

Abstract
Hypothyroidism is a well-described cause of hypomyelination. In addition, thyroid hormone (T3) has recently been shown to enhance remyelination in various animal models of CNS demyelination. What are the ways in which T3 promotes the development and regeneration of healthy myelin? To begin to understand the mechanisms by which T3 drives myelination, we have identified genes regulated specifically by T3 in purified oligodendrocyte precursor cells (OPCs). Among the genes identified by genomic expression analyses were four transcription factors, Kruppel-like factor 9 (KLF9), basic helix-loop-helix family member e22 (BHLHe22), Hairless (Hr), and Albumin D box-binding protein (DBP), all of which were induced in OPCs by both brief and long term exposure to T3. To begin to investigate the role of these genes in myelination, we focused on the most rapidly and robustly induced of these, KLF9, and found it is both necessary and sufficient to promote oligodendrocyte differentiation in vitro. Surprisingly, we found that loss of KLF9 in vivo negligibly affects the formation of CNS myelin during development, but does significantly delay remyelination in cuprizone-induced demyelinated lesions. These experiments indicate that KLF9 is likely a novel integral component of the T3-driven signaling cascade that promotes the regeneration of lost myelin. Future analyses of the roles of KLF9 and other identified T3-induced genes in myelination may lead to novel insights into how to enhance the regeneration of myelin in demyelinating diseases such as multiple sclerosis.
Keywords: Oligodendrocyte, Thyroid hormone, KLF9, BTEB1, Myelin, Cuprizone
Introduction
Myelination has evolved in vertebrates to insulate axons and thereby promote rapid, energy efficient action potential propagation. Loss of myelin sheaths produces a wide variety of neurological symptoms, as observed in multiple sclerosis and other demyelinating diseases. In the central nervous system (CNS), myelin sheaths are produced by oligodendrocytes (OLs). The location and timing of CNS myelination is controlled largely by regulating the onset of OL differentiation, as the expression of markers of OL maturity is rapidly followed by the initiation of myelination in proximal axon tracts (Baumann and Pham-Dinh, 2001). Therefore, better understanding of the mechanisms that regulate OL differentiation should provide novel insight into how myelin formation is regulated in vivo, both during development and also during remyelination of demyelinated lesions produced by disease or injury.
Interestingly, OL differentiation can be stimulated by distinct pathways. Mitogen signaling by trophic factors such as platelet derived growth factor (PDGF) and fibroblast growth factor (FGF) maintains OL precursor cells (OPCs) in a proliferative, undifferentiated state, and mitogen withdrawal leads to rapid OL differentiation (Barres et al., 1993; Baumann and Pham-Dinh, 2001). Alternatively, thyroid hormone (T3) can override mitogen signaling to promote OL differentiation even in the presence of saturating amounts of mitogens (Barres et al., 1994; Temple and Raff, 1986). These different environmental cues apparently induce OL differentiation through distinct molecular pathways, as evidenced by differential regulation of several cell cycle control genes in response to T3 versus mitogen withdrawal (Tokumoto et al., 2001). The importance of T3 in myelin development is illustrated by the reduced CNS myelination observed in hypothyroid rodents (Leung et al., 1992; Malone et al., 1975; Walters and Morell, 1981) and human patients (Gupta et al., 1995; Jagannathan et al., 1998; Mussa et al., 2001). Hypothyroidism also results in the reduced expression of several myelin genes in vivo (Barradas et al., 2001; Ibarrola and Rodriguez-Pena, 1997; Noguchi and Sugisaki, 1984; Pombo et al., 1998; Rodriguez-Pena et al., 1993). Conversely, increasing T3 levels in vivo accelerates both myelin gene expression and myelination during development (Figueiredo et al., 1993; Marta et al., 1998; Pombo et al., 1998; Walters and Morell, 1981), and also myelin regeneration after demyelination (Calza et al., 2005; Fernandez et al., 2004; Franco et al., 2008; Harsan et al., 2008).
To better understand how T3 promotes myelination, we wanted to identify candidate downstream genes that could transduce the promyelinating effects of T3. Utilizing genomics, we identified a select set of genes that are immediately induced by T3 in OPCs. To begin to characterize these genes, we focused on the most robustly induced of these, KLF9. Our finding that KLF9 is induced by T3 in OPCs is consistent with previous reports looking at KLF9 regulation in the CNS (Denver et al., 1999; Furlow and Kanamori, 2002; Hoopfer et al., 2002; Martel et al., 2002). KLF9 has previously been found to regulate development in a number of different tissues, including the uterus, intestine, adipocytes, and myocytes (Mitchell and DiMario, 2010; Pei et al., 2011; Simmen et al., 2004, 2007; Velarde et al., 2005). Consistent with a pro-differentiation role, KLF9 levels are reduced in endometrial tumors, and KLF9 expression can induce glioblastoma tumor cell differentiation (Simmen et al., 2010; Ying et al., 2011). We found that KLF9 is both sufficient to induce OL differentiation and required for normal T3-induced OL differentiation in vitro. Interestingly, loss of KLF9 in vivo negligibly impacts the initial development of myelin, but does significantly disrupt CNS remyelination in cuprizone-induced demyelinated lesions. Cumulatively, these data support the hypothesis that KLF9 is a novel integral component of the T3-driven signaling cascade that regulates the timing of OL differentiation and myelination regeneration. These experiments indicate that KLF9, and therefore potentially T3, may play a more crucial role in promoting remyelination after injury or disease than in the initial development of myelin, and support the concept of targeting the T3-signaling pathway to promote remyelination.
Discussion
Identification of KLF9 as a downstream effector of T3 in OL differentiation
OL differentiation is an obligate step in the generation of CNS myelin. Therefore, a deeper understanding of the mechanisms that regulate OL differentiation should yield insights into how myelin formation is regulated both during development and in myelin regeneration. T3 has been identified as a prominent inducer of OL differentiation and myelination both in vitro and in vivo (Barres et al., 1994; Figueiredo et al., 1993; Marta et al., 1998; Temple and Raff, 1986; Walters and Morell, 1981), yet the downstream molecular components via which T3 promotes OL differentiation remain poorly characterized. Therefore, to identify genes involved in transducing the effects of T3 in OL differentiation, we utilized genomics to produce a concise list of genes that are rapidly induced by T3 in immature OPCs. It is important to note that these analyses were carried out in the presence of saturating amounts of OPC mitogens, which allowed us to focus specifically on genes induced in T3 (a.k.a. “clock”) mediated differentiation, as opposed to genes induced by reduced mitogen signaling that stimulates immediate OL differentiation. By focusing on genes that were rapidly and robustly induced specifically by T3 exposure, we were able to identify a concise list of seven genes that may play a role in mediating T3-induced OL differentiation.
To begin to functionally validate whether any of these identified genes could impact OL differentiation, we focused on the most rapidly and robustly induced gene identified, the transcription factor gene KLF9. We found that this gene was more highly induced by T3 than by reduced mitogen signaling, and also demonstrated that KLF9 is both necessary for normal T3-induced differentiation, and sufficient to initiate the initial stages of OL differentiation in immature OPCs. The fact that KLF9 is robustly induced by T3 in OPCs is consistent with previous findings that the KLF9 promoter contains functional T3 responsive elements (Denver and Williamson, 2009), and in fact, although previously unreported, an examination of data presented in Denver et al. (1999) shows that T3-induced KLF9 expression has been documented in cultured OLs to an extent similar to that reported here. In addition, KLF9 has been implicated in promoting the normal development of various other cell types, often in response to hormonal signaling, including intestinal crypt cells, uterine endometrial cells, adipocytes, myocytes, and neurons (Bonett et al., 2009; Denver et al., 1999; Mitchell and DiMario, 2010; Pei et al., 2011; Simmen et al., 2004, 2007; Velarde et al., 2005; Zeng et al., 2008). KLF9 has even been implicated as a potential effector of changes occurring during Xenopus metamorphosis (Hoopfer et al., 2002). These data, along with the widespread detected expression of KLF9 in various somatic tissues (Morita et al., 2003), indicate that KLF9 function is likely not specific to promoting OL differentiation, but instead is involved in transducing widespread effects of hormonal signaling throughout the body, including T3 promotion of CNS remyelination. We were surprised to find that KLF9 overexpression could specifically induce genes normally expressed early in OL differentiation (CNP and MBP), but not a gene normally expressed in the final stages of OL differentiation (MOG). These data are consistent with our previous findings that the early and late stages of OL differentiation can be independently regulated (Dugas et al., 2006). Interestingly, although KLF9 is not sufficient to induce MOG expression, siRNA knockdown of KLF9 expression indicates that KLF9, or a gene induced by KLF9, is required for T3-regulated MOG expression. The fact that reducing KLF9 levels impacts the expression of every marker gene tested in OPCs exposed to T3 indicates that, whereas KLF9 may not be sufficient to induce all aspects of OL differentiation on its own, KLF9 does in fact play a crucial role in mediating the ability of T3 to promote OL differentiation. It is also worth noting that we observed BHLHB5 and Hr overexpression could also induce the expression of CNP and MBP, but not MOG, yet knockdown of these genes via siRNA did not produce any significant reduction in myelin gene expression (data not shown). While it is possible that the lack of phenotype may have resulted from insufficient reduction in BLHLB5 or Hr expression, at minimum these findings illustrate that T3 likely acts through the induction of several regulatory genes, of which KLF9 is an important, but not sole, downstream effector.
