapies (DMT). Acute MS relapses are often treated with high-doses of intravenous corticosteroids such as methylprednisolone. This is effective for short-term treatment, but it does not have any effect on long-term recovery. Mitoxantrone (Novantrone®), interferon β-1a (IFNβ-1a or Avonex®/Rebif®/CinnoVex®), IFNβ-1b (marketed as Betaseron®/Betaferon®), glatiramer acetate (GA, Copaxone®), and natalizumab (Tysabri®) are current DMT drugs. Mitoxantrone will not cure the disease, but it is effective in slowing the progression of SPMS and delaying the relapse interval time in RRMS.149 The mechanism of action of mitoxantrone involves disrupting both DNA synthesis and DNA repair. The side effects of mitoxantrone are irreversible cardiomyopathy, acute leukemia, and bone-marrow suppression; patients receiving this treatment are advised to have regular echocardiograms.
IFNβ-1b was first introduced in 1993 for RRMS treatment. In the clinical trials, patients treated with IFNβ-1b were free from relapse at the end of 2 years compared with a placebo-treated group.150 IFNβ-1b acts as an anti-inflammatory agent by reducing the production of IFN-γ and TNF-α and inhibits T-cell activation, clonal expansion, and migration into the CNS.151 The mechanism of action of IFNβ-1a is thought to be similar to that of IFNβ-1b.
Copaxone® (glatiramer acetate) is a synthetic peptide polymer (40–100 residues) that consists of Glu, Lys, Ala, and Tyr amino acids in a ratio of 3:7:9:2; it is designed as a decoy for MBP. Copaxone® has been suggested to alter the immune response from TH1- to TH2-cell differentiation.
Finally, natalizumab (Tysabri®) is the first recombinant human mAb approved for the treatment of MS. It inhibits leukocyte adhesion to vascular endothelium by binding to the α4-subunit of α4β1 and α4β7 integrins on the surface of leukocytes to prevent leukocyte infiltration into the CNS.152 Natalizumab has been shown to significantly slow the progress of disability with increasing numbers of disease-free individuals compared with placebo group. It can also reduce disease relapse, loss of vision, and number of lesions in patients.153–155 Despite its greater efficacy compared with any other approved MS therapies, its use is limited because of the development of potential fatal adverse effects, such as progressive multifocal leukoencephalopathy (PML), melanoma, hepatotoxicity, opportunistic infections, and primary CNS lymphoma.156,157 Recent studies suggest that patients receiving this drug for more than 1 year are at an increased risk for PML.158 Currently, the FDA has limited the usage of natalizumab to patients not responding to other therapies, and the drug is under a mandatory safety monitoring program.
B. Type-1 Diabetes
Type 1 diabetes (T1D) is marked by insulitis and the destruction of the pancreatic islet of Langerhans β cells.159 Although not all-inclusive, studies carried out in nonobese diabetic (NOD) mice show that CD4+ and CD8+ T cells are partly responsible for the destruction of the β-cells.7 Those afflicted with T1D are genetically predisposed, and the progress toward the disease is initiated after encountering an environmental insult, such as diet or microbial infections.7 Insulin, glutamic acid decarboxylase (GAD), zinc transporter (ZNT8), and insulinoma antigen (IA-2) are the four major antigens (Table I) for activation of immune cells, and they have been used in assays to detect the progress of T1D.160,161 Glutamic acid carboxylase 65 kDa (GAD65) is considered a target for therapy because of its partial role in the activation of certain T cells that recognize it as an antigen.159
The immunotherapy of T1D focuses on two alternative methods of treatment: prevention of T1D or treating it at its onset. Ever since its discovery, insulin has been considered to be the gold standard of treatment for patients with Type 1 diabetes. Similar to GAD65,159 insulin has also received attention as a therapeutic method to suppress the activation of T cells.161 By suppressing the activation of T cells, β-cells may be regenerated, and this may lead to the remission of diabetes.7 The administration of autoantigens (GAD65 or insulin) and their peptides has been shown to suppress β-cell autoimmunity.7,159
The role of insulin as an autoantigen is still ambiguous. To date, many epitopes of insulin and proinsulin have been identified.161 Recent findings have shown that insulin auto-antibodies can be induced by insulin B9–23 peptide. NOD mice (H-2d) were immunized with B9–23 peptides, which led to the development of MHC-activated autoantibodies toward insulin.162 However, it has been shown that the antibodies toward insulin react only with insulin and not with NOD B9-23 peptides, suggesting that insulin antibodies recognize the whole molecule rather than the peptide sequence itself.161 Proinsulin, the precursor of insulin, also has a sequence of amino acids (B24–C36) that are recognized by NOD mice CD4+ T cells.163 B9–23 has been hypothesized to play an important role in humans as it did in NOD mice because the amino acid sequences are similar in both species. Higashide et al. have shown that the most frequent epitopes recognized by T cells are B10–24, B1–15, and B11–25, with B9–23, B4–18, and B12–26 being identified in some patients.164 Alternatively, it has been observed in NOD that intranasal vaccines containing proinsulin in a plasmid DNA increased the number of Treg cells and effectively prevented the onset of diabetes.165
GAD65 is an enzyme that is targeted as an autoantigen by T cells. Anti-GAD65 antibodies can be observed in NOD mice before the onset of T1D. Injection of GAD65, intravenously or intrathymically, to young NOD female mice before any signs of disease has been shown to prevent diabetes and induce CD4+ Treg cells.166 Controlling the activation and increasing the induction of Treg cells by GAD65 antigen may help to suppress T1D.159 Tisch et al. revealed that peptides derived from GAD65 are able to suppress the progression of T1D in NOD mice before the onset of insulitis. The peptides used include amino acids 217–236, 247–265, 290–309, and 524–543.167
C. Rheumatoid Arthritis
RA is a chronic idiopathic disease characterized by persistent inflammation and local destruction in the synovial tissues including the synovium, bone, and cartilage. Although the nature of the antigens responsible for RA pathogenesis has not been clearly elucidated, there is evidence that the disease-associated HLA DR (B1*0401, 0404, 0405, and 0101) molecules, which reside in the MHC and participate in antigen presentation, are associated with the disease.168 Viral proteins, such as cytomegalovirus and Epstein-Barr virus, as well as several autologous proteins normally expressed in the joints, including gp39, proteoglycan, and type II collagen, have been implicated in the generation of the pathogenic T-cell response in RA (Table I). The driving force behind rheumatoid inflammation is believed to be CD4+ T cells responding to an antigenic epitope in the synovium in an HLA-DR-restricted manner. Increased numbers of “memory” CD4+ and decreased numbers of “naïve” CD4+ T cells in the synovial tissues of RA patients169,170 suggest that CD4+ T cells play a crucial role in the pathogenesis of RA. Synovial tissue T cells are enriched with CD4+ T cells that do not express the Leu 8 antigen, now known to be the human equivalent of the murine homing receptor.171–173 Loss of this molecule is a characteristic of activated T cells. Synovial fluid also contains augmented expression of the activated antigens HLA-DR, CD9, CD38, CD49a/CD29 (VLA-1), and adhesion molecule CD54 (ICAM-1).174–177
In addition to CD4+ T cells, a number of other inflammatory mediators produced in the rheumatoid synovium, including arachidonic acid metabolites, vasoactive amines, platelet-activating factor, and complement cleavage products, contribute to the inflammation. Recently, IL-17 produced primarily by the TH17 cells has also been implicated in human autoimmune inflammation. A potential key role in diseases such as RA has only recently been attributed to IL-17, although it was discovered decades ago. Elevated levels of IL-17 in blood and synovium of RA patients have been correlated with synovial levels and joint damage.178–180 Recent advancement in research toward new and better-tolerated therapies to attenuate the inflammation and pain associated with RA as well as halt the progression of erosive joint damage has led to the development of strategies for modulating stimulation of immune cells or blocking cytokines as potential therapeutic strategies.
Injections of soluble antigens have been shown to induce antigen-specific immune tolerance. It is thought that tolerance could be achieved through anergy/deletion of CD4+ T cells or the induction of CD4+ Treg cells that produce IL-10 or TGF-β.181 Oral, and more recently nasal, tolerance resulting from administration of antigens has attracted attention as a potential treatment of autoimmune diseases. Oral administration of type II collagen (CII) has been shown to suppress arthritis in mice182 and rats.183 Oral administration of chicken CII to patients with RA provided significantly less progression of the disease compared with placebo-treated patients.184
In another study, peripheral blood mononuclear cells of RA patients responded well in vitro to CII256–271 epitope and its overlapping variants.185 Similarly, oral administration of this peptide has been shown to suppress arthritis in a CIA mouse model.185 Specific T-cell activity, proliferation, and secretion of IFN-γ in spleen cells were actively suppressed in CII250–270-fed mice, and the serum anti-CII, anti-CII250–270 antibody activities and frequency of specific antibody-forming spleen cells were significantly lower in CII250–270-fed mice than in controls. Moreover, IL-4-producing cells (TH2 cells) were upregulated when CIA mice were treated with type II collagen (CII250–270), suggesting that oral administration of CII250–270 can suppress the cellular and humoral immune response in collagen-induced arthritis. Although the majority of animal studies have yielded positive results with the oral tolerance regimen, under some circumstances, mucosal application of antigen may instead exacerbate the disease process.186
Therapeutic effects of murine MHC class II (I-Aq)-derived recombinant T-cell receptor ligands (RTL), consisting of the α1 and β1 domains of MHC class II molecules that are covalently linked to the CII immunodominant peptide (CII257–270), were also found to reduce the incidence and clinical and histological signs of CIA.187 Treatment with RTL resulted in increased CII-specific IgG1and a reduction in IgG2a isotype that is normally associated with a pathogenic response. The mechanism of RTL in CIA appears to be, due to an increased expression of the FoxP3 gene, an upregulation in anti-inflammatory cytokines (IL-10 and IL-13) and a downregulation in proinflammatory cytokines (IL-17 and IFN-γ).187
Abatacept, also known as CTLA4-Ig, was approved in 2005 by the FDA for the treatment of patients with moderate-to-severe active RA who have had an inadequate response to disease-modifying antirheumatic drugs (DMARDS), including methotrexate (MTX) and TNF antagonists. It is a fusion protein composed of the extracellular domain of the CTLA4 molecule complexed to the Fc domain of human IgG1. The IgG1 solubilizes the fusion protein; thus, unlike the membrane-bound CTLA4 molecule, its binding to CD80/CD86 does not induce a negative signal to T cells. Its binding blocks the engagement of the CD28 costimulatory signal required for T-cell activation, resulting in T-cell anergy.188 Patients receiving abatacept responded better than those receiving placebo. Quality of life was also improved with abatacept and benefits were sustained at 12 months, these included significant reduction in disease activity as well as improved physical function.189
Infliximab is a chimeric monoclonal antibody that binds to circulating and transmembrane TNF-α and thereby neutralizes its biological effect. Circulating TNF-α is a form of TNF-α generated by cleavage from the transmembrane precursor. Both forms of TNF-α have been shown to sustain the inflammation process. Transgenic mice overexpressing TNF-α were observed to develop a destructive arthritis resembling RA. Moreover, high levels of TNF-α detected in synovial fluid and in the tissues of patients with RA190 suggest that TNF-α plays a pivotal role in physiologic and pathogenic response.191 In addition to infliximab, a number of agents that block TNF-α have been developed for clinical use, including etanercept, adalimumab, and certolizumab. Etanercept is a conjugate between a human TNF-α receptor and the Fc-portion of the human IgG1. Adalimumab is a recombinant human IgG1 monoclonal antibody specific for TNF-α; it not only inhibits binding of TNF-α to its receptors but also lyses cells expressing membrane-bound TNF-α. Certolizumab, on the other hand, is a polyethylene glycol (PEG)-conjugated humanized anti-TNF Fab fragment. Although anti-TNF-α drugs have been shown to be effective and relatively safe for use in treating RA, blockade of this cytokine may have effects beyond the suppression of synovial inflammation, which can influence mortality of these patients. Mycobacterial infections have been reported among patients undergoing therapy with infliximab.192,193 Therefore, tuberculin skin tests and a chest radiograph are usually recommended before initiation of anti-TNF treatment. Also, avoidance of anti-TNF therapy is recommended for patients with severe chronic heart failure.194 Aside from TNF-α, cytokines appear to be of particular importance in the development of RA and, therefore, have been investigated as therapeutic targets. Kineret (Anakinra) is a recombinant, non-glycosylated form of the IL-1 receptor antagonist (IL-1Ra), a naturally occurring receptor antagonist known to counteract the proinflammatory effects of interleukins. Patients taking background MTX with anakinra had significant clinical improvement as compared with the placebo group.195,196 Simultaneous blockade of TNF-α and IL-1 receptor had very promising results in experimental murine arthritis;197 however, this did not translate well to clinical outcome. Moreover, the risk of mycobacterial infections under combination therapy increased significantly; it is therefore not recommended for RA treatment.198 Tocilizumab (MRA) is a humanized monoclonal antibody against IL-6 receptor (IL-6R). Randomized phase II studies showed that patients receiving tocilizumab had reduced disease activity in a dose-dependent manner.
B lymphocytes play several critical roles in the pathogenesis of RA. They are the source of rheumatoid factors and anticitrullinated protein antibodies, which contribute to the immune complex and complement of T-cell activation in the joints. It is therefore assumed that the depletion of B lymphocytes provides a potential therapeutic approach to controlling disease activity and inducing remission. This hypothesis has been proven in an open-label study of rituximab, a chimeric monoclonal antibody against the protein CD20. CD20 is widely expressed on B cells from early pre-B-cells to later stages in differentiation, but it is absent on terminally differentiated plasma cells. Rituximab was significantly superior to placebo when added to MTX.199 Safety and tolerability of rituximab were found to be comparable to that of the placebo. Moreover, there have been no higher infection rates under B-cell depletion therapy.199
3. BLOCKING SIGNAL-2
Blocking Signal-2 formation has been shown to alter the immune response via induction or anergy of a certain T-cell phenotype (i.e., TH1 or TH2). Inhibition of Signal-2 has been used for the treatment of autoimmune diseases. Monoclonal antibodies and peptides (Table II) that inhibit Signal-2 have been shown to suppress allograft rejection,200,201 type-1-diabetes,202,203 RA,204,205 and psoriasis.206,207 Despite the great promise of mAb, these drugs are costly to produce and difficult to formulate or deliver. Peptides and peptidomimetics are a better alternative to mAbs. Despite progress in designing small molecule inhibitors, a limited number of small molecules have been designed to target Signal-2, but none has yet reached the clinical setting.
Table II.
Antibodies and Peptides That Have Been Investigated for Blocking Signal-2 in Autoimmune Diseases, Organ Transplantation, and Cancer
| Signal-2 Receptor | Peptide/protein ligand | Target disease | References |
|---|---|---|---|
| LFA-1/ICAM-1 | Anti-LFA-1 mAb (Odulimomab, Efalizumab) | Psoriasis, Type 1 diabetes, and transplantation | 203,214,216–219,237 |
| LFA-1/ICAM-1 | Anti-ICAM-1 mAb (Enlimomab) | Rheumatoid arthritis, Type 1 diabetes, and transplantation | 226,233,234 |
| LFA-1/ICAM-1 | Cyclo(1,12)-PenPRGGSVLVTGC (cIBR) | Rheumatoid arthritis, inflammation, and immunosuppression | 246,250 |
| LFA-1/ICAM-1 | Cyclo(1,12)-PenITDGEATDSGC (cLABL) | Rheumatoid arthritis, inflammation, and immunosuppression | 250,251,317,318 |
| LFA-1/ICAM-1 | Cyclo(1,9)-CLLRMRSIC | Inflammation | 319 |
| B7/CD28 | Anti-CD28 mAb | Transplantation | 320 |
| B7/CTLA-4 | Anti-CTLA4 mAb (Ipilimumab and Tremelimumab) | Melanoma and other types of malignancies | 287 |
| B7/CTLA-4 | Abatacept (CTLA4-Ig) | Rheumatoid arthritis | 321 |
CTLA, cytotoxic T-lymphocyte antigen; mAb, monoclonal antibody; ICAM, intercellular adhesionmolecule.
A. LFA-1/ICAM-1 Interaction
Studies performed in vivo and in vitro have demonstrated that T-cell activation requires interaction of adhesion receptors. Initially, when T cell, encounter APC, adhesion receptors physically anchor and provide feedback to the T cells about the surface milieu of the APC. This may indeed be followed by TCR signaling and tightening of the distance between the two membranes through translocation of the receptors.8,208 Adhesion receptors such as ICAM-1 are considered to be involved in intracellular signaling, leading to the accumulation of the immune receptors along with MHC molecules at the contact area for the efficient presentation of the MHC–p complex to the T cells.209 Thus, adhesion is considered a costimulatory signal required to generate TCR signaling and, further, immune response. There are several adhesion receptors on T cells that can deliver a costimulatory signal including LFA-1. LFA-1 interacts with ICAM-1, -2, and -3.209,210 The expression of ICAM-1 on DC is necessary for longstanding interaction between CD8+ T cells and DC for activating effector CD8+ T cells and their survival.211 Knocking out ICAM-1 expression resulted in elimination of the long-lasting T-cell interaction with DC for memory induction and lowering of the production of IFN-γ cytokine.211 This result is attributed to the inability to form the IS in the absence of ICAM-1.211 There is a prolonged polarized synapse upon DC–T cell interactions before asymmetry T-cell division during mitosis to produce two different cells with distinctive functions.212
Efforts made toward understanding the structure, function and mechanisms of interaction between LFA-1 and ICAM-1 have produced possibilities of new therapies. Currently, several strategies have been developed using LFA-1- and ICAM-1-targeted therapeutics, which include antibodies, peptides, peptidomimetics, small molecules, and antisense oligo-nucleotides to suppress LFA-1/ICAM-1 costimulatory signal for the treatment of in-flammation,213 autoimmune diseases,214 allograft rejection,200,215–219 and cancer.220 Other than blocking ICAM-1/LFA-1 interaction, the mechanisms of action of these antagonists could involve the downregulation of surface expression of these receptors as well as the prevention of the formation of active conformation of these molecules.
