

Abstract
Objective
While it is accepted that macrophage glycolysis is up-regulated under hypoxic conditions, it is not known whether this is linked to a similar increase in macrophage pro-inflammatory activation and whether specific energy demands regulate cell viability in the atheromatous plaque.
Approach and Results
We studied the interplay between macrophage energy metabolism, polarization and viability in the context of atherosclerosis. Cultured human and murine macrophages and an in vivo murine model of atherosclerosis were used to evaluate the mechanisms underlying metabolic and inflammatory activity of macrophages in the different atherosclerotic conditions analyzed. We observed that macrophage energetics and inflammatory activation are closely and linearly related, resulting in dynamic calibration of glycolysis to keep pace with inflammatory activity. Additionally, we show that macrophage glycolysis and proinflammatory activation mainly depend on hypoxia-inducible factor (HIF) and on its impact on glucose uptake, and on the expression of hexokinase II and ubiquitous 6-phosphofructo-2-kinase (PFKFB3). As a consequence, hypoxia potentiates inflammation and glycolysis mainly via these pathways. Moreover, when macrophages’ ability to increase glycolysis through PFKFB3 is experimentally attenuated, cell viability is reduced if subjected to proinflammatory and/or hypoxic conditions, but unaffected under control conditions. In addition to this, GM-CSF enhances anaerobic glycolysis while exerting a mild pro-inflammatory activation.
Conclusions
These findings, in human and murine cells and in an animal model, show that hypoxia potentiates macrophage glycolytic flux in concert with a proportional up-regulation of pro-inflammatory activity, in a manner that is dependent on both HIF-1α and PFKFB3.
Keywords: hypoxia, atherogenesis, PET, energy metabolism, glycolysis, macrophage viability
Introduction
A body of research extending over three-quarters of a century shows that myeloid cells including macrophages are highly dependent on glycolysis for energy metabolism 1. Subsequently, several lines of evidence have linked macrophage pro-inflammatory activation with cellular energetics 2–4. However, the precise nature of this link remains poorly understood. It was recently shown that hypoxic macrophages up-regulate the expression of glucose transporters, hexokinase II and the ubiquitous form of 6-phosphofructo-2-kinase (encoded by PFKFB3) in order to substantially increase the glycolytic flux 5. However, it remains unknown if this increased glycolytic flux occurs in order to simply maintain constant energy delivery in an anaerobic environment or it parallels a similar increase in pro-inflammatory activation. Moreover, the relationship between macrophage pro-inflammatory activation and energetics is not well understood in the setting of an atherogenic milieu. Accordingly, in this series of investigations, we sought to investigate the relationship between two fundamental processes: energy metabolism and inflammatory activation of macrophages in an atherogenic environment.
Inflammation plays an important role in a wide range of diseases. Atherosclerosis is a chronic inflammatory condition accounting for the largest share of mortality in the developed world 6 and is one of the most devastating of inflammatory conditions. Consequently, there is a critical need for greater understanding of the biological mechanisms underlying atherosclerotic inflammation and its complications, including plaque rupture and intraplaque hemorrhage 7, 8. Atherosclerosis is characterized by macrophage-predominated inflammation existing in the context of pro-inflammatory cytokines, oxidized low-density lipoproteins (oxLDL), and hypoxia within the arterial wall 9–12. Despite mounting evidence that hypoxia promotes inflammation in cancer, obesity and other inflammatory diseases13, in atherosclerosis, the mechanism and precise contribution of hypoxia to other plaque constituents (e.g. cytokines [CK] and oxLDL) on the pro-inflammatory milieu remains to be established and a recent study questioned whether pro-inflammatory stimuli present in atheroma up-regulate macrophage glucose metabolism at all 14.
We studied macrophage responses to a range of pro-inflammatory stimuli relevant to atherosclerosis, and observed that pro-inflammatory stimuli substantially increased both the rate of glycolysis (flux) and pro-inflammatory activation (e.g. TNF-α production). Moreover, we found that glycolytic flux and pro-inflammatory activation show a significant correlation over a range of physiological inflammatory stimuli and across normoxic and hypoxic conditions. We also noted that hypoxia substantially potentiates the effect of cytokines on both glycolytic flux and pro-inflammatory activation (in similar proportions). We then demonstrated that blocking hypoxia sensing or enzymes responsible for high glycolytic flux substantially reduced both the pro-inflammatory activation and the glycolytic flux of macrophages. Moreover, inhibition of glycolysis after PFKFB3 expression has a profound effect on murine and human macrophage viability, leading to enhanced necrotic and apoptotic death under pro-inflammatory conditions. Subsequently, we confirmed the in vivo relevance of our findings in an animal model.
Materails and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Hypoxia Potentiates Macrophage Pro-Inflammatory Activation and Glycolysis
In atherosclerosis, as with other inflamed tissues, the microenvironment plays a key role in determining the activation state of macrophages. Cytokines are an important constituent of the inflammatory milieu influencing macrophage commitment along a classical (M1 pro-inflammatory) or alternative (M2 anti-inflammatory) polarization 15. To establish a firm basis for the series of experiments in this study, we sought to confirm the known effects of various M1 vs. M2 activators on elicited peritoneal murine macrophage polarization. Additionally, we evaluated the relative action of GM-CSF (GM), a cytokine that is relevant to the atheroma and a potent macrophage regulator 16, which served as a physiological classical stimulus given its ubiquitous, endogenous expression in rodents and humans 16–22. Despite the relative importance of GM-CSF to macrophage biology in vivo, the metabolic impact of GM-CSF on macrophage metabolism is not well understood. Accordingly, we examined macrophages after exposure to CK (classical stimuli: TNF-α, IL-1β, IFN-γ), LPS (innate stimulus), IL4/IL13 (alternative stimulus) as well as GM-CSF. Using complementary techniques to evaluate the polarization (Fig. 1A–D), we observed as expected that M1 markers increased after classic or innate activation (with LPS and CK), while M2 markers increased after stimulation with IL4/IL13. We furthermore observed that GM-CSF exerts a M1-like polarization, resulting in mild increases in NO and TNF-α accumulation, (Fig. 1B,C), well-accepted signatures of the M1 activation 23–25. Further, under either classic or innate stimuli, we observed a shift in the gene expression of the bifunctional enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB), from the liver type-PFK2 (PFKFB1), which has a low net kinase activity, to the more active inducible form PFKFB3 (Fig. 1D). This response was further exacerbated under hypoxic conditions, as suggested by the increased HIF-1α levels (Fig. 1E,F). It is well established that PFKFB3 has greater kinase than bisphosphatase activity 26 and thus favors the rise in fructose-2,6-bisphosphate (Fru-2,6-P2), a potent regulator of upper glycolysis that is required for high glycolytic rates 5, 26, 27.
