

Abstract
Objective
Esophageal adenocarcinoma (EAC) is associated with a dismal prognosis. The identification of cancer biomarkers advances the possibility for early detection and better monitoring of tumor progression and/or response to therapy. The current study presents results of the development of a serum based four-protein (biglycan, myeloperoxidase, annexin-A6, and protein S100-A9) biomarker-panel for EAC.
Design
A vertically integrated proteomics-based biomarker discovery approach was used to identify candidate serum biomarkers for detection of EAC. Liquid chromatography-mass spectrometry (LC-MS/MS) analysis was performed on FFPE tissue samples that were collected from across the Barrett's esophagus (BE)-EAC disease spectrum. The MS-based spectral count data was used to guide the selection of candidate serum biomarkers. The serum ELISA data was validated in an independent cohort and used to develop a multi-parametric risk assessment model to predict the presence of disease.
Results
With a minimum threshold of 10 spectral counts, 351 proteins were identified as differentially abundant along the spectrum of BE, HGD and EAC (p < 0.05). Eleven proteins from this dataset were then tested using ELISAs in serum samples of which five proteins were significantly elevated in abundance in the EAC patients compared to normal controls, which mirrored trends across the disease spectrum present in the tissue data. Using serum data a Bayesian Rule Learning predictive model with four biomarkers was developed to accurately classify disease class; the cross-validation results for the merged dataset yielded accuracy of 87% and AUROC of 93 %.
Conclusion
Serum biomarkers hold significant promise for early non-invasive detection of EAC.
Introduction
The incidence of esophageal adenocarcinoma (EAC) is rapidly rising; outpacing the rate of increase of all other cancers. The number of patients affected per year is up to 600% higher than in the 1970s'.1, 2 Additionally, EAC is associated with a dismal prognosis, with a five-year survival of less than 15%. Although survival and prognosis depend on the stage of the disease, unfortunately, because the esophagus is a distensible organ, the majority of patients who develop EAC do not sense difficulty swallowing until the tumor is advanced.3 Accordingly, there is an urgent need for improved risk stratification to facilitate early detection and thereby reducing the mortality due to EAC.4
Currently without clinical risk factors that signal the early development of EAC, identification of early-stage and curable disease is only possible through endoscopic Barrett's esophagus (BE) screening in patients with symptoms of gastroesophageal reflux disease (GERD).5, 6 Those diagnosed with BE then typically undergo lifetime endoscopic surveillance for the development of malignancy.7 However, 95% of patients who develop EAC have never undergone BE screening prior to cancer diagnosis and up to 57% of patients who develop EAC do not report antecedent GERD symptoms.8, 9
The identification of cancer biomarkers raises the possibility for early detection, better monitoring of tumor progression and/or response to therapy. Protein biomarkers that have been identified and are in regular clinical use for different tumors include carcinoembryonic antigen (CEA), prostate specific antigen (PSA), alpha-fetoprotein (AFP) and cancer antigen 125 (CA-125). Development of biomarkers is even more important for cancers like EAC that are typically diagnosed at advanced stages of disease and have poor long-term survival rates with the currently employed clinical management paradigm.10
Towards this goal, the current study presents results of a serum based four-protein biomarker-panel for EAC [B-AMP© - comprising biglycan, annexin-A6 (ANXA6), myeloperoxidase (MPO) and protein S100-A9]; that were identified using a vertically integrated proteomics-based biomarker discovery approach to initially identify candidate tissue and then serum biomarkers. These proteins were noted to be clinically relevant and followed a distinct pattern of expression along the sequence of disease progression. These data were subsequently used to develop a multi-parametric risk assessment model to predict the presence of disease.
Materials and Methods
Figure 1 outlines the overall study schema with the patient populations and methods used. The study was initiated in 2009 and completed in 2013. Institutional Review Board approval was obtained prior to the initiation of the study and informed consent was obtained at the time of tissue collection.
Figure 1.
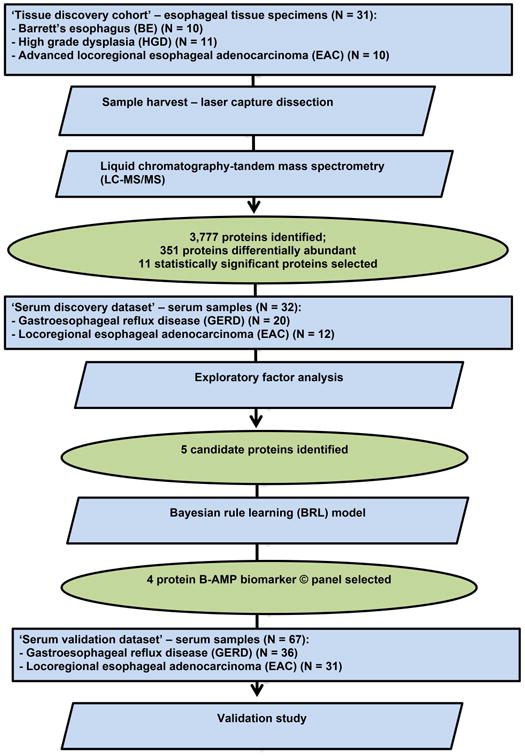
Study schema with the patient populations and methods used. As noted, serum protein biomarker discovery was guided by tissue-based proteomics followed by analysis and evaluation in serum samples using ELISAs.
Proteomic biomarker identification from Barrett's esophagus (BE), high-grade dysplasia (HGD) and esophageal adenocarcinoma (EAC) tissues
To identify candidate protein biomarkers associated with disease progression, we performed a mass spectrometry based proteomics discovery study using appropriate pathologically-defined esophageal tissue specimens. The tissue discovery data was generated from archival de-identified formalin-fixed, paraffin-embedded (FFPE) blocks obtained from the Department of Pathology at the University of Pittsburgh. This cohort consisted of 10 BE, 11 HGD and 10 advanced locoregional EAC unpaired patient samples (‘Tissue discovery cohort’- Figure 1).
