

Abstract
In the context of most induced pluripotent stem (iPS) cell reprogramming methods, heterogeneous populations of nonproductive and staggered productive intermediates arise at different reprogramming time points1–11. Despite recent reports claiming substantially increased reprogramming efficiencies using genetically modified donor cells12,13 prospectively isolating distinct reprogramming intermediates remains an important goal to decipher reprogramming mechanisms. Previous attempts to identify surface markers of intermediate cell populations were based on the assumption that during reprogramming cells progressively lose donor cell identity and gradually acquire iPS cell properties1,2,7,8,10. Here, we report that iPS cell and epithelial markers, such as SSEA1 and EpCAM, respectively, are not predictive of reprogramming during early phases. Instead, in a systematic functional surface marker screen we find that early reprogramming-prone cells express a unique set of surface markers, including CD73, CD49d and CD200 that are absent in fibroblasts and iPS cells. Single cell mass cytometry and prospective isolation show that these distinct intermediates are transient and bridge the gap between donor cell silencing and pluripotency marker acquisition during the early, presumably stochastic reprogramming phase2. Expression profiling revealed early upregulation of the transcriptional regulators Nr0b1 and Etv5 in this reprogramming state, preceding activation of key pluripotency regulators such as Rex1, Dppa2, Nanog and Sox2. Both factors are required for the generation of the early intermediate state and fully reprogrammed iPS cells, and thus mark some of the earliest known regulators of iPS cell induction. Our study deconvolutes the first steps in a hierarchical series of events that lead to pluripotency acquisition.
Reprogramming somatic cells to a pluripotent state by forced transcription factor expression is typically an inefficient process involving heterogeneous populations that impede molecular analysis of productive reprogramming1–11,14. Previous studies have shown that reprogramming is a multi-stage process involving a presumably early stochastic and late deterministic phase2,3,5. Progress has been made characterizing intermediates of the late phase given the appearance of well-known pluripotency markers at that time1,2,7,8,10. In contrast, not much is known about the early stochastic phase except the consistent observation that downregulation of donor cell markers is an early feature of successful reprogramming1,2,7,8,10,15–19.
To identify surface markers of early reprogramming stages, we screened 176 antibodies on cells representing three stages of the reprogramming process: 1) mouse embryonic fibroblasts (MEFs), 2) a previously characterized partially reprogrammed cell (PRC) line18,20 and 3) embryonic stem (ES) cells. We identified 21 markers enriched or shared between these cell types and characterized their co-expression by single cell mass cytometry using spanning-tree progression analysis of density-normalized events (SPADE), which groups similar cells into a defined number of clusters21,22. (Fig. 1a,b and Extended Data Fig. 1). Next, we characterized their expression by mass cytometry during Oct4, Sox2, Klf4 and c-Myc-driven MEF reprogramming. By day 3, downregulation of the fibroblast expression program was evident (Fig. 1c and Extended Data Fig. 2 and 3). At day 6, major branches were delineated by the PRC marker CD73 and ESC markers CD54, CD326 and SSEA1. Little co-expression was observed between these markers suggesting several intermediates arise during early reprogramming or early expression of some of these markers may not be indicative of productive reprogramming. By day 9, CD326 and SSEA1 expression converged in a subpopulation and persisted on days 12 and 16 (Extended Data Fig. 2b). These clusters were heterogeneous for CD73, suggesting they may be derivatives of separate populations or a CD73high subpopulation becomes CD326high, SSEA1high. Over our time course, the ESC marker CD54 largely localized to fibroblast branches and did not cluster with CD326high and SSEA1high clusters, suggesting CD54 expression in pluripotent cells is a late event7.
Figure 1. Reprogramming surface marker profiling by mass cytometry.

a, Histogram overlays show enrichment levels for the three cell populations analyzed: mouse embryonic fibroblasts (MEF, black), partially reprogrammed cells (PRC, red) and embryonic stem cells (ESC, green). b, SPADE analysis of combined MEF, PRC and ESC datasets. Color bars represent absolute percentages (top row) and ArcSinh-transformed counts for each marker. c, SPADE analysis of infected reprogramming MEF populations at days 0, 3, 6 and 9. For each marker, the same color scale was applied to every sample, allowing direct comparison between time points. Color bars represent absolute percentages (left) and ArcSinh-transformed counts.
Hypothesizing that cells destined to successfully reprogram, acquire surface markers in a stepwise, non-stochastic manner during early reprogramming, we assessed reprogramming efficiencies for cells with high or low expression of the above-characterized surface markers at early time points (Fig. 2a–c and Extended Data Fig. 4a). As mass culture experiments can be misleading since a single proliferative population can seed multiple secondary colonies, we conducted 96-well assays to assess unique reprogramming events. Based on the current literature it would be expected that reprogramming cells would be enriched by (i) low levels of fibroblast markers and (ii) high levels of ESC markers independent of the time and state of reprogramming2,7,8,10. Indeed, by day 3, populations expressing low levels of all fibroblast markers except CD47 enriched for reprogramming populations when compared to highly expressing populations (Fig. 2a). Surprisingly though, at these early timepoints, cells with high levels of the ESC markers SSEA1, CD54, CD326, or CD71 did not show significantly increased reprogramming (Fig. 2a–c). Day 9 fractions expressing high levels of CD326 or SSEA1 began to show greater but insignificant enrichment for reprogramming populations. We confirmed previous reports that SSEA1-sorted cells produce more iPS cell colonies in mass culture1,8,10 emphasizing the critical importance of the 96-well assay (Extended Data Fig. 4b,c). Unlike previously assumed, our findings demonstrate that acquisition of markers that define the pluripotent state is a late event and early expression of ESC markers has little predictive value for successful reprogramming. Additionally these data support that the mesenchymal-to-epithelial transition (MET) as judged by the epithelial marker CD326 (EpCAM) is a late event2,15–17,19.
Figure 2. A surface marker screen identifies an early CD73high CD49dhigh reprogramming intermediate.

a–c, 96 well reprogramming assays on days 3, 6 and 9. 20 cells/well sorted at days 3, 6 and 9. Sox2-EGFP+ colonies were assayed on day 24. Asterisks indicate two-sided t-test P-value<0.05. d, Plating efficiencies for GFP expressing MEFs sorted on day 6 for CD73high or CD49dhigh (assayed 24 hours post-sort). e, Single cell reprogramming efficiencies for day 6 CD73high or CD49dhigh fractions. f, Reprogramming efficiencies (e) adjusted for plating efficiencies (d). Error bars represent standard deviation. n=3 independent experiments for all assays.
Our surface marker screen identified several specific markers for stable partially reprogrammed cells4,9,18,20. Though thought to be ‘stuck’ during reprogramming4,18,20, we hypothesized a productive intermediate might arise between fibroblasts and iPS cells that share a subset of these markers. Indeed, day 6 fractions expressing high levels of the PRC markers CD73, CD49d and CD200 significantly enriched for reprogramming populations (Fig. 2b). Also on day 9 the CD73high and CD49dhigh populations contained higher reprogramming activity (Fig. 2c). In agreement with these results, the day 6 SPADE analysis showed CD73high, CD49dhigh and CD200high branches largely clustering independently from branches enriched for ESC and MEF markers (Fig. 1c). These results demonstrate that distinct intermediate populations arise after fibroblast program repression but before ESC marker acquisition.
We next focused on the markers CD73 and CD49d. When corrected for plating efficiency both CD73high and CD49dhigh populations showed remarkably high reprogramming efficiencies of 9.5% +/− 3.5 and 12.5% +/− 5.7, respectively (Fig. 2d–f). Similar enrichment of a reprogramming-prone population was observed in reprogramming tail tip fibroblasts (TTFs) and glial-restricted neural precursor cells suggesting CD73 and CD49d may be universal markers of intermediate reprogramming stages (Fig. 3a–d). Finally, we explored their potential functional implications during reprogramming, and observed that adenosine, the enzymatic product of CD73, has a negative effect throughout and CD49d activity is necessary during late reprogramming (Extended Data Fig. 4e).
Figure 3. Characterization of CD73high and CD49dhigh intermediates.

a–c, Percent of wells with Nanog expressing cells on day 24 for reprogramming tail tip fibroblasts (TTF) (a) (n=3) and glia (b,c) (n=1, independent primary cells and infections). d, Representative Nanog immunostaining. Scale bar is 200µm. e, Heterogeneously expressed markers in day 6 CD73high population. f, Single cell 96-well assays for day 6 CD73high fraction with additional surface markers (n=3). g, Refined poised signature. Clusters are low for mesenchymal markers. h, Continuation analysis shows SSEA1high CD326high branch unique to poised populations (boxed). All experiments represent independent biological replicates. Error bars represent standard deviation.