Fig. 1. Characterization of the interplay between macrophage polarization and glycolytic enzymes.

Peritoneal murine macrophages were incubated with GM-CSF (20 ng/ml), LPS (200 ng/ml), CK (TNF-α, IL-1β and IFN-γ; 20 ng/ml each) or IL4+IL13 (20 ng/ml). Cells were maintained under normoxic or hypoxic conditions (1% O2) for 18h prior to challenge. (A) left, Western blot of phospho-STAT-5 and phospho-ERK to ensure a selective GM-CSF response; right, expression of M1 and M2 polarization markers in response to the indicated stimuli. (B) The time-course of the accumulation of nitrate plus nitrite in the culture medium was measured. (C) The accumulation of TNF-α was measured as an index of macrophage activation at 8h. (D) mRNA levels of M1 (TNF-α, IL-6, PFKFB3) and M2 (Arg-1, IL-10) polarization markers. (E–F) Western blot analysis of HK-II, PFKFB3, PFKFB1 and HIF-1α under normoxic and hypoxic conditions (left). Relative band intensity ratios levels of HK-II, PFKFB3 and PFKFB1 were determined (F). Results show a representative blot out of three (A,E). Results show the mean±SD of four experiments (B,C,F).*P<0.05; **P<0.01 vs. non stimulated cells.
Observations under Atherogenic Conditions
Next, we evaluated the actions of key atherosclerotic constituents (oxLDL, LDL, and hypoxia) 7, 28 on murine macrophage pro-inflammatory activation and glycolysis. We found that oxLDL and GM-CSF (but not LDL) each resulted in pro-inflammatory activation of murine macrophages (Fig. 2A). Notably, hypoxia on its own did not increase pro-inflammatory stimulation. However, in cells exposed to GM-CSF and/or oxLDL, hypoxia substantially potentiated pro-inflammatory activation (Fig. 2A, right). The effects on glycolysis of these atherosclerotic constituents were similar to those on TNF-α production: oxLDL, GM-CSF, and CK each resulted in substantial increases in Fru-2,6-P2 concentration (Fig. 2B). Moreover, while hypoxia alone caused a minimal increase in glycolysis, hypoxia markedly potentiated the glycolytic flux in the presence of proatherogenic mediators (cytokines and oxLDL) (Fig. 2B, right; Fig. 2C). Furthermore, while hypoxia alone was not associated with an increase in glucose consumption in non-activated macrophages, hypoxia substantially potentiated the effect of CK and GM-CSF (Fig. 2D). These results agree with the expression levels of genes encoding the glucose transporter Glut-1 as well as the genes encoding HK-II and PFKFB3, the enzyme responsible for the high-throughput synthesis of Fru-2,6-P2. We observed a similar action of hypoxia on the expression of those genes: hypoxia had a minor impact on its own, but substantially potentiated the actions of CK and GM-CSF (Fig. 2E).
Fig. 2. Hypoxia potentiates murine macrophage pro-inflammatory activation and glycolytic flux.

Murine macrophages were incubated with GM-CSF (20 ng/ml), CK (TNF-α, IL-1β and IFN-γ; 20 ng/ml each) or treated with LDL or oxLDL (50 μg/ml). Cells were maintained under normoxic (20% O2) or hypoxic (1% O2) conditions for 18h prior to challenge. (A) TNF-α measurement (8h) served as an index of macrophage activation (note, TNF-α was not assessed after CK, since CK contains TNF-α; N.D.= not determined). (B) Intracellular levels of Fru-2,6-P2 were measured at the end of the experiment (24h). (C) Lactate accumulation in the culture medium (24h), and (D) glucose consumption from 8 to 24h across oxygen tensions after cytokine challenge. (E) Time-course of indicated mRNA levels under normoxia (top) and hypoxia (bottom) after cytokine treatment. Results show the mean±SD of four experiments. #P<0.05; ##P<0.01 vs. the same O2 in the absence of LDL or oxLDL (A,B); *P<0.05; **P<0.01 vs. the same condition under normoxia (A–C), vs. the same condition in the absence of cytokine (D), vs. the same condition at 0h (E). N.D=not etermined
Close Interrelationship between Glycolysis and Pro-Inflammatory Activation. Role of TNF-α
Because we observed that both macrophage pro-inflammatory activation and energetics increase in the presence of atherosclerotic mediators, we sought to define the strength of the interrelationship between these two processes. We observed a strong linear correlation (R=0.97; P<0.001) between glycolytic flux (as Fru-2,6-P2 levels) and pro-inflammatory activity (measured as TNF-α production) under both hypoxic and normoxic conditions Fig. 3A. Hence, we demonstrate that macrophage glycolysis keeps pace with the pro-inflammatory activated state of the cell regardless of oxygen tension. Moreover, since GM-CSF promotes TNF-α synthesis, we sought to determine its contribution to the glycolytic phenotype. As Fig. 3B shows, the presence of neutralizing antibodies against TNF-α in the culture medium significantly attenuated the expression of PFKFB3, the rise in Fru-2,6-P2 levels and the release of lactate, providing a link for TNF-α between the inflammatory and glycolytic profiles.