A single-tube experimental protocol was used to digest proteins from FFPE tissue sections with trypsin and the resultant tryptic peptides were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for an exploratory proteomic analysis of BE, HGD and EAC. Approximately 40,000 cells per sample were collected by laser capture micro dissection (LCM); the tryptic digests were analyzed in duplicate by nanoflow LC-MS/MS using a hybrid linear ion trap-Orbitrap® mass spectrometer (MS). The primary tandem MS/MS data were searched with SEQUEST® against the human proteome database for peptide identification and against a “decoy” human proteome database where the protein sequences are reversed to maintain a false discovery rate of less than 1%.11 We then integrated the resulting peptide lists using a suite of in-house MATLAB® enabled relational database tools yielding spectral counts for identified proteins.
A quantitative estimate of the relative abundance of the identified proteins from these data sets was obtained by comparison of their spectral count values between BE-, HGD-, and EAC-derived cells. To determine statistically significant differentially abundant proteins from each tissue type, we applied a Kruskal-Wallis non-parametric analysis of variance test (Kruskal-Wallis test is a one-way analysis of variance by ranks and determines statistically significant differences between two or more groups of an independent variable on a continuous or ordinal dependent variable); proteins with significant differences were utilized for further hierarchical clustering analysis.
Digestion of laser capture microdissected (LCM) FFPE tissues
Heat-induced trypsin digestion was applied to the LCM cells to extract peptides as described.12 Samples were resuspended in 100μl of 100mM NH4HCO3/20% acetonitrile and then heated at 90°C for 1 hour, followed by 65°C for 2 hours. Trypsin digestion was carried out by adding 500ng sequencing grade modified trypsin (Promega, Madison, WI) followed by overnight incubation at 37°C. After a rapid spin, the aqueous solution was transferred to a new Eppendorf tube, lyophilized and then resuspended in 100μl 0.1% trifluoroacetic acid (TFA), followed by desalting with PepClean C-18 Spin Columns (PIERCE, Rockford, IL), vacuum-dried and resuspended in 25μl 0.1% TFA. The BCA assay (PIERCE, Rockford, IL) was used to determine peptide concentration.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis of peptides
The tryptic digests were analyzed in duplicate (1μg for each injection) by reverse-phase LC-MS/MS using a nanoflow LC (Dionex Ultimate 3000, Dionex Corporation, Sunnyvale, CA) coupled online to an LTQ/Orbitrap XL hybrid mass spectrometer (ThermoFisher Scientific Inc., San Jose, CA). Peptide separation was performed using 75μm inner diameter × 360μm outer diameter × 20cm long fused silica capillary columns (Polymicro Technologies, Phoenix, AZ) slurry packed in house with 5μm, 300Å pore size C-18 silica-bonded stationary phase (Jupiter, Phenomenex, Torrance, CA). Following sample injection onto a C-18 trap column (Dionex), the column was washed for 3min with mobile phase A (2% acetonitrile, 0.1% formic acid) at a flow rate of 30μl/min. Peptides were eluted using a linear gradient of 0.30% mobile phase B (0.1% formic acid in acetonitrile)/minute for 130 min, then to 95% B in an additional 10 min, all at a constant flow rate of 250nl/min. Column washing was performed at 95% B for 20min, after which the column was re-equilibrated in mobile phase A prior to subsequent injections. The LTQ/Orbitrap XL MS was configured to collect high resolution (R = 60,000 at m/z 400) broadband mass spectra (m/z 375-1800) from which the 7 most abundant peptide molecular ions dynamically determined from the MS scan were selected for MS/MS using a 30% normalized collision-induced dissociation energy. Dynamic exclusion was utilized to minimize redundant selection of peptides for MS/MS analysis.
MS and MS/MS data analysis
For relative quantitation using spectral count, tandem mass spectra were searched against the UniProt human proteome database (06/2009 release) from the European Bioinformatics Institute (http://www.ebi.ac.uk/integr8) using SEQUEST (ThermoFisher Scientific Inc.) with variable modification of methionine (oxidation, +15.9949 Da). The mass tolerance for the precursor ions and fragment ions were set to 20 ppm and 1 Da respectively. Peptides were considered legitimately identified if they achieved specific charge state and proteolytic cleavage-dependent cross-correlation (Xcorr) scores of 1.9 for [M+H]1+, 2.2 for [M+2H]2+, and 3.5 for [M+3H]3+, and a minimum delta correlation score (ΔCn) of 0.08. An in-house MATLAB® script was used to combine the total number of CID spectra that resulted in positive identification of any peptides for a given protein (spectral count).
Hierarchical clustering was carried out using MATLAB®. The values for spectral counts were standardized for each protein so that each had a mean of 0 and a standard deviation of 1. Both sample distance and protein feature distance were calculated using Pearson’s correlation and average linkage was used for the clustering of both samples and protein features.
Patient study populations and serum sample collection, processing and storage
For initial evaluation of the abundances of the candidate protein biomarkers in serum, a ‘serum discovery dataset’ of samples was collected from a total of 32 patients: 20 with a clinical diagnosis of gastroesophageal reflux disease (GERD) and 12 with advanced loco-regional EAC (T2N1 to T3N0) from the Esophageal Risk Registry at the University of Pittsburgh in 2010.
For this purpose, venous blood samples (4 ml) from normal control GERD and EAC patients were drawn using standard venipuncture into red/yellow top Vacuette® Serum Clot Activator with Gel Separator Blood Collection Tubes (Greiner-Bio-One #454067, Monroe, NC, USA) and placed upright 30 to 60 minutes until clot formation. The tubes were centrifuged in a swinging bucket rotor (1,300 g × 20 min) and the serum was pipetted and distributed as 200 μl aliquots in 1.5 ml cryovials for storage at -80°C. To ensure consistency and reliability in the subsequent analyses, no more than one freeze–thaw cycle was allowed for any sample.