KLF9 loss does not affect initial myelin formation
In our initial functional experiments, we found that KLF9 expression was required for normal levels of T3-induced OL differentiation in vitro. Yet when we examined developmental myelination in KLF9−/− mice, we detected no delay in the formation of CNS myelin, nor in the initial differentiation of OLs, despite data indicating that KLF9 is normally expressed by mature OLs in vivo (Figs. 1G–I; Cahoy et al. (2008)). How can we reconcile these disparate findings? The role of T3 in promoting CNS myelin formation has been extensively demonstrated: increasing T3 levels early in development accelerates OL differentiation and myelination, and conversely, hypothyroid rodents and humans have reduced levels of myelination and myelin gene expression (Barradas et al., 2001; Figueiredo et al., 1993; Gupta et al., 1995; Ibarrola and Rodriguez-Pena, 1997; Jagannathan et al., 1998; Leung et al., 1992; Malone et al., 1975; Marta et al., 1998; Mussa et al., 2001; Noguchi and Sugisaki, 1984; Pombo et al., 1998; Rodriguez-Pena et al., 1993; Walters and Morell, 1981). Yet the effects of T3 loss on CNS myelination are not nearly as severe as mutations that directly and completely impair OL differentiation, such as ablation of SOX10, OLIG1/2, or MRF (Emery et al., 2009; Stolt et al., 2002; Zhou and Anderson, 2002). In addition, the observed disruptions in OL differentiation and myelination are regionally heterogeneous (Ibarrola and Rodriguez-Pena, 1997). These data indicate that factors in addition to T3 contribute to the stimulation of myelin formation in vivo. Indeed, reduced mitogen signaling is another potent stimulator of OL differentiation, and manipulating mitogen levels in vivo can alter the timing of OL differentiation (Barres et al., 1992; Calver et al., 1998; Fruttiger et al., 1999). These previous studies may indicate that large numbers of proliferating OPCs produced during CNS development may exhaust limited supplies of local mitogens, and that this reduction in mitogen signaling may be the predominant inducer of initial OL differentiation. This would explain why loss of T3 leads to at worst a slight delay in the production of mature OLs and CNS myelination. By extension, in the absence of the T3-induced gene KLF9 during development, limiting levels of mitogen signaling would still be the predominant driver of OL differentiation and myelination, leading to the negligible developmental phenotype observed. In addition, KLF9 loss may not perfectly recapitulate hypothyroidism in vivo, because KLF9 is only one of several genes rapidly induced by T3 in OPCs, and one of many more genes induced over a longer time scale by T3 exposure. These other genes may contribute to promoting OL differentiation and, if so, they could help to compensate for KLF9 loss.
Fig. 1.
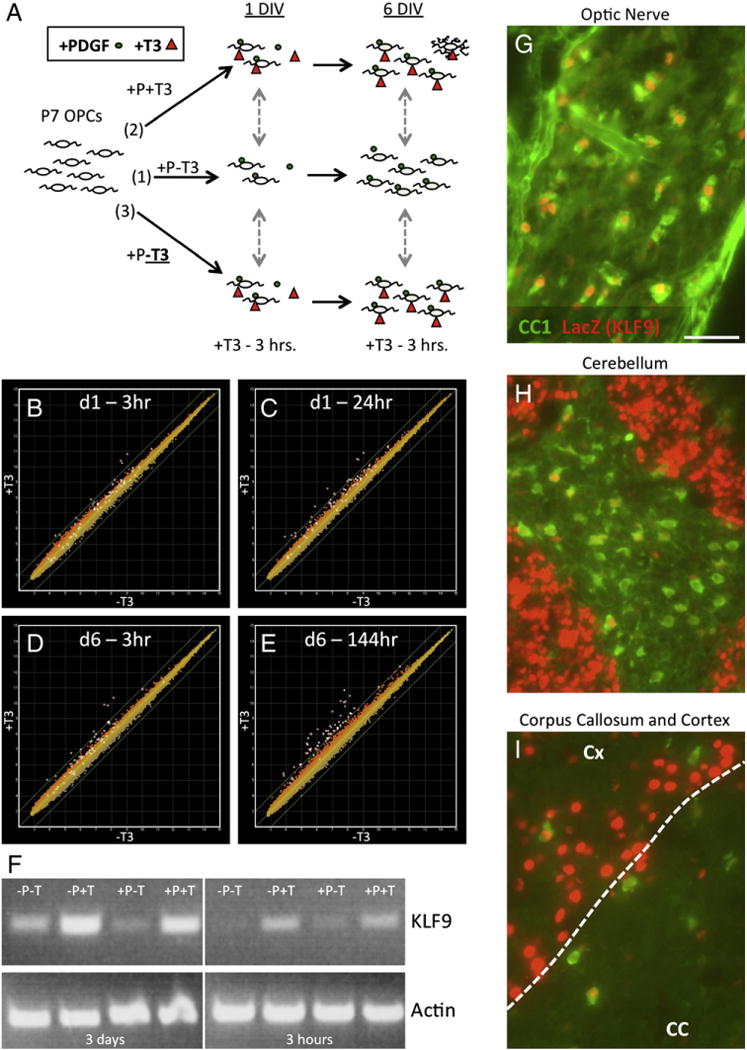
The T3-induced gene KLF9 is expressed by OLs in vitro and in vivo. A. Analysis of gene expression in cultured OPCs. (1) Acutely purified P7 OPCs were cultured in + PDGF−T3 media (control condition) for either 1 or 6 DIV. (2) OPCs were cultured in + PDGF + T3 media for either 1 or 6 DIV to observe the effects of long-term exposure to T3 on gene expression. (3) OPCswereculturedin + PDGF–T3 media for1 or 6 DIV, and T3 was added 3 hours prior tosample collection toanalyze genes rapidly regulated by T3. B–E. Graphs showing gene expression data in OPCs cultured in + PDGF–T3 media (horizontal axes) vs. OPCs cultured in + PDGF−T3 21 h, then + PDGF + T3 3 h (B), + PDGF + T3 24 h (C), +PDGF−T3 6 days (141 h), then +PDGF+T3 3 h (D), or +PDGF+T3 6 days (144 h) (E), all on vertical axes. All data plotted on a log2 scale. Green diagonal lines indicate 2× relative change in − T3 vs. + T3 samples. All probe sets listed in Table 1 are highlighted by white boxes in all graphs. F. RT-PCR (32 cycles) to determine KLF9 expression levels in OPCs cultured in media +/−PDGF and +/−T3 for either 3 days, or first for 6 days in +PDGF−T3 media, then 3 h in the indicated media. RT-PCR (24 cycles) to detect actin levels to confirm equivalent starting material also shown. G–I. Heterozygous P10 KLF9+/LacZ mouse tissue immunostained for CC1 (OLs) and LacZ (indicating KLF9-expressing cells) expression in the optic nerve (G), central region of the cerebellum (H) and medial corpus callosum plus overlying cortex (I). Scale bar=50 μm.
It is also worth noting that although myelination levels appear to be normal in KLF9−/− mice, this is based only on gross assessment of the extent of myelin sheath formation and myelin protein expression. Other labs have detected more subtle yet persistent and widespread deficits in myelin sheath structure and protein distribution in the absence of T3 signaling (Barradas et al., 2001; Ferreira et al., 2004, 2007). It remains to be seen whether KLF9 is required for more subtle aspects of myelin formation, such as the maturation and maintenance of fully functional compact myelin sheaths.
Importance of KLF9 and T3 in myelin regeneration
In contrast to the negligible role of KLF9 in promoting developmental myelination, loss of KLF9 appeared to significantly impair the ability of the mature CNS to regenerate lost myelin in both the cortex and corpus callosum. Previous data also indicate a potentially prominent role for T3 in promoting remyelination, as increasing T3 levels in vivo after cuprizone administration or experimental autoimmune encephalomyelitis induction accelerates myelin regeneration (Calza et al., 2005; Franco et al., 2008; Harsan et al., 2008). In addition, the identification of mutants that have a more prominent role in remyelination than initial myelin development is not unprecedented, as mutations in the Olig1 locus have been described that do not disrupt initial myelin development but do specifically impair myelin regeneration in adult mice (Arnett et al., 2004). What might account for a larger reliance on KLF9, and therefore potentially T3, in the regeneration of lost myelin in adult rodents?