Fig. 3. Close interrelationship between murine macrophage pro-inflammatory activation (TNF-α) and Fru-2,6-P2 concentration as indicator of glycolysis.

(A) Correlation between the accumulation of TNF-α in the culture medium and intracellular Fru-2,6-P2 concentrations under the experimental conditions described in Figs. 1–2. (B) Neutralizing goat anti-mouse TNF-α Abs (10 μg/ml) in the culture medium prevent PFKFB3 induction, the rise in Fru-2,6-P2 concentration and the release of lactate in macrophages treated for 24h with GM-CSF (20 ng/ml) under normoxic and hypoxic (1% O2) conditions. Goat IgG (10 μg/ml) was used as control immunoglobulin. Results show a representative blot and the mean±SD of three experiments. **P<0.01 vs. the same IgG condition.
PFKFB3 Regulates both Glycolytic Metabolism and Inflammation
Given the strong linear association between macrophage glycolysis and inflammatory activity in murine cells, we next sought to evaluate whether: 1) a high glycolytic rate was necessary for inflammatory activation, and 2) this relationship was maintained in human macrophages. To investigate this human macrophages were separately treated with two inhibitors of PFKFB3: i) a human small interfering RNA (siRNA) targeting PFKFB3, and ii) 3PO, a selective inhibitor of PFKFB3 29. In the first experiment, we observed that silencing PFKFB3 effectively attenuated PFKFB3 without impacting the constitutively expressed PFKFB1 or HK-II (Fig. 4A), and thus did not affect basal glycolysis (Fig. 4B). However, we observed that inhibition of PFKFB3 activity under stimulatory conditions results in a significant reduction in glycolytic flux (measured as Fru-2,6-P2 levels and lactate release; Fig. 4B and Fig. S1A–B, respectively). In parallel to these findings, we found that blocking PFKFB3 resulted in a significant reduction in pro-inflammatory activation (measured by TNF-α production; Fig. 4C) across oxygen tensions and under a range of stimuli.
Fig. 4. Inhibition of PFKFB3 attenuates energy metabolism, pro-inflammatory activation and viability in activated human macrophages.

Human macrophages were maintained under normoxia or hypoxia, and transfected for 18h with a specific pool of siRNA (5 nM) to silence PFKFB3, or inactive RNA control (scRNA) and then activated with human GM-CSF and CK (used at 20 ng/ml each). (A) PFKFB3, PFKFB1, HK-II and β-actin were assessed by Western blot. (B,C) Glycolytic flux (as Fru-2,6-P2 concentration) and pro-inflammatory activation (as TNF-α accumulation) were determined after PFKFB3 silencing with siRNA or inhibition with 3PO (10 μM). (D) Cellular viability was assessed in the absence/presence of PFKFB3 inhibition (by 3PO). Results show a representative blot out of three (A) or the mean±SD of 3–4 experiments (B–D). *P<0.05; **P<0.01 vs. the same condition with fully active PFKFB3; #P<0.05; ##P<0.01 vs. the same condition under normoxia (B,C). N.D.= not determined.
Thereafter, we tested the hypothesis that high-flux glycolysis is required to maintain macrophage viability under conditions of pro-inflammatory stimulation. In this experiment, we observed that blocking PFKFB3 did not reduce the viability of resting macrophages; however, under conditions of pro-inflammatory stimulation, the viability of activated macrophages decreased when PFKFB3 was inhibited (Fig. 4D). Taken together, these observations provide evidence that the ability to up-regulate PFKFB3 activity is necessary to maintain the viability of activated (but not resting) macrophages.
HIF-1α also Regulates both Glycolytic Metabolism and Inflammation
Since we observed that hypoxia potentiates macrophage pro-inflammatory activation, we hypothesized that interruption of hypoxia-sensing might attenuate inflammation in an atherosclerotic environment. Indeed, within the context of cancer, it has recently been shown that hypoxia-induced over-expression of HIF-1α results in metabolic adaptation (a shift to glycolysis from Krebs cycle and oxidative phosphorylation) which in turn facilitates cell survival 30. We sought to evaluate whether a similar relationship exists in atherosclerotic macrophages. We observed that silencing HIF-1α substantially reduced glycolytic flux (as Fru-2,6-P2 levels and lactate; Fig. 5A,B) across oxygen tensions and under various pro-inflammatory conditions. In parallel, blocking HIF-1α substantially reduced pro-inflammatory activation of macrophages (as TNF-α production; Fig. 5C). Further, we observed that inhibition of either PFKFB3 and/or HIF-1α, resulted in proportionate reductions in both glycolysis (as Fru-2,6-P2) and pro-inflammatory activation (R=0.68, P=0.007; Fig. 5D). In addition to this, we evaluated the role of hypoxia on the inducibility of PFKFB3 in response to GM-CSF. As Fig. 5E shows, macrophages transfected with a plasmid carrying the HRE sequence of the human pfkfb3 promoter (29 nucleotides, from −1297 to −1269) linked to a luciferase reporter 31–33 exhibited an increase in activity due to hypoxia, but also by the action of GM-CSF, a response that was enhanced under hypoxic conditions. Collectively, these observations suggest that both up-regulation of upper glycolysis and pro-inflammatory activation are dependent on HIF-1α across oxygen tensions, highlighting the crosstalk between hypoxic and non-hypoxic signaling pathways in the regulation of metabolic adaptation in response to atherosclerotic stimuli. Indeed, silencing of HIF-1α provoked a loss in viability with increased rates of apoptosis and necrosis compared to controls (Fig. 5F). This was especially evident in the presence of oxLDL or under hypoxic conditions. This finding reaffirms that an intact HIF-1α system is required for macrophage survival in an atherosclerotic microenvironment and that survival of activated macrophages is also dependent on high glycolytic flux, through up-regulation of PFKFB3. In addition to this, O2 consumption decreased in M1 macrophages. Moreover, under low glucose availability, forcing respiration with FCCP resulted in cell death of M1 cells, but not in resting or M2 counterparts (Fig. 6A). Moreover, GM-CSF depressed mitochondrial ATP synthesis (Fig. 6B), but enhanced non-mitochondrial O2 consumption in agreement with ROS production, in particular in the presence of oxLDL (Fig. 6C,D).