We then used our day 6 SPADE analysis to identify heterogeneously expressed markers that could subdivide the CD73high reprogramming-prone population and conducted single cell efficiency assays (Fig. 3e,f). Within the CD73-positive population the CD44high, CD71low and CD326high fractions failed to reprogram, while CD49dhigh and CD326low fractions enriched for a reprogramming population. Thus, a CD73high CD49dhigh CD326low CD44low signature best describes the population undergoing productive reprogramming on day 6. Overlaying this signature onto the day 6 SPADE tree allowed for determination of the exact cellular clusters most similar to this reprogramming-prone signature (Fig. 3g). As expected, MEF markers were low in these poised populations. This suggests this intermediate arises after loss of mesenchymal markers, but before MET completion as indicated by reprogramming enrichment in the CD326low fraction.
To identify subsequent reprogramming stages we conducted continuation analysis where cells were sorted on day 6 and characterized by mass cytometry on day 16 (Fig. 3h, Extended Data Fig. 4–6). By day 10, reprogramming-prone populations formed distinct colonies with ESC-like morphology, while CD73low cells were highly proliferative but failed to develop into mature colonies (Extended Data Fig. 5b). Continuation analysis on day 16 revealed that while reprogramming-prone and non-prone populations contained CD326 expressing cells, broad overlap between CD326high and SSEA1high clusters was only in mature reprogramming-prone populations (Fig. 3h, Extended Data Fig. 6). These clusters did not overlap with the ESC marker CD54, and were heterogeneous for CD73 and CD49d. We conclude a distinct CD326high, SSEA1high, CD54low intermediate arises after the CD73high/CD49dhigh intermediate and before pluripotency acquisition.
We then used the intermediates stages to gain molecular insights into transcriptional regulation of early reprogramming. Gene expression analysis intriguingly showed these intermediates precede activation of the majority of transcription factors thought to be predictive markers for pluripotency induction (Fig. 4a and Extended Data Fig. 7)2,7. The observation that these intermediates arise prior to key pluripotency regulators suggests a separate combination of early transcription factors must be induced to generate early intermediates and poise them for pluripotency acquisition. We found the transcription factors Nr0b1 and Etv5 preferentially expressed in reprogramming-prone populations and highly expressed in ESCs suggesting a functional role in poising early reprogramming (Fig. 4a, Extended Data Fig. 7b–d).
Figure 4. Reprogramming regulators identified with CD73high/CD49dhigh intermediates.

a, Day 6 and 9 reprogramming-prone and non-prone pluripotency associated gene differential expression. Dotted line represents value of 1(no difference). b–c, Day 9 CD73high/CD49dhigh quantification for knockdown (b, n=3 independent experiments) and rescue (c) experiments. Gating shown in Extended Data Fig. 7g. Asterisks indicate two-sided t-test P-value<0.05. d–e, Day 24 Sox2-EGFP+(d, n=1) and Nanog+ colonies (e, n=2 independent experiments) from Rosa-rtTA+/−, Sox2-EGFP+/− and Rosa-rtTA+/− MEFs, respectively. f, Early and late reprogramming model. Dotted red boxes distinguish CD104 observed in the Mbd3fl/− system. Error bars indicate standard deviation.
To assess whether these genes were necessary to induce the early intermediate populations we generated three short hairpins against each gene (Extended Data Fig. 7e,f). We then assessed the ability of reprogramming MEFs infected with these short hairpins to induce CD73high/CD49dhigh intermediates 9 days after reprogramming induction (Fig. 4b and Extended Data Fig. 7g). Surprisingly, while MEFs infected with a control short hairpin were able to induce the CD73high/CD49high intermediate (6.70 +/− 2.27%), reprogramming MEFs infected with short hairpins targeting Etv5 or Nr0b1 were significantly impaired (Fig. 4b). This phenotype could be rescued by cDNA overexpression in combination with a hairpin targeting the UTR for the gene of interest (Fig. 4c). Further, when Nanog+ colonies or Sox2-EGFP+ colonies were assessed 24 days after reprogramming induction, a dramatic decrease in reprogramming efficiencies was observed (Fig. 4d,e). In contrast, knock-down of either gene in ESCs did not affect survival or proliferation (Extended Data Fig. 7h). While this paper was under review an independent report confirmed Nr0b1 as necessary for reprogramming23. These data indicate that Etv5 and Nr0b1 are required for the generation of the CD73high/CD49dhigh poised intermediate necessary to induce the canonical pluripotency program and definitive iPS cell formation.
We then wondered whether a similar intermediate population arises in high efficiency reprogramming systems12,13. Published expression analysis of two high efficiency systems showed transient CD73 upregulation, suggesting the presence of a similar intermediate (Extended Data Fig. 8a,b). We then characterized one of these systems, the Mbd3fl/− secondary MEF system, in greater detail12. After confirming reported reprogramming efficiencies, we analyzed this system by mass spectrometry (Extended Data Figs. 8–10). By day 3, fibroblast marker repression was evident, and CD73 was upregulated within this population (Extended Data Fig. 8e and 9b). Within the CD73high/MEFSK4low population, CD49d (itga4) upregulation was not apparent, but we noticed the emergence of a separate integrin, CD104 (itgb4). By day 4, the major CD73high branch clearly overlapped with the CD104high branch and persisted into day 5. SSEA1high and CD326high expression was present on day 4, but clear coexpression was not seen until day 5. By day 9, CD73 and CD104 expression was dramatically reduced while CD326 and SSEA1 expression remained high. These data demonstrate a transient CD73high/CD104high population arises after donor cell program repression and prior to ESC marker acquisition even in a highly efficient reprogramming system. Similar to CD49d and CD73, CD104 is not highly expressed in ESCs (Fig. 1a). Similar to viral reprogramming, adenosine treatment abolished reprogramming in the Mbd3 reprogramming system, albeit only at late stages, whereas compounds affecting CD49d function had little effect (Extended Data Fig. 4f).
The stage-specific framework provided in this study bridges the previously unexplored gap between donor program silencing and pluripotent marker acquisition (Fig. 4f). We demonstrate a transient, ‘poised’ intermediate present across multiple reprogramming systems suggesting a general property of iPSC reprogramming. We note similar to SSEA1, TRA-1-60 enriches for reprogramming-prone intermediates at later time points during human reprogramming11,24, and we speculate a similar transient intermediate arises in the human system.
Methods
Cell Culture
Embryonic fibroblasts were isolated from E13.5 embryos derived from B6;129S-Sox2EGFP/+ mated with B6.Cg-Gt(ROSA)26Sortm1(rtTA*M2)Jae (allele referred to as Rosa-rtTA) as previously described25,26. Tail tip fibroblasts were derived from 1 week old B6 mice. This was done by chilling the animals on ice for 5 minutes, cutting the tail, and mincing in a 6cm dish in 1ml 0.25% trypsin. Another 1ml of 0.25% trypsin was added after mincing and this was incubated at 37C for 10 minutes. The mixture was then resuspended in 15ml MEF medium (described below) and plated on a 0.2% gelatinized 15cm plate. Glial cultures were prepared from CD1 mice as previously described25. MEFs, TTFs and glial cells were cultured in MEF medium which consisted of 10% cosmic calf serum (Thermo Scientific) in DMEM (Invitrogen) supplemented with non-essential amino acids (Invitrogen), penicillin-streptomycin (Invitrogen), sodium pyruvate (Invitrogen) and 2-mercaptoethanol (Invitrogen). Primary Mbd3fl/−, Oct4-GFP secondary MEFs12 were a generous gift from Jacob Hanna and were grown in MEF medium. We verified modification of the Mbd3 locus by Western blot analysis (described below under ‘Western blotting’, Extended Data Figure 8f). Our partially reprogrammed line was previously characterized20. These were grown in MEF medium supplemented with 15% cosmic calf serum. Oct4-neoR knock-in mouse ES cells18,20 were grown in mESC medium which consisted of 12% knockout replacement serum (Invitrogen), 3% cosmic calf serum and supplemented with non-essential amino acids, penicillin-streptomycin, sodium pyruvate, 2-mercaptoethanol and LIF. MEFs, PRCs and mESCs were routinely tested for mycoplasma contamination.
Surface marker screening
We screened mouse surface markers using the “Mouse cell surface marker screening panel” Lyoplate (BD Biosciences, material number 562208) according to the manufacturer’s instructions. In brief, 150×10^6 cells were used for our partially reprogrammed cells (passage 15) and Oct4-neoR mES cells. Cells were plated on 0.2% gelatin coated plates and treated with neomycin three days before screening. 175×10^6 cells were used for passage 4 Sox2-EGFP MEFs. Cells were washed with PBS-EDTA, dissociated in 10× TrypLE (Invitrogen) for 5 minutes, washed once in PBS before staining for 30 minutes in primary antibody in staining medium (PBS-EDTA supplemented with 0.5% BSA) according to manufacturer’s instructions, washing once in PBS, staining for 30 minutes in fluorophore conjugated secondary antibody in staining medium, washing once in PBS and analyzed on a BD LSR Fortessa FACS analyzer in in staining medium. We used a high concentration of TrypLE to verify that all identified markers would not be cleaved by our dissociation reagent. We further validated a subset of these identified makers with our control populations dissociated in 1× TrypLE and stained with fluorophore conjugated antibodies as described below.