Proximal to lesions where mature myelin is lost, the small numbers of residual, undifferentiated adult OPCs nearby begin to proliferate, enabling the production of newly generated OLs that subsequently remyelinate denuded axons (McTigue and Tripathi, 2008; Redwine and Armstrong, 1998; Reynolds et al., 2002). In some cases, newly generated OPCs that arise from the subventricular zone also contribute to the production of replacement OLs (Nait-Oumesmar et al., 2008). Although the exact nature of the signals that stimulate OPC generation and proliferation following demyelination are not fully characterized, reactive astrocytes that arise in the area of the lesion likely contribute by locally secreting large amounts of mitogens, including PDGF and FGF-2 (Albrecht et al., 2003; Hinks and Franklin, 1999; Moore et al., 2011; Redwine and Armstrong, 1998). These mitogens then stimulate proliferation of the limited numbers of nearby adult OPCs, and also stimulate migration of OPCs into the demyelinated lesion (McTigue and Tripathi, 2008). As adult OPCs proliferate more slowly than OPCs in the postnatal animal, the environment in the lesion may therefore be one of relatively low OPC numbers and high mitogen levels. In such an environment, OL differentiation may be triggered primarily not by reduced mitogen signaling, but by T3 activation of clock-mediated differentiation through induction of genes such as KLF9. Also, as T3 only activates OL differentiation in sufficiently “mature” OPCs, reliance on clock-mediated, T3/KLF9-induced differentiation could allow for sufficient initial expansion of the newly generated OPC pool, followed by timely differentiation to allow remyelination of demyelinated lesions.
The data from this and previous studies are consistent with a model in which T3 plays a prominent role in promoting remyelination after injury or disease. By utilizing genomics, we have identified at least one functional downstream effector of T3, KLF9, which contributes to this remyelination. As KLF9 is only one of several genes identified in our initial genomic analyses, it will be of great interest to more thoroughly investigate the roles of the additional T3-induced genes in OL differentiation and myelination in future studies.
Experimental methods
Complete protocols available upon request from jcdugas@alum.mit.edu.
OPC purification and culturing
All purification and culturing of OPCs from P7–8 Sprague-Dawley rat or C57/Bl6 mouse brains (Charles Rivers) were carried out as described previously (Dugas et al., 2006, 2010). Mouse OPCs were utilized for genomic analyses; rat OPCs were utilized for all other in vitro experiments. Throughout the text, “+PDGF” refers to addition of the OPC mitogens platelet derived growth factor AA (10 ng/ml) + 1 ng/ml neurotrophin 3 (1 ng/ml) to the media (both Peprotech, Rocky Hill, NJ), and “+T3” refers to addition of 40 ng/ml triiodothyronine (Sigma-Aldrich, St. Louis, MO) to the media. OPCs plated in −PDGF±T3 media were plated at a density of 30,000 cells/coverslip in 24-well plates (for immunostaining) or 1.5× 106 cells into 10 cm tissue culture plates (for RNA collection). OPCs plated in +PDGF ±T3 media were plated at 1500 cells/coverslip in 24-well plates or 300,000–400,000 cells into 10 cm tissue culture plates.
Gene chip hybridization and analysis
Total RNA was isolated from cultured mouse OPCs and amplified prior to chip hybridization as described previously (Dugas et al., 2006). Four independent biological samples were collected for each condition (Fig. 1A). Final amplified and labeled samples were hybridized to Affymetrix Mouse 430 2.0 arrays (Santa Clara, CA) at the Stanford Protein and Nucleic Acid Biotechnology (PAN) Facility according to standard Affymetrix protocols. Output CEL files obtained via MAS 5.0 (Affymetrix) were analyzed using DNAStar ArrayStar (Madison, WI) to obtain expression values and comparisons between control (−PDGF −T3) and experimental (+T3) conditions.
RT-PCR and qRT-PCR
RNA from cultured rat OPCs was collected using Qiagen RNeasy kit (Valencia, CA) and 400 ng/sample was reverse transcribed using Super-ScriptIII (Invitrogen, Carlsbad, CA), both according to manufacturers’ instructions. For standard RT-PCR reactions, equivalent volumes of each sample were then amplified using Platinum Taq (Invitrogen): 94°C 30″, 55°C 30″, 68°C 45″ cycles, number of cycles indicated in figure legends. Real time qRT-PCR reactions were carried out and analyzed by the ddCt method using an ABI StepOne Plus (Carlsbad, CA) instrument and associated software at the Stanford PAN facility.
- Actin:
- Forward–GCATTGTCACCAACTGGGACG
- Reverse–ACCGCTCATTGCCGATAGTG
- KLF9:
- Forward–TCCGGAACTTTCAAACCTTG
- Reverse–GTGTGCCAAACAGAATGTCG
- GAPDH: (for internal normalization of qRT-PCR reactions)
- Forward–TCCGGAACTTTCAAACCTTG
- Reverse–GTGTGCCAAACAGAATGTCG
OPC transfection protocol
Cultured OPCs were transfected using the Lonza oligodendrocyte transfection kit (Basel, Switzerland) as described previously (Dugas et al., 2006). To identify transfected cells, OPCs were always co-transfected with 1.5–2.5 μg pC1-eGFP (CMV-promoter driven eGFP expression; Clontech 6084–1, Mountain View, CA). To drive expression of KLF9, OPCs were transfected with 2.5 μg pSport6-KLF9 (full length human KLF9 from OpenBiosystems (Huntsville, AL) clone MHS1768-9143875, excised with SacI (then blunted) and KpnI, then cloned into the pSPORT6 expression vector, Open Biosystems MMM1013-63856, behind the CMV promoter after excising the existing insert with ApaI (then blunted) and KpnI). Individual and pooled siRNA was obtained from Dharmacon (Lafayette, CO): siControl non-targeting siRNA pool (targets firefly luciferase; D-001206-13), siKLF9 siGenome SMARTpool (rat; M-091801-00), and individual siKLF9 siGenome #1 – #4 (rat; D-091801-01-01, −02 −03, −04); all used at 0.2–0.4 nmol/transfection.
Tissue collection and cuprizone treatment
KLF9LacZ/LacZ or age-matched littermate control KLF9+/+ or KLF9+/LacZ mice, originally obtained from Dr. Rosalia Simmen (University of Arkansas, Little Rock, AR), were genotyped as described previously (Morita et al., 2003). For cuprizone treatment, starting at 8 weeks of age mice were supplied exclusively with food containing 0.2% cuprizone (cuprizone obtained from Sigma-Aldrich, and milled into chow by Harlan, Indianapolis, IN) for 6 weeks, with fresh food replaced every 2–3 days to maintain potency of cuprizone. To allow recovery from cuprizone treatment, food was replaced with standard chow for an additional 1–2 weeks. At ages/time points described, perfusion-fixed tissue was collected as described previously (Dugas et al., 2010). All fixed brains were cut along the midline and mounted for sagittal sectioning, and 10 μm thick sections were collected. Similar depth sections, as determined by number of sections from the midline and anatomical markers, were used in all quantitative analyses, with 2 different depths of tissue approximately 200–300 μm apart analyzed/animal. For optic nerves, fixed tissue was mounted for longitudinal sectioning/staining.
Immunostaining and histology
Immunostaining of OPC and OL cultures for NG2, CNP, MBP, MOG, and GFP expression was performed as described previously (Dugas et al., 2006, 2010). Immunostaining with the CC1 antibody to detect mature OLs was performed with 1/50 mouse-anti-APC (CC-1) antibody (Calbiochem/EMD Chemicals OP-80, Gibbstown, NJ).
Staining of tissue sections with anti-MBP antibodies or Fluoromyelin stain was performed as described previously (Dugas et al., 2010). The same CC1 antibody described above was used at 1/50 for tissue section staining, staining for LacZ expression was carried out with 1/5000 rabbit-anti-beta-galactosidase antibody (MP Biochemicals 55976, Solon, OH).
Quantification of staining
For cell culture staining quantification, all coverslips were scored blind, with >150 GFP+ cells (in transfection experiments) or >500 cells (in cortex vs. cerebellar OPC experiments) with healthy nuclei being classified as positive or negative for marker protein (NG2, CC1, CNP, MBP, or MOG) expression per coverslip. Three or four individual, independently cultured coverslips were generated from each transfection. For quantification of CNS myelination, either cerebellar arms and corpora callosa were outlined, or 700×700 μm boxes were placed above the caudal, mid, and rostral corpus callosum to select standardized cortical areas, and the percentage area myelinated within selected regions was determined either as the number of pixels +10 arbitrary units (0–255 scale in 8-bit images) brighter than background (for MBP staining) or the number of pixels +50 arbitrary units brighter than background (for Fluoromyelin staining to detect dense myelin)/the total number of pixels. For each animal, two sagittal sections at different depths (~200–300 μm apart) were analyzed; the two depths analyzed were the same across all littermates.