Fig. 5. Silencing HIF-1α decreases human macrophage energetics, pro-inflammatory activation and viability.

Human macrophages were maintained under normoxia or hypoxia and silenced with siRNA for HIF-1α (5 nM). (A,B) Effect of siRNA for HIF-1α on the levels of Fru-2,6-P2 and lactate release after stimulation with GM-CSF or CK (20 ng/ml each). (C) Time course of TNF-α accumulation under hypoxia and after silencing HIF-1α. (D) Interrelationship between TNF-α and Fru-2,6-P2 levels under the different experimental conditions described in Fig. 4 and 5 (including normoxia, hypoxia, LDL and oxLDL and silencing PFKFB3 and HIF-1α), and inhibition of PFKFB3 (3PO; 10 μM). (E) After transfection of macrophages with a luciferase reporter plasmid containing the HRE domain of the pfkfb3 promoter, or the mutated sequence lacking HRE binding (pHREwt.Luc and pHREmut.Luc, respectively at 600 ng/ml each) and a β-galactosidase plasmid (60 ng/ml) cells were submitted for 4h to normoxia or hypoxia (1% O2) and then activated for 6h with GM-CSF. The ratio between the luciferase and β-galactosidase activities was determined for in the same cell extracts. (F) After activation for 60h with GM-CSF or CK, the percentage of apoptotic/necrotic cells was determined. Results show the mean±SD (n=3) (A,B,D) or a representative experiment out of three (C). *P<0.05; **P<0.01 vs. the same condition with scRNA (A,B), vs. the basal condition under normoxia (E) or vs. the corresponding LDL condition (F). #P<0.05; ##P<0.01 vs. the same condition under normoxia (A,B).
Fig. 6. Real time changes in the oxygen consumption rate (OCR) and ROS production in human macrophages under atherogenic conditions.
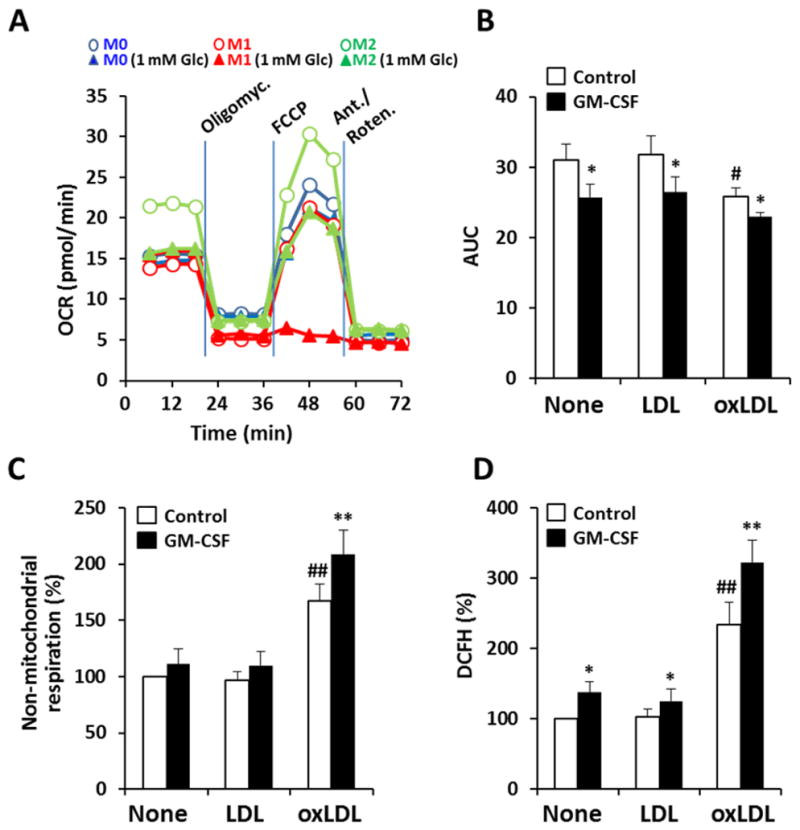
Cells were maintained for 18h with complete medium or medium with 1 mM glucose and activated with CK (20 ng/ml), GM-CSF (20 ng/ml), LDL and oxLDL (50 μg/ml) or combinations of these. (A) The OCR was measured and then cells were treated with oligomycin (inhibitor of ATP synthase), FCCP (ionophore to ‘force’ O2 consumption) and rotenone+antimycin A (inhibitors of the electron-transport chain). (B) Basal O2 consumption (area under the curve, AUC) as measured during the early 20 min in A. (C) Non-mitochondrial respiration, corresponding to cells treated with antimycin A plus rotenone. (D) ROS production by macrophages measured by the oxidation of DCFH. Results show the mean (A) or the mean±SD (n=4) (B–D). *P<0.05; **P<0.01 vs. the same condition without GM-CSF; #P<0.05; ##P<0.01 vs. the none condition.