1. Antibodies
Antibodies have been used to determine the functions of ICAM-1 and LFA-1 in disease pathologies, and these antibodies have therapeutic activities in preventing organ transplant rejection221–224 and suppressing autoimmune diseases, including arthritis,225 insulin-dependent diabetes mellitus,202,203,226 multiples sclerosis,227–231 and lupus.232 The first LFA-1/ICAM-1-targeted monoclonal antibody tested clinically was anti-LFA-1 antibody Odulimomab (antibody 25.3) in bone marrow216,219 and kidney transplant,217,218 while anti-ICAM-1 antibody Enlimomab (antibody R6.5 or BIRR1) has been investigated in kidney transplant233 and RA.234 Anti-ICAM-1 antibody is progressing through phase I and II trials for the treatment of RA.205 Blocking ICAM-1/LFA-1 interaction using Enlimomab failed to show any benefit in a randomized renal transplantation study.235 Efalizumab, a recombinant humanized monoclonal antibody that binds to CD11a (the α-subunit of LFA-1), has been successfully used in the treatment of psoriasis.207,214,236–242 Recently, a crystal structure of the efalizumab Fab in complex with the LFA-1 I-domain reveals that the antibody binds to the I-domain distinct from the ICAM-1 binding site and blocks the binding of LFA-1 to ICAM-1 via steric hindrance.243 Efalizumab, under the trade name Raptiva® has been approved by the FDA in 2003 for the treatment of moderate-to-severe plaque psoriasis; it has been withdrawn from the market because of increased patient susceptibility to PML.244 Although monoclonal antibodies for adhesion molecules have successfully demonstrated effectiveness in suppressing autoimmune diseases in patients, the risk of secondary infections and immunogenic response has been shown to increase. Therefore, peptides and small molecules are being investigated as immunotherapeutics, and these molecules offer distinct advantages in terms of development and treatment costs.
2. Peptides
The Siahaan group has discovered several cell adhesion peptides that block LFA-1/ICAM-1 interaction (Signal-2). These peptides were derived either from domain-1 of ICAM-1 or from α- and β-subunits of LFA-1 (Table II).159,245–252 Studies carried out using in vitro cellular models, such as homotypic or heterotypic T cell adhesion or mixed lymphocyte reaction, have clearly demonstrated the ability of these peptides to inhibit the binding of T cells to APC by blocking the binding of adhesion receptors to their natural ligands.245,247–252 It should be noted that although the suppression of autoimmune diseases can be accomplished by delivering Signal-2 blockers, inhibiting Signal-2 may also suppress the general immune response to fight infections.
3. Antisense oligonucleotides
In addition to expression on immune cells, ICAM-1 is also constitutively expressed on endothelial and epithelial cells, and during inflammation ICAM-1 expression is upregulated. Antisense oligonuclotide (Alicaforsen, ISIS 2302) binds to RNA and blocks the translation of ICAM-1 protein within the cell; this molecule was investigated in phase II and III clinical trials for the treatment of Crohn’s disease and ulcerative colitis.253,254 Although the initial trials with a small group of subjects was promising, later larger trials in both diseases failed to show efficacies.255–261 The utility of ISIS 2302 was also evaluated for treating RA, and this drug was not efficacious in the clinical trials.262
B. CD28/B7 Interaction
Interactions between CD28 and B7-1 (or CD80) or B7-2 (or CD86) have a role in T-cell differentiation and proliferation. Inhibition of CD28 signal prevents recruitment and activation of naïve T cells as well as interfering with epitope spreading. This inhibition is being investigated for development of treatments for autoimmune diseases and organ transplantations.93,263–269 CD28-deficient mice have impaired immune responses to viral antigens or autoantigens,270,271 suggesting that T-cell responses to autoantigens or alloantigens are dependent on costimulation through CD28.272 Antibody-induced modulation of a costimulatory signal via the CD28 pathway has been shown to prolong allograft survival in rats.273\
Administration of anti-CD28 Fab fragments efficiently led to suppression as well as reversal in the induction of EAE268 and uveoretinitis274 in mice. The activity of anti-CD28 Fab suppresses EAE due to ablation of TNF-α production and prevention of T-cell infiltration into the CNS without stimulating CD28.268,269,275,276 Unfortunately, the Fab fragment is difficult to develop as a drug because it is rapidly cleared from the body.277 To solve the clearance problems, the Fab fragment has conjugated to polyethylene glycol or fused with serum albumin, and these conjugates have been shown to have longer half-lives in the body.277 Although CD28 blockade using mAb attenuated EAE and prevented subsequent relapses, it did not completely eliminate the encephalitogenic response, suggesting that the activation of encephalitogenic T cells independent of CD28 costimulation may not have been completely abolished. This observation is consistent with findings in CD28-deficient mice. CD28−/− mice developed autoimmune heart disease, although it was less severe than that observed in heterozygous littermates.270 Similarly, breeding NOD mice with CD28−/− was found to exacerbate autoimmunity.278
C. CTLA-4/B7 Interaction
Blocking the CTLA-4/B7 negative signal has been proposed to improve the response of cytotoxic T cells to antigen presentation by APC, thus enhancing their activation and proliferation.279 The signal transmitted through CTLA-4 leads to dephosphorylation of the second messengers in the CD3 complex and subsequently leads to the control of the production of various cytokines produced by TH1 and TH2 cells.280,281 It has been suggested that CTLA-4 is involved in the development of Treg cells in organ transplantation models,282 and CTLA-4-deficient animals have been shown to be resistant to immunotolerance.283 A single injection of anti-CTLA-4 2 days after immunization in mice resulted in a mild increase in severity and incidence of EAE.279 When anti-CTLA-4 mAb was administered on the second day after clinical signs of EAE were observed followed by its administration every other day for the following 6 days, a marked increase in the disease score was observed and there was a considerable increase in the production of IL-2, TNF-α, and IFN-γ.284 Histological evaluation of mice treated with anti-CTLA-4 mAb showed more inflammation in the brain and the spinal cord compared with those treated with control antibody, suggesting that inhibition of CTLA-4 signal enhances the activation and proliferation of antigen-reactive T cells.285 Blocking the CTLA-4 signal is not a good approach for developing therapeutic agents for autoimmune diseases. In contrast, inhibition of CTLA-4 signal can activate T cells for fighting tumors.286 Several clinical trials have been conducted using the anti-CTLA-4 antibody to treat melanoma and other types of malignancies (Table II). Two of these antibodies, ipilimumab and tremelimumab, have reached phase III clinical trials.287
4. BIFUNCTIONAL PEPTIDE INHIBITORS
Recently, we have discovered a novel and selective method to suppress autoimmune diseases using bifunctional peptide inhibitor (BPI) molecules that simultaneously target both Signal-1 and Signal-2 (Fig. 3). Results from our studies have demonstrated that two different BPI molecules (i.e., PLP-BPI and GAD-BPI) can induce immunotolerance in autoimmune diseases in two different animal models, EAE288,289 and NOD mice.159 In addition to these two BPI molecules, we also have ongoing and promising studies toward developing similar molecules to treat collagen-induced arthritis in DBA/1J mice, a mouse model for RA. In the case of PLP-BPI, the antigenic-peptide epitope derived from the PLP (PLP139–151) was conjugated to LABL peptide derived from αL integrin (CD11a237–246) to make BPI molecules.288,289 GAD-BPI is made from GAD208–217 and LABL peptides. We hypothesize that the antigenic-peptide fragment (i.e., PLP139–151 and GAD208–217) binds to MHC-II and the LFA-1 peptide fragment (LABL) binds to ICAM-1 on the surface of APC. Because both peptides are conjugated via a linker, simultaneous binding of BPI to MHC-II and ICAM-1 will prevent the translocation between TCR:MHC-II-peptide (Signal-1) and ICAM-1/LFA-1 complexes (Signal-2) that forms the IS. Inhibition of IS formation selectively alters the activation of T cells from TH1 to TH2 phenotypes and/or induces the production of Treg cells. Because these BPI molecules contain specific antigenic peptides, we hope that we can target only a specific subpopulation of T cells involved in the onset and progression of autoimmune diseases without affecting the general immune response.
Figure 3.
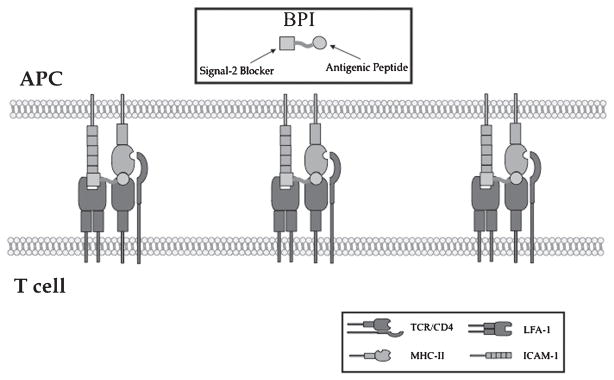
The proposed binding of the BPI molecule to MHC-II and ICAM-1, which inhibits the translocation of Signal-1 and Signal-2. ICAM-1, intercellular adhesion molecule-1; MHC, major histocompatibility complex; BPI, bifunctional peptide inhibitor.
Studies with experimental models of autoimmune disease and allergy have shown that the administration of soluble peptides based on known T-cell epitopes leads to suppression of the specific response, induction of bystander suppression, and prevention and treatment of hypersensitivity. Injections of antigenic peptides, such as PLP139–151266,290 and GAD208–217167 in saline, have been shown to suppress the progression of EAE and T1D, respectively, in mice models. However, treatment with antigenic peptides is potentially dangerous and can lead to fatal anaphylactic reactions. Many researchers have modified these antigenic peptides via mutation291 or development of altered peptide ligands (APL)292 in an effort to improve their therapeutic potential in treating autoimmune diseases as well as address their safety concerns.
Our data showed that PLP-BPI has better activity in suppressing EAE than do other peptides, including VP2-BPI (a BPI with an epitope peptide derived from Theiler’s encephalomyelitis virus capsid protein, VP274–86), which is known to bind to MHC-II in SJL/J mice,293 PLP-BPIsLABL (PLP-BPI with a scrambled sequence of LABL), OVA-BPI (BPI in which PLP139–151 has been replaced with OVA326–337 derived from ovalbumin), and the unlinked mixture of PLP139–151 and LABL. PLP-BPI-treated mice had very low EAE clinical scores and minimal loss in body weight compared with other groups. In addition, some of the PLP-BPI-treated mice did not develop the disease.288,289 Similarly, dosing regimens designed for therapeutic treatment instead of prophylactic treatment showed that BPI was able to reverse disease severity very quickly.289 Some of our unpublished work shows that PLP-BPIsPLP (PLP-BPI with a scrambled sequence of PLP139–151) has no EAE- suppressing activity, suggesting the significance of a unique structure in PLP-BPI such as the need for both PLP139–151 and LABL peptides and covalent linking of these two peptides in the same molecule. Also, more importantly, we found out that these molecules are highly antigen-specific. We observed that splenocytes isolated from PLP139–151-immunized mice responded to in vitro re-stimulation with PLP139–151 but not MBP87–89 and vice versa.289 It was also interesting to find that splenocytes isolated from Ac-PLP-BPI-NH2 (a modified form of PLP-BPI) showed significantly less proliferation in re-call to PLP139–151, suggesting that injection of Ac-PLP-BPI-NH2 reduces and/or suppresses the number of PLP139–151- responsive populations. In a parallel study, GAD-BPI had the capacity to suppress the progression of T1D in NOD mice as demonstrated by significantly less insulitis and lower blood glucose levels in GAD-BPI-treated mice compared with control.159
Another important issue we looked at is the safety of these BPI molecules. As with any other peptide-based therapy, there is an associated risk involved in the use of BPI such as the possibility of an anaphylactic response, a life-threatening immediate hypersensitivity reaction caused by injections of the antigen-related peptides. Owing to hypersensitivity reactions in patients during phase II clinical trials, development of an MS-targeted peptide drug was suspended.294,295 In our studies, i.v. injection of PLP-BPI at 4–5 weeks after immunization caused anaphylactic reaction to a much lower number of mice than does PLP139–151. Additionally, a modified form of PLP-BPI (i.e., Ac-PLP-BPI-NH2-2 with a longer linker and its N- and C-terminal acetylated and amidated, respectively) was found to be even less aggressive in inducing anaphylactic reactions.289 We speculate that the lower incidence of anaphylaxis in BPI is due to the presence of LABL peptide that inhibits LFA-1/ICAM-1 interactions at the site of antigen recognition or the addition of another moiety at the N- or C-terminal to the parental allergic peptide PLP139–151. Involvement of LFA-1/ICAM-1- mediated heterotypic aggregation of activated T cells to mast cells has been implicated in augmenting mast cell degranulation and histamine release.296
5. PEPTIDE SAFETY
A potential problem that can arise when treating diseases with multiple injections of antigen-related peptides is the possibility of anaphylactic shock.291,297 As a result of the fact that peptides are smaller in size than proteins, it is thought that they are safer and less likely to induce an anaphylactic response in the immune system. Despite the numerous advantages of peptides over proteins, anaphylactic reactions during treatments involving peptides have been widely described in the literature.288,291,297
In general, the mechanism of anaphylactic response is thought to occur when an antigen crosslinks with IgE bound to FcεRI on mast cells, which leads to degranulation and the release of histamine and cytokines, both of which are inflammatory mediators.296 The administration of anti-IgE antibodies along with myelin-specific peptides during the treatment of EAE has been shown to inhibit anaphylaxis, which suggests that IgE plays a large role in anaphylaxis when treating with peptides.298
APL have been shown to suppress anaphylaxis in EAE without affecting its activity toward myelin-reactive T cells.292 Short linear peptide sequences generally lack the ability to crosslink adjacent IgE molecules on mast cells and basophils.299 Because a nonimmunogenic peptide sequence can become immunogenic when only one of its amino acid residues is altered, care must be exercised in designing an APL so that it retains its activity toward its target and does not become more antigenic.
The avoidance of IgE activation is necessary to circumvent allergic response. It is optimal to use peptides shorter than 20 amino acids, as longer peptides have been associated with more adverse events compared with their shorter counterparts.299 Five-to-six amino acids have been shown to be sufficient minimal antigenic determinants.300 Alternatively, APL that can bypass the body’s ability to create an anaphylactic response against a peptide without affecting its activity toward its target have been shown to work and must be utilized in creating effective therapies.
6. MECHANISM OF PEPTIDES THAT TREAT AUTOIMMUNE DISEASES
It has been described before that the immune system functions as a balance between pathogenic effector cells and regulatory cells.299 The suppression and activation of these two will lead to either an immunogenic response or an immunotolerant response (Fig. 4). Autoimmune diseases typically arise due to an imbalance between these two responses. Therefore, to combat autoimmune diseases, we must try to restore balance in the immune system. One way to restore the balance is by either inducing the suppressive cells or activating the regulatory cells.
Figure 4.

The proposed mechanism of activation and suppression of immune response during autoimmune diseases. On the left, interaction between mature/activated DC and naïve T cells is mediated by MHC-II antigen/TCR complex (Signal-1) in the presence of the costimulatory signal (Signal-2) that leads to differentiation intoTH1 and TH17 cells. On the right, interaction between immature/steady-state DC and naïve T cells is mediated by MHC-II antigen/TCR complex (Signal-1) in the absence of the costi-mulatory signal (Signal-2) that leads to differentiation intoTreg cells. Treg cells produce IL-10 that suppresses the proliferation of TH1 and TH17. MHC, major histocompatibility complex; TCR, T-cell receptors; DC, dendritic cells.
DC are considered to be the most prominent APC in inducing an autoimmune disease; Yamazaki et al.301 and Steinman et al.302 provided excellent reviews on the function of DC in tolerance and immune responses. They are believed to send a “danger” signal to induce an immunogenic response, but that is not always the case.303 The function of the DC depends on whether it is in a mature (activated) or immature state.304
The maturation of DC into a T-cell stimulatory mode is activated by an inflammatory stimulus that is initiated by the uptake of an insoluble antigen (Fig. 4, left side).299,304 Once the DC is in the mature state, its phenotype changes and there is an upregulation of costi-mulatory and adhesion molecule expression on its cell surface. Presentation of antigen in this way leads to the differentiation of proinflammatory cellular responses.299 Such an inflammatory cellular response triggered by a mature DC (mDC) includes the induction of TH1 cells that produce IFN-γ and IL-2.304 Other proinflammatory responses include the induction of TH17 and the release of IL-17.128 These inflammatory responses triggered toward self tissues usually lead to an autoimmune disease such as MS.