ELISA testing for the candidate serum protein biomarker panel
Commercial ELISA kits were used to quantitate the abundance of the candidate biomarkers in serum (Supplementary Table 1). The following ELISA kits were used: alpha-1-antitrypsin (A1A), myeloperoxidase (MPO), apolipoprotein A-I (APO A1) (Immunology Consultants Laboratory, Inc., #E-80A, # E-80PX, E80AP1, Portland, OR), resistin (R and D Systems, #DRSN00, Minneapolis, MN), isoform 1 of fibronectin (Bioxys, #EF1045, Brussels, Belgium), lymphocyte cytosolic protein 1 (LCP1) (Antibodies-online.com, #ABIN415176, Atlanta GA), cathespin B (USCNK, #SEC964Hu Houston, TX), protein S-100A9/MRP14, biglycan, annexin A6, (CedarLane #CY-8062, SE9822HU, SE92345HU, Burlington, NC) and cellular fibronectin (Biohit #6030010, Helsinki, Finland). Briefly, each assay comprised a single-plex sandwich ELISA with primary antibody specific for the selected protein pre-coated in planar arrays in 96-well microtiter plates. After serum incubation and washing, a second biotinylated antibody to a different site on the protein from the capture epitope was introduced and streptavidin-horseradish peroxidase (HRP) subsequently bound to the biotinylated detection antibody. Chromagen-substrate reagent was added and the absorbance (OD) was read according to manufacturer instructions on a SpectraMax M2e plate reader (Molecular Devices, Sunnyvale, CA) The OD values were acquired and processed using a four-parameter curve fit to compare the experimental samples to the recombinant protein calibration curve run in parallel wells to derive absolute protein concentrations adjusted for dilution.
Validation study of the selected serum biomarkers
A second stage study of 67 independent serum samples (‘serum validation dataset’) from 36 non-BE GERD and 31 EAC patients from RPCI and AHN was run in order to validate the previous discovery findings from the 32 sample ‘serum discovery dataset’ from the University of Pittsburgh patients. The sample preparation and quality control (QC) methodological protocols and serum ELISAs were performed as before for the ‘serum discovery dataset’ using the final five candidate biomarkers that demonstrated statistical discrimination between the 2 patient groups in the initial dataset.
Development of a serum biomarker panel and predictive model for EAC
Serum ELISA data from the discovery and validation datasets were subsequently used to develop and test predictive biomarker rule models using a new bioinformatics method called the Bayesian Rule Learning (BRL) system.13 The BRL is a set of classification algorithms that we have previously successfully applied to biomarker discovery and validation from serum proteomic datasets for early detection of ALS and lung cancer.14, 15
A rule model consists of a set of IF-THEN rules, as an example:
| IF (BGN > 245 μg/ml) AND (S100A9 > 3 ng/ml) AND (MPO > 120 ng/ml) |
| THEN (Class = EAC) |
| Posterior Probability=0.917, P=0.0, TP=21, FP=1 |
This rule states that if a patient sample has biomarkers BGN, S100-A9, and MPO with serum levels greater than 245 μg/ml, 3 ng/ml, and 120 ng/ml, respectively, (defined in the IF part of the rule) then the patient has EAC (defined in the THEN part of the rule). The posterior probability gives the probability of a true positive for EAC, given all positive matches from the rule. The p-value (P) of the rule is obtained from Fisher's exact test. Fisher's exact test16 is a significance test appropriate for categorical count data such as number of true positives and number of positives corresponding to each rule.
The BRL system first learns a Bayesian network (BN)17 constrained to the target node (EAC or non-EAC), and subsequently biomarkers are added as potential parents to this node. It learns the BN from the training data and evaluates it using an extension of the K2 score13, 18 assuming all models are equally probable a priori (uniform prior distribution). The details of the BRL algorithms are presented in Lustgarten, 2009. 13, 18, 19
Since the BRL method can handle only discrete variables, we discretized the continuous-valued ELISA data for each biomarker into a small number of intervals using a method called Efficient Bayesian Discretization (EBD) that we have developed.20 For each biomarker, EBD identifies a small number of intervals in the range of values for that biomarker that is optimal in terms of a Bayesian measure (based on the K2 score18). Using EBD to discretize variables has been shown to yield better classification performance on a range of biomedical datasets.21
We generated predictive rule models from the ‘discovery dataset’ and applied them to the ‘validation dataset’, using different values for the user-defined λ parameter, which is the mean of a Poisson distribution that represents the expected number of cut-points between the ranges of continuous values for each biomarker. We found that, λ values of 0.5 and 1 yielded models with the highest predictive accuracies. In order to use the validation data as a test set for predictive rule models, it was first necessary to normalize the quantities for each biomarker in the discovery and validation datasets together using Equation 1.
| (1) |
Here, F is the factor of normalization computed for each biomarker, N+ and N- refer to the total number of cases and controls in each of the datasets: training (DT) and test (DV), respectively. The variables p and q iterate over instances with a specific class value (EAC or non-EAC) in the training dataset (p) and validation dataset (q), respectively.
With the normalized dataset values as determined above, we generated predictive rules from the discovery and validation datasets, using each one independently as the training dataset and the test dataset, respectively. We further appended the discovery and validation datasets to create a merged dataset to which we then applied ten-fold stratified cross validation. Herein, we randomized and divided the combined dataset into ten almost equal portions. We then learned a predictive model from 9 portions of the data, designated as training data, and tested the remaining set-aside portion. This was done ten times by applying BRL to learn a predictive model over each fold and test that model to obtain performance metrics. Finally, the average accuracy, balanced accuracy (average of sensitivity and specificity) (BACC) and area under the ROC (AUROC) metrics are reported over this ten-fold cross fold validation. To develop the final predictive model that we report in this manuscript, we applied BRL to the complete merged dataset.
Sample size and statistical analysis
The serum validation study required 26 patients per group for an anticipated effect size of 0.8 with a calculated study power of 80% and a target alpha of 0.05. Statistical analyses were performed using SPSS software (IBM, Armonk, NY, Version 20). A p-value <0.05 was considered statistically significant.
Results
Tissue based LC-MS/MS proteomics biomarker identification
A total of 3,777 proteins were identified from 62 LC-MS/MS analyses (duplicate analyses for each of the 31 tissue samples). The range of total spectral counts obtained in each sample analysis ranged from 2,759 to 5,181 and was significantly associated with the patient groups (Kruskal-Wallis test, p=0.0364). With a minimum threshold of 10 spectral counts, 351 proteins were observed to be differentially abundant along the spectrum of BE, HGD and EAC (Kruskal – Wallis test, p<0.05) (Supplementary Table 1; Figure 2). These results showed nearly perfect clustering of relative protein abundance from BE to EAC (Figure 2).