Western blotting
Tissue was acutely collected from P12–P13 or P18 KLF9−/− or littermate control mice brain regions noted, then flash frozen in liquid nitrogen. Protein samples were collectedby grinding and sonicating samples in RIPA buffer+Complete Protease Inhibitor (Roche) at 4°C. Western blots were then performed as described previously (Dugas et al., 2010), with the addition of 1/1000 sheep-anti-Transferrin (Trf) (Novus NB100-1947). Blots were quantified with FluorChemQ software, and normalized to in-lane actin levels as loading control.
In situ hybridization
In situ hybridizations were performed as described previously (Dugas et al., 2010) with both the previously described PLP gene probe, and also a probe for Trf: 1593 (EcoRI site) to 2293 (3′ end) of the mouse Trf insert in pSport6 obtained from OpenBiosystems/ThermoScientific (clone 3594705). Brains acutely collected from P12 or P17 KLF9−/− or control littermate mice were mounted for sagittal sectioning then flash frozen in OCT embedding medium prior to being sectioned for in situs.
Results
Genomic analysis of genes induced by T3 in OPCs
Several experiments have demonstrated that T3 is both able to promote OL differentiation in vitro and required for normal myelin development in vivo (Barres et al., 1994; Jagannathan et al., 1998; Leung et al., 1992; Malone et al., 1975; Mussa et al., 2001; Noguchi and Sugisaki, 1984; Temple and Raff, 1986; Walters and Morell, 1981). T3 generally acts by binding to thyroid hormone receptors, after which the T3-hormone receptor complexes translocate to the nucleus and activate gene transcription (Harvey and Williams, 2002; Oetting and Yen, 2007). Therefore, to investigate how T3 promotes OL differentiation and myelination, we began by asking which genes are induced by T3 in immature OPCs.
To address this question, we utilized previously established protocols to highly purify and culture immature OPCs from postnatal day 7 (P7) mouse brains, thereby allowing us to precisely control both the T3 and mitogen levels to which the immature OPCs are exposed. Mitogen withdrawal alone triggers rapid and robust differentiation of OPCs (Barres et al., 1993, 1994); therefore, to specifically isolate the effects of T3 on OPCs, all cultures were maintained in saturating amounts of mitogens. We then compared gene expression between OPCs cultured in the presence or absence of T3 at both 1 and 6 days in vitro (DIV) (Fig. 1A). To increase the likelihood of identifying genes that are directly induced by T3, we also analyzed gene expression in OPCs cultured in T3-free media for 1 or 6 DIV, then exposed to T3 for just 3 h prior to sample collection. In this way, we were able to identify genes induced in OPCs at 1 DIV by either 3 h or 24 h of T3 exposure, and also at 6 DIV by 3 h or 6 full days of T3 exposure. Surprisingly, we found very few genes that were induced ≥ 2-fold specifically by T3, especially with brief T3 exposure (Figs. 1B–E). In fact, by focusing on genes induced by both rapid (3–24 h) and long-term (6 day) T3 exposure, we identified only seven genes that are induced both rapidly and persistently by T3 (Table 1). Of these, four genes are transcription factors that could potentially serve as global transducers of the downstream effects of T3 on OL differentiation: kruppel-like factor 9 (KLF9), hairless (Hr), D site albumin promoter binding protein (DBP), and basic helix-loop-helix family, member e22 (BHLHe22). To determine whether any of these genes could functionally promote OL differentiation, we chose to focus on KLF9, the most consistently and robustly induced candidate gene identified.
Table 1.
Genes induced by T3 in OPCs. Genes induced ≥2-fold by T3 in OPCs in at least one experimental condition (see Fig. 1), as identified on Affymetrix Mouse Genome 430 2.0 arrays. Probe sets changing ≥2-fold in more than one condition are listed above the dashed line. No probe sets were repressed ≥2-fold by T3 exposure in any analyzed time point. Note that the unannotated probe set 1456053_at likely represents Bhlhe22, as it aligns with mouse genomic sequence <2 kb 3′ of the identified Bhlhe22 locus. P-values from DNAStar ArrayStar analysis. The bold numbers indicate conditions where there was a ≥2-fold increase in expression.
| Probe set ID | Gene symbol | 1 DIV
|
6 DIV
|
P value | ||
|---|---|---|---|---|---|---|
| 3 h/cont | T3/cont | 3 h/cont | T3/cont | |||
| 1456341_a_at | Klf9 | 3.5 | 3.8 | 5.3 | 6.2 | 0.2490 |
| 1428288_at | Klf9 | 3.5 | 3.7 | 4.7 | 5.2 | 0.0001 |
| 1428289_at | Klf9 | 3.0 | 3.5 | 4.4 | 4.9 | 0.0032 |
| 1435950_at | Hr | 2.4 | 3.9 | 1.9 | 2.9 | 0.0001 |
| 1438211_s_at | Dbp | 1.7 | 2.1 | 2.5 | 3.2 | 0.1860 |
| 1418174_at | Dbp | 1.8 | 2.0 | 2.0 | 2.4 | 0.0000 |
| 1418271_at | Bhlhe22 | 1.1 | 1.2 | 2.0 | 8.6 | 0.5810 |
| 1437230_at | Kcna1 | 1.3 | 1.7 | 2.0 | 5.6 | 0.0000 |
| 1455785_at | Kcna1 | 1.5 | 1.6 | 2.0 | 4.9 | 0.0000 |
| 1417416_at | Kcna1 | 1.4 | 1.6 | 2.0 | 4.0 | 0.0011 |
| 1428547_at | Nt5e | 1.7 | 1.9 | 2.7 | 3.9 | 0.0007 |
| 1448998_at | Lpo | 1.3 | 2.1 | 1.8 | 2.8 | 0.0006 |
|
| ||||||
| 1422428_at | Acsbg1 | 1.3 | 1.7 | 1.6 | 3.2 | 0.5510 |
| 1422573_at | Ampd3 | 1.5 | 1.5 | 1.2 | 2.3 | 0.0757 |
| 1428303_at | Btbd17 | 1.2 | 1.3 | 1.8 | 2.2 | 0.6750 |
| 1451620_at | C1ql3 | 1.1 | 1.2 | 1.1 | 2.1 | 0.0000 |
| 1434785_at | Cacng5 | 0.9 | 1.1 | 1.1 | 2.2 | 0.1140 |
| 1428466_at | Chd3 | 1.1 | 1.4 | 1.4 | 2.2 | 0.0341 |
| 1433781_a_at | Cldn12 | 1.5 | 1.6 | 1.9 | 2.1 | 0.1230 |
| 1433782_at | Cldn12 | 1.9 | 1.9 | 1.8 | 2.0 | 0.6100 |
| 1430855_at | Col20a1 | 1.1 | 1.1 | 1.3 | 3.1 | 0.0228 |
| 1452035_at | Col4a1 | 1.1 | 1.3 | 1.1 | 2.4 | 0.0005 |
| 1424051_at | Col4a2 | 1.1 | 1.3 | 1.0 | 2.2 | 0.0005 |
| 1421074_at | Cyp7b1 | 1.1 | 1.5 | 1.6 | 5.4 | 0.0017 |
| 1421075_s_at | Cyp7b1 | 1.1 | 1.3 | 1.4 | 4.8 | 0.0218 |
| 1456069_at | Dtna | 1.2 | 1.2 | 1.2 | 2.0 | 0.9360 |
| 1436293_x_at | Ildr2 | 1.2 | 1.4 | 1.5 | 2.7 | 0.1140 |
| 1436221_at | Ildr2 | 1.1 | 1.4 | 1.3 | 2.5 | 0.0013 |
| 1434129_s_at | Lhfpl2 | 1.1 | 1.2 | 1.3 | 2.1 | 1.0000 |
| 1433532_a_at | Mbp | 1.0 | 1.0 | 1.0 | 2.0 | 0.0152 |
| 1451527_at | Pcolce2 | 1.3 | 1.7 | 1.5 | 2.2 | 0.2540 |
| 1447623_s_at | Prkd1 | 1.0 | 1.2 | 1.0 | 2.0 | 0.2050 |
| 1448754_at | Rbp1 | 1.1 | 0.9 | 1.0 | 2.1 | 1.0000 |
| 1437598_at | Zbtb20 | 1.4 | 1.6 | 1.2 | 3.0 | 0.0246 |
| 1438443_at | Zbtb20 | 1.2 | 1.4 | 1.2 | 2.7 | 0.0001 |
| 1437066_at | Zbtb20 | 1.2 | 1.4 | 1.3 | 2.4 | 0.0003 |
| 1439278_at | Zbtb20 | 1.4 | 1.5 | 1.3 | 2.3 | 0.0494 |
| 1439128_at | Zbtb20 | 1.2 | 1.4 | 1.2 | 2.2 | 0.0007 |
| 1451577_at | Zbtb20 | 1.3 | 1.5 | 1.2 | 2.0 | 0.8520 |
| 1456053_at | (Bhlhe22) | 0.9 | 1.3 | 1.4 | 5.1 | 0.0119 |
| 1452065_at | Vstm2a | 1.4 | 2.2 | 1.0 | 1.6 | 0.0000 |
KLF9 expression in OLs in vitro and in vivo
Previous reports have identified a T3 response element (TRE) in the 5′ flanking region of the KLF9 gene, and have also demonstrated that T3 can drive KLF9 expression (Denver and Williamson, 2009; Denver et al., 1999; Hoopfer et al., 2002). To confirm that T3 induces KLF9 expression in OPCs, we exposed purified rat OPCs to T3 for either 3 h or 3 days. We also wanted to determine whether mitogen withdrawal could similarly induce the expression of KLF9, as mitogen withdrawal is a potent stimulator of rapid OL differentiation in OPCs isolated from neonatal rodents. Interestingly, we found that in the briefest exposure condition (3 h), only T3 was able to induce KLF9 expression, whereas mitogen withdrawal had no effect on KLF9 expression levels (Fig. 1F). In fact, T3 similarly induced KLF9 expression regardless of the presence (+PDGF+T3) or absence (−PDGF+T3) of mitogens in the culture medium. T3 induction of KLF9 expression was robustly maintained at 3 DIV. We did find that mitogen withdrawal alone was able to somewhat induce KLF9 expression by 3 DIV (−PDGF−T3), but this induction was weaker that that produced solely by T3 exposure (+PDGF+T3), and we also observed that T3 was able to further boost KLF9 expression in mitogen-free media (−PDGF−T3 vs. −PDGF+T3 media at 3 DIV). These data were confirmed by qRT-PCR, which revealed a 4-fold increase in KLF9 expression within purified OPCs in response to T3 after 3 DIV (n=8; p < 0.00001). Cumulatively, these data indicate that KLF9 expression is rapidly induced by T3 in OPCs, and that, relative to mitogen withdrawal, T3 is the predominant inducer of KLF9 expression in immature OPCs.