On the other hand, an immature DC (iDC) is one that did not take up and process any antigens. It has been shown, using antibodies specific for peptide-free MHC-II molecules, that iDC express empty MHC-II molecules on their surface and can bind antigenic peptides in solution. These bound antigens can be presented to T cells without internalization and processing.305 Soluble peptides that are presented by iDC lead to a differentiation of Treg cells (Fig. 4, right side).299 Activation of regulatory T cells leads to the production of regulatory cytokines such as IL-10 and TGF-β. Such cytokines have been shown to have an important role in ameliorating autoimmune diseases in animal experimental models.306–308 In the case of some autoimmune diseases, expanding the “regulatory pool” leads to a downregulation of the effector response (TH1) and enhancement of the immunosuppressive response (TH2). 299
7. PROPOSED BPI MECHANISMS
There are several possible mechanisms that could explain how BPI molecules suppress the activation of T cells and result in suppression of autoimmune diseases. The first of these is that only the antigenic fragment of BPI molecule binds to MHC-II on the iDC (steady-state DC) and is presented to naïve T cells for differentiation to produce Treg cells that generate IL-10 (Fig. 5, right side). This proposed mechanism, which is similar to the mechanism of action of soluble antigenic peptide as a therapeutic vaccine,299 ignores the role of cell adhesion peptide (e.g., cIBR or LABL) on the BPI molecule. It has been shown that injection of the BPI molecules generates IL-10;288 however, the kinetics and the amount of IL-10 production upon BPI and PLP peptide injections have not been compared. Because the BPI molecule has better efficacy than the parent antigenic peptide (PLP),288 it is predicted that the amount of IL-10 produced by injecting BPI molecules will be higher than that produced by injecting PLP peptide. The second possible mechanism is that the BPI molecule binds not only to MHC-II but also to adhesion molecules (e.g., LFA-1 or ICAM-1) on iDC. Thus, during the interaction between BPI-loaded iDC and naïve T cells, the BPI molecule blocks the IS formation to induce differentiation of naïve T cells to Treg cells. The presence of cell adhesion peptide on BPI makes it a more efficient modulator for naïve T cell differentiation than the antigenic peptide alone. The third possible mechanism is that the BPI molecule simultaneously binds to MHC-II and LFA-1 to block the IS formation during the interaction between naïve T cells and mDC (Fig. 5, left side) to suppress the differentiation of naïve T cells to TH1 and TH17 cells. There is some indication that the BPI molecule induces the generation of IL-4-producing TH2 cells, which may tip the balance to lower the production of TH17 and TH1 cells. The increase in IL-4 production was observed after BPI injection into the EAE mouse model.288 The mechanism of action of BPI molecules may also be a combination of these three proposed mechanisms.
Figure 5.

Potential mechanism of BPI molecules in altering the differentiation of naïve T cells upon interaction with immature and mature DC. On the right, interaction of steady-state/immature DC with BPI molecule produces differentiation of naïve T cells to IL-10-producingTreg cells. IL-10 induces the proliferation of immunosuppressant TH2 cells. On the left, interaction of BPI molecule with mature/activated DC induces immunosuppressant TH2 cell differentiation. MHC, major histocompatibility complex; TCR, T-cell receptors; DC, dendritic cells; BPI, bifunctional peptide inhibitor.
8. CONCLUSIONS
Immunomodulating compounds have been explored as potential treatments for autoimmune diseases, such as MS, T1D, and RA among others. However, most of the agents developed thus far aim at broad modulation of the immune response and, therefore, may present some undesirable side effects. Targeted drug delivery is a more attractive strategy to improve the efficacy and reduce the side effects of these immunomodulating drugs. In our studies, we hope to design BPIs that target only a subpopulation of T cells responsible for the progression of the disease without affecting the general immune response. Although it has not been clearly elucidated, BPI-based therapies work either by inhibiting the formation of the IS by blocking both Signal-1 and Signal-2 or by shifting the T-cell subpopulation into Tregs and/or TH2-like phenotype. The detailed mechanisms of action of BPI-type molecules, including the antigen-specific immunosuppressive activity as well as its safety, are being investigated in depth in our current studies. Additionally, we are looking at ways of improving the structure and sequence of BPI to provide a more efficient and well-tolerated immunotherapy.
Biographies
Prakash Manikwar is a postdoctoral researcher working for Dr. David B. Volkin in the Department of Pharmaceutical Chemistry at The University of Kansas, Lawrence. He received his B. Pharmacy degree from Rajiv Gandhi University of Health Sciences in India, M.S. in Biological Sciences from South Dakota State University, and M.S. and Ph.D. degrees in Pharmaceutical Chemistry from The University of Kansas. He is currently working on analytical characterization, stabilization, and formulation of biopharmaceutical.
Paul Kiptoo is a postdoctoral researcher working for Dr. Teruna J. Siahaan in the Department of Pharmaceutical Chemistry at the University of Kansas. He received his Ph.D. in Pharmaceutical Sciences from the University of Kentucky. He is currently working on enhancing the absorption of anticancer drugs into the brain using E-cadherin peptides that inhibit E-cadherin interactions at the intercellular tight junctions and also targeting antigenic peptides to leukocytes in order to modulate the formation of the immunological synapse for the suppression of autoimmune diseases such as MS and RA.
Ahmed H. Badawi received his Bachelor of Science degree from the University of Missouri –Kansas City in Pharmaceutical Science and Chemistry. Ahmed then moved to the University of Kansas where he earned his Master of Science degree in Pharmaceutical Chemistry with honors. Currently, Ahmed is a doctoral student working for Dr. Teruna J. Siahaan in the Department of Pharmaceutical Chemistry at The University of Kansas. His project entails developing peptides for the treatment of autoimmune diseases.
Barlas Büyüktimkin is a Ph.D. candidate working for Dr. Teruna J. Siahaan in the Pharmaceutical Chemistry Department at The University of Kansas, Lawrence. He received his Doctor of Pharmacy degree from the University of Kansas, and M.S. in Pharmaceutical Chemistry from The University of Kansas. He is currently working on peptide and protein-mediated drug delivery.
Teruna J. Siahaan is a Professor and the Associate Chair of the Department of Pharmaceutical Chemistry at The University of Kansas. His research has explored the utilization and modulation of cell adhesion molecules on the cell surface for targeted drug delivery to a specific cell type, and for enhancing drug permeation through the intestinal mucosa and blood– brain barrier (BBB). His research group is very well known in the study of peptide-mediated drug delivery. He has more than 20 years of experience in both academia and industry and is a recipient of numerous awards. He has given many invited presentations and has published more than 120 research and review articles. He serves in the Editorial board of several journals and in grant review panels for National Institutes of Health (NIH), Department of Defense (DOD), and various Research Foundations. He has received several grants from NIH, National MS Society, Alzheimer’s Association, Arthritis Foundation, and American Heart Association.
References
- 1.Unanue ER. Antigen-presenting function of the macrophage. Annu Rev Immunol. 1984;2:395–428. doi: 10.1146/annurev.iy.02.040184.002143. [DOI] [PubMed] [Google Scholar]
- 2.Unanue ER. Perspective on antigen processing and presentation. Immunol Rev. 2002;185:86–102. doi: 10.1034/j.1600-065x.2002.18510.x. [DOI] [PubMed] [Google Scholar]
- 3.Monks CR, Freiberg BA, Kupfer H, Sciaky N, Kupfer A. Three-dimensional segregation of supramolecular activation clusters in T cells. Nature. 1998;395:82–86. doi: 10.1038/25764. [DOI] [PubMed] [Google Scholar]
- 4.Paul WE, Seder RA. Lymphocyte responses and cytokines. Cell. 1994;76:241–251. doi: 10.1016/0092-8674(94)90332-8. [DOI] [PubMed] [Google Scholar]
- 5.Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: A molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- 6.Tseng SY, Dustin ML. T-cell activation: A multidimensional signaling network. Curr Opin Cell Biol. 2002;14:575–580. doi: 10.1016/s0955-0674(02)00370-8. [DOI] [PubMed] [Google Scholar]
- 7.Paul WE. Fundamental immunology. 6. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins; 2008. pp. 1603–1616. [Google Scholar]
- 8.Delon J, Germain RN. Information transfer at the immunological synapse. Curr Biol. 2000;10:R923–R933. doi: 10.1016/s0960-9822(00)00870-8. [DOI] [PubMed] [Google Scholar]
- 9.Blanchard N, Hivroz C. The immunological synapse: The more you look the less you know. Biol Cell. 2002;94:345–354. doi: 10.1016/s0248-4900(02)00007-2. [DOI] [PubMed] [Google Scholar]
- 10.Lee KH, Holdorf AD, Dustin ML, Chan AC, Allen PM, Shaw AS. T cell receptor signaling precedes immunological synapse formation. Science. 2002;295:1539–1542. doi: 10.1126/science.1067710. [DOI] [PubMed] [Google Scholar]
- 11.van der Merwe PA. Formation and function of the immunological synapse. Curr Opin Immunol. 2002;14:293–298. doi: 10.1016/s0952-7915(02)00350-3. [DOI] [PubMed] [Google Scholar]
- 12.Stinchcombe JC, Bossi G, Booth S, Griffiths GM. The immunological synapse of CTL contains a secretory domain and membrane bridges. Immunity. 2001;15:751–761. doi: 10.1016/s1074-7613(01)00234-5. [DOI] [PubMed] [Google Scholar]
- 13.Bossi G, Trambas C, Booth S, Clark R, Stinchcombe J, Griffiths GM. The secretory synapse: The secrets of a serial killer. Immunol Rev. 2002;189:152–160. doi: 10.1034/j.1600-065x.2002.18913.x. [DOI] [PubMed] [Google Scholar]
- 14.Kupfer H, Monks CR, Kupfer A. Small splenic B cells that bind to antigen-specific T helper (Th) cells and face the site of cytokine production in the Th cells selectively proliferate: Immunofluorescence microscopic studies of Th-B antigen-presenting cell interactions. J Exp Med. 1994;179:1507–1515. doi: 10.1084/jem.179.5.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stinchcombe JC, Griffiths GM. The role of the secretory immunological synapse in killing by CD8+ CTL. Semin Immunol. 2003;15:301–305. doi: 10.1016/j.smim.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 16.Davis DM, Dustin ML. What is the importance of the immunological synapse? Trends Immunol. 2004;25:323–327. doi: 10.1016/j.it.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 17.Iezzi G, Karjalainen K, Lanzavecchia A. The duration of antigenic stimulation determines the fate of naive and effector T cells. Immunity. 1998;8:89–95. doi: 10.1016/s1074-7613(00)80461-6. [DOI] [PubMed] [Google Scholar]
- 18.Celli S, Lemaitre F, Bousso P. Real-time manipulation of T cell-dendritic cell interactions in vivo reveals the importance of prolonged contacts for CD4+ T cell activation. Immunity. 2007;27:625–634. doi: 10.1016/j.immuni.2007.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Alcover A, Alarcon B. Internalization and intracellular fate of TCR-CD3 complexes. Crit Rev Immunol. 2000;20:325–346. [PubMed] [Google Scholar]
- 20.Lee KH, Dinner AR, Tu C, Campi G, Raychaudhuri S, Varma R, Sims TN, Burack WR, Wu H, Wang J, Kanagawa O, Markiewicz M, Allen PM, Dustin ML, Chakraborty AK, Shaw AS. The immunological synapse balances T cell receptor signaling and degradation. Science. 2003;302:1218–1222. doi: 10.1126/science.1086507. [DOI] [PubMed] [Google Scholar]
- 21.Crabtree GR. Contingent genetic regulatory events in T lymphocyte activation. Science. 1989;243:355–361. doi: 10.1126/science.2783497. [DOI] [PubMed] [Google Scholar]
- 22.Jacobelli J, Andres PG, Boisvert J, Krummel MF. New views of the immunological synapse: Variations in assembly and function. Curr Opin Immunol. 2004;16:345–352. doi: 10.1016/j.coi.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Wetzel SA, Parker DC. MHC transfer from APC to T cells following antigen recognition. Crit Rev Immunol. 2006;26:1–21. doi: 10.1615/critrevimmunol.v26.i1.10. [DOI] [PubMed] [Google Scholar]
- 24.Allen PM, Matsueda GR, Evans RJ, Dunbar JB, Jr, Marshall GR, Unanue ER. Identification of the T-cell and Ia contact residues of a T-cell antigenic epitope. Nature. 1987;327:713–715. doi: 10.1038/327713a0. [DOI] [PubMed] [Google Scholar]
- 25.Sette A, Buus S, Colon S, Smith JA, Miles C, Grey HM. Structural characteristics of an antigen required for its interaction with Ia and recognition by T cells. Nature. 1987;328:395–399. doi: 10.1038/328395a0. [DOI] [PubMed] [Google Scholar]
- 26.Bouvier M. Accessory proteins and the assembly of human class I MHC molecules: A molecular and structural perspective. Mol Immunol. 2003;39:697–706. doi: 10.1016/s0161-5890(02)00261-4. [DOI] [PubMed] [Google Scholar]
- 27.Flutter B, Gao B. MHC class I antigen presentation—recently trimmed and well presented. Cell Mol Immunol. 2004;1:22–30. [PubMed] [Google Scholar]
- 28.Trombetta ES, Mellman I. Cell biology of antigen processing in vitro and in vivo. Annu Rev Immunol. 2005;23:975–1028. doi: 10.1146/annurev.immunol.22.012703.104538. [DOI] [PubMed] [Google Scholar]
- 29.Bricard G, Porcelli SA. Antigen presentation by CD1 molecules and the generation of lipid-specific T cell immunity. Cell Mol Life Sci. 2007;64:1824–1840. doi: 10.1007/s00018-007-7007-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brigl M, Brenner MB. CD1: Antigen presentation and T cell function. Annu Rev Immunol. 2004;22:817–890. doi: 10.1146/annurev.immunol.22.012703.104608. [DOI] [PubMed] [Google Scholar]
- 31.Dessen A, Lawrence CM, Cupo S, Zaller DM, Wiley DC. X-ray crystal structure of HLA-DR4 (DRA*0101, DRB1*0401) complexed with a peptide from human collagen II. Immunity. 1997;7:473–481. doi: 10.1016/s1074-7613(00)80369-6. [DOI] [PubMed] [Google Scholar]
- 32.Hennecke J, Wiley DC. T cell receptor-MHC interactions up close. Cell. 2001;104:1–4. doi: 10.1016/s0092-8674(01)00185-4. [DOI] [PubMed] [Google Scholar]
- 33.Jardetzky TS, Brown JH, Gorga JC, Stern LJ, Urban RG, Chi YI, Stauffacher C, Strominger JL, Wiley DC. Three-dimensional structure of a human class II histocompatibility molecule complexed with superantigen. Nature. 1994;368:711–718. doi: 10.1038/368711a0. [DOI] [PubMed] [Google Scholar]
- 34.Reinherz EL, Tan K, Tang L, Kern P, Liu J, Xiong Y, Hussey RE, Smolyar A, Hare B, Zhang R, Joachimiak A, Chang HC, Wagner G, Wang J. The crystal structure of a T cell receptor in complex with peptide and MHC class II. Science. 1999;286:1913–1921. doi: 10.1126/science.286.5446.1913. [DOI] [PubMed] [Google Scholar]