Figure 2.
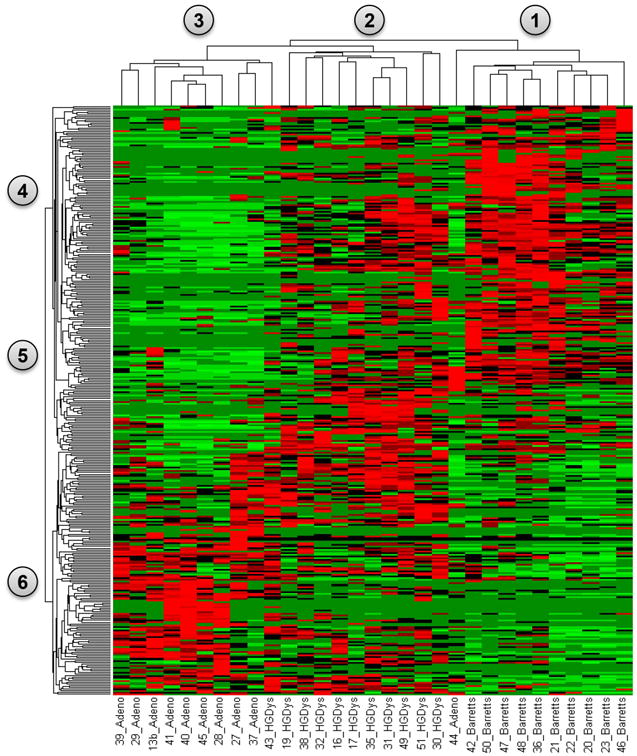
A heat map representation from a supervised clustering analysis of significantly differentially abundant proteins identified from BE in column grouping 1 (n=10), HGD in column grouping 2 (n=11) and EAC tissues in column grouping 3 (n=10). Individually significant proteins are represented in row groups 4, 5 and 6. Protein abundances are plotted as mean observed spectral counts for each tissue type where red represents proteins with a normalized spectral count value greater than 1.5 and green represents values less than 1.5. Significance was determined by the Kruskal-Wallis test. The results demonstrate clear patterns of protein abundance that can be seen correlating with BE (nodes 1&4), HGD (nodes 2&5) and EAC (nodes 3&6).
B-AMP© biomarker ELISA
Eleven of these 351 differentially abundant proteins were selected for evaluation in serum using ELISAs on discovery sample sets based on their functional relevance and availability of commercial ELISA kits. The serum ELISA results obtained from these selected tissue-based candidate biomarkers demonstrated significantly elevated serum levels for five of the eleven proteins tested in the samples from EAC patients compared to the samples collected from the non-BE GERD patients in the ‘serum discovery dataset’ (Figure 3). These included ANXA6, BGN, S100A9, MPO and resistin. The serum levels followed similar trends across the disease spectrum as observed in the corresponding tissue samples (Table 1; Figure 1).
Figure 3.
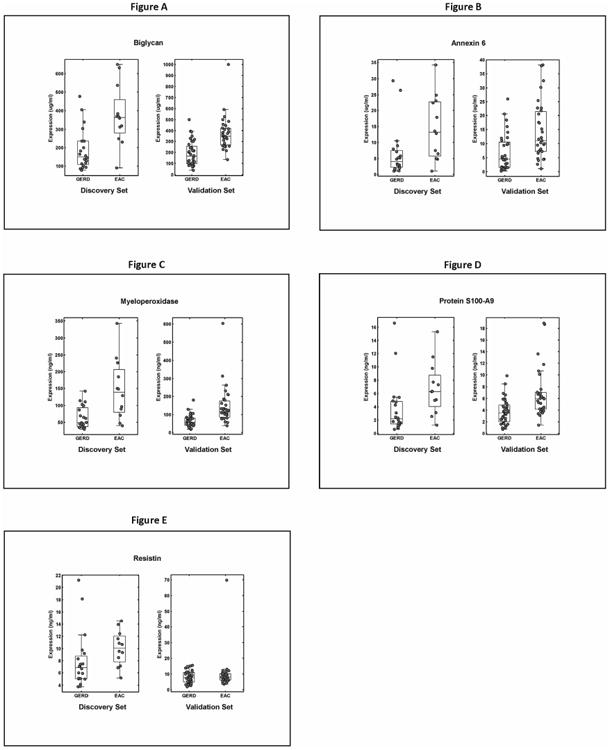
Boxplot distributions (A-E) of candidate protein abundance in serum samples of EAC vs. GERD patients using ELISAs. Scatter plots are overlaid on top of the box plots to visualize the individual data points [ANXA6 (dilution factor (df) =1600×), BGN (df=200×), S100A9 (df=25×), MPO (df=10×) and resistin (df=5×). For each candidate serum biomarker, on the left are results from the discovery set comprising 20 GERD and 12 EAC samples and on the right are results from the validation set consisting of 36 GERD and 31 EAC samples, respectively. The bottom and top of the boxplots indicate the first and third quartiles of the data, the line within each boxplot is the median value. The length of the boxplot whiskers is specified as 1.5 times the (25thto 75th) interquartile range of the data. For the candidate biomarkers, t-test was used to compare the means of EAC vs. GERD in each set, with a p value < 0.05 considered significant. All markers except resistin were significant.
Table 1.