Inhibition of PFKFB3 and HIF-1α Attenuates Glycolytic Flux in ApoE−/− Mice
We then sought to evaluate the impact of selective inhibition of PFKFB3 and HIF-1α on glycolytic flux in atherosclerotic mice (ApoE−/− mice fed a high-fat diet). Glycolysis in the arterial wall was assessed using FDG PET/CT imaging before and after impairing PFKFB3 (using 3PO or PFKFB3 siRNA) and HIF-1α (using HIF-1α siRNA). Subsequently, we removed the arterial segments and measured tissue levels of PFKFB3, Fru-2,6-P2, TNF-α and CCL2. We observed substantial attenuation of glycolysis (measured as a target-to-background ratio of FDG uptake; TBR) after inhibition of either PFKFB3 and/or HIF-1 α (Fig. 7A). Moreover, after removal of the arterial tissues, we measured the tissue quantities of PFKFB3 and Fru-2,6-P2, and found that their tissue levels mirrored the measurements of FDG uptake across the tested conditions (Fig. 7B,C). Similarly, the tissue measurements of pro-inflammatory mediators TNF-α and CCL2 were similarly affected by the inhibitors (Fig. 7D). Also, silencing or inhibition of PFKFB3 resulted in an enhancement of active caspase 3 in the plaque (Fig. 7E) confirming the loss in cell viability under these conditions. Interestingly, all animals were alive and stable after treatment courses. Given the tenacious relationship between glycolysis and inflammation, these in vivo observations raise the possibility that antagonism of HIF-1α and/or modulation of glycolysis may provide a pharmacologic approach to alleviate the inflammatory burden in atherosclerotic disease.
Fig. 7. In vivo glycolytic flux measurement with 2GD and FDG-PET in atherosclerotic ApoE-deficient mice.

(A) TBR in male ApoE deficient mice fed a high fat diet for three weeks and silenced for HIF-1α, PFKFB3, or treated i.p. with the PFKFB3 inhibitor 3PO (50 mg/kg in Solutol) at days 3, 7, 10, 14 and 17 prior to FDG study. (B) Intra-plaque PFKFB3 levels were determined by Western blot and normalized for β-actin levels. (C) Intra-plaque Fru-2,6-P2 concentration and (D) TNF-α and CCL2 levels. (E) Intra-plaque active caspase 3 protein levels (normalized with β-actin) and enzymatic activity, using N-acetyl-DEVD-7-amino-4-trifluoromethylcoumarin as substrate. C+= positive control of macrophages treated for 24h with 0.5 mM GSNO. Results show the mean±SD (n=4–9 animals; 30 animals in total). *P<0.05; **P<0.01 vs. the absence of treatment (A,C,D) or expressed as percentage (B,E).
Discussion
The findings of this study highlight the basis for and importance of macrophage metabolic adaptation within atherosclerosis. We show that hypoxia potentiates macrophage glycolytic flux along with a proportional up-regulation of pro-inflammatory activity (in a manner that is dependent on both HIF-1α and PFKFB3). Moreover, we show that antagonizing either HIF-1α or PFKFB3 activities simultaneously impairs both glycolysis and pro-inflammatory activation in vitro, and we provide translational evidence of this phenomenon in vivo. Further, we show that impairing either HIF-1α or PFKFB3 in the setting of pro-inflammatory stimulation results in loss of monocyte viability after activation of caspase cascades. The potential impact of reduced monocyte/macrophage viability in atherosclerotic plaques is unclear. Macrophage proliferation within plaque has been described 34, and reduced viability of such cells, as well perhaps of other macrophages may indeed be beneficial. However, there is also the possibility of detrimental effects of enhancing cell death with possible post-apoptotic necrosis if efferocytosis is deficient. Thus, further work is needed to better understand the implications of such modulation of macrophage viability within the atheroma.
The observation that hypoxia increases glycolysis is a phenomenon referred to, in cancer cells, as the Warburg Effect 35, and appears to be involved in macrophage physiology at different points of their maturation and function 36. One proposed explanation for Warburg’s observation is that hypoxia obligates a shift in metabolism, from the tricarboxylic acid cycle to glycolysis simply to compensate for loss of aerobic metabolism 37. However, our observations provide an alternate explanation for the Warburg Effect, at least as far as it relates to atherosclerotic macrophages, namely that 1) hypoxia also increases the pro-inflammatory activation of macrophages and, that 2) the glycolytic rate is increased in a manner that keeps pace with this increased inflammatory and proatherogenic activity, namely due to the contribution of oxLDL to the enhanced glucose consumption and ROS production 10, 11. Collectively, these observations give rise to a paradigm wherein hypoxia can act to exacerbate a cycle of inflammation (Fig. 8). Within this paradigm, cytokines trigger up-regulation of HIF-1α, which is stabilized under hypoxic conditions 38, 39. HIF-1α, after transcriptional induction of PFKFB3 leads to increased pro-inflammatory activation as well as increased glycolysis 5, 29, 32, 33, 40, 41. The activated macrophages then produce additional cytokines to perpetuate this vicious inflammatory cycle.
Fig. 8. Hypoxia potentiates macrophage pro-inflammatory activation, thereby strengthening a vicious cycle.

(A) Cytokines trigger up-regulation of HIF-1α, which is further stabilized by hypoxic conditions. HIF, after binding to the promoter region of PFKFB3 leads to increased glycolysis as well as to increased pro-inflammatory activation and maintained viability. The viable activated macrophages then produce additional cytokines to perpetuate this vicious inflammatory cycle. Antagonism of HIF-1α and/or PFKFB3 results in decreased pro-inflammatory activation and metabolism as well as reduced viability.