- 35.Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 36.Latek RR, Suri A, Petzold SJ, Nelson CA, Kanagawa O, Unanue ER, Fremont DH. Structural basis of peptide binding and presentation by the type I diabetes-associated MHC class II molecule of NOD mice. Immunity. 2000;12:699–710. doi: 10.1016/s1074-7613(00)80220-4. [DOI] [PubMed] [Google Scholar]
- 37.Garboczi DN, Ghosh P, Utz U, Fan QR, Biddison WE, Wiley DC. Structure of the complex between human T-cell receptor, viral peptide and HLA-A2. Nature. 1996;384:134–141. doi: 10.1038/384134a0. [DOI] [PubMed] [Google Scholar]
- 38.Yuan F, Georgiou T, Hillon T, Gostick E, Price DA, Sewell AK, Moysey R, Gavarret J, Vuidepot A, Sami M, Bell JI, Gao GF, Rizkallah PJ, Jakobsen BK. Crystallization and preliminary X-ray structural studies of a Melan-A pMHC-TCR complex. Acta Crystallogr F. 2007;63:758–760. doi: 10.1107/S1744309107037244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cole DK, Rizkallah PJ, Sami M, Lissin NM, Gao F, Bell JI, Boulter JM, Glick M, Vuidepot AL, Jakobsen BK, Gao GF. Crystallization and preliminary X-ray structural studies of a high-affinity CD8alphaalpha co-receptor to pMHC. Acta Crystallogr F. 2005;61:285–287. doi: 10.1107/S1744309105002988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dustin ML. Membrane domains and the immunological synapse: Keeping T cells resting and ready. J Clin Invest. 2002;109:155–160. doi: 10.1172/JCI14842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bromley SK, Burack WR, Johnson KG, Somersalo K, Sims TN, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse. Annu Rev Immunol. 2001;19:375–396. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 42.Rudolph MG, Luz JG, Wilson IA. Structural and thermodynamic correlates of T cell signaling. Annu Rev Biophys Biomol Struct. 2002;31:121–149. doi: 10.1146/annurev.biophys.31.082901.134423. [DOI] [PubMed] [Google Scholar]
- 43.Kabat EA, Wu TT, Bilofsky H. Evidence indicating independent assortment of framework and complementarity-determining segments of the variable regions of rabbit light chains. Delineation of a possible J minigene. J Exp Med. 1980;152:72–84. doi: 10.1084/jem.152.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davis MM, Bjorkman PJ. T-cell antigen receptor genes and T-cell recognition. Nature. 1988;334:395–402. doi: 10.1038/334395a0. [DOI] [PubMed] [Google Scholar]
- 45.Hava DL, Brigl M, van den Elzen P, Zajonc DM, Wilson IA, Brenner MB. CD1 assembly and the formation of CD1-antigen complexes. Curr Opin Immunol. 2005;17:88–94. doi: 10.1016/j.coi.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 46.Mosmann TR, Coffman RL. TH1 and TH2 cells: Different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 47.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 48.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 49.Tate KM, Lee C, Edelman S, Carswell-Crumpton C, Liblau R, Jones PP. Interactions among polymorphic and conserved residues in MHC class II proteins affect MHC-peptide conformation and T cell recognition. Int Immunol. 1995;7:747–761. doi: 10.1093/intimm/7.5.747. [DOI] [PubMed] [Google Scholar]
- 50.Wade WF, Khrebtukova I, Schreiber KL, McKean DJ, Wade TK. Truncated MHC class II cytoplasmic and transmembrane domains: Effect on plasma membrane expression. Mol Immunol. 1995;32:433–446. doi: 10.1016/0161-5890(94)00165-w. [DOI] [PubMed] [Google Scholar]
- 51.Harton JA, Van Hagen AE, Bishop GA. The cytoplasmic and transmembrane domains of MHC class II beta chains deliver distinct signals required for MHC class II-mediated B cell activation. Immunity. 1995;3:349–358. doi: 10.1016/1074-7613(95)90119-1. [DOI] [PubMed] [Google Scholar]
- 52.Griffith IJ, Ghogawala Z, Nabavi N, Golan DE, Myer A, McKean DJ, Glimcher LH. Cytoplasmic domain affects membrane expression and function of an Ia molecule. Proc Natl Acad Sci USA. 1988;85:4847–4851. doi: 10.1073/pnas.85.13.4847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.St Pierre Y, Watts TH. Characterization of the signaling function of MHC class II molecules during antigen presentation by B cells. J Immunol. 1991;147:2875–2882. [PubMed] [Google Scholar]
- 54.Wade WF, Freed JH, Edidin M. Translational diffusion of class II major histocompatibility complex molecules is constrained by their cytoplasmic domains. J Cell Biol. 1989;109:3325–3331. doi: 10.1083/jcb.109.6.3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Steinman RM, Witmer MD. Lymphoid dendritic cells are potent stimulators of the primary mixed leukocyte reaction in mice. Proc Natl Acad Sci USA. 1978;75:5132–5136. doi: 10.1073/pnas.75.10.5132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sallusto F, Cella M, Danieli C, Lanzavecchia A. Dendritic cells use macropinocytosis and the mannose receptor to concentrate macromolecules in the major histocompatibility complex class II compartment: Downregulation by cytokines and bacterial products. J Exp Med. 1995;182:389–400. doi: 10.1084/jem.182.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Berthiaume EP, Medina C, Swanson JA. Molecular size-fractionation during endocytosis in macrophages. J Cell Biol. 1995;129:989–998. doi: 10.1083/jcb.129.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang W, Swiggard WJ, Heufler C, Peng M, Mirza A, Steinman RM, Nussenzweig MC. The receptor DEC-205 expressed by dendritic cells and thymic epithelial cells is involved in antigen processing. Nature. 1995;375:151–155. doi: 10.1038/375151a0. [DOI] [PubMed] [Google Scholar]
- 59.Allen PM, Strydom DJ, Unanue ER. Processing of lysozyme by macrophages: Identification of the determinant recognized by two T-cell hybridomas. Proc Natl Acad Sci USA. 1984;81:2489–2493. doi: 10.1073/pnas.81.8.2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Allen PM, Unanue ER. Differential requirements for antigen processing by macrophages for lysozyme-specific T cell hybridomas. J Immunol. 1984;132:1077–1079. [PubMed] [Google Scholar]
- 61.Chesnut RW, Colon SM, Grey HM. Requirements for the processing of antigens by antigen-presenting B cells. I. Functional comparison of B cell tumors and macrophages. J Immunol. 1982;129:2382–2388. [PubMed] [Google Scholar]
- 62.Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell. 1988;52:329–341. doi: 10.1016/s0092-8674(88)80026-6. [DOI] [PubMed] [Google Scholar]
- 63.Mellman I. Endocytosis and molecular sorting. Annu Rev Cell Dev Biol. 1996;12:575–625. doi: 10.1146/annurev.cellbio.12.1.575. [DOI] [PubMed] [Google Scholar]
- 64.Hudson AW, Ploegh HL. The cell biology of antigen presentation. Exp Cell Res. 2002;272:1–7. doi: 10.1006/excr.2001.5402. [DOI] [PubMed] [Google Scholar]
- 65.Neefjes JJ, Stollorz V, Peters PJ, Geuze HJ, Ploegh HL. The biosynthetic pathway of MHC class II but not class I molecules intersects the endocytic route. Cell. 1990;61:171–183. doi: 10.1016/0092-8674(90)90224-3. [DOI] [PubMed] [Google Scholar]
- 66.Pieters J. MHC class II-restricted antigen processing and presentation. Adv Immunol. 2000;75:159–208. doi: 10.1016/s0065-2776(00)75004-8. [DOI] [PubMed] [Google Scholar]
- 67.Bijlmakers MJ, Benaroch P, Ploegh HL. Assembly of HLA DR1 molecules translated in vitro: Binding of peptide in the endoplasmic reticulum precludes association with invariant chain. EMBO J. 1994;13:2699–2707. doi: 10.1002/j.1460-2075.1994.tb06560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Miller J, Germain RN. Efficient cell surface expression of class II MHC molecules in the absence of associated invariant chain. J Exp Med. 1986;164:1478–1489. doi: 10.1084/jem.164.5.1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nijenhuis M, Calafat J, Kuijpers KC, Janssen H, de Haas M, Nordeng TW, Bakke O, Neefjes JJ. Targeting major histocompatibility complex class II molecules to the cell surface by invariant chain allows antigen presentation upon recycling. Eur J Immunol. 1994;24:873–883. doi: 10.1002/eji.1830240415. [DOI] [PubMed] [Google Scholar]
- 70.Sekaly RP, Tonnelle C, Strubin M, Mach B, Long EO. Cell surface expression of class II histocompatibility antigens occurs in the absence of the invariant chain. J Exp Med. 1986;164:1490–1504. doi: 10.1084/jem.164.5.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Danese S, Sans M, Fiocchi C. The CD40/CD40L costimulatory pathway in inflammatory bowel disease. Gut. 2004;53:1035–1043. doi: 10.1136/gut.2003.026278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nat Rev Immunol. 2004;4:336–347. doi: 10.1038/nri1349. [DOI] [PubMed] [Google Scholar]
- 73.Brerscher P. The two-signal model of lymphocyte activation twenty-one years later. Immunol Today. 1992;13:74–76. doi: 10.1016/0167-5699(92)90138-W. [DOI] [PubMed] [Google Scholar]
- 74.Jenkins MK, Johnson JG. Molecules involved in T-cell costimulation. Curr Opin Immunol. 1993;5:351–367. doi: 10.1016/0952-7915(93)90054-v. [DOI] [PubMed] [Google Scholar]
- 75.June CH, Bluestone JA, Nadler LM, Thompson CB. The B7 and CD28 receptor families. Immunol Today. 1994;15:321–331. doi: 10.1016/0167-5699(94)90080-9. [DOI] [PubMed] [Google Scholar]
- 76.June CH, Ledbetter JA, Linsley PS, Thompson CB. Role of the CD28 receptor in T-cell activation. Immunol Today. 1990;11:211–216. doi: 10.1016/0167-5699(90)90085-n. [DOI] [PubMed] [Google Scholar]
- 77.Linsley PS, Ledbetter JA. The role of the CD28 receptor during T cell responses to antigen. Annu Rev Immunol. 1993;11:191–212. doi: 10.1146/annurev.iy.11.040193.001203. [DOI] [PubMed] [Google Scholar]
- 78.Cerdan C, Martin Y, Courcoul M, Mawas C, Birg F, Olive D. CD28 costimulation up-regulates long-term IL-2R beta expression in human T cells through combined transcriptional and post-transcriptional regulation. J Immunol. 1995;154:1007–1013. [PubMed] [Google Scholar]
- 79.Freeman GJ, Boussiotis VA, Anumanthan A, Bernstein GM, Ke XY, Rennert PD, Gray GS, Gribben JG, Nadler LM. B7-1 and B7-2 do not deliver identical costimulatory signals, since B7-2 but not B7-1 preferentially costimulates the initial production of IL-4. Immunity. 1995;2:523–532. doi: 10.1016/1074-7613(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 80.Cerdan C, Martin Y, Courcoul M, Brailly H, Mawas C, Birg F, Olive D. Prolonged IL-2 receptor alpha/CD25 expression after T cell activation via the adhesion molecules CD2 and CD28. Demonstration of combined transcriptional and post-transcriptional regulation. J Immunol. 1992;149:2255–2261. [PubMed] [Google Scholar]
- 81.Reiser H, Freeman GJ, Razi-Wolf Z, Gimmi CD, Benacerraf B, Nadler LM. Murine B7 antigen provides an efficient costimulatory signal for activation of murine T lymphocytes via the T-cell receptor/CD3 complex. Proc Natl Acad Sci USA. 1992;89:271–275. doi: 10.1073/pnas.89.1.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boise LH, Minn AJ, Noel PJ, June CH, Accavitti MA, Lindsten T, Thompson CB. CD28 costimulation can promote T cell survival by enhancing the expression of Bcl-XL. Immunity. 1995;3:87–98. doi: 10.1016/1074-7613(95)90161-2. [DOI] [PubMed] [Google Scholar]
- 83.Civil A, Verweil CL. Regulation of IL2 gene transcription by the accessory molecule CD28. Res Immunol. 1995;146:158–164. doi: 10.1016/0923-2494(96)80250-1. [DOI] [PubMed] [Google Scholar]
- 84.Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of lymphokine messenger RNA stability by a surface-mediated T cell activation pathway. Science. 1989;244:339–343. doi: 10.1126/science.2540528. [DOI] [PubMed] [Google Scholar]
- 85.Gimmi CD, Freeman GJ, Gribben JG, Sugita K, Freedman AS, Morimoto C, Nadler LM. B-cell surface antigen B7 provides a costimulatory signal that induces T cells to proliferate and secrete interleukin 2. Proc Natl Acad Sci USA. 1991;88:6575–6579. doi: 10.1073/pnas.88.15.6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Linsley PS, Wallace PM, Johnson J, Gibson MG, Greene JL, Ledbetter JA, Singh C, Tepper MA. Immunosuppression in vivo by a soluble form of the CTLA-4 T cell activation molecule. Science. 1992;257:792–795. doi: 10.1126/science.1496399. [DOI] [PubMed] [Google Scholar]
- 87.Linsley PS, Greene JL, Tan P, Bradshaw J, Ledbetter JA, Anasetti C, Damle NK. Coexpression and functional cooperation of CTLA-4 and CD28 on activated T lymphocytes. J Exp Med. 1992;176:1595–1604. doi: 10.1084/jem.176.6.1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16:23–35. doi: 10.1016/s1074-7613(01)00259-x. [DOI] [PubMed] [Google Scholar]
- 89.Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, Straus D, Samelson LE, Thompson CB, Bluestone JA. Molecular basis of T cell inactivation by CTLA-4. Science. 1998;282:2263–2266. doi: 10.1126/science.282.5397.2263. [DOI] [PubMed] [Google Scholar]
- 90.Girvin AM, Dal Canto MC, Rhee L, Salomon B, Sharpe A, Bluestone JA, Miller SD. A critical role for B7/CD28 costimulation in experimental autoimmune encephalomyelitis: A comparative study using costimulatory molecule-deficient mice and monoclonal antibody blockade. J Immunol. 2000;164:136–143. doi: 10.4049/jimmunol.164.1.136. [DOI] [PubMed] [Google Scholar]
- 91.Salomon B, Bluestone JA. Complexities of CD28/B7: CTLA-4 costimulatory pathways in autoimmunity and transplantation. Annu Rev Immunol. 2001;19:225–252. doi: 10.1146/annurev.immunol.19.1.225. [DOI] [PubMed] [Google Scholar]
- 92.Salomon B, Lenschow DJ, Rhee L, Ashourian N, Singh B, Sharpe A, Bluestone JA. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 2000;12:431–440. doi: 10.1016/s1074-7613(00)80195-8. [DOI] [PubMed] [Google Scholar]
- 93.Salomon B, Rhee L, Bour-Jordan H, Hsin H, Montag A, Soliven B, Arcella J, Girvin AM, Padilla J, Miller SD, Bluestone JA. Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice. J Exp Med. 2001;194:677–684. doi: 10.1084/jem.194.5.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Inaba K, Witmer-Pack M, Inaba M, Hathcock KS, Sakuta H, Azuma M, Yagita H, Okumura K, Linsley PS, Ikehara S, Muramatsu S, Hodes RJ, Steinman RM. The tissue distribution of the B7-2 costimulator in mice: Abundant expression on dendritic cells in situ and during maturation in vitro. J Exp Med. 1994;180:1849–1860. doi: 10.1084/jem.180.5.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Linsley PS, Greene JL, Brady W, Bajorath J, Ledbetter JA, Peach R. Human B7-1 (CD80) and B7-2 (CD86) bind with similar avidities but distinct kinetics to CD28 and CTLA-4 receptors. Immunity. 1994;1:793–801. doi: 10.1016/s1074-7613(94)80021-9. [DOI] [PubMed] [Google Scholar]
- 96.van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7-1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997;185:393–403. doi: 10.1084/jem.185.3.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yoshinaga SK, Whoriskey JS, Khare SD, Sarmiento U, Guo J, Horan T, Shih G, Zhang M, Coccia MA, Kohno T, Tafuri-Bladt A, Brankow D, Campbell P, Chang D, Chiu L, Dai T, Duncan G, Elliott GS, Hui A, McCabe SM, Scully S, Shahinian A, Shaklee CL, Van G, Mak TW, Senaldi G. T-cell co-stimulation through B7RP-1 and ICOS. Nature. 1999;402:827–832. doi: 10.1038/45582. [DOI] [PubMed] [Google Scholar]
- 98.Ling V, Wu PW, Finnerty HF, Bean KM, Spaulding V, Fouser LA, Leonard JP, Hunter SE, Zollner R, Thomas JL, Miyashiro JS, Jacobs KA, Collins M. Cutting edge: Identification of GL50, a novel B7-like protein that functionally binds to ICOS receptor. J Immunol. 2000;164:1653–1657. doi: 10.4049/jimmunol.164.4.1653. [DOI] [PubMed] [Google Scholar]