Correlation of tissue expression (upregulated proteins represented by red signal in Group 6 of Figure 2) determined by LC-MS/MS and spectral counting with serum abundance by ELISA for the final five candidate protein biomarkers.
| SERUM RESULTS (UNNORMALIZED MEAN) | TISSUE RESULTS (MEAN SPECTRAL COUNTS) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| PROTEIN | GERD | EAC | Unit | pValue (Ttest) | pValue (Rank) | BE | HGD | EAC | pValue |
| MYELOPEROXIDASE | 64.36 | 147.23 | ng/ml | 0.00073 | 0.00256 | 0.1 | 3.18 | 7.7 | 0.01257 |
| RESISTIN | 7.93 | 10.05 | ng/ml | 0.15967 | 0.01852 | 0 | 0 | 0.8 | 0.0094 |
| PROTEIN S100-A9 | 3.74 | 6.77 | ng/ml | 0.04708 | 0.00964 | 4 | 6.36 | 12.2 | 0.03244 |
| BIGLYCAN | 190.11 | 375.24 | μg/ml | 0.00065 | 0.00256 | 0 | 0.36 | 1.1 | 0.01151 |
| ANNEXIN A6 | 6.64 | 14.48 | μg/ml | 0.01956 | 0.02278 | 0.7 | 1.45 | 6.4 | 0.00265 |
Table 1 summarizes the observed differences in candidate biomarker protein abundance measured by LC-MS/MS and spectral counting along the disease spectrum in FFPE-derived tissue samples and their corresponding concentrations in the serum samples determined by ELISA. Largely consistent with the results in the ‘serum discovery set’, the concentrations of all of the biomarkers were significantly higher in the EAC patients' samples in the ‘serum validation set’ with the exception of resistin (Figure 3).
Final rule model
The rule model that was obtained by applying BRL to the merged discovery and validation ELISA datasets is shown visually in Figure 4. In the tree, the interior nodes (shown as ellipses) represent predictor biomarkers, the leaf nodes (shown as rectangles) show the patient counts for the number of EAC cases and controls respectively, and the labels on the arcs represent the serum biomarker levels. The rules which consist of combinations of individual biomarkers at specific cutoff concentrations produced by BRL are shown below (Table 2). Each rule has a posterior probability associated with it, along with the p-value (P) obtained from Fisher's exact test.13, 16 Fisher's exact test is applicable in situations wherein the number of samples is fairly small, as in our case, which leads to small numbers of counts for positives covered by a rule. The number of true positives (TP) and false positives (FP) covered by the rule are also presented. This set of rules constitutes the predictive model that can be applied to a future patient for whom these serum biomarker levels have been measured and we want to use the matching rule from among this set of mutually exclusive and exhaustive rules to provide an estimate of the probability that the patient has EAC.
Figure 4.
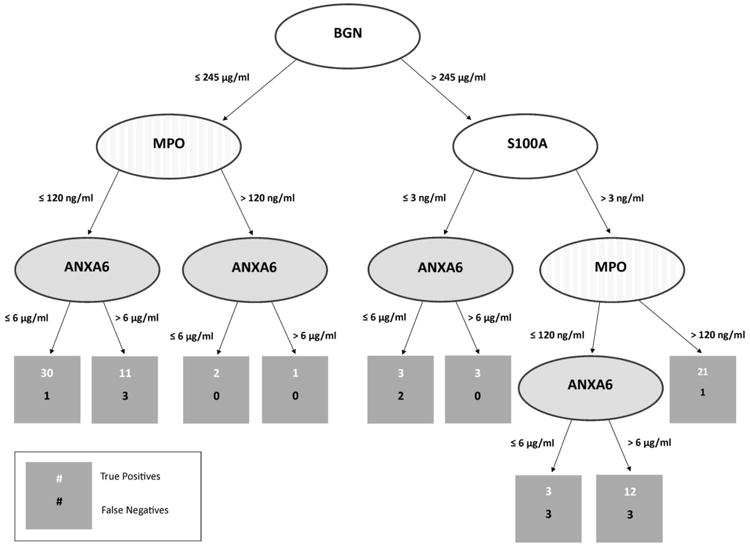
Visual representation of the final BRL rule model derived from the merged serum ELISA data.
Table 2.
| 1. IF (BGN ≤ 245 μg/ml)&(MPO ≤ 120 ng/ml)&(ANXA6 ≤ 6 μg/ml) THEN (Class = Normal) |
| Posterior Probability=0.939, P=0.0, TP=30, FP=1 |
| 2. IF (BGN ≤ 245 μg/ml)&(MPO ≤ 120 ng/ml)&(ANXA6 > 6 μg/ml) THEN (Class = Normal) |
| Posterior Probability=0.75, P=0.064, TP=11, FP=3 |
| 3. IF (BGN ≤ 245 μg/ml)&(MPO > 120 ng/ml)&(ANXA6 ≤ 6 μg/ml) THEN (Class = Normal) |
| Posterior Probability=0.75, P=0.317, TP=2, FP=0 |
| 4. IF (BGN ≤ 245 μg/ml)&(MPO > 120 ng/ml)&(ANXA6 > 6 μg/ml) THEN (Class = Cancer) |
| Posterior Probability=0.667, P=0.434, TP=1, FP=0 |
| 5. IF (BGN > 245 μg/ml)&(S100A9 ≤ 3 ng/ml)&(ANXA6 ≤ 6 μg/ml) THEN (Class = Normal) |
| Posterior Probability=0.571, P=0.624, TP=3, FP=2 |
| 6. IF (BGN > 245 μg/ml)&(S100A9 ≤ 3 ng/ml)&(ANXA6 > 6 μg/ml) THEN (Class = Normal) |
| Posterior Probability=0.8, P=0.177, TP=3, FP=0 |
| 7. IF (BGN > 245 μg/ml)&(S100A9 > 3 ng/ml)&(MPO ≤ 120 ng/ml)&(ANXA6 ≤ 6 μg/ml) |
| THEN (Class = Normal) |
| Posterior Probability=0.5, P=0.777, TP=3, FP=3 |
| 8. IF (BGN > 245 μg/ml)&(S100A9 > 3 ng/ml)&(MPO ≤ 120 ng/ml)&(ANXA6 > 6 μg/ml) |
| THEN (Class = Cancer) |
| Posterior Probability=0.765, P=0.002, TP=12, FP=3 |
| 9. IF (BGN > 245 μg/ml)& (S100A9 > 3 ng/ml)&(MPO > 120 ng/ml) THEN (Class = Cancer) |
| Posterior Probability=0.917, P=0.0, TP=21, FP=1 |
Serum protein biomarkers used (4): S100A9, ANXA6, BGN, MPO
Evaluation of alternative rule models
The highest accuracy that we obtained when a BRL predictive model was learned using the ‘discovery dataset’ and applied to the ‘validation dataset’ was 76%, with a balanced accuracy (BACC) of 74% and the area under the receiver operating characteristic curve (AUROC) of 86%. The highest accuracy that we obtained when a BRL predictive model was learned using the ‘validation dataset’ and applied to the ‘discovery dataset’ was 75%, with a BACC of 73% and AUROC of 84%.