The rapid and relatively specific induction of KLF9 in vitro by T3 potentially implicates KLF9 as a downstream effector of T3-induced OL differentiation. But if KLF9 plays a role in promoting OL differentiation within the CNS, it should also be expressed by OLs in vivo. To investigate this, we utilized mutant mice in which the KLF9 gene has been disrupted by insertion of a LacZ gene (Morita et al., 2003). By immunostaining for LacZ expression in heterozygous KLF9+/LacZ mice, we were able to reveal the endogenous pattern of KLF9 expression. By co-staining with CC1 antibody, which stains mature OLs, we were able to identify OLs expressing KLF9 in the P10 optic nerve, cerebellum, corpus callosum, and cortex (Figs. 1G–I).
KLF9 promotes the initiation of OL differentiation in vitro
Having ascertained that KLF9 is both induced by T3 in OPCs in vitro and expressed by OLs in vivo, we next wanted to investigate whether KLF9 plays a functional role in promoting OL differentiation. Initially, we overexpressed KLF9 in immature OPCs to determine whether KLF9 could promote OL differentiation. Cultured OPCs were co-transfected with a plasmid that constitutively expresses full-length KLF9 (pSp-KLF9) and pC1-eGFP to allow identification of transfected cells, and control cells were transfected with pC1-eGFP alone. Transfected OPCs were then cultured in saturating mitogens in the absence of T3 for 4–6 DIV, and subsequently immunostained to analyze the expression of various markers of OL differentiation. We found that overexpression of KLF9 in OPCs more than doubled the number of cells expressing two different markers of earlier-stage OL differentiation, 2′,3′-cyclic nucleotide 3′ phosphodiesterase (CNP) and myelin basic protein (MBP), after 4–6 DIV (Figs. 2A–E), and this increase was statistically similar to the increase induced by T3 exposure (Fig. 2E). Interestingly, whereas KLF9 was able to induce early myelin gene expression, it was not similarly able to induce the expression of myelin oligodendrocyte glycoprotein (MOG), a marker of late-stage OL differentiation (Baumann and Pham-Dinh, 2001; Dugas et al., 2006), despite the fact that MOG is induced by T3 exposure (Fig. 2E). It is perhaps not surprising that a single transcription factor, KLF9, is not able to completely recapitulate the complex effects of T3 on OL differentiation. Nonetheless, these data indicate that KLF9, on its own, is able to initiate the process of OL differentiation at least as robustly as T3 when expressed in immature OPCs.
Fig. 2.
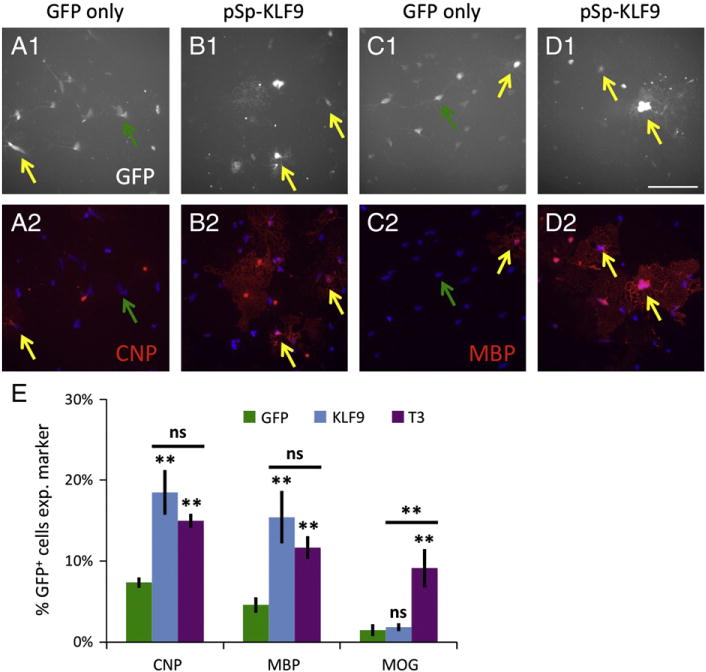
KLF9 promotes early myelin gene expression. A–D. OPCs transfected with pC1-eGFP alone (A, C) or plus pSp-KLF9 (B, D) were incubated 4–6 DIV in +PDGF−T3 media, then stained for GFP (A1–D1) (white) and CNP (A2–B2) or MBP (C2–D2) (red). Nuclei stained with DAPI (A2–D2) (blue). Yellow arrows denote transfected GFP+ cells expressing CNP or MBP, green arrows denote transfected GFP+ cells not expressing CNP or MBP; scale bar=100 μm. E. Percentages of transfected, GFP+ cells expressing CNP, MBP, or MOG; GFP and KLF9=data from transfected cells cultured in +PDGF−T3 media, T3 =GFP-only transfected cells cultured in +PDGF + T3 media. All graphs mean±S.E.M., n= 12 samples/condition. ** p< 0.01, ns p> 0.05 post-hoc Holm–Sidak test all pairwise comparisons (below bar comparisons to GFP+PDGF−T3 control, above bar KLF9+PDGF−T3 compared to GFP+PDGF+ T3).
KLF9 is required for normal OL differentiation in vitro
To further determine whether KLF9 is necessary for T3-induced OL differentiation, we next utilized siRNA to knock down KLF9 expression in cultured OPCs. OPCs were transfected with pC1-eGFP to mark transfected cells, and co-transfected with either negative control non-targeting siRNAs (siControl), or a pool of siRNAs targeting KLF9 expression (siKLF9). Transfected cells were then cultured in mitogen-rich media plus T3 for 7 DIV, and subsequently immunostained for markers of OL differentiation (Figs. 3A–H). We found that knock down of KLF9 produced a ~50% reduction in the expression of every OL gene examined, from a marker of initial OL differentiation, CC1 (an antibody against adenomatosis polyposis coli), to the late stage marker MOG (Figs. 3C–I). In addition, we found that knock down of KLF9 not only repressed mature OL gene expression, but also significantly increased the number of transfected cells that expressed chondroitin sulfate proteoglycan 4 (CSPG4, stained by NG2 antibody), a marker of undifferentiated OPCs (Figs. 3A–B, I). Cumulatively, these data indicate that blocking the induction of KLF9 significantly impairs the ability of T3 to induce OL differentiation.
Fig. 3.