Our data have potentially important implications for the treatment of atherosclerotic inflammation. We found that selective inhibition (or silencing) of PFKFB3, HIF-1α, or both results in a substantial reduction in pro-inflammatory activation. Along these lines, PFKFB3 has been recently shown to play a regulatory role in proliferation 34, 41 and angiogenesis 40. Accordingly, drugs that target PFKFB3 or hypoxia sensing, which are also currently in development as cancer therapeutics 42–45, should be further studied in the context of atherosclerosis. Moreover, studies should assess whether targeting PFKFB3 or reducing hypoxia sensing within plaques will decrease atherosclerotic inflammation and its clinical consequences. Indeed, in a previous work using macrophages deficient for HIF-1α in the myeloid lineage 5 we were surprised by the observation that glycolysis remained increased and PFKFB3 was also overexpressed, perhaps due to the fact that HIF-2α assumed part of the transcriptional control due to HIF-1α 46. Therefore, the rationale for the use of PFKFB3 inhibitors offers many advantages since this enzyme is only expressed under hypoxic and pro-inflammatory conditions.
The study’s findings also have important implications for imaging of atherosclerosis. While the bulk of prior evidence suggests that pro-inflammatory stimulation is associated with increased glycolysis, one recent study suggested that hypoxia and not pro-inflammatory activation augments macrophage glucose metabolism 14. That report proposed that imaging measures of glucose metabolism (e.g. with labeled 2-deoxyglucose), may provide an indirectly assessment tissue hypoxia in vivo 14. However, the findings of the current study suggest that measures of glycolysis are not well suited for assessing hypoxia. Specifically, we observe, across oxygen tensions, that glycolytic flux remains tightly related to the state of macrophage pro-inflammatory activity, (e.g. TNF-α production, see Fig. 3), thus supporting the use of FDG to image inflammatory cell activity within atheromatous plaques. On the other hand, we found that oxygen tension per se is not a good predictor of the degree of inflammation, since pro-inflammatory activity varies quite markedly within both normoxic and hypoxic conditions, depending on the availability of other pro-inflammatory factors such as oxidized LDL and cytokines (see Fig. 2A). Accordingly, hypoxia-directed probes (such as FMISO), while likely co-localize to sites of inflammation, may not be so useful for reporting on the level of pro-inflammatory activation that exists in those lesions.
The central importance of inflammation to atherosclerosis, warrants pursuit of a deeper understanding of the interconnection between hypoxia, glycolysis and inflammatory activation within an atheroma. This study demonstrates that a surprisingly tight inter-connection exists between macrophage glycolysis and pro-inflammatory activity, that the link requires HIF-1α and transcriptional induction of PFKFB3, and that inhibition of either under pro-inflammatory conditions results in a reduction in inflammatory activity. Hence, modulation of macrophage metabolic adaptation may provide an opportunity to develop novel treatments against atherosclerotic inflammation.
Supplementary Material
SIGNIFICANCE.
The present data show a connection between macrophage viability, activation and oxygen availability that impacts macrophage function in an atherosclerotic environment. This tight inter-connection between macrophage glycolysis and pro-inflammatory activity, requiring HIF-1α and transcriptional induction of PFKFB3, is susceptible to be modulated by pharmacological inhibitors of PFKFB3 (and HIF-1α). Therefore, selective inhibition of glycolytic activity may result in a loss of macrophage function and a potential stabilization of atherosclerotic lesions, with relevance in the pharmacologic management of atherogenesis.
Acknowledgments
Detailed procedures are available in Supplementary Materials. We thank M. Teresa Macías and Diana Hernández from the PET/SPECT service, IIBm (Madrid).
SOURCES OF FUNDING
JHFR is part supported by the Cambridge Biomedical Research Centre, the British Heart Foundation and HEFCE. ZAF is supported by NIH/NHLBI R01 HL071021, R01 HL078667; NIH/NBIB R01 EB009638 and NIH/NHLBI Program of Excellence in Nanotechnology (PEN) Award, Contract #HHSN268201000045C. PS was supported by a grant from the NHLBI (5T32 HL076136). LB was supported by BFU2011-24760, SAF2014-52492, IPT2012-1331-060000 and FIS-RIC RD12/0042/0019 from MINECO and ISCIII, respectively.
Nonstandard abbreviations and acronyms
- 3PO
(3- (3-pyridinyl)-1- (4-pyridinyl)-2-propen-1-one)
- CK
IL-1β+TNF-α+IFN-γ
- SUV
standardized uptake values
- TBR
target-to-background ratio
Footnotes
DISCLOSURES
The authors declare no conflict of interest
References
- 1.Fleischmann W, Kubowitz F. Über den Stoffwechsel der Leukocyten. Boiochem Z. 1927;181:385. [Google Scholar]
- 2.Murray HW, Cohn ZA. Macrophage oxygen-dependent antimicrobial activity. III. Enhanced oxidative metabolism as an expression of macrophage activation. The Journal of experimental medicine. 1980;152:1596–609. doi: 10.1084/jem.152.6.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godfrey RW, Wilder MS. Relationships between oxidative metabolism, macrophage activation, and antilisterial activity. Journal of leukocyte biology. 1984;36:533–43. doi: 10.1002/jlb.36.4.533. [DOI] [PubMed] [Google Scholar]