- 99.Swallow MM, Wallin JJ, Sha WC. B7h, a novel costimulatory homolog of B7.1 and B7. 2, is induced by TNF-alpha. Immunity. 1999;11:423–432. doi: 10.1016/s1074-7613(00)80117-x. [DOI] [PubMed] [Google Scholar]
- 100.Hutloff A, Dittrich AM, Beier KC, Eljaschewitsch B, Kraft R, Anagnostopoulos I, Kroczek RA. ICOS is an inducible T-cell co-stimulator structurally and functionally related to CD28. Nature. 1999;397:263–266. doi: 10.1038/16717. [DOI] [PubMed] [Google Scholar]
- 101.Mages HW, Hutloff A, Heuck C, Buchner K, Himmelbauer H, Oliveri F, Kroczek RA. Molecular cloning and characterization of murine ICOS and identification of B7h as ICOS ligand. Eur J Immunol. 2000;30:1040–1047. doi: 10.1002/(SICI)1521-4141(200004)30:4<1040::AID-IMMU1040>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 102.McAdam AJ, Chang TT, Lumelsky AE, Greenfield EA, Boussiotis VA, Duke-Cohan JS, Chernova T, Malenkovich N, Jabs C, Kuchroo VK, Ling V, Collins M, Sharpe AH, Freeman GJ. Mouse inducible costimulatory molecule (ICOS) expression is enhanced by CD28 costimulation and regulates differentiation of CD4+ T cells. J Immunol. 2000;165:5035–5040. doi: 10.4049/jimmunol.165.9.5035. [DOI] [PubMed] [Google Scholar]
- 103.Parry RV, Rumbley CA, Vandenberghe LH, June CH, Riley JL. CD28 and inducible costimulatory protein Src homology 2 binding domains show distinct regulation of phosphatidylinositol 3-kinase, Bcl-xL, and IL-2 expression in primary human CD4 T lymphocytes. J Immunol. 2003;171:166–174. doi: 10.4049/jimmunol.171.1.166. [DOI] [PubMed] [Google Scholar]
- 104.Probst HC, McCoy K, Okazaki T, Honjo T, van den Broek M. Resting dendritic cells induce peripheral CD8+ T cell tolerance through PD-1 and CTLA-4. Nat Immunol. 2005;6:280–286. doi: 10.1038/ni1165. [DOI] [PubMed] [Google Scholar]
- 105.Nishimura H, Honjo T, Minato N. Facilitation of beta selection and modification of positive selection in the thymus of PD-1-deficient mice. J Exp Med. 2000;191:891–898. doi: 10.1084/jem.191.5.891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, Iwai Y, Long AJ, Brown JA, Nunes R, Greenfield EA, Bourque K, Boussiotis VA, Carter LL, Carreno BM, Malenkovich N, Nishimura H, Okazaki T, Honjo T, Sharpe AH, Freeman GJ. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–268. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 107.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, Sasayama S, Mizoguchi A, Hiai H, Minato N, Honjo T. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–322. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 108.Valitutti S, Dessing M, Aktories K, Gallati H, Lanzavecchia A. Sustained signaling leading to T cell activation results from prolonged T cell receptor occupancy. Role of T cell actin cytoskeleton. J Exp Med. 1995;181:577–584. doi: 10.1084/jem.181.2.577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Randolph GJ, Furie MB. Mononuclear phagocytes egress from an in vitro model of the vascular wall by migrating across endothelium in the basal to apical direction: Role of intercellular adhesion molecule 1 and the CD11/CD18 integrins. J Exp Med. 1996;183:451–462. doi: 10.1084/jem.183.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Weber C, Lu C, Casasnovas J, Springer T. Role of aLb2 integrin avidity in transendothelial chemotaxis of mononuclear cells. J Immunol. 1997;159:3968–3975. [PubMed] [Google Scholar]
- 111.Warnock RA, Askari S, Butcher EC, von Andrian UH. Molecular mechanisms of lymphocyte homing to peripheral lymph nodes. J Exp Med. 1998;187:205–216. doi: 10.1084/jem.187.2.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kuhlman P, Moy VT, Lollo BA, Brian AA. The accessory function of murine intercellular adhesion molecule-1 in T lymphocyte activation. Contributions of adhesion and co-activation. J Immunol. 1991;146:1773–1782. [PubMed] [Google Scholar]
- 113.Bachmann MF, McKall-Faienza K, Schmits R, Bouchard D, Beach J, Speiser DE, Mak TW, Ohashi PS. Distinct roles for LFA-1 and CD28 during activation of naive T cells: Adhesion versus costimulation. Immunity. 1997;7:549–557. doi: 10.1016/s1074-7613(00)80376-3. [DOI] [PubMed] [Google Scholar]
- 114.Fischer H, Gjorloff A, Hedlund G, Hedman H, Lundgren E, Kalland T, Sjogren HO, Dohlsten M. Stimulation of human naive and memory T helper cells with bacterial superantigen. Naive CD4+45RA+ T cells require a costimulatory signal mediated through the LFA-1/ICAM-1 pathway. J Immunol. 1992;148:1993–1998. [PubMed] [Google Scholar]
- 115.Curtsinger JM, Schmidt CS, Mondino A, Lins DC, Kedl RM, Jenkins MK, Mescher MF. Inflammatory cytokines provide a third signal for activation of naive CD4+ and CD8+ T cells. J Immunol. 1999;162:3256–3262. [PubMed] [Google Scholar]
- 116.Halloran PF. Immunosuppressive drugs for kidney transplantation. N Engl J Med. 2004;351:2715–2729. doi: 10.1056/NEJMra033540. [DOI] [PubMed] [Google Scholar]
- 117.Kalinski P, Hilkens CM, Wierenga EA, Kapsenberg ML. T-cell priming by type-1 and type-2 polarized dendritic cells: The concept of a third signal. Immunol Today. 1999;20:561–567. doi: 10.1016/s0167-5699(99)01547-9. [DOI] [PubMed] [Google Scholar]
- 118.Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 119.Janeway CA, Jr, Goodnow CC, Medzhitov R. Danger—pathogen on the premises! Immunological tolerance. Curr Biol. 1996;6:519–522. doi: 10.1016/s0960-9822(02)00531-6. [DOI] [PubMed] [Google Scholar]
- 120.Wang D, Matsumoto R, You Y, Che T, Lin XY, Gaffen SL, Lin X. CD3/CD28 costimulation-induced NF-kappaB activation is mediated by recruitment of protein kinase C-theta, Bcl10, and IkappaB kinase beta to the immunological synapse through CARMA1. Mol Cell Biol. 2004;24:164–171. doi: 10.1128/MCB.24.1.164-171.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hafler DA. Multiple sclerosis. J Clin Invest. 2004;113:788–794. doi: 10.1172/JCI21357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 123.Smorodchenko A, Wuerfel J, Pohl EE, Vogt J, Tysiak E, Glumm R, Hendrix S, Nitsch R, Zipp F, Infante-Duarte C. CNS-irrelevant T-cells enter the brain, cause blood-brain barrier disruption but no glial pathology. Eur J Neurosci. 2007;26:1387–1398. doi: 10.1111/j.1460-9568.2007.05792.x. [DOI] [PubMed] [Google Scholar]
- 124.Wohler JE, Smith SS, Zinn KR, Bullard DC, Barnum SR. Gammadelta T cells in EAE: Early trafficking events and cytokine requirements. Eur J Immunol. 2009;39:1516–1526. doi: 10.1002/eji.200839176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. 1986. J Immunol. 2005;175:5–14. [PubMed] [Google Scholar]
- 126.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL. Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem. 2003;278:1910–1914. doi: 10.1074/jbc.M207577200. [DOI] [PubMed] [Google Scholar]
- 127.Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hedegaard CJ, Krakauer M, Bendtzen K, Lund H, Sellebjerg F, Nielsen CH. T helper cell type 1 (Th1), Th2 and Th17 responses to myelin basic protein and disease activity in multiple sclerosis. Immunology. 2008;125:161–169. doi: 10.1111/j.1365-2567.2008.02837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Matusevicius D, Kivisakk P, He B, Kostulas N, Ozenci V, Fredrikson S, Link H. Interleukin-17 mRNA expression in blood and CSF mononuclear cells is augmented in multiple sclerosis. Mult Scler. 1999;5:101–104. doi: 10.1177/135245859900500206. [DOI] [PubMed] [Google Scholar]
- 130.Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol. 2005;6:1133–1141. doi: 10.1038/ni1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Haak S, Gyulveszi G, Becher B. Th17 cells in autoimmune disease: Changing the verdict. Immunotherapy. 2009;1:199–203. doi: 10.2217/1750743X.1.2.199. [DOI] [PubMed] [Google Scholar]
- 132.Bitar DM, Whitacre CC. Suppression of experimental autoimmune encephalomyelitis by the oral administration of myelin basic protein. Cell Immunol. 1988;112:364–370. doi: 10.1016/0008-8749(88)90305-x. [DOI] [PubMed] [Google Scholar]
- 133.Higgins PJ, Weiner HL. Suppression of experimental autoimmune encephalomyelitis by oral administration of myelin basic protein and its fragments. J Immunol. 1988;140:440–445. [PubMed] [Google Scholar]
- 134.Weiner HL, Friedman A, Miller A, Khoury SJ, al-Sabbagh A, Santos L, Sayegh M, Nussenblatt RB, Trentham DE, Hafler DA. Oral tolerance: Immunologic mechanisms and treatment of animal and human organ-specific autoimmune diseases by oral administration of autoantigens. Annu Rev Immunol. 1994;12:809–837. doi: 10.1146/annurev.iy.12.040194.004113. [DOI] [PubMed] [Google Scholar]
- 135.Whitacre CC, Gienapp IE, Orosz CG, Bitar DM. Oral tolerance in experimental autoimmune encephalomyelitis. III. Evidence for clonal anergy. J Immunol. 1991;147:2155–2163. [PubMed] [Google Scholar]
- 136.Weiner HL, Mackin GA, Matsui M, Orav EJ, Khoury SJ, Dawson DM, Hafler DA. Double-blind pilot trial of oral tolerization with myelin antigens in multiple sclerosis. Science. 1993;259:1321–1324. doi: 10.1126/science.7680493. [DOI] [PubMed] [Google Scholar]
- 137.Warren KG, Catz I. Administration of myelin basic protein synthetic peptides to multiple sclerosis patients. J Neurol Sci. 1995;133:85–94. doi: 10.1016/0022-510x(95)00187-7. [DOI] [PubMed] [Google Scholar]
- 138.Warren KG, Catz I, Wucherpfennig KW. Tolerance induction to myelin basic protein by intravenous synthetic peptides containing epitope P85 VVHFFKNIVTP96 in chronic progressive multiple sclerosis. J Neurol Sci. 1997;152:31–38. doi: 10.1016/s0022-510x(97)00130-5. [DOI] [PubMed] [Google Scholar]
- 139.Metzler B, Wraith DC. Inhibition of experimental autoimmune encephalomyelitis by inhalation but not oral administration of the encephalitogenic peptide: Influence of MHC binding affinity. Int Immunol. 1993;5:1159–1165. doi: 10.1093/intimm/5.9.1159. [DOI] [PubMed] [Google Scholar]
- 140.Metzler B, Wraith DC. Mucosal tolerance in a murine model of experimental autoimmune encephalomyelitis. Ann N Y Acad Sci. 1996;778:228–242. doi: 10.1111/j.1749-6632.1996.tb21131.x. [DOI] [PubMed] [Google Scholar]
- 141.Bai XF, Shi FD, Xiao BG, Li HL, van der Meide PH, Link H. Nasal administration of myelin basic protein prevents relapsing experimental autoimmune encephalomyelitis in DA rats by activating regulatory cells expressing IL-4 and TGF-beta mRNA. J Neuroimmunol. 1997;80:65–75. doi: 10.1016/s0165-5728(97)00133-1. [DOI] [PubMed] [Google Scholar]
- 142.Bai XF, Li HL, Shi FD, Liu JQ, Xiao BG, Van der Meide PH, Link H. Complexities of applying nasal tolerance induction as a therapy for ongoing relapsing experimental autoimmune encephalomyelitis (EAE) in DA rats. Clin Exp Immunol. 1998;111:205–210. doi: 10.1046/j.1365-2249.1998.00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Irla M, Kupfer N, Suter T, Lissilaa R, Benkhoucha M, Skupsky J, Lalive PH, Fontana A, Reith W, Hugues S. MHC class II-restricted antigen presentation by plasmacytoid dendritic cells inhibits T cell-mediated autoimmunity. J Exp Med. 2010;207:1891–1905. doi: 10.1084/jem.20092627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Burkhart C, Liu GY, Anderton SM, Metzler B, Wraith DC. Peptide-induced T cell regulation of experimental autoimmune encephalomyelitis: A role for IL-10. Int Immunol. 1999;11:1625–1634. doi: 10.1093/intimm/11.10.1625. [DOI] [PubMed] [Google Scholar]
- 145.Platten M, Ho PP, Youssef S, Fontoura P, Garren H, Hur EM, Gupta R, Lee LY, Kidd BA, Robinson WH, Sobel RA, Selley ML, Steinman L. Treatment of autoimmune neuroinflammation with a synthetic tryptophan metabolite. Science. 2005;310:850–855. doi: 10.1126/science.1117634. [DOI] [PubMed] [Google Scholar]
- 146.Sinha S, Subramanian S, Proctor TM, Kaler LJ, Grafe M, Dahan R, Huan J, Vandenbark AA, Burrows GG, Offner H. A promising therapeutic approach for multiple sclerosis: Recombinant T-cell receptor ligands modulate experimental autoimmune encephalomyelitis by reducing interleukin-17 production and inhibiting migration of encephalitogenic cells into the CNS. J Neurosci. 2007;27:12531–12539. doi: 10.1523/JNEUROSCI.3599-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Offner H, Sinha S, Burrows GG, Ferro AJ, Vandenbark AA. RTL therapy for multiple sclerosis: A phase I clinical study. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.09.013. E-publication October 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sinha S, Subramanian S, Miller L, Proctor TM, Roberts C, Burrows GG, Vandenbark AA, Offner H. Cytokine switch and bystander suppression of autoimmune responses to multiple antigens in experimental autoimmune encephalomyelitis by a single recombinant T-cell receptor ligand. J Neurosci. 2009;29:3816–3823. doi: 10.1523/JNEUROSCI.5812-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Scott LJ, Figgitt DP. Mitoxantrone: A review of its use in multiple sclerosis. CNS Drugs. 2004;18:379–396. doi: 10.2165/00023210-200418060-00010. [DOI] [PubMed] [Google Scholar]
- 150.The IFNB Multiple Sclerosis Study Group. Interferon beta-1b is effective in relapsing-remitting multiple sclerosis. I. Clinical results of a multicenter, randomized, double-blind, placebo-controlled trial. Neurology. 1993;43:655–661. doi: 10.1212/wnl.43.4.655. [DOI] [PubMed] [Google Scholar]
- 151.Yong VW, Chabot S, Stuve O, Williams G. Interferon beta in the treatment of multiple sclerosis: Mechanisms of action. Neurology. 1998;51:682–689. doi: 10.1212/wnl.51.3.682. [DOI] [PubMed] [Google Scholar]
- 152.Ransohoff RM. Natalizumab for multiple sclerosis. N Engl J Med. 2007;356:2622–2629. doi: 10.1056/NEJMct071462. [DOI] [PubMed] [Google Scholar]
- 153.Miller DH, Khan OA, Sheremata WA, Blumhardt LD, Rice GP, Libonati MA, Willmer-Hulme AJ, Dalton CM, Miszkiel KA, O’Connor PW. A controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2003;348:15–23. doi: 10.1056/NEJMoa020696. [DOI] [PubMed] [Google Scholar]
- 154.Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, Phillips JT, Lublin FD, Giovannoni G, Wajgt A, Toal M, Lynn F, Panzara MA, Sandrock AW. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354:899–910. doi: 10.1056/NEJMoa044397. [DOI] [PubMed] [Google Scholar]
- 155.Balcer LJ, Galetta SL, Calabresi PA, Confavreux C, Giovannoni G, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Miller DH, O’Connor PW, Phillips JT, Polman CH, Radue EW, Rudick RA, Stuart WH, Wajgt A, Weinstock-Guttman B, Wynn DR, Lynn F, Panzara MA. Natalizumab reduces visual loss in patients with relapsing multiple sclerosis. Neurology. 2007;68:1299–1304. doi: 10.1212/01.wnl.0000259521.14704.a8. [DOI] [PubMed] [Google Scholar]
- 156.Comi G. Treatment of multiple sclerosis: Role of natalizumab. Neurol Sci. 2009;30:S155–S158. doi: 10.1007/s10072-009-0147-2. [DOI] [PubMed] [Google Scholar]
- 157.Schweikert A, Kremer M, Ringel F, Liebig T, Duyster J, Stuve O, Hemmer B, Berthele A. Primary central nervous system lymphoma in a patient treated with natalizumab. Ann Neurol. 2009;66:403–406. doi: 10.1002/ana.21782. [DOI] [PubMed] [Google Scholar]