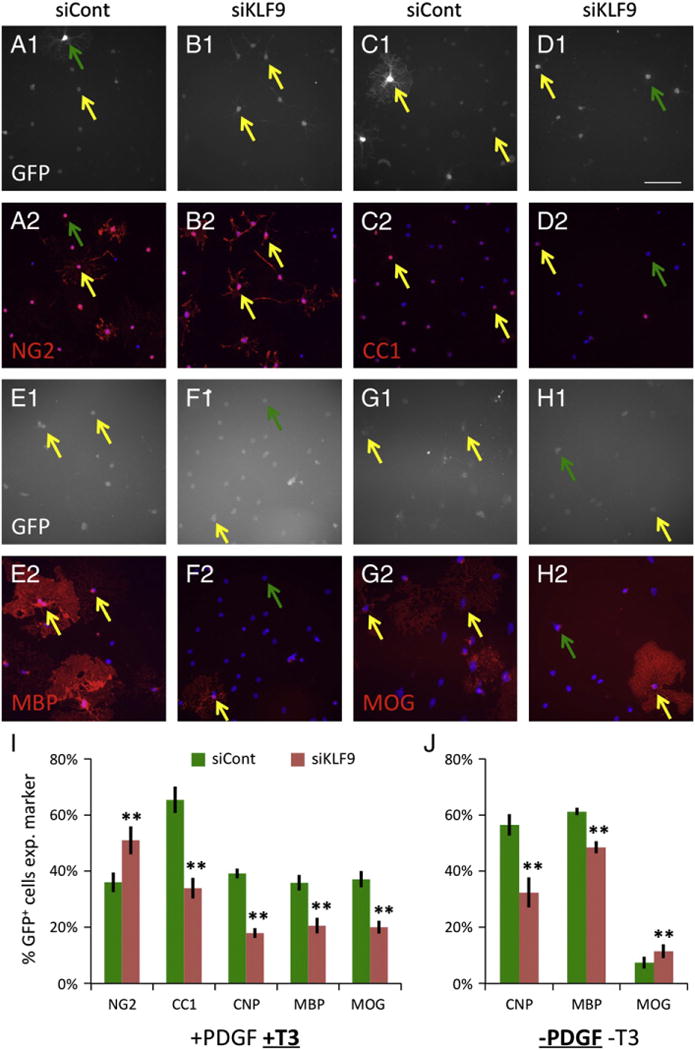
KLF9 is required for T3-induced OL differentiation. A–H. OPCs transfected with pC1-eGFP plus siControl (A, C, E, G) or plus siKLF9 pool (B, D, F, H) were incubated7 DIVin +PDGF+T3 media, then stained for GFP (A1–H1) (white) and NG2 (A2–B2), CC1 (C2–D2), MBP (E2–F2), or MOG (G2–H2) (red). Nuclei stained with DAPI (A2–H2) (blue). Note that NG2+ cells are identified by staining of processes, as the antibody stains the perinuclear regions of both immature OPCs and mature OLs. Yellow arrows denote transfected GFP+ cells co-expressing indicated markers, green arrows denote transfected GFP+ cells not co-expressing indicated markers; scale bar=100 μm. I–J. Percentages of transfected, GFP+ cells expressing indicated markers after 7 DIV in +PDGF+T3 media (I) or 3–4 DIV in −PDGF−T3 media (J). All graphs mean±S.E.M., n=6–14 samples/condition. ** p < 0.001 paired T-test siKLF9 vs. control.
To confirm that the pool of siRNAs targeting KLF9 was not repressing OL differentiation by inappropriate knock down of off-target genes, we transfected OPCs with the individual siRNAs designed to repress KLF9 expression. We found that two of the individual components of the siKLF9 pool, siKLF9 #1 and #3, were both able to repress KLF9 expression, and both significantly repressed the ability of T3 to induce OL differentiation (Fig. 4). As siKLF9 #1 and #3 target distinct, non-overlapping regions of KLF9, it is unlikely that they would have any commonly repressed targets other than KLF9. These data therefore suggest that the reduced OL differentiation phenotypes we observe result directly from repression of KLF9 expression by siKLF9.
Fig. 4.
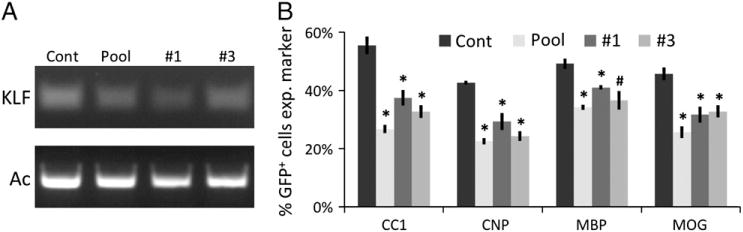
Individual siRNAs repress KLF9 expression and OL differentiation. A. RT-PCR (30 cycles) to determine KLF9 expression levels in OPCs transfected with siControl, pooled siKLF9 #1–4, only siKLF9 #1, or only siKLF9 #3 cultured in +PDGF+T3 media 3 DIV. RT-PCR (27 cycles) to detect actin levels to confirm equivalent starting material also shown. B. Percentages of transfected cells expressing indicated markers after 7 DIV in +PDGF+T3 media. All graphs mean±S.E.M., n=3 samples/condition. * p < 0.01, # p < 0.05 Holm–Sidak post-hoc test vs. control.
Finally, although we found that KLF9 was most prominently induced by T3 exposure, we did note that mitogen withdrawal was also able to induce KLF9 expression, albeit weakly (Fig. 1F). We were therefore interested in determining whether repression of KLF9 expression could also impair OL differentiation induced by mitogen withdrawal. OPCs were transfected as described above, with either siControl or siKLF9, and cultured in mitogen-free media (−PDGF−T3) for 3 DIV. We observed that siKLF9 significantly repressed CNP and MBP expression induced by mitogen withdrawal, although the extent of repression was weaker than seen during T3-induced differentiation: CNP/MBP were 54%/43% repressed by siKLF9 in +PDGF+T3 media, and only 42%/21% repressed by siKLF9 in −PDGF−T3 media (Fig. 4J). We also found that siKLF9 failed to repress expression of the late-stage marker MOG in mitogen-free media. Altogether, these data indicate that KLF9 is a pro-differentiation factor in OLs, and that KLF plays a more prominent role in promoting T3-induced differentiation than mitogen-withdrawal induced differentiation, consistent with the relatively stronger upregulation of KLF9 expression by T3.
KLF9 is not required for normal myelin development in vivo
Having found that the T3-induced gene KLF9 is both necessary to transduce the pro-differentiation effects of T3 in OPCs and sufficient to initiate the normal program of OL differentiation in vitro, we next wanted to determine whether KLF9 is also required for the normal development of myelin in vivo. To address this question, we utilized previously generated KLF9 knockout mice, in which a LacZ insertion disrupts the KLF9 coding region (Morita et al., 2003). KLF9−/− mice are viable and fertile, with no overt morphological defects, but subtle behavioral, uterine, intestinal, and hippocampal defects have been reported in these mice (Morita et al., 2003; Scobie et al., 2009; Simmen et al., 2004, 2007; Velarde et al., 2005). However, the state of myelination in these animals had not previously been assessed. To determine whether loss of KLF9 impairs normal myelin development, we compared the levels of myelination in various CNS structures in KLF9−/− and wild type littermate controls at P10–12, P17– 18, and P30–35. Surprisingly, we were unable to detect a significant delay in the production of myelin in mutant mice (Fig. 5). By staining for the expression of MBP, which is concentrated in mature myelin in vivo, we observed that myelination appeared to proceed normally in the densely myelinated corpus callosum and cerebellum. Similar results were obtained when myelination levels were quantified by analyzing Fluoromyelin staining, which detects fully compacted myelin sheaths (data not shown). To further confirm this result, we analyzed mature myelin protein expression by western blot, and although some myelin genes demonstrated a slightly reduced expression in the KLF9−/− CNS, no significant differences in the expression of CNP, MBP, Transferrin (Trf), or MOG in either the cerebellum or cortices of mutant vs. control littermate animals were detected at either P12 or P18 (Figs. 6A–C). Similarly, quantification of differentiated OL cell bodies, identified by in situ hybridization for Proteolipid protein (PLP) or Trf expression, also failed to detect a significant reduction in mature OL generation in KLF9−/− mice at P12 or P17 (Figs. 6D–F). Cumulatively, these data indicate that KLF9 does not appear to play a prominent role in the generation of mature OLs or myelin during normal development.
Fig. 5.