Reprogramming
Reprogramming assays were conducted with passage 4 Rosa-rtTA+/−; Sox2EGFP/+ or Rosa-rtTA+/− (derived from the same litter – see mating scheme above) mouse embryonic fibroblasts, passage 2 B6 tail tip fibroblasts or passage 3 B6 glia as indicated. FUW-tetO lentiviral vectors and lentiviral packaging were used as previously described25. Passage 3 MEFs and passage 2 glial cells were split onto 10cm 0.2% gelatin coated plates at 250,000 cells per plate one day before infection. Passage 1 TTFs were split onto 10cm 0.2% gelatin coated plates at 100,000 cells per plate one day before infection. Cells were infected in MEF medium supplemented with polybrene (8 µg/mL; Sigma). One day after infection, MEF medium supplemented with doxycycline was added and this was considered to be day 0. Media was replaced every 2 days. On day 16, medium was replaced with mES cell medium without doxycycline. Due to the complication of getting a large number of glial cells, we performed the reprogramming efficiency assays once per time point (day 6 and 9) but both were done with independently derived primary cells from different animals, independently derived virus and sorts.
The following compounds were also added to reprogramming medium on the days indicated in the text: BIO 1211 (Tocris, 4nM), adenosine (Sigma, 20µM or 2 µM as indicated), fibronectin (Sigma, 4µg/ml), AMP-CP [Adenosine 5′-(α,β-methylene)diphosphate] (Sigma, 20µM). These were chosen as CD73 (5’-nucleotidase, ecto) converts AMP to adenosine, CD49d (integrin α4) is an integrin whose substrates are VCAM1 and fibronectin, α,β-Methyleneadenosine 5′-diphosphate (AMP-CP) is a CD73 inhibitor and BIO1211 is CD49d inhibitor
Hairpins for Nr0b1 and Etv5 were designed with the pSico oligomaker (Supplementary Table 6) and cloned into the lentiviral pSico-puro vector27 (http://web.mit.edu/jacks-lab/protocols/pSico.html). We assessed knockdown efficiencies in partially reprogrammed cells as they express these genes and grow well in MEF medium supplemented with 15% serum alone. To assess knockdown efficiencies, 50,000 PRCs were plated onto a 6 well gelatin coated well one day before transduction. Cells were transduced with the hairpin of interest in MEF medium supplemented with 15% serum and polybrene (8 µg/mL). The medium was exchanged the following day with MEF medium supplemented with 15% serum and puromycin (2µg/ml). Cells were cultured for a further three days before RNA extraction (for a total of four days after infection). RNA was prepared with an RNeasy purification kit as described below.
To assess the effects of knockdown of these genes on the day 9 CD73high/CD49dhigh intermediate, 100,000 P4 Rosa-rtTA MEFs were plated on a 10cm gelatin coated plate. These were transduced the following day with the doxycycline-inducible FUW-tetO-Klf4, Oct-3/4, Sox2 and c-Myc vectors and indicated hairpins as described above. Medium was supplemented with doxycycline one day after infection (day 0). We note that because Rosa-rtTA MEFs contain a PGK-puromycin cassette, they were not selected with puromycin. On day 9, CD73-Alexa 488 (1:50) and CD49d-Alexa 750 (1:50) were analyzed by FACS (FACS staining described below). To assess reprogramming efficiencies for these genes, 30,000 P4 Rosa-rtTA MEFs were plated on a 6cm gelatin coated plate or 15,000 P4 Rosa-rtTA, Sox2-EGFP MEFs were plated on a 6 well gelatin coated well and transduced the following day in the same manner. At day 16, the culture medium was switched to mES cell medium without doxycycline. 24 days after reprogramming induction, Nanog+ colonies were assessed for Rosa-rtTA MEFs (immunofluorescence staining described below) or Sox2-EGFP+ colonies for Rosa-rtTA, Sox2-EGFP MEFs. In total, three independent reprogramming efficient experiments were conducted across the two different MEF lines. To further validate the specificity of the effects, we have performed a "rescue" experiment to demonstrate that upon re-expression of the cDNA (under knock-down conditions) the effect is eliminated. To this end, the cDNAs for Nr0b1 and Etv5 were cloned into the FUW-tetO vector. 15,000 P4 Rosa-rtTA MEFs on 6 well gelatin coated plates were then transduced the doxycycline-inducible FUW-tetO-Klf4, Oct-3/4, Sox2 and c-Myc vectors and indicated hairpins with the FUW-tetO-cDNA (Etv5 or Nr0b1) or an empty vector. Medium was supplemented with doxycycline one day after infection (day 0). On day 9, CD73-Alexa 488 (1:50) and CD49d-Alexa 750 (1:50) were analyzed by FACS (FACS staining described below). As the rescue experiment was supplementary to and consistent with the main knockdown experiments, we conducted only one rescue experiment for one hairpin.
To verify that mouse ESCs could survive and proliferate after knock-down of Etv5 or Nr0b1, we infected 30,000 ESCs per well in gelatinized 6-well plates in mESC medium and replaced the medium the following day with mESC medium supplemented with puromycin to select for successful transduction. These were then cultured for 3 days, dissociated with 0.25% trypsin and replated onto gelatinized 6 well plates. These were then cultured for 3 days, fixed and stained for Oct4 (described below).
To reprogram Mbd3fl/−, Oct4-GFP secondary MEFs (the Oct4-GFP allele in these cells is not a targeted, but well-characterized, transgenic reporter12), we first assayed reprogramming conditions with 2000 Mbd3fl/− secondary MEFs on 2×106 mitomycin (Sigma) treated B6 feeders, 105 Mbd3fl/− secondary MEFs without feeders and 2×105 Mbd3fl/− secondary MEFs without feeders on 10cm dishes coated with 0.2% gelatin. We found optimal reprogramming efficiencies with 2000 Mbd3fl/− secondary MEFs and 2×106 feeders as previously reported12. All reprogramming assays were done in non-hypoxic conditions. To reprogram cells, cells were cultured in media 1 [recombinant human LIF (10ng/ml, Peprotech), doxycycline (1µg/ml) and ascorbic acid (10µg/ml, Sigma) in 15% cosmic calf serum (Thermo Scientific) in DMEM (Invitrogen) supplemented with non-essential amino acids (Invitrogen), penicillin-streptomycin (Invitrogen), sodium pyruvate (Invitrogen) and 2-mercaptoethanol (Invitrogen)] for 3 days and then media 2 [recombinant human LIF (10ng/ml), doxycycline (1µg/ml), PD0325901 (1µM, Cell Signaling) and CHIR99021 (3µM, Cayman) in 15% knockout replacement serum (Invitrogen) in DMEM (Invitrogen) supplemented with non-essential amino acids (Invitrogen), penicillin-streptomycin (Invitrogen), sodium pyruvate (Invitrogen) and 2-mercaptoethanol (Invitrogen),] until the completion of the experiment. Media was exchanged every 2 days.
Mass Cytometry Analysis
On designated days the reprogramming cultures were treated with 1× TrypLE (Invitrogen) for 5 minutes at 37C, dissociated into single cell suspension by trituration, and then washed twice with PBS. The cell samples were then incubated with metal-conjugated antibodies (Supplementary Table 1) in PBS containing 5% FBS (Omega Scientific) for 30 minutes on ice, washed once with PBS containing 5% FBS, treated with 25 µM cisplatin for 1 minute on ice for live-dead cell discrimination, washed once with PBS containing 5% FBS, and then fixed with 1.6% paraformaldehyde at room temperature for 10 minutes. Formaldehyde-fixed cell samples were then permeabilized with methanol on ice for 15 minutes, washed one time with PBS containing 0.5% BSA, and then incubated at room temperature for 15 minutes with an Irridium-containing DNA intercalator (DVS Sciences/Fluidigm) in PBS containing 1.6% paraformaldehyde. After intercalation/fixation, the cell samples were washed once with PBS containing 0.5% BSA and twice with water before measurement on a CyTOF mass cytometer (DVS Sciences/Fluidigm). Normalization for detector sensitivity was performed as previously described28, using polystyrene normalization beads containing Lanthanum-139, Praseodymium-141, Terbium-159, Thulium-169, and Lutetium-175.
SPADE Analysis
Density-dependent downsampling, hierarchical clustering, cluster upsampling, and extraction of parameter medians was performed by the SPADE package (www.cytospade.org) as described in the main text and as previously described22,29. All assayed surface markers were used in the clustering step unless otherwise indicated, and the parameters for downsampling percentile and target number of clusters were set to 5% and 500, respectively.
The refined poised signature shown in figure 3g was determined by calculating the similarity of each SPADE cluster to the hand-gated CD73high CD49dhigh CD44low CD326low population. Similarity was calculated by the Manhattan distance metric using all measured surface markers, and is indicated by the color scale bar (low distance equals high similarity).
Immunofluorescence
Plates were fixed in 4% PFA for 10 minutes, washed 3 times with PBS, blocked and permeabilized in PBS supplemented with 5% CCS and 0.1% Triton-X 100 (Sigma) (blocking solution) for 10 minutes. 96 well plates were then incubated with mouse anti-Nanog (1:500, BD) or mouse anti-Oct4 (1:200, Santa Cruz Biotechnology) in blocking solution for 30 minutes, washed 3 times with PBS, incubated with donkey anti-mouse Alexa-555 (1:1000, Invitrogen) or anti-mouse Alexa-488 (1:1000, Invitrogen) in blocking solution for 30 minutes, washed three times with PBS and stained with DAPI for three minutes. Cells were then washed with PBS and visualized.