- 158.Chen Y, Bord E, Tompkins T, Miller J, Tan CS, Kinkel RP, Stein MC, Viscidi RP, Ngo LH, Koralnik IJ. Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Engl J Med. 2009;361:1067–1074. doi: 10.1056/NEJMoa0904267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Murray JS, Oney S, Page JE, Kratochvil-Stava A, Hu Y, Makagiansar IT, Brown JC, Kobayashi N, Siahaan TJ. Suppression of type 1 diabetes in NOD mice by bifunctional peptide inhibitor: Modulation of the immunological synapse formation. Chem Biol Drug Des. 2007;70:227–236. doi: 10.1111/j.1747-0285.2007.00552.x. [DOI] [PubMed] [Google Scholar]
- 160.Pihoker C, Gilliam LK, Hampe CS, Lernmark A. Autoantibodies in diabetes. Diabetes. 2005;54:S52–S61. doi: 10.2337/diabetes.54.suppl_2.s52. [DOI] [PubMed] [Google Scholar]
- 161.Zhang L, Nakayama M, Eisenbarth GS. Insulin as an autoantigen in NOD/human diabetes. Curr Opin Immunol. 2008;20:111–118. doi: 10.1016/j.coi.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Abiru N, Maniatis AK, Yu L, Miao D, Moriyama H, Wegmann D, Eisenbarth GS. Peptide and major histocompatibility complex-specific breaking of humoral tolerance to native insulin with the B9-23 peptide in diabetes-prone and normal mice. Diabetes. 2001;50:1274–1281. doi: 10.2337/diabetes.50.6.1274. [DOI] [PubMed] [Google Scholar]
- 163.Chen W, Bergerot I, Elliott JF, Harrison LC, Abiru N, Eisenbarth GS, Delovitch TL. Evidence that a peptide spanning the B-C junction of proinsulin is an early Autoantigen epitope in the pathogenesis of type 1 diabetes. J Immunol. 2001;167:4926–4935. doi: 10.4049/jimmunol.167.9.4926. [DOI] [PubMed] [Google Scholar]
- 164.Higashide T, Kawamura T, Nagata M, Kotani R, Kimura K, Hirose M, Inada H, Niihira S, Yamano T. T cell epitope mapping study with insulin overlapping peptides using ELISPOT assay in Japanese children and adolescents with type 1 diabetes. Pediatr Res. 2006;59:445–450. doi: 10.1203/01.pdr.0000200803.72985.3c. [DOI] [PubMed] [Google Scholar]
- 165.Chen J, Liu X. The role of interferon gamma in regulation of CD4+ T-cells and its clinical implications. Cell Immunol. 2009;254:85–90. doi: 10.1016/j.cellimm.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 166.Tisch R, Liblau RS, Yang XD, Liblau P, McDevitt HO. Induction of GAD65-specific regulatory T-cells inhibits ongoing autoimmune diabetes in nonobese diabetic mice. Diabetes. 1998;47:894–899. doi: 10.2337/diabetes.47.6.894. [DOI] [PubMed] [Google Scholar]
- 167.Tisch R, Wang B, Serreze DV. Induction of glutamic acid decarboxylase 65-specific Th2 cells and suppression of autoimmune diabetes at late stages of disease is epitope dependent. J Immunol. 1999;163:1178–1187. [PubMed] [Google Scholar]
- 168.Winchester R. The molecular basis of susceptibility to rheumatoid arthritis. Adv Immunol. 1994;56:389–466. doi: 10.1016/s0065-2776(08)60456-3. [DOI] [PubMed] [Google Scholar]
- 169.Pitzalis C, Kingsley G, Murphy J, Panayi G. Abnormal distribution of the helper-inducer and suppressor-inducer T-lymphocyte subsets in the rheumatoid joint. Clin Immunol Immunopathol. 1987;45:252–258. doi: 10.1016/0090-1229(87)90040-7. [DOI] [PubMed] [Google Scholar]
- 170.Reme T, Portier M, Frayssinoux F, Combe B, Miossec P, Favier F, Sany J. T cell receptor expression and activation of synovial lymphocyte subsets in patients with rheumatoid arthritis. Phenotyping of multiple synovial sites. Arthritis Rheum. 1990;33:485–492. doi: 10.1002/art.1780330404. [DOI] [PubMed] [Google Scholar]
- 171.Butcher EC. Warner-Lambert/Parke-Davis Award lecture. Cellular and molecular mechanisms that direct leukocyte traffic. Am J Pathol. 1990;136:3–11. [PMC free article] [PubMed] [Google Scholar]
- 172.Goto M, Miyamoto T, Nishioka K, Uchida S. T cytotoxic and helper cells are markedly increased, and T suppressor and inducer cells are markedly decreased, in rheumatoid synovial fluids. Arthritis Rheum. 1987;30:737–743. doi: 10.1002/art.1780300703. [DOI] [PubMed] [Google Scholar]
- 173.Poulter LW, Duke O, Panayi GS, Hobbs S, Raftery MJ, Janossy G. Activated T lymphocytes of the synovial membrane in rheumatoid arthritis and other arthropathies. Scand J Immunol. 1985;22:683–690. doi: 10.1111/j.1365-3083.1985.tb01931.x. [DOI] [PubMed] [Google Scholar]
- 174.Forre O, Dobloug JH, Natvig JB. Augmented numbers of HLA-DR-positive T lymphocytes in the synovial fluid and synovial tissue of patients with rheumatoid arthritis and juvenile rheumatoid arthritis: In vivo-activated T lymphocytes are potent stimulators in the mixed lymphocyte reaction. Scand J Immunol. 1982;15:227–231. doi: 10.1111/j.1365-3083.1982.tb00643.x. [DOI] [PubMed] [Google Scholar]
- 175.Fox RI, Fong S, Sabharwal N, Carstens SA, Kung PC, Vaughan JH. Synovial fluid lymphocytes differ from peripheral blood lymphocytes in patients with rheumatoid arthritis. J Immunol. 1982;128:351–354. [PubMed] [Google Scholar]
- 176.Hale LP, Martin ME, McCollum DE, Nunley JA, Springer TA, Singer KH, Haynes BF. Immunohistologic analysis of the distribution of cell adhesion molecules within the inflammatory synovial microenvironment. Arthritis Rheum. 1989;32:22–30. doi: 10.1002/anr.1780320105. [DOI] [PubMed] [Google Scholar]
- 177.Hemler ME, Glass D, Coblyn JS, Jacobson JG. Very late activation antigens on rheumatoid synovial fluid T lymphocytes. Association with stages of T cell activation. J Clin Invest. 1986;78:696–702. doi: 10.1172/JCI112629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Lipsky PE, Davis LS, Cush JJ, Oppenheimer-Marks N. The role of cytokines in the pathogenesis of rheumatoid arthritis. Springer Semin Immunopathol. 1989;11:123–162. doi: 10.1007/BF00197186. [DOI] [PubMed] [Google Scholar]
- 179.Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS, Shnier R, Portek IJ. Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: A two-year prospective study (the DAMAGE study cohort) Arthritis Rheum. 2006;54:1122–1131. doi: 10.1002/art.21749. [DOI] [PubMed] [Google Scholar]
- 180.Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W. High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol. 2000;164:2832–2838. doi: 10.4049/jimmunol.164.5.2832. [DOI] [PubMed] [Google Scholar]
- 181.Meyer O. Oral immunomodulation therapy in rheumatoid arthritis. Joint Bone Spine. 2000;67:384–392. [PubMed] [Google Scholar]
- 182.Nagler-Anderson C, Bober LA, Robinson ME, Siskind GW, Thorbecke GJ. Suppression of type II collagen-induced arthritis by intragastric administration of soluble type II collagen. Proc Natl Acad Sci USA. 1986;83:7443–7446. doi: 10.1073/pnas.83.19.7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Zhang ZY, Lee CS, Lider O, Weiner HL. Suppression of adjuvant arthritis in Lewis rats by oral administration of type II collagen. J Immunol. 1990;145:2489–2493. [PubMed] [Google Scholar]
- 184.Trentham DE, Dynesius-Trentham RA, Orav EJ, Combitchi D, Lorenzo C, Sewell KL, Hafler DA, Weiner HL. Effects of oral administration of type II collagen on rheumatoid arthritis. Science. 1993;261:1727–1730. doi: 10.1126/science.8378772. [DOI] [PubMed] [Google Scholar]
- 185.Zhu P, Li XY, Wang HK, Jia JF, Zheng ZH, Ding J, Fan CM. Oral administration of type-II collagen peptide 250–270 suppresses specific cellular and humoral immune response in collagen-induced arthritis. Clin Immunol. 2007;122:75–84. doi: 10.1016/j.clim.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 186.Blanas E, Heath WR. Oral administration of antigen can lead to the onset of autoimmune disease. Int Rev Immunol. 1999;18:217–228. doi: 10.3109/08830189909043026. [DOI] [PubMed] [Google Scholar]
- 187.Huan J, Kaler LJ, Mooney JL, Subramanian S, Hopke C, Vandenbark AA, Rosloniec EF, Burrows GG, Offner H. MHC class II derived recombinant T cell receptor ligands protect DBA/1LacJ mice from collagen-induced arthritis. J Immunol. 2008;180:1249–1257. doi: 10.4049/jimmunol.180.2.1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Tan P, Anasetti C, Hansen JA, Melrose J, Brunvand M, Bradshaw J, Ledbetter JA, Linsley PS. Induction of alloantigen-specific hyporesponsiveness in human T lymphocytes by blocking interaction of CD28 with its natural ligand B7/BB1. J Exp Med. 1993;177:165–173. doi: 10.1084/jem.177.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Kremer JM, Dougados M, Emery P, Durez P, Sibilia J, Shergy W, Steinfeld S, Tindall E, Becker JC, Li T, Nuamah IF, Aranda R, Moreland LW. Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: Twelve-month results of a phase IIb, double-blind, randomized, placebo-controlled trial. Arthritis Rheum. 2005;52:2263–2271. doi: 10.1002/art.21201. [DOI] [PubMed] [Google Scholar]
- 190.Futterer A, Mink K, Luz A, Kosco-Vilbois MH, Pfeffer K. The lymphotoxin beta receptor controls organogenesis and affinity maturation in peripheral lymphoid tissues. Immunity. 1998;9:59–70. doi: 10.1016/s1074-7613(00)80588-9. [DOI] [PubMed] [Google Scholar]
- 191.Jelinek DF, Lipsky PE. Enhancement of human B cell proliferation and differentiation by tumor necrosis factor-alpha and interleukin 1. J Immunol. 1987;139:2970–2976. [PubMed] [Google Scholar]
- 192.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM. Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. N Engl J Med. 2001;345:1098–1104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 193.Wolfe F, Michaud K, Anderson J, Urbansky K. Tuberculosis infection in patients with rheumatoid arthritis and the effect of infliximab therapy. Arthritis Rheum. 2004;50:372–379. doi: 10.1002/art.20009. [DOI] [PubMed] [Google Scholar]
- 194.Khanna D, McMahon M, Furst DE. Anti-tumor necrosis factor alpha therapy and heart failure: What have we learned and where do we go from here? Arthritis Rheum. 2004;50:1040–1050. doi: 10.1002/art.20164. [DOI] [PubMed] [Google Scholar]
- 195.Cohen S, Hurd E, Cush J, Schiff M, Weinblatt ME, Moreland LW, Kremer J, Bear MB, Rich WJ, McCabe D. Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate: Results of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:614–624. doi: 10.1002/art.10141. [DOI] [PubMed] [Google Scholar]
- 196.Cohen SB, Moreland LW, Cush JJ, Greenwald MW, Block S, Shergy WJ, Hanrahan PS, Kraishi MM, Patel A, Sun G, Bear MB. A multicentre, double-blind, randomized, placebo-controlled trial of anakinra (Kineret), a recombinant interleukin 1 receptor antagonist, in patients with rheumatoid arthritis treated with background methotrexate. Ann Rheum Dis. 2004;63:1062–1068. doi: 10.1136/ard.2003.016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeiser H, Redlich K, Feige U, Dunstan C, Kollias G, Steiner G, Smolen J, Schett G. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: Effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. 2004;50:277–290. doi: 10.1002/art.11487. [DOI] [PubMed] [Google Scholar]
- 198.Genovese MC, Cohen S, moreland L, Lium D, Robbins S, Newmark R, Bekker P. Combination therapy with etanercept and anakinra in the treatment of patients with rheumatoid arthritis who have been treated unsuccessfully with methotrexate. Arthritis Rheum. 2004;50:1412–1419. doi: 10.1002/art.20221. [DOI] [PubMed] [Google Scholar]
- 199.Edwards JC, Szczepanski L, Szechinski J, Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM, Shaw T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N Engl J Med. 2004;350:2572–2581. doi: 10.1056/NEJMoa032534. [DOI] [PubMed] [Google Scholar]
- 200.Isobe M, Yagita H, Okumura K, Ihara A. Specific acceptance of cardiac allograft after treatment with antibodies to ICAM-1 and LFA-1. Science. 1992;255:1125–1127. doi: 10.1126/science.1347662. [DOI] [PubMed] [Google Scholar]
- 201.Takazawa K, Hosoda Y, Bashuda H, Senio K, Yagita H, Tamatani T, Miyasaka M, Okumura K. Synergistic effects of mycophenolate mofetil (MMF, RS-61443) and anti-LFA-1/ICAM-1 monoclonal antibodies on the prolongation of heart allograft survival in rats. Transplant Proc. 1996;28:1980–1981. [PubMed] [Google Scholar]
- 202.Barouch FC, Miyamoto K, Allport JR, Fujita K, Bursell SE, Aiello LP, Luscinskas FW, Adamis AP. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Invest Ophthalmol Vis Sci. 2000;41:1153–1158. [PubMed] [Google Scholar]
- 203.Moriyama H, Yokono K, Amano K, Nagata M, Hasegawa Y, Okamoto N, Tsukamoto K, Miki M, Yoneda R, Yagi N, Tominaga Y, Kikutani H, Hioki K, Okumura K, Yagita H, Kasuga M. Induction of tolerance in murine autoimmune diabetes by transient blockade of leukocyte function-associated antigen-1/intercellular adhesion molecule-1 pathway. J Immunol. 1996;157:3737–3743. [PubMed] [Google Scholar]
- 204.Schulze-Koops H, Lipsky PE, Kavanaugh AF, Davis LS. Elevated Th1- or Th0-like cytokine mRNA in peripheral circulation of patients with rheumatoid arthritis. Modulation by treatment with anti-ICAM-1 correlates with clinical benefit. J Immunol. 1995;155:5029–5037. [PubMed] [Google Scholar]
- 205.Kavanaugh AF, Davis LS, Jain RI, Nichols LA, Norris SH, Lipsky PE. A phase I/II open label study of the safety and efficacy of an anti-ICAM-1 (intercellular adhesion molecule-1; CD54) monoclonal antibody in early rheumatoid arthritis. J Rheumatol. 1996;23:1338–1344. [PubMed] [Google Scholar]
- 206.Papp K, Bissonnette R, Krueger JG, Carey W, Gratton D, Gulliver WP, Lui H, Lynde CW, Magee A, Minier D, Ouellet JP, Patel P, Shapiro J, Shear NH, Kramer S, Walicke P, Bauer R, Dedrick RL, Kim SS, White M, Garovoy MR. The treatment of moderate to severe psoriasis with a new anti-CD11a monoclonal antibody. J Am Acad Dermatol. 2001;45:665–674. doi: 10.1067/mjd.2001.117850. [DOI] [PubMed] [Google Scholar]
- 207.Gottlieb AB, Krueger JG, Wittkowski K, Dedrick R, Walicke PA, Garovoy M. Psoriasis as a model for T-cell-mediated disease: Immunobiologic and clinical effects of treatment with multiple doses of efalizumab, an anti-CD11a antibody. Arch Dermatol. 2002;138:591–600. doi: 10.1001/archderm.138.5.591. [DOI] [PubMed] [Google Scholar]
- 208.Dustin ML, Chan AC. Signaling takes shape in the immune system. Cell. 2000;103:283–294. doi: 10.1016/s0092-8674(00)00120-3. [DOI] [PubMed] [Google Scholar]
- 209.Lebedeva T, Dustin ML, Sykulev Y. ICAM-1 co-stimulates target cells to facilitate antigen presentation. Curr Opin Immunol. 2005;17:251–258. doi: 10.1016/j.coi.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 210.Sims TN, Dustin ML. The immunological synapse: Integrins take the stage. Immunol Rev. 2002;186:100–117. doi: 10.1034/j.1600-065x.2002.18610.x. [DOI] [PubMed] [Google Scholar]
- 211.Scholer A, Hugues S, Boissonnas A, Fetler L, Amigorena S. Intercellular adhesion molecule-1-dependent stable interactions between T cells and dendritic cells determine CD8+ T cell memory. Immunity. 2008;28:258–270. doi: 10.1016/j.immuni.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 212.Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, Killeen N, Orange JS, Russell SM, Weninger W, Reiner SL. Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science. 2007;315:1687–1691. doi: 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- 213.Kaser A, Tilg H. Novel therapeutic targets in the treatment of IBD. Expert Opin Ther Targets. 2008;12:553–563. doi: 10.1517/14728222.12.5.553. [DOI] [PubMed] [Google Scholar]
- 214.Gottlieb A, Krueger JG, Bright R, Ling M, Lebwohl M, Kang S, Feldman S, Spellman M, Wittkowski K, Ochs HD, Jardieu P, Bauer R, White M, Dedrick R, Garovoy M. Effects of administration of a single dose of a humanized monoclonal antibody to CD11a on the immunobiology and clinical activity of psoriasis. J Am Acad Dermatol. 2000;42:428–435. doi: 10.1016/s0190-9622(00)90214-7. [DOI] [PubMed] [Google Scholar]