- 4.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber HP, Ferrara N, Johnson RS. HIF-1alpha is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–57. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, Bosca L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. Journal of immunology. 2010;185:605–14. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 6.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB on behalf of the American Heart Association Statistics C, Stroke Statistics S. Executive Summary: Heart Disease and Stroke Statistics--2013 Update: A Report From the American Heart Association. Circulation. 2013;127:143–152. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 7.Sluimer JC, Daemen MJ. Novel concepts in atherogenesis: angiogenesis and hypoxia in atherosclerosis. The Journal of pathology. 2009;218:7–29. doi: 10.1002/path.2518. [DOI] [PubMed] [Google Scholar]
- 8.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature reviews Immunology. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 10.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: a dynamic balance. Nature reviews Immunology. 2013;13:709–21. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parathath S, Yang Y, Mick S, Fisher EA. Hypoxia in murine atherosclerotic plaques and its adverse effects on macrophages. Trends in cardiovascular medicine. 2013;23:80–4. doi: 10.1016/j.tcm.2012.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramkhelawon B, Yang Y, van Gils JM, Hewing B, Rayner KJ, Parathath S, Guo L, Oldebeken S, Feig JL, Fisher EA, Moore KJ. Hypoxia induces netrin-1 and Unc5b in atherosclerotic plaques: mechanism for macrophage retention and survival. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1180–8. doi: 10.1161/ATVBAHA.112.301008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. The New England journal of medicine. 2011;364:656–65. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Folco EJ, Sheikine Y, Rocha VZ, Christen T, Shvartz E, Sukhova GK, Di Carli MF, Libby P. Hypoxia but not inflammation augments glucose uptake in human macrophages: Implications for imaging atherosclerosis with 18fluorine-labeled 2-deoxy-D-glucose positron emission tomography. Journal of the American College of Cardiology. 2011;58:603–14. doi: 10.1016/j.jacc.2011.03.044. [DOI] [PubMed] [Google Scholar]
- 15.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–55. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fejer G, Wegner MD, Gyory I, Cohen I, Engelhard P, Voronov E, Manke T, Ruzsics Z, Dolken L, Prazeres da Costa O, Branzk N, Huber M, Prasse A, Schneider R, Apte RN, Galanos C, Freudenberg MA. Nontransformed, GM-CSF-dependent macrophage lines are a unique model to study tissue macrophage functions. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:E2191–8. doi: 10.1073/pnas.1302877110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hamze M, Desmetz C, Berthe ML, Roger P, Boulle N, Brancherau P, Picard E, Guzman C, Tolza C, Guglielmi P. Characterization of resident B cells of vascular walls in human atherosclerotic patients. Journal of immunology (Baltimore, Md: 1950) 2013;191:3006–16. doi: 10.4049/jimmunol.1202870. [DOI] [PubMed] [Google Scholar]
- 18.Kim HJ, Oh JS, An SS, Pennant WA, Gwak SJ, Kim AN, Han PK, Yoon DH, Kim KN, Ha Y. Hypoxia-specific GM-CSF-overexpressing neural stem cells improve graft survival and functional recovery in spinal cord injury. Gene therapy. 2012;19:513–21. doi: 10.1038/gt.2011.137. [DOI] [PubMed] [Google Scholar]
- 19.Lacey DC, Achuthan A, Fleetwood AJ, Dinh H, Roiniotis J, Scholz GM, Chang MW, Beckman SK, Cook AD, Hamilton JA. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. Journal of immunology (Baltimore, Md: 1950) 2012;188:5752–65. doi: 10.4049/jimmunol.1103426. [DOI] [PubMed] [Google Scholar]
- 20.Lilly MB, Zemskova M, Frankel AE, Salo J, Kraft AS. Distinct domains of the human granulocyte-macrophage colony-stimulating factor receptor alpha subunit mediate activation of Jak/Stat signaling and differentiation. Blood. 2001;97:1662–70. doi: 10.1182/blood.v97.6.1662. [DOI] [PubMed] [Google Scholar]
- 21.Min L, Isa SA, Fam WN, Sze SK, Beretta O, Mortellaro A, Ruedl C. Synergism between curdlan and GM-CSF confers a strong inflammatory signature to dendritic cells. Journal of immunology (Baltimore, Md: 1950) 2012;188:1789–98. doi: 10.4049/jimmunol.1101755. [DOI] [PubMed] [Google Scholar]
- 22.Roda JM, Sumner LA, Evans R, Phillips GS, Marsh CB, Eubank TD. Hypoxia-inducible factor-2alpha regulates GM-CSF-derived soluble vascular endothelial growth factor receptor 1 production from macrophages and inhibits tumor growth and angiogenesis. Journal of immunology (Baltimore, Md: 1950) 2011;187:1970–6. doi: 10.4049/jimmunol.1100841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sullivan KE, Cutilli J, Piliero LM, Ghavimi-Alagha D, Starr SE, Campbell DE, Douglas SD. Measurement of cytokine secretion, intracellular protein expression, and mRNA in resting and stimulated peripheral blood mononuclear cells. Clinical and diagnostic laboratory immunology. 2000;7:920–4. doi: 10.1128/cdli.7.6.920-924.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rhoades KL, Golub SH, Economou JS. The regulation of the human tumor necrosis factor alpha promoter region in macrophage, T cell, and B cell lines. The Journal of biological chemistry. 1992;267:22102–7. [PubMed] [Google Scholar]
- 25.Castrillo A, Pennington DJ, Otto F, Parker PJ, Owen MJ, Bosca L. Protein kinase Cepsilon is required for macrophage activation and defense against bacterial infection. The Journal of experimental medicine. 2001;194:1231–42. doi: 10.1084/jem.194.9.1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yalcin A, Telang S, Clem B, Chesney J. Regulation of glucose metabolism by 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatases in cancer. Experimental and molecular pathology. 2009;86:174–9. doi: 10.1016/j.yexmp.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Traves PG, de Atauri P, Marin S, Pimentel-Santillana M, Rodriguez-Prados JC, Marin de Mas I, Selivanov VA, Martin-Sanz P, Bosca L, Cascante M. Relevance of the MEK/ERK signaling pathway in the metabolism of activated macrophages: a metabolomic approach. Journal of immunology. 2012;188:1402–10. doi: 10.4049/jimmunol.1101781. [DOI] [PubMed] [Google Scholar]