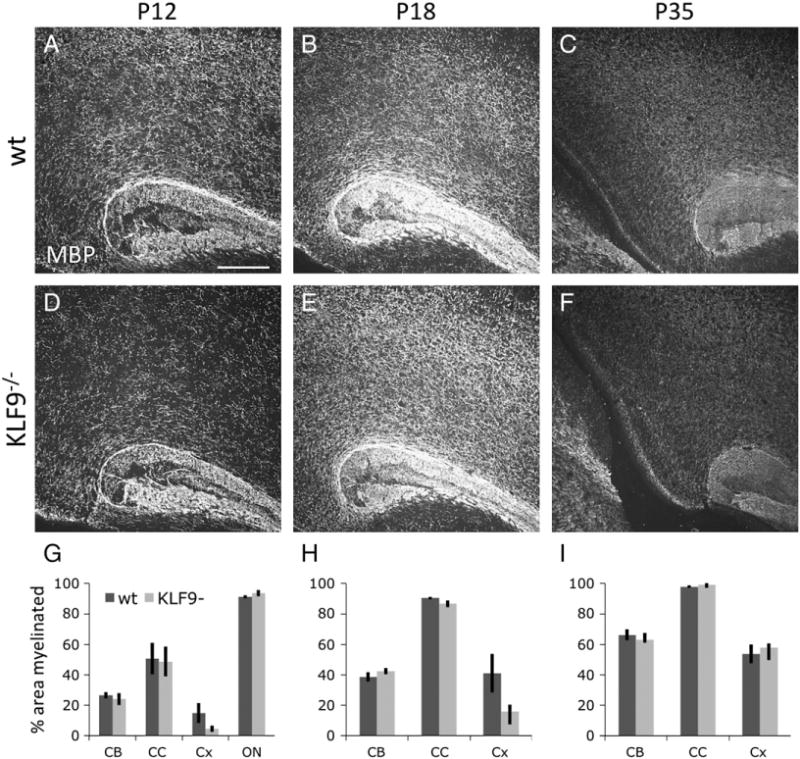
KLF9−/− mice do not show delayed cortical myelination. A–F. Similar depth sagittal brain sections from P12 (A, D), P18 (B, E), and P35 (C, F) KLF9−/− (D–F) and wt littermate controls (A–C), showing the caudal corpus callosum and overlying cortex, stained for MBP expression; scale bar=250 μm. G–I. Quantification of percentage area myelinated (by MBP staining) in P10–12 (G), P17–18 (H) and P30–35 (I) KLF9−/− and matched littermate control cerebellar arms (CB), corpus callosum (CC), cortex (Cx), and optic nerve (ON). All graphs mean±S.E.M., n=4–5 mice/condition. No significant differences detected by T-tests in any of the comparisons between wt and KLF9−/− groups.
Fig. 6.
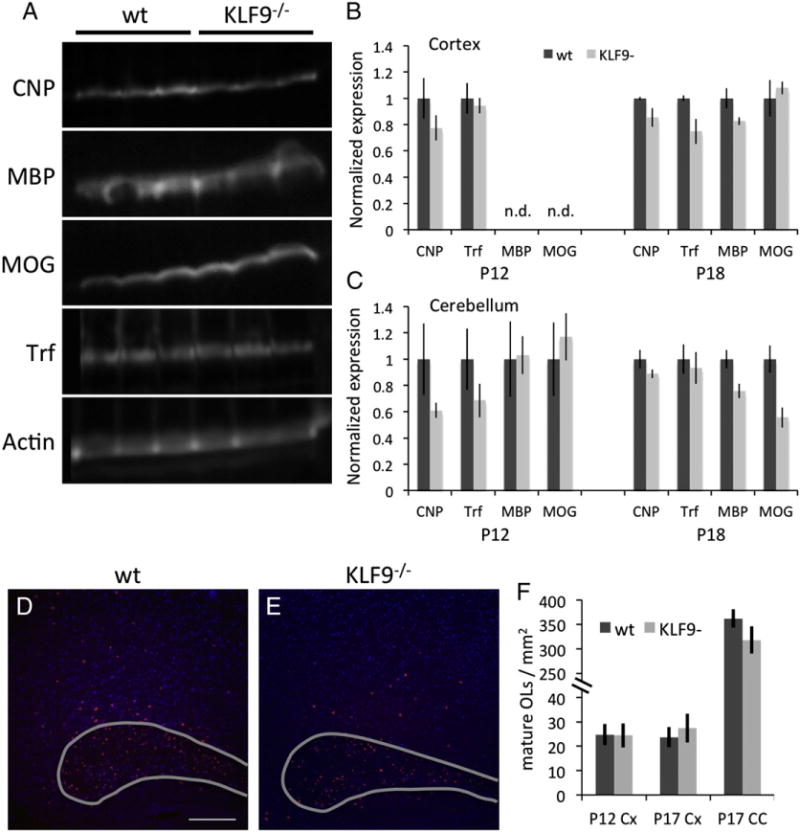
KLF9−/− mice do not show delayed OL differentiation in vivo. A. Western blots showing cortical expression of CNP, MBP, MOG, Trf, and Actin in 3 × P18 KLF9−/− or wt littermate control mice. B–C. Quantification of indicated protein expression from the cortex (B) or cerebellum (C) of P12 and P18 KLF9−/− or wt littermate control mice. Expression levels expressed as ratios to loading control actin expression in the same lane for each sample, then normalized to average wt expression levels in the indicated age and tissue. All graphs mean±S.E.M.; n=3–5 samples/condition; n.d. = not detected. No significant differences detected by T-tests followed by Sidak correction for multiple comaparisons in any of the comparisons between wt and KLF9−/− groups. E–F. In situs to detect Trf expression (red) reveal mature OL cell bodies in the caudal corpus callosum (outlined in grey) and overlying cortex in P17 KLF9−/− or wt littermate control sagittal brain sections. Nuclei stained by DAPI (blue); scale bar=250 μm. F. Quantification of mature OLs/mm2 by PLP expression (P12) or Trf expression (P17) in the cortex (Cx) or corpus callosum (CC) of KLF9−/− or wt littermate control mice. All graphs mean±S.E.M.; n=4–5 mice/condition. No significant differences detected by T-tests in any of the comparisons between wt and KLF9−/− groups. Similarly, no significant differences were observed in the P12 corpus callosum with PLP in situ probe or the P12 corpus callosum with the Trf in situ probe, data not shown.
KLF9 is required for normal myelin regeneration in vivo
A key component to the etiology of demyelinating diseases such as MS is a failure of the CNS’s normally robust ability to regenerate lost myelin. As several labs have recently observed that artificially increasing T3 levels can enhance myelin regeneration in demyelinating disease models in rodents (Calza et al., 2005; Franco et al., 2008; Harsan et al., 2008), we wanted to as certain whether T3-induced KLF9 expression was required for normal levels of remyelination after myelin loss. To examine this, we utilized the widely used cuprizone model of demyelination, wherein supplying a low level of cuprizone in the diet for 6 weeks produces a loss of myelin in the corpus callosum and overlying cortex (Franco et al., 2008; Matsushima and Morell, 2001; Skripuletz et al., 2008). Subsequent to this, withdrawal of cuprizone from the diet allows regeneration of lost myelin to commence over a period of several weeks. To assess the role of KLF9 in promoting remyelination, we compared the ability of KLF9−/− and wt littermate mice to recover from cuprizone-induced demyelination. We found that a 6 week course of cuprizone feeding produced a similar level of demyelination in the cortex (Figs. 7A–C, H) and corpus callosum (Figs. 8A–C, H) in KLF9−/− and wt mice. To assess the extent of remyelination in the cortex, we quantified the area immunostained for MBP expression. These analyses revealed a significant delay in cortical remyelination at both 1 and 2 weeks following removal of cuprizone from the diet (Figs. 7D–H). To assess remyelination capacity in the densely myelinated corpus callosum, we quantified the area that was robustly stained by the compact myelin stain Fluoromyelin. Consistent with the observation in the cortex, loss of KLF9−/− produced a robust delay in the regeneration of myelin in the corpus callosum (Figs. 8D–H). These data indicate that KLF9, and by extension T3, may play a more prominent role in promoting the regeneration of lost myelin than in inducing the developmental production of myelin.
Fig. 7.
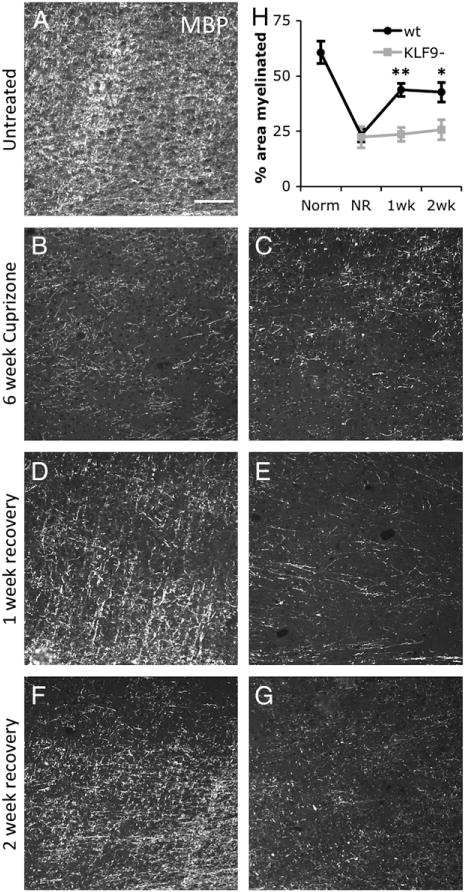
KLF9−/− mice show delayed myelin regeneration in the cortex. A–G. Cortex immediately dorsal to the mid corpus callosum/fornix in sagittal brain sections from wt (A, B, D, F) and KLF9−/− (C, E, G) 3 month old untreated mice (A), 6 week cuprizone treated (B–C), and 6 week cuprizone +1 week (D–E) or +2 week (F–G) recovery. All stained with MBP; scale bar=100 μm. H. Quantification of percentage area of the cortex myelinated (by MBP staining) in untreated (Norm), 6-week cuprizone treated (NR=no recovery), and 1 or 2 week post-cuprizone recovery in wt and KLF9−/− mice. All graphs mean±S.E.M., n=4–5 mice/condition. ** p<0.0005, * p<0.02 T-test KLF9−/− vs. control.