Fluorescent activated cell sorting and efficiency assays
Cells were washed with PBS-EDTA, dissociated in 1× TrypLE for 5 minutes, washed with PBS, and incubated on ice with a fluorophore conjugated antibody and DAPI for 30 minutes in PBS-EDTA supplemented with 0.5% BSA. The sources and detailed descriptions of all antibodies used are listed in Supplementary Table 2. For 96 well assays, single or 20 DAPI− cells per well were double sorted on the indicated day into gelatinized 96 well plates supplemented with 400,000 feeders per plate in MEF medium supplemented with doxycycline. For the primary sort, cells were sorted into PBS supplemented with 0.5% BSA, and for the secondary sort, cells were sorted directly into 96 well plates. Efficiency assays were conducted 24 days after transgene induction and determined by the number of wells with Sox2-EGFP+ colonies. For mass culture reprogramming, 10,000 SSEA1high or SSEA1low cells were double sorted 6 days after transgene induction onto 3cm gelatinized plates supplemented with feeders and Sox2-EGFP+ colonies were assayed 24 days after transgene induction. We note that we used the same SSEA1 clone and vendor as previously used for determining reprogramming efficiencies8,10. TTF and glial reprogramming efficiencies were determined by double sorting on the indicated days into 96 wells (as described above) and assaying for Nanog by immunofluorescence. Plating efficiencies were determined by infecting Sox2-EGFP MEFs with FUW-tetO-hygroB-T2A-EGFP, selecting for 5 days in hygromycin, and counting the number of wells with GFP+ cells 24 hours after sorting. We note that for all assays where CD73 was used for sorting, CD73-Alexa 647 was used (gating shown in Extended Data Fig. 4). For CD73xCD49d analysis, CD73-488 was used (Extended Data Fig. 7g).
RNA Preparation and Expression Analysis
RNA of reprogramming populations for microarray analysis was prepared from Rosa-rtTA+/− day 6 and day 9 reprogramming cultures double sorted for CD73 or CD49d (as described above). RNA of control populations for microarray analysis was prepared from Rosa-rtTA+/− MEFs (passage 4), partially reprogrammed cells (passage 10) and V6.5 mouse ESCs (passage 11). RNA was prepared with RNeasy Mini Kit (Qiagen) and DNA was removed by on-column RNase-Free DNase treatment (Qiagen) according to the manufacturer’s instructions. Mouse Gene 2.0 ST Arrays (Affymetrix) were prepared by the Stanford Protein and Nucleic Acid (PAN) Facility. Data was normalized and gene names were assigned by Partek Genomic Suite. For all analysis, non-coding transcripts were removed. Preprocessing (floor = 100, ceiling = 20000, min fold change = 2), K-means clustering (k=5, seed value = 12345) and hierarchical clustering of k-means clusters (Pearson correlation, pairwise complete-linkage), and heat maps were generated by Gene Pattern (http://www.broadinstitute.org/cancer/software/genepattern/). Microarray data can be accessed with accession number GSE62957 from the National Center for Biotechnology Information database.
For figure 4a, genes were selected based on pluripotency associated genes characterized in previous studies2,7 or from differential expression of sorted populations and ESCs. Oct4 is not shown as the probe failed to detect expression in mESCs.
For qPCR analysis, cDNA was generated with SuperScript First-Strand Synthesis System (Invitrogen). Data was generated with the 7900HT Real-Time PCR System (Applied Biosystems). 6µL reactions were prepared with SYBR Green Real-Time PCR Master Mix (Life Technologies) under the following conditions: 50 °C for 2 min, 95 °C for 10 min, and 40 cycles of 15 s at 95 °C and 1 min at 60 °C. All expression was normalized to GAPDH before comparing to control hairpin expression levels. Refer to Supplementary Table 6 for primer sequences.
Western blotting
Passage 3 Rosa-rtTA+/− MEFs or secondary Mbd3fl/−, Rosa26-CreER MEFs were grown in 10-cm tissue culture plates. Secondary Mbd3fl/− MEFs were treated with 1µM 4OH-tamoxifen for 24 hours, then samples were cultured for a further 48 hours and dissociated with 0.25% Trypsin. The cell pellet was then lysed with Cell Lysis Buffer (200mM NaCL, 50mM Tris pH8.0, 1% Trion X-100, 5% glycerol). 20ug of soluble protein was run on a 4–12% gradient Bis-Tris gel (Life Technologies) and blotted onto PVDF membrane. After blocking, membrane was incubated with primary antibody against Mbd3 (1:1000, Bethyl Laboratories, A302-528A) for an hour at RT, washed 3 times with PBS with 0.1% Tween-20, and incubated with secondary antibody anti-rabbit HRP (1: 5000, Jackson Immuno, A5441) for an hour at RT.
Extended Data
Extended Data Figure 1. Results from surface marker screen.
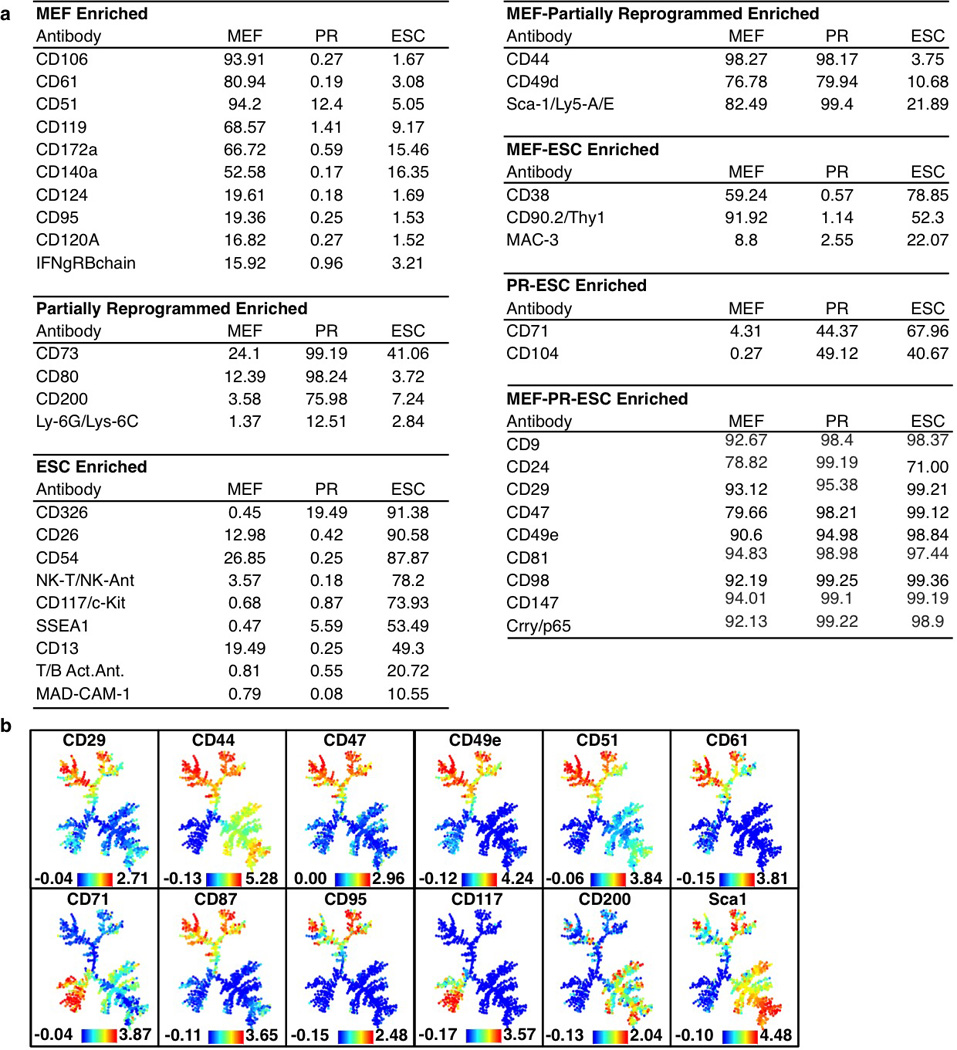
a, Shown are surface markers detected in mouse embryonic fibroblasts (MEF), partially reprogrammed cells (PR) or mouse embryonic stem cells (ESC). Numbers indicate the percentage of each population positive for the marker of interest, relative to isotype control samples. Percentages are colored from green to red to signify high to low values. Markers are grouped for enrichment in single populations or shared between multiple populations. b, SPADE analysis for MEFs, mESCs and PRCs for surface markers analyzed (continued from Fig. 1b). Colors bars (bottom) represent ArcSinh transformed counts for each marker.
Extended Data Figure 2. SPADE and biaxial analysis for MEF reprogramming.