- 28.Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–6. doi: 10.1126/science.1230719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Clem B, Telang S, Clem A, Yalcin A, Meier J, Simmons A, Rasku MA, Arumugam S, Dean WL, Eaton J, Lane A, Trent JO, Chesney J. Small-molecule inhibition of 6-phosphofructo-2-kinase activity suppresses glycolytic flux and tumor growth. Molecular cancer therapeutics. 2008;7:110–20. doi: 10.1158/1535-7163.MCT-07-0482. [DOI] [PubMed] [Google Scholar]
- 30.Zhdanov AV, Dmitriev RI, Golubeva AV, Gavrilova SA, Papkovsky DB. Chronic hypoxia leads to a glycolytic phenotype and suppressed HIF-2 signaling in PC12 cells. Biochimica et biophysica acta. 2013;1830:3553–69. doi: 10.1016/j.bbagen.2013.02.016. [DOI] [PubMed] [Google Scholar]
- 31.Bando H, Atsumi T, Nishio T, Niwa H, Mishima S, Shimizu C, Yoshioka N, Bucala R, Koike T. Phosphorylation of the 6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase/PFKFB3 family of glycolytic regulators in human cancer. Clin Cancer Res. 2005;11:5784–92. doi: 10.1158/1078-0432.CCR-05-0149. [DOI] [PubMed] [Google Scholar]
- 32.Calvo MN, Bartrons R, Castano E, Perales JC, Navarro-Sabate A, Manzano A. PFKFB3 gene silencing decreases glycolysis, induces cell-cycle delay and inhibits anchorage-independent growth in HeLa cells. FEBS Lett. 2006;580:3308–14. doi: 10.1016/j.febslet.2006.04.093. [DOI] [PubMed] [Google Scholar]
- 33.Obach M, Navarro-Sabate A, Caro J, Kong X, Duran J, Gomez M, Perales JC, Ventura F, Rosa JL, Bartrons R. 6-Phosphofructo-2-kinase (pfkfb3) gene promoter contains hypoxia-inducible factor-1 binding sites necessary for transactivation in response to hypoxia. The Journal of biological chemistry. 2004;279:53562–70. doi: 10.1074/jbc.M406096200. [DOI] [PubMed] [Google Scholar]
- 34.Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. 2013;19:1166–72. doi: 10.1038/nm.3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. The Journal of general physiology. 1927;8:519–30. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Roiniotis J, Dinh H, Masendycz P, Turner A, Elsegood CL, Scholz GM, Hamilton JA. Hypoxia prolongs monocyte/macrophage survival and enhanced glycolysis is associated with their maturation under aerobic conditions. Journal of immunology (Baltimore, Md: 1950) 2009;182:7974–81. doi: 10.4049/jimmunol.0804216. [DOI] [PubMed] [Google Scholar]
- 37.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nature reviews Cancer. 2004;4:891–9. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 38.Peyssonnaux C, Cejudo-Martin P, Doedens A, Zinkernagel AS, Johnson RS, Nizet V. Cutting edge: Essential role of hypoxia inducible factor-1alpha in development of lipopolysaccharide-induced sepsis. Journal of immunology. 2007;178:7516–9. doi: 10.4049/jimmunol.178.12.7516. [DOI] [PubMed] [Google Scholar]
- 39.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–15. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong BW, Cantelmo AR, Quaegebeur A, Ghesquiere B, Cauwenberghs S, Eelen G, Phng LK, Betz I, Tembuyser B, Brepoels K, Welti J, Geudens I, Segura I, Cruys B, Bifari F, Decimo I, Blanco R, Wyns S, Vangindertael J, Rocha S, Collins RT, Munck S, Daelemans D, Imamura H, Devlieger R, Rider M, Van Veldhoven PP, Schuit F, Bartrons R, Hofkens J, Fraisl P, Telang S, Deberardinis RJ, Schoonjans L, Vinckier S, Chesney J, Gerhardt H, Dewerchin M, Carmeliet P. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell. 2013;154:651–63. doi: 10.1016/j.cell.2013.06.037. [DOI] [PubMed] [Google Scholar]
- 41.Colombo SL, Palacios-Callender M, Frakich N, Carcamo S, Kovacs I, Tudzarova S, Moncada S. Molecular basis for the differential use of glucose and glutamine in cell proliferation as revealed by synchronized HeLa cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:21069–74. doi: 10.1073/pnas.1117500108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Akter S, Clem BF, Lee HJ, Chesney J, Bae Y. Block copolymer micelles for controlled delivery of glycolytic enzyme inhibitors. Pharmaceutical research. 2012;29:847–55. doi: 10.1007/s11095-011-0613-4. [DOI] [PubMed] [Google Scholar]
- 43.Clem BF, O’Neal J, Tapolsky G, Clem AL, Imbert-Fernandez Y, Kerr DA, 2nd, Klarer AC, Redman R, Miller DM, Trent JO, Telang S, Chesney J. Targeting 6-phosphofructo-2-kinase (PFKFB3) as a therapeutic strategy against cancer. Molecular cancer therapeutics. 2013;12:1461–70. doi: 10.1158/1535-7163.MCT-13-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Telang S, Clem BF, Klarer AC, Clem AL, Trent JO, Bucala R, Chesney J. Small molecule inhibition of 6-phosphofructo-2-kinase suppresses t cell activation. Journal of translational medicine. 2012;10:95. doi: 10.1186/1479-5876-10-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miranda E, Nordgren IK, Male AL, Lawrence CE, Hoakwie F, Cuda F, Court W, Fox KR, Townsend PA, Packham GK, Eccles SA, Tavassoli A. A cyclic peptide inhibitor of HIF-1 heterodimerization that inhibits hypoxia signaling in cancer cells. Journal of the American Chemical Society. 2013;135:10418–25. doi: 10.1021/ja402993u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Takeda N, O’Dea EL, Doedens A, Kim JW, Weidemann A, Stockmann C, Asagiri M, Simon MC, Hoffmann A, Johnson RS. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.