- 215.Kvist M, Kanje M, Ekberg H, Corbascio M, Dahlin LB. Costimulation blockade in transplantation of nerve allografts: Long-term effects. J Peripher Nerv Syst. 2008;13:200–207. doi: 10.1111/j.1529-8027.2008.00178.x. [DOI] [PubMed] [Google Scholar]
- 216.Fischer A, Griscelli C, Blanche S, Le Deist F, Veber F, Lopez M, Delaage M, Olive D, Mawas C, Janossy G. Prevention of graft failure by an anti-HLFA-1 monoclonal antibody in HLA-mismatched bone-marrow transplantation. Lancet. 1986;2:1058–1061. doi: 10.1016/s0140-6736(86)90465-4. [DOI] [PubMed] [Google Scholar]
- 217.Hourmant M, Le Mauff B, Le Meur Y, Dantal J, Cantarovich D, Giral M, Caudrelier P, Albericci G, Soulillou JP. Administration of an anti-CD11a monoclonal antibody in recipients of kidney transplantation. A pilot study. Transplantation. 1994;58:377–380. [PubMed] [Google Scholar]
- 218.Le Mauff B, Hourmant M, Rougier JP, Hirn M, Dantal J, Baatard R, Cantarovich D, Jacques Y, Soulillou JP. Effect of anti-LFA1 (CD11a) monoclonal antibodies in acute rejection in human kidney transplantation. Transplantation. 1991;52:291–296. doi: 10.1097/00007890-199108000-00020. [DOI] [PubMed] [Google Scholar]
- 219.Stoppa AM, Maraninchi D, Blaise D, Viens P, Hirn M, Olive D, Reiffers J, Milpied N, Gaspard MH, Mawas C. Anti-LFA1 monoclonal antibody (25. 3) for treatment of steroid-resistant grade III-IV acute graft-versus-host disease. Transpl Int. 1991;4:3–7. doi: 10.1007/BF00335508. [DOI] [PubMed] [Google Scholar]
- 220.Schmidmaier R, Baumann P. Anti-adhesion evolves to a promising therapeutic concept in oncology. Curr Med Chem. 2008;15:978–990. doi: 10.2174/092986708784049667. [DOI] [PubMed] [Google Scholar]
- 221.Arai K, Sunamura M, Wada Y, Takahashi M, Kobari M, Kato K, Yagita H, Okumura K, Matsuno S. Preventing effect of anti-ICAM-1 and anti-LFA-1 monoclonal antibodies on murine islet allograft rejection. Int J Pancreatol. 1999;26:23–31. doi: 10.1385/IJGC:26:1:23. [DOI] [PubMed] [Google Scholar]
- 222.Bowles MJ, Pockley AG, Wood RF. Effect of anti-LFA-1 monoclonal antibody on rat small bowel allograft survival and circulating leukocyte populations. Transpl Immunol. 2000;8:75–80. doi: 10.1016/s0966-3274(00)00007-1. [DOI] [PubMed] [Google Scholar]
- 223.Hori J, Isobe M, Yamagami S, Mizuochi T, Tsuru T. Specific immunosuppression of corneal allograft rejection by combination of anti-VLA-4 and anti-LFA-1 monoclonal antibodies in mice. Exp Eye Res. 1997;65:89–98. doi: 10.1006/exer.1997.0316. [DOI] [PubMed] [Google Scholar]
- 224.Kato Y, Yamataka A, Yagita H, Okumura K, Fujiwara T, Miyano T. Specific acceptance of fetal bowel allograft in mice after combined treatment with anti-intercellular adhesion molecule-1 and leukocyte function-associated antigen-1 antibodies. Ann Surg. 1996;223:94–100. doi: 10.1097/00000658-199601000-00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kakimoto K, Nakamura T, Ishii K, Takashi T, Iigou H, Yagita H, Okumura K, Onoue K. The effect of anti-adhesion molecule antibody on the development of collagen-induced arthritis. Cell Immunol. 1992;142:326–337. doi: 10.1016/0008-8749(92)90294-y. [DOI] [PubMed] [Google Scholar]
- 226.Chowdhury SA, Nagata M, Yamada K, Nakayama M, Chakrabarty S, Jin Z, Kotani R, Yokono K. Tolerance mechanisms in murine autoimmune diabetes induced by anti-ICAM-1/LFA-1 mAb and anti-CD8 mAb. Kobe J Med Sci. 2002;48:167–175. [PubMed] [Google Scholar]
- 227.Gordon EJ, Myers KJ, Dougherty JP, Rosen H, Ron Y. Both anti-CD11a (LFA-1) and anti-CD11b (MAC-1) therapy delay the onset and diminish the severity of experimental autoimmune encephalomyelitis. J Neuroimmunol. 1995;62:153–160. doi: 10.1016/0165-5728(95)00120-2. [DOI] [PubMed] [Google Scholar]
- 228.Inoue A, Koh CS, Yamazaki M, Ichikawa M, Isobe M, Ishihara Y, Yagita H, Kim BS. Anti-adhesion molecule therapy in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. Int Immunol. 1997;9:1837–1847. doi: 10.1093/intimm/9.12.1837. [DOI] [PubMed] [Google Scholar]
- 229.Kawai K, Kobayashi Y, Shiratori M, Sobue G, Tamatani T, Miyasaka M, Yoshikai Y. Intrathecal administration of antibodies against LFA-1 and against ICAM-1 suppresses experimental allergic encephalomyelitis in rats. Cell Immunol. 1996;171:262–268. doi: 10.1006/cimm.1996.0202. [DOI] [PubMed] [Google Scholar]
- 230.Kobayashi Y, Kawai K, Honda H, Tomida S, Niimi N, Tamatani T, Miyasaka M, Yoshikai Y. Antibodies against leukocyte function-associated antigen-1 and against intercellular adhesion molecule-1 together suppress the progression of experimental allergic encephalomyelitis. Cell Immunol. 1995;164:295–305. doi: 10.1006/cimm.1995.1173. [DOI] [PubMed] [Google Scholar]
- 231.Willenborg DO, Staykova MA, Miyasaka M. Short term treatment with soluble neuroantigen and anti-CD11a (LFA-1) protects rats against autoimmune encephalomyelitis: Treatment abrogates autoimmune disease but not autoimmunity. J Immunol. 1996;157:1973–1980. [PubMed] [Google Scholar]
- 232.Connolly MK, Kitchens EA, Chan B, Jardieu P, Wofsy D. Treatment of murine lupus with monoclonal antibodies to lymphocyte function-associated antigen-1: Dose-dependent inhibition of autoantibody production and blockade of the immune response to therapy. Clin Immunol Immunopathol. 1994;72:198–203. doi: 10.1006/clin.1994.1130. [DOI] [PubMed] [Google Scholar]
- 233.Haug CE, Colvin RB, Delmonico FL, Auchincloss H, Jr, Tolkoff-Rubin N, Preffer FI, Rothlein R, Norris S, Scharschmidt L, Cosimi AB. A phase I trial of immunosuppression with anti-ICAM-1 (CD54) mAb in renal allograft recipients. Transplantation. 1993;55:766–772. doi: 10.1097/00007890-199304000-00016. discussion 772–763. [DOI] [PubMed] [Google Scholar]
- 234.Kavanaugh AF, Davis LS, Nichols LA, Norris SH, Rothlein R, Scharschmidt LA, Lipsky PE. Treatment of refractory rheumatoid arthritis with a monoclonal antibody to intercellular adhesion molecule 1. Arthritis Rheum. 1994;37:992–999. doi: 10.1002/art.1780370703. [DOI] [PubMed] [Google Scholar]
- 235.Salmela K, Wramner L, Ekberg H, Hauser I, Bentdal O, Lins LE, Isoniemi H, Backman L, Persson N, Neumayer HH, Jorgensen PF, Spieker C, Hendry B, Nicholls A, Kirste G, Hasche G. A randomized multicenter trial of the anti-ICAM-1 monoclonal antibody (enlimomab) for the prevention of acute rejection and delayed onset of graft function in cadaveric renal transplantation: A report of the European Anti-ICAM-1 Renal Transplant Study Group. Transplantation. 1999;67:729–736. doi: 10.1097/00007890-199903150-00015. [DOI] [PubMed] [Google Scholar]
- 236.Lebwohl M, Tyring SK, Hamilton TK, Toth D, Glazer S, Tawfik NH, Walicke P, Dummer W, Wang X, Garovoy MR, Pariser D. A novel targeted T-cell modulator, efalizumab, for plaque psoriasis. N Engl J Med. 2003;349:2004–2013. doi: 10.1056/NEJMoa030002. [DOI] [PubMed] [Google Scholar]
- 237.Gordon KB, Papp KA, Hamilton TK, Walicke PA, Dummer W, Li N, Bresnahan BW, Menter A. Efalizumab for patients with moderate to severe plaque psoriasis: A randomized controlled trial. J Am Med Assoc. 2003;290:3073–3080. doi: 10.1001/jama.290.23.3073. [DOI] [PubMed] [Google Scholar]
- 238.Vugmeyster Y, Kikuchi T, Lowes MA, Chamian F, Kagen M, Gilleaudeau P, Lee E, Howell K, Bodary S, Dummer W, Krueger JG. Efalizumab (anti-CD11a)-induced increase in peripheral blood leukocytes in psoriasis patients is preferentially mediated by altered trafficking of memory CD8+ T cells into lesional skin. Clin Immunol. 2004;113:38–46. doi: 10.1016/j.clim.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 239.Leonardi CL, Papp KA, Gordon KB, Menter A, Feldman SR, Caro I, Walicke PA, Compton PG, Gottlieb AB. Extended efalizumab therapy improves chronic plaque psoriasis: Results from a randomized phase III trial. J Am Acad Dermatol. 2005;52:425–433. doi: 10.1016/j.jaad.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 240.Menter A, Gordon K, Carey W, Hamilton T, Glazer S, Caro I, Li N, Gulliver W. Efficacy and safety observed during 24 weeks of efalizumab therapy in patients with moderate to severe plaque psoriasis. Arch Dermatol. 2005;141:31–38. doi: 10.1001/archderm.141.1.31. [DOI] [PubMed] [Google Scholar]
- 241.Toth DP, Papp K, Gratton D. Long-term efficacy of up to 15 months’ efalizumab therapy in patients with moderate-to-severe chronic plaque psoriasis. Dermatol Ther. 2008;21:S6–S14. doi: 10.1111/j.1529-8019.2008.00235.x. [DOI] [PubMed] [Google Scholar]
- 242.Leonardi C, Menter A, Hamilton T, Caro I, Xing B, Gottlieb AB. Efalizumab: Results of a 3-year continuous dosing study for the long-term control of psoriasis. Br J Dermatol. 2008;158:1107–1116. doi: 10.1111/j.1365-2133.2008.08548.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Li S, Wang H, Peng B, Zhang M, Zhang D, Hou S, Guo Y, Ding J. Efalizumab binding to the LFA-1 alphaL I domain blocks ICAM-1 binding via steric hindrance. Proc Natl Acad Sci USA. 2009;106:4349–4354. doi: 10.1073/pnas.0810844106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Marecki S, Kirkpatrick P. Efalizumab. Nat Rev Drug Discov. 2004;3:473–474. doi: 10.1038/nrd1420. [DOI] [PubMed] [Google Scholar]
- 245.Anderson ME, Yakovleva T, Hu Y, Siahaan TJ. Inhibition of ICAM-1/LFA-1-mediated heterotypic T-cell adhesion to epithelial cells: Design of ICAM-1 cyclic peptides. Bioorg Med Chem Lett. 2004;14:1399–1402. doi: 10.1016/j.bmcl.2003.09.100. [DOI] [PubMed] [Google Scholar]
- 246.Gursoy RN, Siahaan TJ. Binding and internalization of an ICAM-1 peptide by the surface receptors of T cells. J Pept Res. 1999;53:414–421. doi: 10.1034/j.1399-3011.1999.00079.x. [DOI] [PubMed] [Google Scholar]
- 247.Iskandarsyah, Tejo BA, Tambunan US, Verkhivker G, Siahaan TJ. Structural modifications of ICAM-1 cyclic peptides to improve the activity to inhibit heterotypic adhesion of T cells. Chem Biol Drug Des. 2008;72:27–33. doi: 10.1111/j.1747-0285.2008.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Jois DS, Pal D, Tibbetts SA, Chan MA, Benedict SH, Siahaan TJ. Inhibition of homotypic adhesion of T-cells: Secondary structure of an ICAM-1-derived cyclic peptide. J Pept Res. 1997;49:517–526. doi: 10.1111/j.1399-3011.1997.tb01159.x. [DOI] [PubMed] [Google Scholar]
- 249.Tibbetts SA, Chirathaworn C, Nakashima M, Jois DS, Siahaan TJ, Chan MA, Benedict SH. Peptides derived from ICAM-1 and LFA-1 modulate T cell adhesion and immune function in a mixed lymphocyte culture. Transplantation. 1999;68:685–692. doi: 10.1097/00007890-199909150-00015. [DOI] [PubMed] [Google Scholar]
- 250.Tibbetts SA, Seetharama Jois D, Siahaan TJ, Benedict SH, Chan MA. Linear and cyclic LFA-1 and ICAM-1 peptides inhibit T cell adhesion and function. Peptides. 2000;21:1161–1167. doi: 10.1016/s0196-9781(00)00255-2. [DOI] [PubMed] [Google Scholar]
- 251.Yusuf-Makagiansar H, Makagiansar IT, Siahaan TJ. Inhibition of the adherence of T-lymphocytes to epithelial cells by a cyclic peptide derived from inserted domain of lymphocyte function-associated antigen-1. Inflammation. 2001;25:203–214. doi: 10.1023/a:1011044616170. [DOI] [PubMed] [Google Scholar]
- 252.Yusuf-Makagiansar H, Yakovleva TV, Tejo BA, Jones K, Hu Y, Verkhivker GM, Audus KL, Siahaan TJ. Sequence recognition of alpha-LFA-1-derived peptides by ICAM-1 cell receptors: Inhibitors of T-cell adhesion. Chem Biol Drug Des. 2007;70:237–246. doi: 10.1111/j.1747-0285.2007.00549.x. [DOI] [PubMed] [Google Scholar]
- 253.Barish CF. Alicaforsen therapy in inflammatory bowel disease. Expert Opin Biol Ther. 2005;5:1387–1391. doi: 10.1517/14712598.5.10.1387. [DOI] [PubMed] [Google Scholar]
- 254.Bennett CF, Condon TP, Grimm S, Chan H, Chiang MY. Inhibition of endothelial cell adhesion molecule expression with antisense oligonucleotides. J Immunol. 1994;152:3530–3540. [PubMed] [Google Scholar]
- 255.Schreiber S, Nikolaus S, Malchow H, Kruis W, Lochs H, Raedler A, Hahn EG, Krummenerl T, Steinmann G. Absence of efficacy of subcutaneous antisense ICAM-1 treatment of chronic active Crohn’s disease. Gastroenterology. 2001;120:1339–1346. doi: 10.1053/gast.2001.24015. [DOI] [PubMed] [Google Scholar]
- 256.van Deventer SJ, Tami JA, Wedel MK. A randomised, controlled, double blind, escalating dose study of alicaforsen enema in active ulcerative colitis. Gut. 2004;53:1646–1651. doi: 10.1136/gut.2003.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.van Deventer SJ, Wedel MK, Baker BF, Xia S, Chuang E, Miner PB., Jr A phase II dose ranging, double-blind, placebo-controlled study of alicaforsen enema in subjects with acute exacerbation of mild to moderate left-sided ulcerative colitis. Aliment Pharmacol Ther. 2006;23:1415–1425. doi: 10.1111/j.1365-2036.2006.02910.x. [DOI] [PubMed] [Google Scholar]
- 258.Yacyshyn B, Chey WY, Wedel MK, Yu RZ, Paul D, Chuang E. A randomized, double-masked, placebo-controlled study of alicaforsen, an antisense inhibitor of intercellular adhesion molecule 1, for the treatment of subjects with active Crohn’s disease. Clin Gastroenterol Hepatol. 2007;5:215–220. doi: 10.1016/j.cgh.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 259.Yacyshyn BR, Barish C, Goff J, Dalke D, Gaspari M, Yu R, Tami J, Dorr FA, Sewell KL. Dose ranging pharmacokinetic trial of high-dose alicaforsen (intercellular adhesion molecule-1 antisense oligodeoxynucleotide) (ISIS 2302) in active Crohn’s disease. Aliment Pharmacol Ther. 2002;16:1761–1770. doi: 10.1046/j.1365-2036.2002.01341.x. [DOI] [PubMed] [Google Scholar]
- 260.Yacyshyn BR, Bowen-Yacyshyn MB, Jewell L, Tami JA, Bennett CF, Kisner DL, Shanahan WR., Jr A placebo-controlled trial of ICAM-1 antisense oligonucleotide in the treatment of Crohn’s disease. Gastroenterology. 1998;114:1133–1142. doi: 10.1016/s0016-5085(98)70418-4. [DOI] [PubMed] [Google Scholar]
- 261.Yacyshyn BR, Chey WY, Goff J, Salzberg B, Baerg R, Buchman AL, Tami J, Yu R, Gibiansky E, Shanahan WR. Double blind, placebo controlled trial of the remission inducing and steroid sparing properties of an ICAM-1 antisense oligodeoxynucleotide, alicaforsen (ISIS 2302), in active steroid dependent Crohn’s disease. Gut. 2002;51:30–36. doi: 10.1136/gut.51.1.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Maksymowych WP, Blackburn WD, Jr, Tami JA, Shanahan WR., Jr A randomized, placebo controlled trial of an antisense oligodeoxynucleotide to intercellular adhesion molecule-1 in the treatment of severe rheumatoid arthritis. J Rheumatol. 2002;29:447–453. [PubMed] [Google Scholar]
- 263.Lenschow DJ, Ho SC, Sattar H, Rhee L, Gray G, Nabavi N, Herold KC, Bluestone JA. Differential effects of anti-B7-1 and anti-B7-2 monoclonal antibody treatment on the development of diabetes in the nonobese diabetic mouse. J Exp Med. 1995;181:1145–1155. doi: 10.1084/jem.181.3.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Bour-Jordan H, Salomon BL, Thompson HL, Santos R, Abbas AK, Bluestone JA. Constitutive expression of B7-1 on B cells uncovers autoimmunity toward the B cell compartment in the nonobese diabetic mouse. J Immunol. 2007;179:1004–1012. doi: 10.4049/jimmunol.179.2.1004. [DOI] [PubMed] [Google Scholar]
- 265.Finck BK, Linsley PS, Wofsy D. Treatment of murine lupus with CTLA4Ig. Science. 1994;265:1225–1227. doi: 10.1126/science.7520604. [DOI] [PubMed] [Google Scholar]