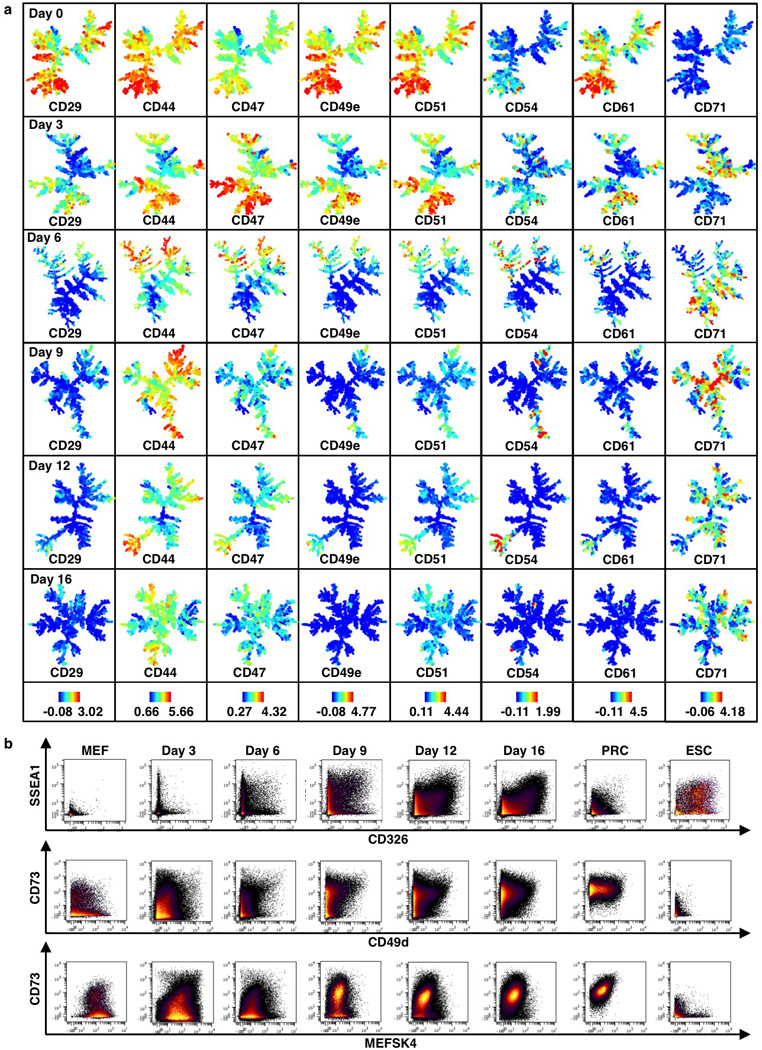
a, SPADE analysis of lentiviral infected MEF reprogramming populations (continued from Fig. 1c). Colors bars (bottom) represent ArcSinh-transformed counts for each marker. b, Biaxial plots for selected markers in control populations and during MEF reprogramming.
Extended Data Figure 3. SPADE analysis for MEF reprogramming.
a, SPADE analysis of lentiviral infected MEF reprogramming populations (continued from Fig. 1c). Colors bars (bottom) represent ArcSinh-transformed counts for each marker. b, Day 12 and 16 time points for markers shown in Fig. 1c). Color bar for % total represents absolute percentages.
Extended Data Figure 4. Details for sorting experiments and chemical treatment assay.
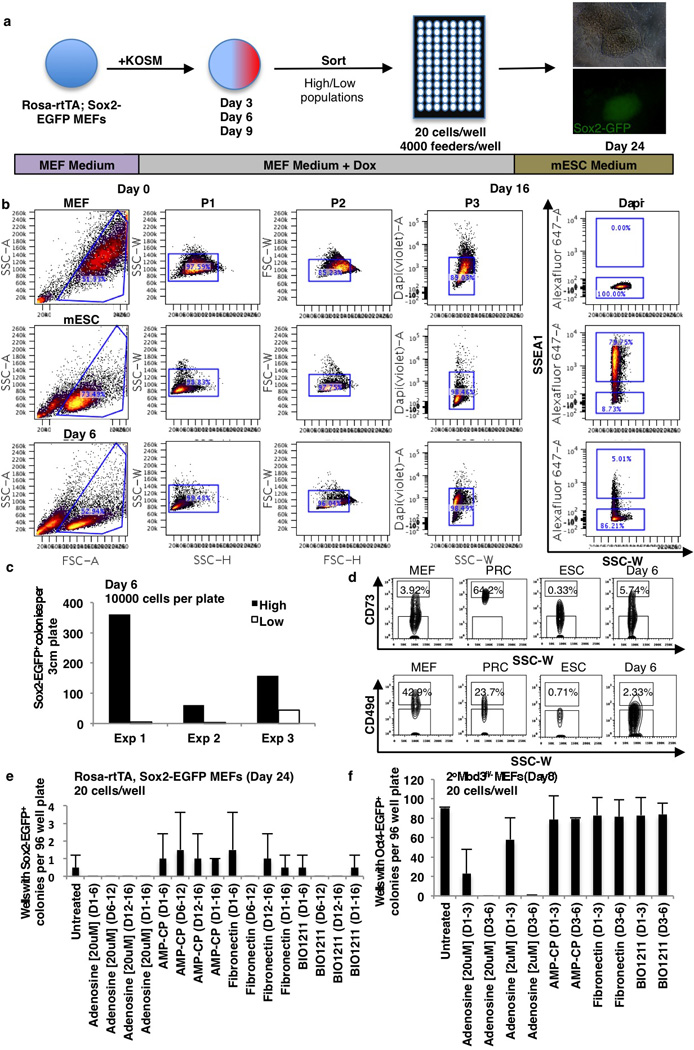
a, 96 well reprogramming assay. 20 cells/well sorted at days 3, 6 and 9. Sox2-EGFP+ colonies were assayed on day 24. b, Gating strategy for SSEA1 in controls and day 6 reprogramming population. High and low-expressing populations were determined based on MEF and ESC control levels. c, 10,000 SSEA1high (black bar) or SSEA1low (white bar) were sorted onto 3cm gelatinized plates with feeders. Sox2-EGFP+ colonies were counted on day 24. d, Gating strategy for CD73 and CD49d in controls and day 6 reprogramming population. High and low-expressing populations were determined based on MEF, PRC and ESC control levels. e–f, Treatment of reprogramming populations with compounds affecting CD73 and CD49d. Shown are 96 well reprogramming efficiencies for infected Rosa-rtTA Sox2-EGFP MEFs (e) or secondary Mbd3fl/− MEFs (f). Y-axis displays wells with Sox2-EGFP+ colonies 24 days after infection (e) or wells with Oct4-EGFP+ colonies 8 days after transgene induction (f) and treated with the indicated compounds for the days (D) indicated (n=2 independent experiments).
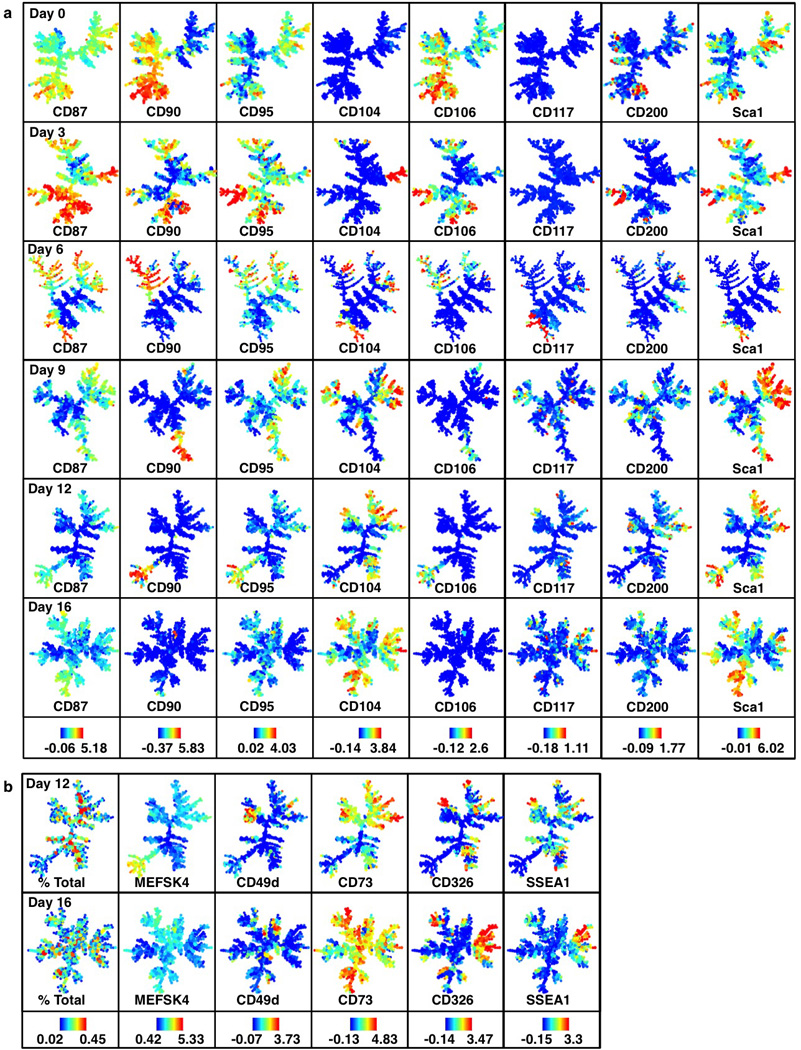
Extended Data Figure 5. Day 6 continuation analysis on day 16.