These two reciprocal results indicate that BRL is accurate in modeling the uncertainty in the validity of the rule models because of the similar results obtained in these two independent datasets for EAC classification. The cross-validation results for the merged discovery and validation datasets analyzed by BRL yielded an overall accuracy of 87%, BACC of 86% and AUROC of 93% (Figure 5).
Figure 5.
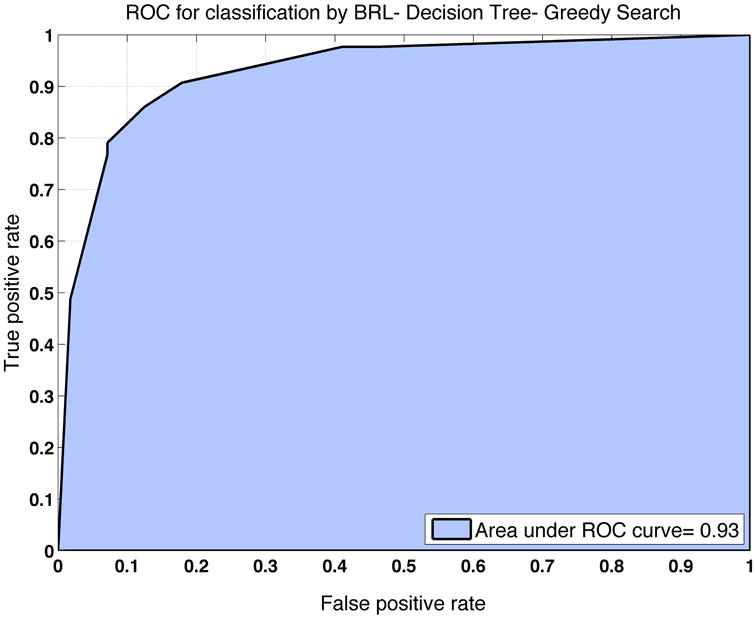
ROC curve generated from ten-fold cross fold validation on the merged datasets to estimate the classification performance of the final BRL predictive model.
Discussion
Serum biomarkers hold significant promise for early non-invasive detection of EAC. However, direct identification of novel biomarkers from serum presents a challenging analytical problem due to the very high dynamic range of protein concentrations present in the complex serum proteome.22 Our study therefore utilized a LC-MS/MS-based tissue proteomics discovery approach to guide the selection of candidate serum biomarkers that were significantly and differentially abundant in the tissue samples along the disease progression pathway from BE to EAC and have clinical and functional relevance.
As summarized in Table 1, these results demonstrate that the observed differences in abundance in the selected proteins along the BE-HGD-EAC disease spectrum in FFPE-derived tissue samples were mirrored by the corresponding protein biomarker concentrations in the corresponding serum samples. The tissue-based results guided targeted serum based biomarker discovery; and with the ease of serum sample collection and relatively low cost, our B-AMP© panel and rule model was able to identify patients with EAC with an overall accuracy of 87%. Of note, even though there were five biomarkers originally noted to be elevated including resistin; the final BRL model excludes it as resistin appears to not add any predictive value to the best scoring models based on the other four biomarkers.
Over the years, a number of biological, IHC- and transcriptomic tissue-based analyses have been performed toward identification of biomarkers of neoplastic progression of BE. As expected, there are many reports citing aberrant biological processes that occur in the development of EAC, such as cell cycle abnormalities and numerous genetic and epigenetic alterations, including loss of heterozygosity, polyploidy and aneuploidy. Although proteins such as TP53 (p53) 23, 24, β-catenin (CTNNB1) 25, p16 24, 26, and cyclin D1 (CCND1)27, have been studied as potential tissue-based immunohistochemical biomarkers of progression 28, none have resulted in widespread clinical adoption or demonstrated adequate clinical utility likely due to the genetic heterogeneity of EAC between patients. Based on these results, it is apparent that a multiple protein panel approach combined with mathematical modeling may offset some of the poor sensitivity associated with a single biomarker “up” or “down” approach.
With respect to the biomarkers identified in our panel, biglycan (BGN) (an extracellular matrix component with a known role in epithelial-to-mesenchymal transdifferentiation central to BE carcinogenesis), annexin A6 (ANXA6) (belonging to the annexin family of calcium and phospholipid binding proteins that is a motility promoting factor), myeloperoxidase (MPO) (an oxidant generating enzyme linked to cancer progression) and protein S100A9 (that promotes tumor growth in inflammation associated cancer development) have been noted to have prognostic significance in several tumor types including esophageal squamous cell carcinoma.29-33
As described earlier, EAC is genetically and phenotypically heterogeneous malignancy and we certainly would be cautious in claiming that these 4 biomarkers will capture all EAC. However, the classification performance of the panel in the 2 clinically independent patient groups does suggest however that it captures the majority of these cases representing the downstream patterns of protein expression of common upstream genetic changes altering these key pathways.
Published studies focused on serum protein biomarkers of EAC, however, are limited to an early report of elevated levels of the squamous cell carcinoma antigen (SCC), carcinoembryonic antigen (CEA), and cytokeratin 19-fragment (CYFRA 21-1) in advanced esophageal cancer patients 34 and more recent evaluations of the circulating lymphocyte antigen 6 complex locus K (LY6K) and elevated serum levels of serum gastrin in patients with a diagnosis of HGD or EAC.35, 36
Similar to our study, tissue- and serum-based protein discovery and proteomic studies have been reported. A 2007 comparative mass spectrometry proteomics analysis identified candidate tissue proteins in surgical specimens that by hierarchical clustering analysis accurately discriminated BE and EAC and identified 38 differentially abundant proteins among which Rho GDP dissociation inhibitor 2, α-enolase, lamin A/C, elongation factor Tu, thioredoxin domain-containing protein 17 and nucleoside-diphosphate kinase A that were noted to be upregulated in both mRNA and protein expression in EAC compared to BE.37 Several of the these proteins or their isoforms along with previously reported progression related proteins were also differentially abundant in our tissue discovery dataset, these included CTNNB1, Rho GDP dissociation inhibitor 2, elongation factor Tu and thioredoxin domain-containing protein 17.