- 266.Kuchroo VK, Das MP, Brown JA, Ranger AM, Zamvil SS, Sobel RA, Weiner HL, Nabavi N, Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: Application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- 267.Vanderlugt CL, Karandikar NJ, Lenschow DJ, Dal Canto MC, Bluestone JA, Miller SD. Treatment with intact anti-B7-1 mAb during disease remission enhances epitope spreading and exacerbates relapses in R-EAE. J Neuroimmunol. 1997;79:113–118. doi: 10.1016/s0165-5728(97)00108-2. [DOI] [PubMed] [Google Scholar]
- 268.Perrin PJ, June CH, Maldonado JH, Ratts RB, Racke MK. Blockade of CD28 during in vitro activation of encephalitogenic T cells or after disease onset ameliorates experimental autoimmune encephalomyelitis. J Immunol. 1999;163:1704–1710. [PubMed] [Google Scholar]
- 269.Vanhove B, Laflamme G, Coulon F, Mougin M, Vusio P, Haspot F, Tiollier J, Soulillou JP. Selective blockade of CD28 and not CTLA-4 with a single-chain Fv-a1-antitrypsin fusion antibody. Blood. 2003;102:564–570. doi: 10.1182/blood-2002-08-2480. [DOI] [PubMed] [Google Scholar]
- 270.Bachmaier K, Pummerer C, Shahinian A, Ionescu J, Neu N, Mak TW, Penninger JM. Induction of autoimmunity in the absence of CD28 costimulation. J Immunol. 1996;157:1752–1757. [PubMed] [Google Scholar]
- 271.Shahinian A, Pfeffer K, Lee KP, Kundig TM, Kishihara K, Wakeham A, Kawai K, Ohashi PS, Thompson CB, Mak TW. Differential T cell costimulatory requirements in CD28-deficient mice. Science. 1993;261:609–612. doi: 10.1126/science.7688139. [DOI] [PubMed] [Google Scholar]
- 272.Wu Y, Zhou Q, Zheng P, Liu Y. CD28-independent induction of T helper cells and immunoglobulin class switches requires costimulation by the heat-stable antigen. J Exp Med. 1998;187:1151–1156. doi: 10.1084/jem.187.7.1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Schwartz RS. The new immunology—the end of immunosuppressive drug therapy? N Engl J Med. 1999;340:1754–1756. doi: 10.1056/NEJM199906033402209. [DOI] [PubMed] [Google Scholar]
- 274.Silver PB, Hathcock KS, Chan CC, Wiggert B, Caspi RR. Blockade of costimulation through B7/CD28 inhibits experimental autoimmune uveoretinitis, but does not induce long-term tolerance. J Immunol. 2000;165:5041–5047. doi: 10.4049/jimmunol.165.9.5041. [DOI] [PubMed] [Google Scholar]
- 275.Issazadeh S, Mustafa M, Ljungdahl A, Hojeberg B, Dagerlind A, Elde R, Olsson T. Interferon gamma, interleukin 4 and transforming growth factor beta in experimental autoimmune encephalomyelitis in Lewis rats: Dynamics of cellular mRNA expression in the central nervous system and lymphoid cells. J Neurosci Res. 1995;40:579–590. doi: 10.1002/jnr.490400503. [DOI] [PubMed] [Google Scholar]
- 276.Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- 277.Damle NK, Doyle LV, Grosmaire LS, Ledbetter JA. Differential regulatory signals delivered by antibody binding to the CD28 (Tp44) molecule during the activation of human T lymphocytes. J Immunol. 1988;140:1753–1761. [PubMed] [Google Scholar]
- 278.Lenschow DJ, Herold KC, Rhee L, Patel B, Koons A, Qin HY, Fuchs E, Singh B, Thompson CB, Bluestone JA. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–293. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- 279.Perrin PJ, Maldonado JH, Davis TA, June CH, Racke MK. CTLA-4 blockade enhances clinical disease and cytokine production during experimental allergic encephalomyelitis. J Immunol. 1996;157:1333–1336. [PubMed] [Google Scholar]
- 280.Alegre ML, Shiels H, Thompson CB, Gajewski TF. Expression and function of CTLA-4 in Th1 and Th2 cells. J Immunol. 1998;161:3347–3356. [PubMed] [Google Scholar]
- 281.Fallarino F, Fields PE, Gajewski TF. B7-1 engagement of cytotoxic T lymphocyte antigen 4 inhibits T cell activation in the absence of CD28. J Exp Med. 1998;188:205–210. doi: 10.1084/jem.188.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Kingsley CI, Karim M, Bushell AR, Wood KJ. CD25+CD4+ regulatory T cells prevent graft rejection: CTLA-4- and IL-10-dependent immunoregulation of alloresponses. J Immunol. 2002;168:1080–1086. doi: 10.4049/jimmunol.168.3.1080. [DOI] [PubMed] [Google Scholar]
- 283.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–155. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 284.Ando DG, Clayton J, Kono D, Urban JL, Sercarz EE. Encephalitogenic T cells in the B10. PL model of experimental allergic encephalomyelitis (EAE) are of the Th-1 lymphokine subtype. Cell Immunol. 1989;124:132–143. doi: 10.1016/0008-8749(89)90117-2. [DOI] [PubMed] [Google Scholar]
- 285.Hurwitz AA, Sullivan TJ, Krummel MF, Sobel RA, Allison JP. Specific blockade of CTLA-4/B7 interactions results in exacerbated clinical and histologic disease in an actively-induced model of experimental allergic encephalomyelitis. J Neuroimmunol. 1997;73:57–62. doi: 10.1016/s0165-5728(96)00168-3. [DOI] [PubMed] [Google Scholar]
- 286.Wolchok JD, Saenger Y. The mechanism of anti-CTLA-4 activity and the negative regulation of T-cell activation. Oncologist. 2008;13:2–9. doi: 10.1634/theoncologist.13-S4-2. [DOI] [PubMed] [Google Scholar]
- 287.O’day SJ, Hamid O, Urba WJ. Targetting Cytotoxic T-Lymphocyte Antigen-4 (CTLA-4) American Cancer Society. 2001;110:2614–2627. doi: 10.1002/cncr.23086. [DOI] [PubMed] [Google Scholar]
- 288.Kobayashi N, Kobayashi H, Gu L, Malefyt T, Siahaan TJ. Antigen-specific suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. J Pharmacol Exp Ther. 2007;322:879–886. doi: 10.1124/jpet.107.123257. [DOI] [PubMed] [Google Scholar]
- 289.Kobayashi N, Kiptoo P, Kobayashi H, Ridwan R, Brocke S, Siahaan TJ. Prophylactic and therapeutic suppression of experimental autoimmune encephalomyelitis by a novel bifunctional peptide inhibitor. Clin Immunol. 2008;129:69–79. doi: 10.1016/j.clim.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Franco A, Southwood S, Arrhenius T, Kuchroo VK, Grey HM, Sette A, Ishioka GY. T cell receptor antagonist peptides are highly effective inhibitors of experimental allergic encephalomyelitis. Eur J Immunol. 1994;24:940–946. doi: 10.1002/eji.1830240424. [DOI] [PubMed] [Google Scholar]
- 291.Pedotti R, Sanna M, Tsai M, DeVoss J, Steinman L, McDevitt H, Galli SJ. Severe anaphylactic reactions to glutamic acid decarboxylase (GAD) self peptides in NOD mice that spontaneously develop autoimmune type 1 diabetes mellitus. BMC Immunol. 2003;4:2. doi: 10.1186/1471-2172-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Leech MD, Chung CY, Culshaw A, Anderton SM. Peptide-based immunotherapy of experimental autoimmune encephalomyelitis without anaphylaxis. Eur J Immunol. 2007;37:3576–3581. doi: 10.1002/eji.200737148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Gerety SJ, Karpus WJ, Cubbon AR, Goswami RG, Rundell MK, Peterson JD, Miller SD. Class II-restricted T cell responses in Theiler’s murine encephalomyelitis virus-induced demyelinating disease. V. Mapping of a dominant immunopathologic VP2 T cell epitope in susceptible SJL/J mice. J Immunol. 1994;152:908–918. [PubMed] [Google Scholar]
- 294.Kappos L, Comi G, Panitch H, Oger J, Antel J, Conlon P, Steinman L. Induction of a non-encephalitogenic type 2 T helper-cell autoimmune response in multiple sclerosis after administration of an altered peptide ligand in a placebo-controlled, randomized phase II trial. The Altered Peptide Ligand in Relapsing MS Study Group. Nat Med. 2000;6:1176–1182. doi: 10.1038/80525. [DOI] [PubMed] [Google Scholar]
- 295.Bielekova B, Goodwin B, Richert N, Cortese I, Kondo T, Afshar G, Gran B, Eaton J, Antel J, Frank JA, McFarland HF, Martin R. Encephalitogenic potential of the myelin basic protein peptide (amino acids 83–99) in multiple sclerosis: Results of a phase II clinical trial with an altered peptide ligand. Nat Med. 2000;6:1167–1175. doi: 10.1038/80516. [DOI] [PubMed] [Google Scholar]
- 296.Inamura N, Mekori YA, Bhattacharyya SP, Bianchine PJ, Metcalfe DD. Induction and enhancement of Fc(epsilon)RI-dependent mast cell degranulation following coculture with activated T cells: Dependency on ICAM-1- and leukocyte function-associated antigen (LFA)-1-mediated heterotypic aggregation. J Immunol. 1998;160:4026–4033. [PubMed] [Google Scholar]
- 297.Pedotti R, Mitchell D, Wedemeyer J, Karpuj M, Chabas D, Hattab EM, Tsai M, Galli SJ, Steinman L. An unexpected version of horror autotoxicus: Anaphylactic shock to a self-peptide. Nat Immunol. 2001;2:216–222. doi: 10.1038/85266. [DOI] [PubMed] [Google Scholar]
- 298.Smith CE, Eagar TN, Strominger JL, Miller SD. Differential induction of IgE-mediated anaphylaxis after soluble vs. cell-bound tolerogenic peptide therapy of autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2005;102:9595–9600. doi: 10.1073/pnas.0504131102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Larche M, Wraith DC. Peptide-based therapeutic vaccines for allergic and autoimmune diseases. Nat Med. 2005;11:S69–S76. doi: 10.1038/nm1226. [DOI] [PubMed] [Google Scholar]
- 300.Kanduc D. Immunogenicity in peptide-immunotherapy: From self/nonself to similar/dissimilar sequences. Adv Exp Med Biol. 2008;640:198–207. doi: 10.1007/978-0-387-09789-3_15. [DOI] [PubMed] [Google Scholar]
- 301.Yamazaki S, Inaba K, Tarbell KV, Steinman RM. Dendritic cells expand antigen-specific Foxp3+ CD25+ CD4+ regulatory T cells including suppressors of alloreactivity. Immunol Rev. 2006;212:314–329. doi: 10.1111/j.0105-2896.2006.00422.x. [DOI] [PubMed] [Google Scholar]
- 302.Steinman RM. Dendritic cells: Understanding immunogenicity. Eur J Immunol. 2007;37:S53–S60. doi: 10.1002/eji.200737400. [DOI] [PubMed] [Google Scholar]
- 303.Ridge JP, Fuchs EJ, Matzinger P. Neonatal tolerance revisited: Turning on newborn T cells with dendritic cells. Science. 1996;271:1723–1726. doi: 10.1126/science.271.5256.1723. [DOI] [PubMed] [Google Scholar]
- 304.Jonuleit H, Schmitt E, Schuler G, Knop J, Enk AH. Induction of interleukin 10-producing, nonproliferating CD4(+) T cells with regulatory properties by repetitive stimulation with allogeneic immature human dendritic cells. J Exp Med. 2000;192:1213–1222. doi: 10.1084/jem.192.9.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Santambrogio L, Sato AK, Fischer FR, Dorf ME, Stern LJ. Abundant empty class II MHC molecules on the surface of immature dendritic cells. Proc Natl Acad Sci USA. 1999;96:15050–15055. doi: 10.1073/pnas.96.26.15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Reddy J, Illes Z, Zhang X, Encinas J, Pyrdol J, Nicholson L, Sobel RA, Wucherpfennig KW, Kuchroo VK. Myelin proteolipid protein-specific CD4+CD25+ regulatory cells mediate genetic resistance to experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2004;101:15434–15439. doi: 10.1073/pnas.0404444101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Yu P, Gregg RK, Bell JJ, Ellis JS, Divekar R, Lee HH, Jain R, Waldner H, Hardaway JC, Collins M, Kuchroo VK, Zaghouani H. Specific T regulatory cells display broad suppressive functions against experimental allergic encephalomyelitis upon activation with cognate antigen. J Immunol. 2005;174:6772–6780. doi: 10.4049/jimmunol.174.11.6772. [DOI] [PubMed] [Google Scholar]
- 308.Zhang X, Reddy J, Ochi H, Frenkel D, Kuchroo VK, Weiner HL. Recovery from experimental allergic encephalomyelitis is TGF-beta dependent and associated with increases in CD4+LAP+ and CD4+CD25+ T cells. Int Immunol. 2006;18:495–503. doi: 10.1093/intimm/dxh390. [DOI] [PubMed] [Google Scholar]
- 309.Schmidt S. Candidate autoantigens in multiple sclerosis. Mult Scler. 1999;5:147–160. doi: 10.1177/135245859900500303. [DOI] [PubMed] [Google Scholar]
- 310.van Lierop MJ, den Hoed L, Houbiers J, Vencovsky J, Ruzickova S, Krystufkova O, van Schaardenburg M, van den Hoogen F, Vandooren B, Baeten D, De Keyser F, Sonderstrup G, Bos E, Boots AM. Endogenous HLA-DR-restricted presentation of the cartilage antigens human cartilage gp-39 and melanoma inhibitory activity in the inflamed rheumatoid joint. Arthritis Rheum. 2007;56:2150–2159. doi: 10.1002/art.22651. [DOI] [PubMed] [Google Scholar]
- 311.Sekine T, Masuko-Hongo K, Matsui T, Asahara H, Takigawa M, Nishioka K, Kato T. Recognition of YKL-39, a human cartilage related protein, as a target antigen in patients with rheumatoid arthritis. Ann Rheum Dis. 2001;60:49–54. doi: 10.1136/ard.60.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Tsuruha J, Masuko-Hongo K, Kato T, Sakata M, Nakamura H, Nishioka K. Implication of cartilage intermediate layer protein in cartilage destruction in subsets of patients with osteoarthritis and rheumatoid arthritis. Arthritis Rheum. 2001;44:838–845. doi: 10.1002/1529-0131(200104)44:4<838::AID-ANR140>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 313.Kim WU, Cho ML, Jung YO, Min SY, Park SW, Min DJ, Yoon JH, Kim HY. Type II collagen autoimmunity in rheumatoid arthritis. Am J Med Sci. 2004;327:202–211. doi: 10.1097/00000441-200404000-00006. [DOI] [PubMed] [Google Scholar]
- 314.Iwaki-Egawa S, Matsuno H, Yudoh K, Nakazawa F, Miyazaki K, Ochiai A, Hirohata S, Shimizu M, Watanabe Y. High diagnostic value of anticalpastatin autoantibodies in rheumatoid arthritis detected by ELISA using human erythrocyte calpastatin as antigen. J Rheumatol. 2004;31:17–22. [PubMed] [Google Scholar]
- 315.van Boekel MA, Vossenaar ER, van den Hoogen FH, van Venrooij WJ. Autoantibody systems in rheumatoid arthritis: specificity, sensitivity and diagnostic value. Arthritis Res. 2002;4:87–93. doi: 10.1186/ar395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Kinloch A, Tatzer V, Wait R, Peston D, Lundberg K, Donatien P, Moyes D, Taylor PC, Venables PJ. Identification of citrullinated alpha-enolase as a candidate autoantigen in rheumatoid arthritis. Arthritis Res Ther. 2005;7:R1421–R1429. doi: 10.1186/ar1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Yusuf-Makagiansar H, Makagiansar IT, Hu Y, Siahaan TJ. Synergistic inhibitory activity of alpha- and beta-LFA-1 peptides on LFA-1/ICAM-1 interaction. Peptides. 2001;22:1955–1962. doi: 10.1016/s0196-9781(01)00546-0. [DOI] [PubMed] [Google Scholar]
- 318.Yusuf-Makagiansar H, Siahaan TJ. Binding and internalization of an LFA-1-derived cyclic peptide by ICAM receptors on activated lymphocyte: a potential ligand for drug targeting to ICAM-1-expressing cells. Pharm Res. 2001;18:329–335. doi: 10.1023/a:1011007014510. [DOI] [PubMed] [Google Scholar]
- 319.Shannon JP, Silva MV, Brown DC, Larson RS. Novel cyclic peptide inhibits intercellular adhesion molecule-1-mediated cell aggregation. J Pept Res. 2001;58:140–150. doi: 10.1034/j.1399-3011.2001.00899.x. [DOI] [PubMed] [Google Scholar]
- 320.Laskowski IA, Pratschke J, Wilhelm MJ, Dong VM, Beato F, Taal M, Gasser M, Hancock WW, Sayegh MH, Tilney NL. Anti-CD28 monoclonal antibody therapy prevents chronic rejection of renal allografts in rats. J Am Soc Nephrol. 2002;13:519–527. doi: 10.1681/ASN.V132519. [DOI] [PubMed] [Google Scholar]
- 321.Tan P, Anasetti C, Hansen JA, Melrose J, Brunvand M, Bradshaw J, Ledbetter JA, Linsley PS. Induction of alloantigen-specific hyporesponsiveness in human T lymphocytes by blocking interaction of CD28 with its natural ligand B7/BB1. J Exp Med. 1993;177:165–173. doi: 10.1084/jem.177.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]