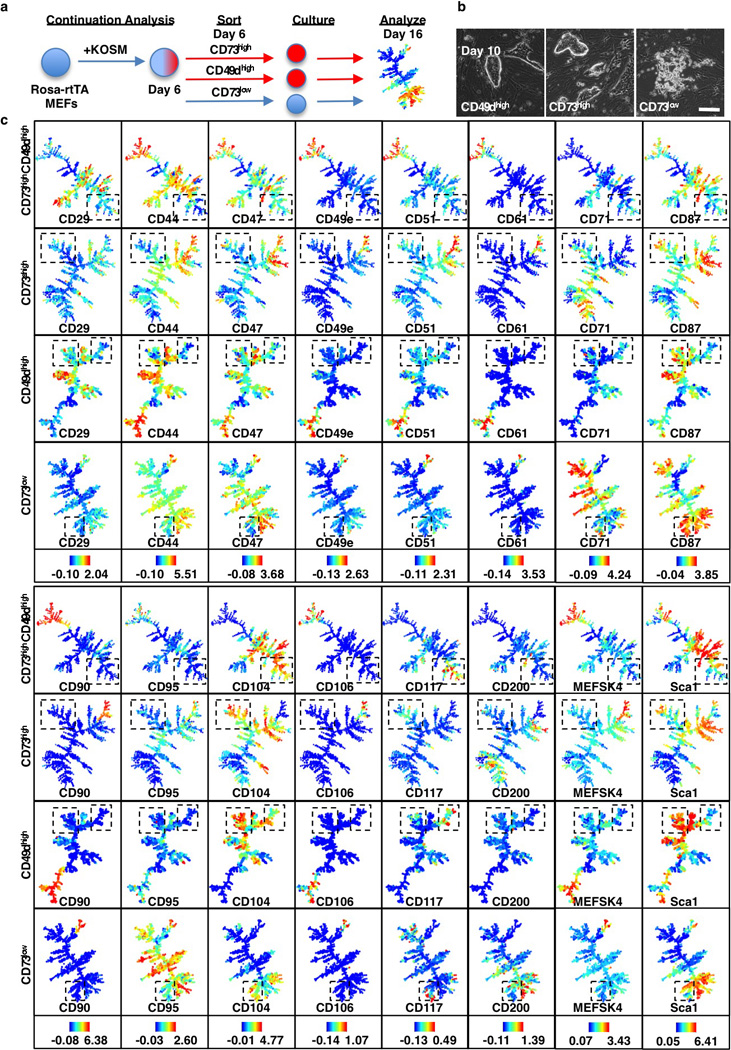
a, Continuation analysis schematic. Reprogramming populations were sorted for poised (CD73high or CD49high) and non-poised (CD73low) populations on day 6, cultured for 10 days on a 3cm plate and analyzed by CyTOF on day 16. b, Morphology of CD49dhigh, CD73high or CD73low cells sorted on day 6 and inspected on day 10. Poised CD49dhigh or CD73high cells form compact colonies within several days of sorting while non-poised CD73low cells fail to do so. c, SPADE analysis (day 16) of cells sorted at day 6 for CD73high/CD49dhigh CD73high, CD49high and CD73low expression (continued from Fig. 3h). Boxes highlight a SSEA1high CD326high branch that is unique to the poised populations. Colors bars (bottom) ArcSinh-transformed counts for each marker.
Extended Data Figure 6. Continuation analysis replicates confirm a SSEA1high CD326high branch that is unique to poised populations.
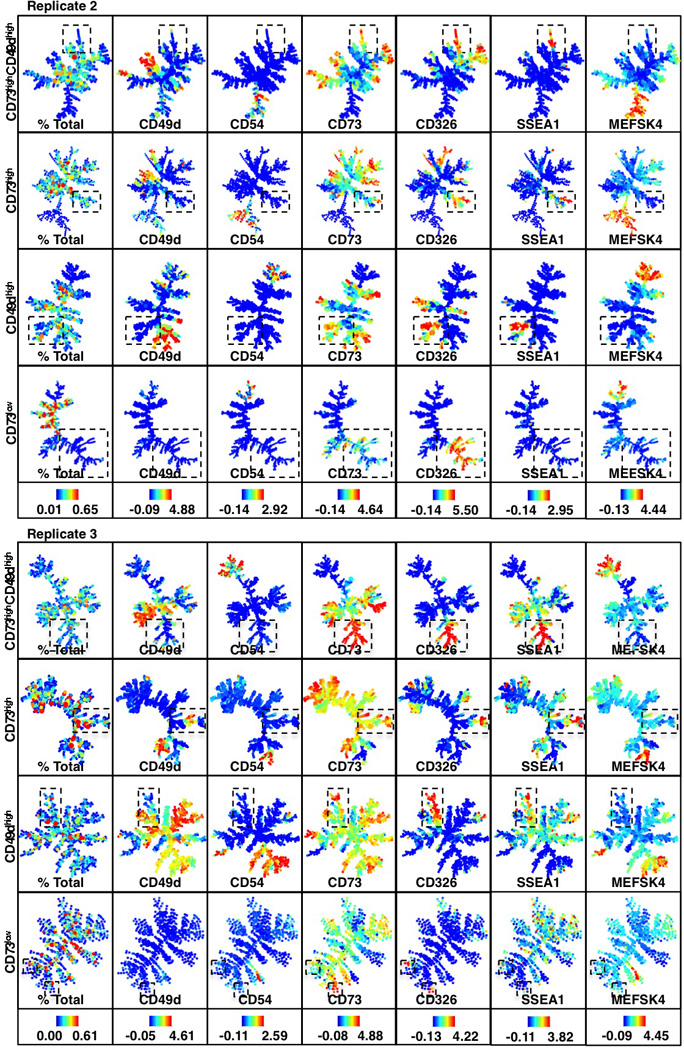
Continuation analysis replicates for reprogramming-prone (CD73high/CD49dhigh, CD73high, CD49dhigh) and non-prone (CD73low) populations. Boxes highlight a SSEA1high CD326high branch that is unique to the poised populations. Colors bars (bottom) represent absolute percentages (left panel) and ArcSinh-transformed counts for each marker.
Extended Data Figure 7. Molecular characterization of reprogramming-prone intermediates.
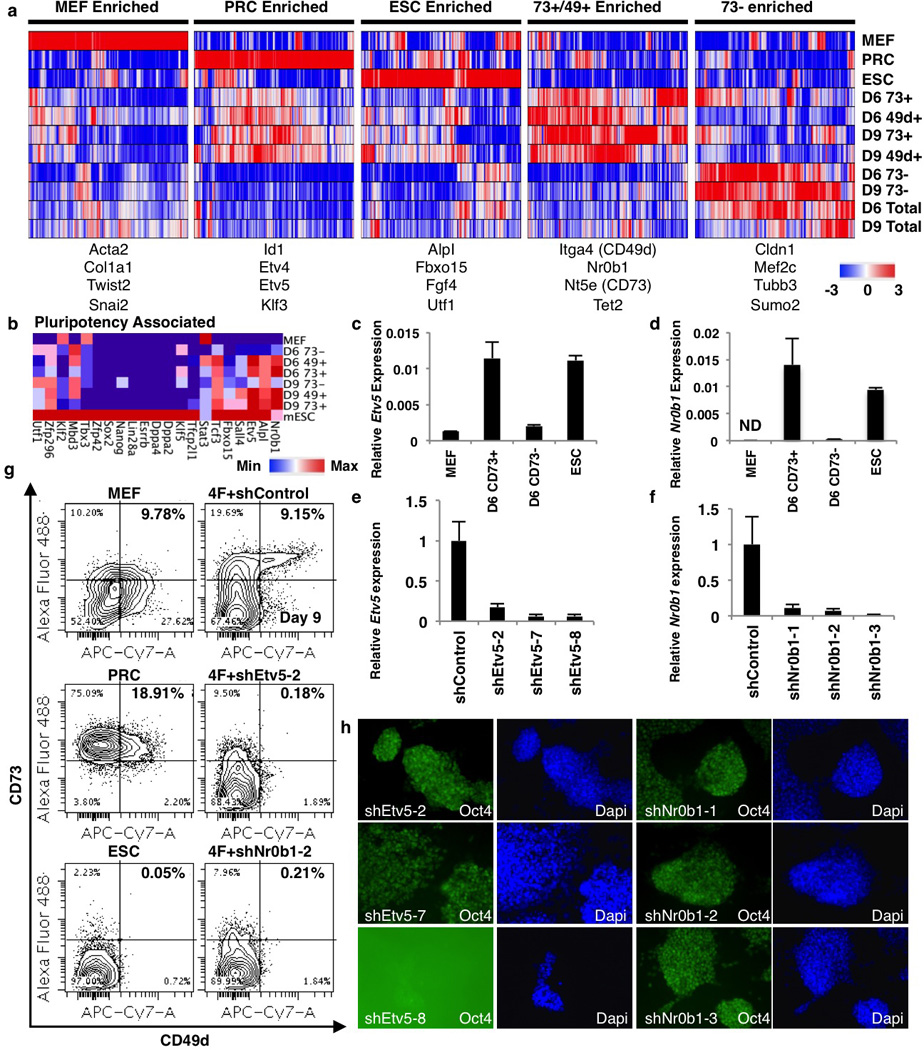
a, Genes differentially expressed between reprogramming-prone (day 6 or 9 CD73high or CD49dhigh) and non-prone (CD73low) populations. Genes with more than 2-fold differential expression between reprogramming-prone and non-prone were selected and k-means clustered (k=5) with control and total reprogramming population expression values. b, Heat map of pluripotency associated genes shown in Fig. 4a (log2). c–d, Quantitative PCR verification of Etv5 (c) and Nr0b1 (d) expression levels (n=3 technical replicates). e–f, Etv5(e) and Nr0b1(f) knockdown qPCRs (n=3 technical replicates). g, Representative FACS plots for day 9 CD73high/CD49dhigh quantification shown in figure 4c. h, Demonstration of ESC self-renewal after infection with Etv5 and Nr0b1 hairpins All infected ESCs continue to express Oct4 after passaging except ESCs infected with shEtv5-8 (n=1).