In a very recent methods publication as well, as noted in our study, tissue-based studies have been extended to demonstrate the feasibility of using LCM and LC-MS/MS analysis of esophageal tissue biopsy specimens for robust proteomic analysis.38
Our study's main weakness is the small numbers of samples available for EAC case-control discrimination. However, we were able to demonstrate the ability of rule learning methods to successfully predict class values accurately using two independent datasets with similar distributions of cases and controls. We present a predictive model learned from the merged dataset that needs to be validated in a larger prospective patient cohort.
The next logical steps, thus, would be to evaluate the classification performance our B-AMP© serum biomarker panel in other larger recently reported case-control patient groups and to test it prospectively to detect and/or monitor progression of EAC. This evaluation follow-up study would necessarily include the measurement of the levels of these serum biomarkers in samples from patients with other solid organ epithelial malignancies as well as clinically relevant non-malignant conditions to evaluate EAC specificity.
Application of the serum-based proteomic biomarker panels and risk prediction models like ours could be extended to other studies to help determine the presence or absence of BE, HGD or EAC. Potential advantages would include: (1) improved clinical resource utilization using blood-based detection as a means of directing effective screening and post-therapy surveillance; (2) detect progression from BE to HGD or EAC in patients undergoing surveillance; (3) prevent death from EAC by employing an assay in high-risk patients to identify BE, HGD and EAC; (4) track therapeutic response and thereby enable tailored therapy based on individual disease biology; and (5) detect subclinical EAC recurrence prior to the development of recurrence as detected by clinical imaging.
Supplementary Material
Acknowledgments
Funding: Partial support award P30CA047904 and R01-LM010950 from NIH
David E. Gold & Irene Blumenkranz and David Scaife Foundations.
Footnotes
Financial disclosure: Nothing to disclose
References
- 1.Dubecz A, Solymosi N, Stadlhuber RJ, Schweigert M, Stein HJ, Peters JH. Does the Incidence of Adenocarcinoma of the Esophagus and Gastric Cardia Continue to Rise in the Twenty-First Century?-a SEER Database Analysis. J Gastrointest Surg. 2013 doi: 10.1007/s11605-013-2345-8. [DOI] [PubMed] [Google Scholar]
- 2.Prasad GA, Bansal A, Sharma P, Wang KK. Predictors of progression in Barrett's esophagus: current knowledge and future directions. Am J Gastroenterol. 2010;105(7):1490–502. doi: 10.1038/ajg.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y. Epidemiology of esophageal cancer. World J Gastroenterol. 2013;19(34):5598–606. doi: 10.3748/wjg.v19.i34.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Spechler SJ. Barrett esophagus and risk of esophageal cancer: a clinical review. JAMA. 2013;310(6):627–36. doi: 10.1001/jama.2013.226450. [DOI] [PubMed] [Google Scholar]
- 5.Spechler SJ. Screening and surveillance for complications related to gastroesophageal reflux disease. Am J Med. 2001;111(Suppl 8A):130S–36S. doi: 10.1016/s0002-9343(01)00851-8. [DOI] [PubMed] [Google Scholar]
- 6.Sampliner RE. Practice guidelines on the diagnosis, surveillance, and therapy of Barrett's esophagus. The Practice Parameters Committee of the American College of Gastroenterology. Am J Gastroenterol. 1998;93(7):1028–32. doi: 10.1111/j.1572-0241.1998.00362.x. [DOI] [PubMed] [Google Scholar]
- 7.Sampliner RE Practice Parameters Committee of the American College of G. Updated guidelines for the diagnosis, surveillance, and therapy of Barrett's esophagus. Am J Gastroenterol. 2002;97(8):1888–95. doi: 10.1111/j.1572-0241.2002.05910.x. [DOI] [PubMed] [Google Scholar]
- 8.Conio M, Cameron AJ, Romero Y, et al. Secular trends in the epidemiology and outcome of Barrett's oesophagus in Olmsted County, Minnesota. Gut. 2001;48(3):304–9. doi: 10.1136/gut.48.3.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reavis KM, Morris CD, Gopal DV, Hunter JG, Jobe BA. Laryngopharyngeal reflux symptoms better predict the presence of esophageal adenocarcinoma than typical gastroesophageal reflux symptoms. Ann Surg. 2004;239(6):849–56. doi: 10.1097/01.sla.0000128303.05898.ee. discussion 56-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reid BJ, Prevo LJ, Galipeau PC, et al. Predictors of progression in Barrett's esophagus II: baseline 17p (p53) loss of heterozygosity identifies a patient subset at increased risk for neoplastic progression. Am J Gastroenterol. 2001;96(10):2839–48. doi: 10.1111/j.1572-0241.2001.04236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207–14. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 12.Teng PN, Rungruang BJ, Hood BL, et al. Assessment of buffer systems for harvesting proteins from tissue interstitial fluid for proteomic analysis. J Proteome Res. 2010;9(8):4161–9. doi: 10.1021/pr100382v. [DOI] [PubMed] [Google Scholar]
- 13.Gopalakrishnan V, Lustgarten JL, Visweswaran S, Cooper GF. Bayesian rule learning for biomedical data mining. Bioinformatics. 2010;26(5):668–75. doi: 10.1093/bioinformatics/btq005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryberg H, An J, Darko S, et al. Discovery and verification of amyotrophic lateral sclerosis biomarkers by proteomics. Muscle Nerve. 2010;42(1):104–11. doi: 10.1002/mus.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bigbee WL, Gopalakrishnan V, Weissfeld JL, et al. A multiplexed serum biomarker immunoassay panel discriminates clinical lung cancer patients from high-risk individuals found to be cancer-free by CT screening. J Thorac Oncol. 2012;7(4):698–708. doi: 10.1097/JTO.0b013e31824ab6b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sokal RR, Rohlf FJ. Biometry: the principles and practice of statistics in biological research. New York: WH Freeman; 1995. [Google Scholar]