Extended Data Figure 8. Characterization of high efficiency reprogramming systems.
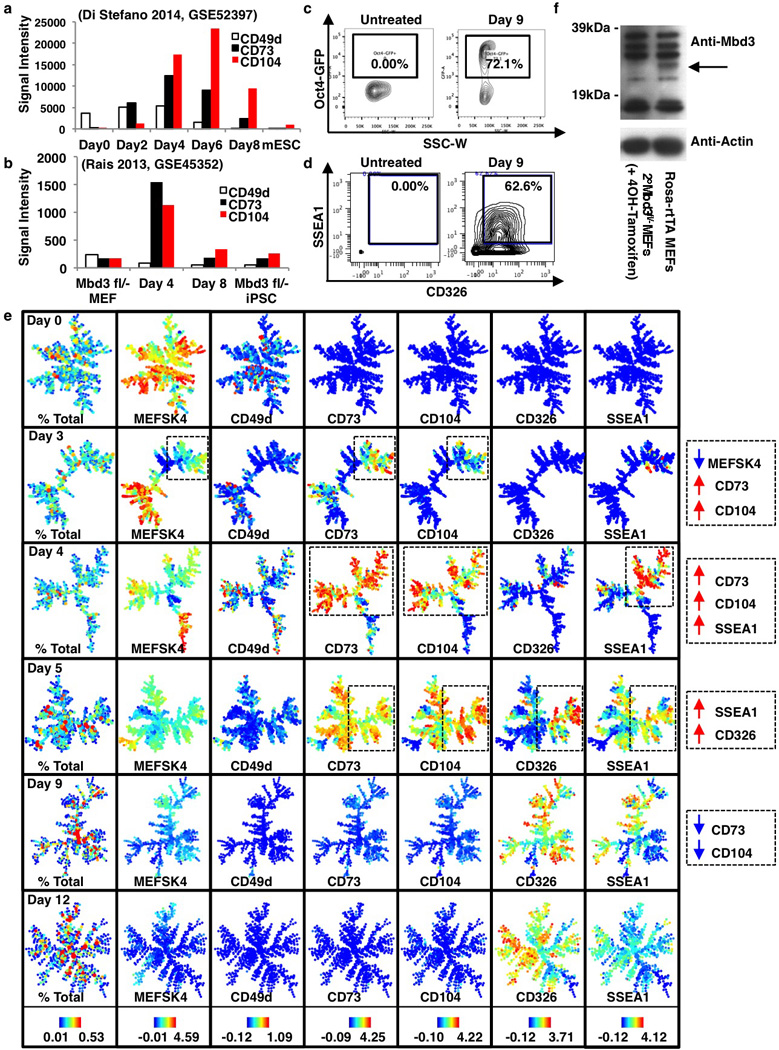
a–b, Expression analysis for CD49d, CD73 and CD104 for previously reported highly efficient reprogramming systems generated by transient expression of C/EBPα13 (a) or Mbd3 depletion12 (b). c, Oct4-GFP transgene reporter signal and d, SSEA1 and CD326 levels for the Mbd3fl/− secondary reprogramming system for untreated (left) and 9 days post induction (right) MEFs. e, SPADE analysis for reprogramming Mbd3fl/− secondary MEFs at days 0, 3, 6, 9 and 12 using all surface markers. Percent total of cells and representative markers are shown for each time point. Remaining markers are shown in Extended Data Figures 9 and 10. Colors bars represent absolute percentages (left panel) and ArcSinh-transformed counts for each marker. f, Verification of Mbd3 loss in passage 3 Rosa26-CreER, Mbd3fl/− secondary MEFs after treatment with 4OH-tamoxifen. Mbd3 levels were compared to passage 3 Rosa-rtTA+/− MEFs. While there are several unspecific bands, there is clearly one band around the expected size of Mbd3 absent in 4OH-tamoxifen-treated cells (arrow).
Extended Data Figure 9. SPADE and biaxial analysis for secondary Mbd3fl/− MEFs.
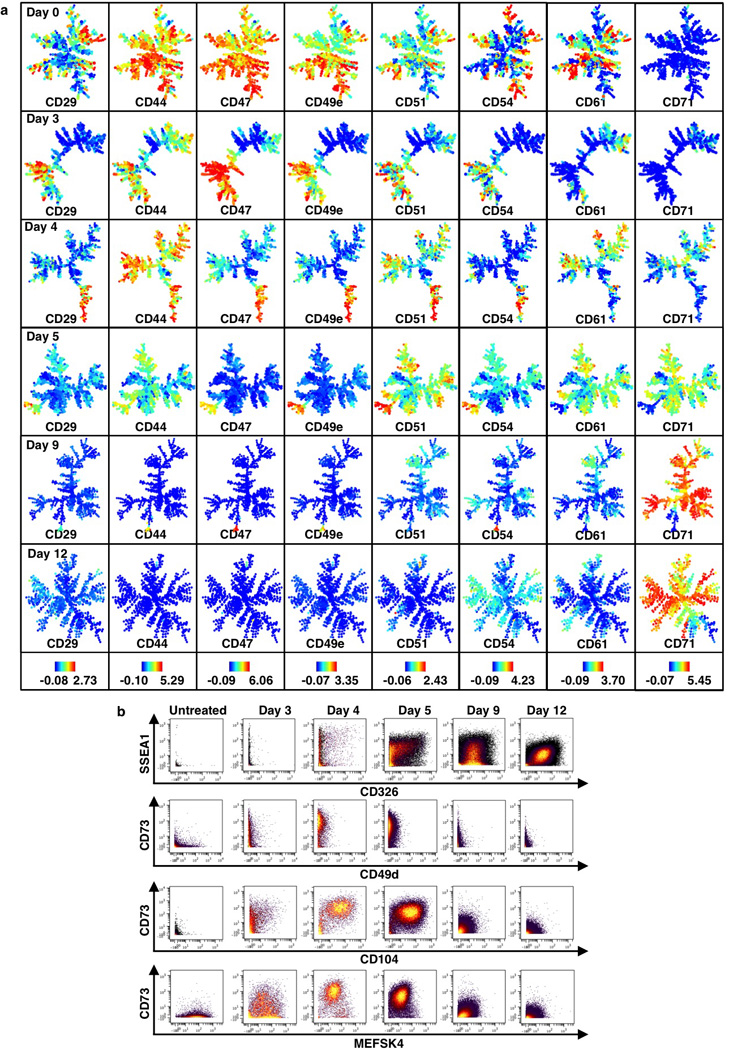
a, SPADE analysis for 2° Mbd3fl/− MEF reprogramming populations (continued from Extended Data Fig. 8e). Colors bars (bottom) represent ArcSinh-transformed counts for each marker. b, Biaxial plots for selected markers.
Extended Data Figure 10. SPADE analysis for secondary Mbd3fl/− MEFs.
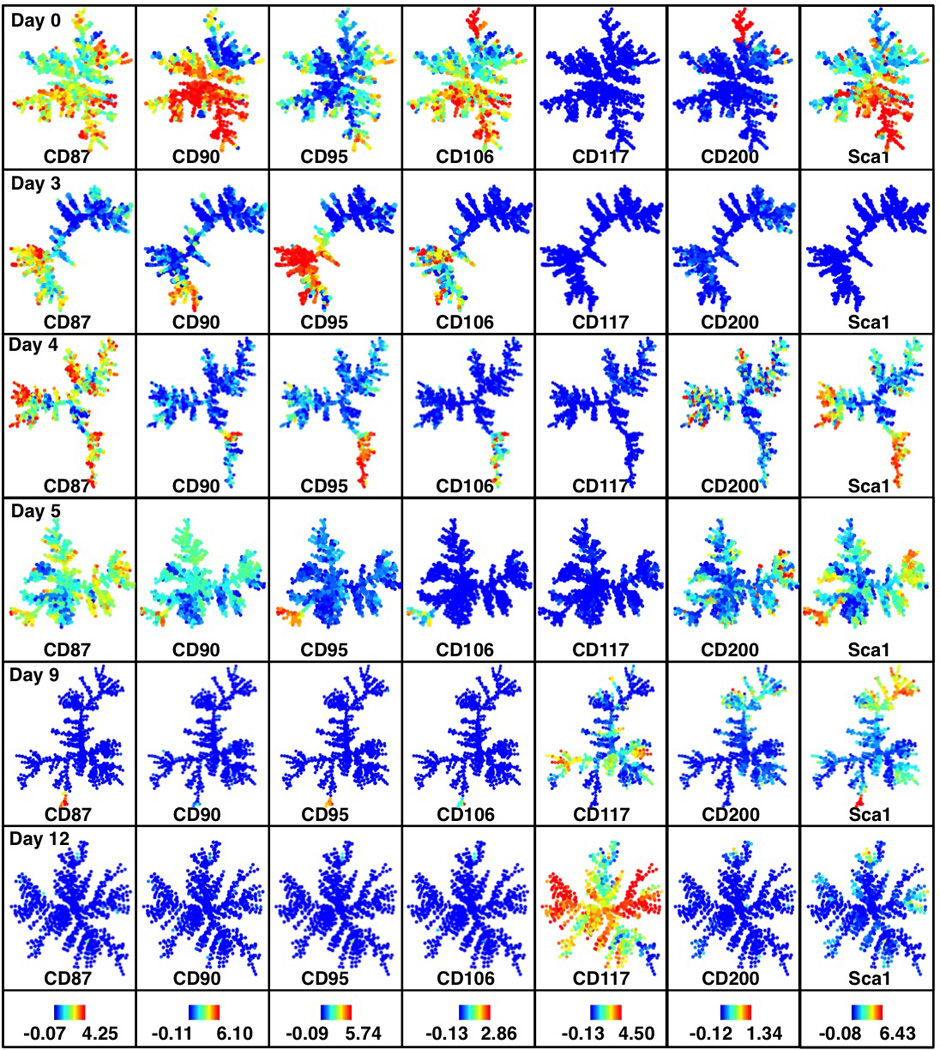
SPADE analysis for 2° Mbd3fl/−reprogramming populations (continued from Extended Data Fig. 8e). Colors bars (bottom) represent ArcSinh-transformed counts for each marker.
Supplementary Material
Acknowledgments
We thank P. Lovelace, R. Finck, K.M. Loh, K. Tanabe and S. Marro for intellectual and technical advice. We would like to thank J. Hanna for his gift of 2° Mbd3fl/− MEFs. We would also like to thank Sebastian Knöbel at Miltenyi for his gift of mEF-SK4 antibody. This work was supported by the California Institute of Regenerative Medicine grant RB2-01592 (G.P.N.). E.L. was supported by the California Institute for Regenerative Medicine Predoctoral Fellowship TG2-01159 and National Science Foundation Graduate Research Fellowship DGE-114747. E.R.Z. was supported by NIH NRSA F32 GM093508-01. M.W. is a New York Stem Cell Foundation-Robertson Investigator and a Tashia and John Morgridge Faculty Scholar at the Child Health Research Institute at Stanford.
Footnotes
Author Contributions
E.L., E.R.Z, G.P.N. and M.W. designed research. E.L. conducted reprogramming and sorting experiments. E.R.Z. conducted CyTOF analysis and data processing. Y.H.N. and I.N.G. assisted with sample processing. E.L., E.R.Z., G.P.N. and M.W. analyzed data. E.L., E.R.Z, G.P.N. and M.W. wrote the paper.
The authors declare no competing financial interests.
References
- 1.Brambrink T, et al. Sequential expression of pluripotency markers during direct reprogramming of mouse somatic cells. Cell stem cell. 2008;2:151–159. doi: 10.1016/j.stem.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buganim Y, et al. Single-cell expression analyses during cellular reprogramming reveal an early stochastic and a late hierarchic phase. Cell. 2012;150:1209–1222. doi: 10.1016/j.cell.2012.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Golipour A, et al. A late transition in somatic cell reprogramming requires regulators distinct from the pluripotency network. Cell stem cell. 2012;11:769–782. doi: 10.1016/j.stem.2012.11.008. [DOI] [PubMed] [Google Scholar]
- 4.Chen J, et al. H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nature genetics. 2013;45:34–42. doi: 10.1038/ng.2491. [DOI] [PubMed] [Google Scholar]
- 5.Hanna J, et al. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ho R, Papp B, Hoffman JA, Merrill BJ, Plath K. Stage-specific regulation of reprogramming to induced pluripotent stem cells by Wnt signaling and T cell factor proteins. Cell reports. 2013;3:2113–2126. doi: 10.1016/j.celrep.2013.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Malley J, et al. High-resolution analysis with novel cell-surface markers identifies routes to iPS cells. Nature. 2013;499:88–91. doi: 10.1038/nature12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Polo JM, et al. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–1632. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sridharan R, et al. Role of the murine reprogramming factors in the induction of pluripotency. Cell. 2009;136:364–377. doi: 10.1016/j.cell.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stadtfeld M, Maherali N, Breault DT, Hochedlinger K. Defining molecular cornerstones during fibroblast to iPS cell reprogramming in mouse. Cell stem cell. 2008;2:230–240. doi: 10.1016/j.stem.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tanabe K, Nakamura M, Narita M, Takahashi K, Yamanaka S. Maturation, not initiation, is the major roadblock during reprogramming toward pluripotency from human fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:12172–12179. doi: 10.1073/pnas.1310291110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rais Y, et al. Deterministic direct reprogramming of somatic cells to pluripotency. Nature. 2013;502:65–70. doi: 10.1038/nature12587. [DOI] [PubMed] [Google Scholar]
- 13.Di Stefano B, et al. C/EBPalpha poises B cells for rapid reprogramming into induced pluripotent stem cells. Nature. 2014;506:235–239. doi: 10.1038/nature12885. [DOI] [PubMed] [Google Scholar]
- 14.Hou P, et al. Pluripotent stem cells induced from mouse somatic cells by small-molecule compounds. Science. 2013;341:651–654. doi: 10.1126/science.1239278. [DOI] [PubMed] [Google Scholar]
- 15.Ichida JK, et al. A small-molecule inhibitor of tgf-Beta signaling replaces sox2 in reprogramming by inducing nanog. Cell stem cell. 2009;5:491–503. doi: 10.1016/j.stem.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li R, et al. A mesenchymal-to-epithelial transition initiates and is required for the nuclear reprogramming of mouse fibroblasts. Cell stem cell. 2010;7:51–63. doi: 10.1016/j.stem.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 17.Maherali N, Hochedlinger K. Tgfbeta signal inhibition cooperates in the induction of iPSCs and replaces Sox2 and cMyc. Current biology : CB. 2009;19:1718–1723. doi: 10.1016/j.cub.2009.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mikkelsen TS, et al. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Samavarchi-Tehrani P, et al. Functional genomics reveals a BMP-driven mesenchymal-to-epithelial transition in the initiation of somatic cell reprogramming. Cell stem cell. 2010;7:64–77. doi: 10.1016/j.stem.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 20.Meissner A, Wernig M, Jaenisch R. Direct reprogramming of genetically unmodified fibroblasts into pluripotent stem cells. Nature biotechnology. 2007;25:1177–1181. doi: 10.1038/nbt1335. [DOI] [PubMed] [Google Scholar]
- 21.Bandura DR, et al. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Analytical chemistry. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- 22.Qiu P, et al. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nature biotechnology. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang J, et al. Dax1 and Nanog act in parallel to stabilize mouse embryonic stem cells and induced pluripotency. Nat Commun. 2014;5:5042. doi: 10.1038/ncomms6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Takahashi K, et al. Induction of pluripotency in human somatic cells via a transient state resembling primitive streak-like mesendoderm. Nature communications. 2014;5:3678. doi: 10.1038/ncomms4678. [DOI] [PubMed] [Google Scholar]
Methods references
- 25.Vierbuchen T, et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature. 2010;463:1035–1041. doi: 10.1038/nature08797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellis P, et al. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Developmental neuroscience. 2004;26:148–165. doi: 10.1159/000082134. [DOI] [PubMed] [Google Scholar]
- 27.Rubinson DA, et al. A lentivirus-based system to functionally silence genes in primary mammalian cells, stem cells and transgenic mice by RNA interference. Nature genetics. 2003;33:401–406. doi: 10.1038/ng1117. [DOI] [PubMed] [Google Scholar]
- 28.Finck R, et al. Normalization of mass cytometry data with bead standards. Cytometry. Part A : the journal of the International Society for Analytical Cytology. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Linderman MD, et al. CytoSPADE: high-performance analysis and visualization of high-dimensional cytometry data. Bioinformatics. 2012;28:2400–2401. doi: 10.1093/bioinformatics/bts425. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.