- 17.Neapolitan RE. Learning bayesian networks. Prentice Hall; Upper Saddle River: 2004. [Google Scholar]
- 18.Cooper GF, Herskovits E. A Bayesian method for the induction of probabilistic networks from data. Machine learning. 1992;9(4):309–47. [Google Scholar]
- 19.Lustgarten JLA. Bayesian Rule Generation Framework for ‘Omic’. Biomedical Data Analysis: University of Pittsburgh; 2009. [Google Scholar]
- 20.Lustgarten JL, Visweswaran S, Gopalakrishnan V, Cooper GF. Application of an efficient Bayesian discretization method to biomedical data. BMC bioinformatics. 2011;12(1):309. doi: 10.1186/1471-2105-12-309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lustgarten JL, Visweswaran S, Grover H, Gopalakrishnan V. An Evaluation of Discretization Methods for Learning Rules from Biomedical Datasets. BIOCOMP. 2008:527–32. [Google Scholar]
- 22.Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol Cell Proteomics. 2002;1(11):845–67. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- 23.Ramel S, Reid BJ, Sanchez CA, et al. Evaluation of p53 protein expression in Barrett's esophagus by two-parameter flow cytometry. Gastroenterology. 1992;102(4 Pt 1):1220–8. [PubMed] [Google Scholar]
- 24.van Dekken H, Hop WC, Tilanus HW, et al. Immunohistochemical evaluation of a panel of tumor cell markers during malignant progression in Barrett esophagus. Am J Clin Pathol. 2008;130(5):745–53. doi: 10.1309/AJCPO31THGVEUIDH. [DOI] [PubMed] [Google Scholar]
- 25.Washington K, Chiappori A, Hamilton K, et al. Expression of beta-catenin, alpha-catenin, and E-cadherin in Barrett's esophagus and esophageal adenocarcinomas. Mod Pathol. 1998;11(9):805–13. [PubMed] [Google Scholar]
- 26.Barrett MT, Sanchez CA, Galipeau PC, Neshat K, Emond M, Reid BJ. Allelic loss of 9p21 and mutation of the CDKN2/p16 gene develop as early lesions during neoplastic progression in Barrett's esophagus. Oncogene. 1996;13(9):1867–73. [PubMed] [Google Scholar]
- 27.Bani-Hani K, Martin IG, Hardie LJ, et al. Prospective study of cyclin D1 overexpression in Barrett's esophagus: association with increased risk of adenocarcinoma. J Natl Cancer Inst. 2000;92(16):1316–21. doi: 10.1093/jnci/92.16.1316. [DOI] [PubMed] [Google Scholar]
- 28.Ong CA, Lao-Sirieix P, Fitzgerald RC. Biomarkers in Barrett's esophagus and esophageal adenocarcinoma: predictors of progression and prognosis. World J Gastroenterol. 2010;16(45):5669–81. doi: 10.3748/wjg.v16.i45.5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Castillo-Tong DC, Pils D, Heinze G, et al. Association of myeloperoxidase with ovarian cancer. Tumour Biol. 2014;35(1):141–8. doi: 10.1007/s13277-013-1017-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fan NJ, Gao CF, Wang CS, et al. Identification of the up-regulation of TP-alpha, collagen alpha-1(VI) chain, and S100A9 in esophageal squamous cell carcinoma by a proteomic method. J Proteomics. 2012;75(13):3977–86. doi: 10.1016/j.jprot.2012.05.008. [DOI] [PubMed] [Google Scholar]
- 31.Zhu YH, Yang F, Zhang SS, Zeng TT, Xie X, Guan XY. High expression of biglycan is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2013;6(11):2497–505. [PMC free article] [PubMed] [Google Scholar]
- 32.Lomnytska MI, Becker S, Bodin I, et al. Differential expression of ANXA6, HSP27, PRDX2, NCF2, and TPM4 during uterine cervix carcinogenesis: diagnostic and prognostic value. Br J Cancer. 2011;104(1):110–9. doi: 10.1038/sj.bjc.6605992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakwe AM, Koumangoye R, Guillory B, Ochieng J. Annexin A6 contributes to the invasiveness of breast carcinoma cells by influencing the organization and localization of functional focal adhesions. Exp Cell Res. 2011;317(6):823–37. doi: 10.1016/j.yexcr.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kawaguchi H, Ohno S, Miyazaki M, et al. CYFRA 21-1 determination in patients with esophageal squamous cell carcinoma: clinical utility for detection of recurrences. Cancer. 2000;89(7):1413–7. doi: 10.1002/1097-0142(20001001)89:7<1413::aid-cncr1>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 35.Ishikawa N, Takano A, Yasui W, et al. Cancer-testis antigen lymphocyte antigen 6 complex locus K is a serologic biomarker and a therapeutic target for lung and esophageal carcinomas. Cancer Res. 2007;67(24):11601–11. doi: 10.1158/0008-5472.CAN-07-3243. [DOI] [PubMed] [Google Scholar]
- 36.Wang JS, Varro A, Lightdale CJ, et al. Elevated serum gastrin is associated with a history of advanced neoplasia in Barrett's esophagus. Am J Gastroenterol. 2010;105(5):1039–45. doi: 10.1038/ajg.2009.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J, Chang AC, Li C, et al. Comparative proteomics analysis of Barrett metaplasia and esophageal adenocarcinoma using two-dimensional liquid mass mapping. Mol Cell Proteomics. 2007;6(6):987–99. doi: 10.1074/mcp.M600175-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Stingl C, van Vilsteren FG, Guzel C, et al. Reproducibility of protein identification of selected cell types in Barrett's esophagus analyzed by combining laser-capture microdissection and mass spectrometry. J Proteome Res. 2011;10(1):288–98. doi: 10.1021/pr100709b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.