Fig. 8.
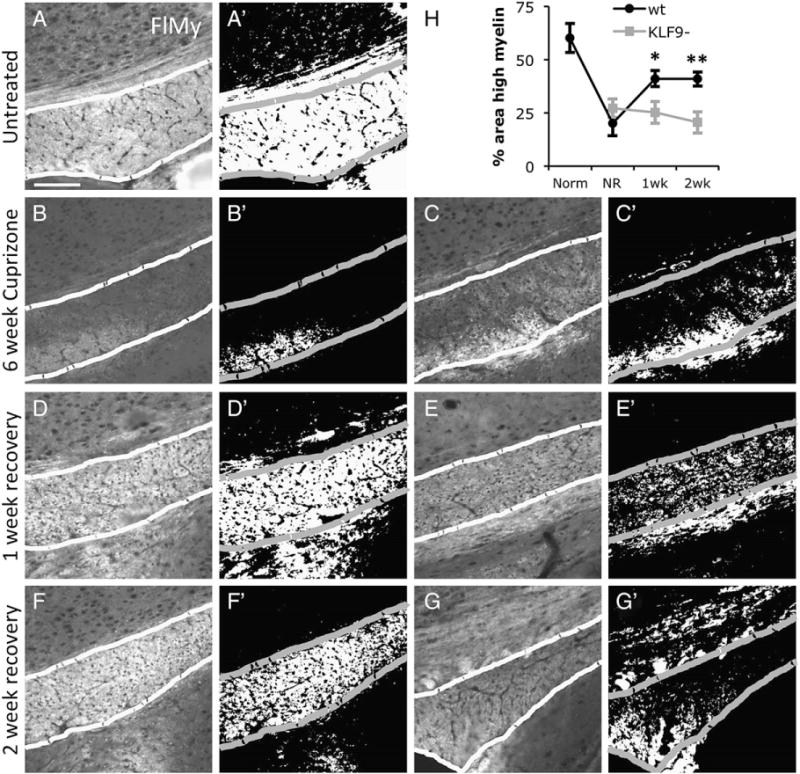
KLF9−/− mice show delayed myelin regeneration in the corpus callosum. A–G. Mid corpus callosum (outlined) in sagittal brain sections from wt (A, B, D, F) and KLF9−/− (C, E, G) 3 month old untreated mice (A), 6 week cuprizone treated (B–C), and 6 week cuprizone +1 week (D–E) or +2 week (F–G) recovery. All stained with Fluoromyelin; scale bar=100 μm. A′–G′. Thresholded images showing pixels >50 units brighter than non-specific tissue background in images A–G (see Experimental methods section). H. Quantification of percentage area of the corpus callosum highly myelinated (by strong Fluoromyelin staining >50 units brighter than background) in untreated (Norm), 6-week cuprizone treated (NR=no recovery), and 1 or 2 week post-cuprizone recovery in wt and KLF9−/− mice. All graphs mean±S.E.M., n=4–9 mice/condition. ** p<0.005, * p<0.02 T-test KLF9−/− vs. control.
Acknowledgments
We would liketothank Dr. Rosalia Simmen for generously providing the KLF9LacZ/+ mice. This work was supported by the Myelin Repair Foundation and the National Multiple Sclerosis Society (RG4059A8).
Abbreviations
- T3
thyroid hormone
- KLF9
Kruppel-like factor 9
- BHLHe22
basic helix-loop-helix family member e22
- Hr
hairless
- DBP
albumin D box-binding protein
- OL
oligodendrocyte
- OPC
oligodendrocyte precursor cell
- PDGF
platelet derived growth factor
- FGF
fibroblast growth factor
- P#
postnatal day (#) (referring to age of mouse or rat pups)
- NG2
nerve/glial antigen 2/chondroitin sulfate proteoglycan 4
- CNP
2′,3′-cyclic nucleotide 3′ phosphodiesterase
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- GFP
green fluorescent protein
- CC1
adenomatosis polyposis coli antibody
- Trf
transferring
- PLP
proteolipid protein
- DIV
days in vitro
- SOX10
SRY-box containing gene 10
- OLIG1/2
oligodendrocyte transcription factor ½
- MRF
myelin gene regulatory factor
References
- Albrecht PJ, Murtie JC, Ness JK, Redwine JM, Enterline JR, Armstrong RC, Levison SW. Astrocytes produce CNTF during the remyelination phase of viral-induced spinal cord demyelination to stimulate FGF-2 production. Neurobiol Dis. 2003;13:89–101. doi: 10.1016/s0969-9961(03)00019-6. [DOI] [PubMed] [Google Scholar]
- Arnett HA, Fancy SP, Alberta JA, Zhao C, Plant SR, Kaing S, Raine CS, Rowitch DH, Franklin RJ, Stiles CD. bHLH transcription factor Olig1 is required to repair demyelinated lesions in the CNS. Science. 2004;306:2111–2115. doi: 10.1126/science.1103709. [DOI] [PubMed] [Google Scholar]
- Barradas PC, Vieira RS, De Freitas MS. Selective effect of hypothyroidism on expression of myelin markers during development. J Neurosci Res. 2001;66:254–261. doi: 10.1002/jnr.1218. [DOI] [PubMed] [Google Scholar]
- Barres BA, Hart IK, Coles HS, Burne JF, Voyvodic JT, Richardson WD, Raff MC. Cell death and control of cell survival in the oligodendrocyte lineage. Cell. 1992;70:31–46. doi: 10.1016/0092-8674(92)90531-g. [DOI] [PubMed] [Google Scholar]
- Barres BA, Schmid R, Sendnter M, Raff MC. Multiple extracellular signals are required for long-term oligodendrocyte survival. Development. 1993;118:283–295. doi: 10.1242/dev.118.1.283. [DOI] [PubMed] [Google Scholar]
- Barres BA, Lazar MA, Raff MC. A novel role for thyroid hormone, glucocorticoids and retinoic acid in timing oligodendrocyte development. Development. 1994;120:1097–1108. doi: 10.1242/dev.120.5.1097. [DOI] [PubMed] [Google Scholar]
- Baumann N, Pham-Dinh D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol Rev. 2001;81:871–927. doi: 10.1152/physrev.2001.81.2.871. [DOI] [PubMed] [Google Scholar]
- Bonett RM, Hu F, Bagamasbad P, Denver RJ. Stressor and glucocorticoid-dependent induction of the immediate early gene kruppel-like factor 9: implications for neural development and plasticity. Endocrinology. 2009;150:1757–1765. doi: 10.1210/en.2008-1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calver AR, Hall AC, Yu WP, Walsh FS, Heath JK, Betsholtz C, Richardson WD. Oligodendrocyte population dynamics and the role of PDGF in vivo. Neuron. 1998;20:869–882. doi: 10.1016/s0896-6273(00)80469-9. [DOI] [PubMed] [Google Scholar]
- Calza L, Fernandez M, Giuliani A, D’Intino G, Pirondi S, Sivilia S, Paradisi M, Desordi N, Giardino L. Thyroid hormone and remyelination in adult central nervous system: a lesson from an inflammatory-demyelinating disease. Brain Res. 2005;48:339–346. doi: 10.1016/j.brainresrev.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Denver RJ, Williamson KE. Identification of a thyroid hormone response element in the mouse Kruppel-like factor 9 gene to explain its postnatal expression in the brain. Endocrinology. 2009;150:3935–3943. doi: 10.1210/en.2009-0050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver RJ, Ouellet L, Furling D, Kobayashi A, Fujii-Kuriyama Y, Puymirat J. Basic transcription element-binding protein (BTEB) is a thyroid hormone-regulated gene in the developing central nervous system. Evidence for a role in neurite outgrowth. J Biol Chem. 1999;274:23128–23134. doi: 10.1074/jbc.274.33.23128. [DOI] [PubMed] [Google Scholar]
- Dugas JC, Tai YC, Speed TP, Ngai J, Barres BA. Functional genomic analysis of oligodendrocyte differentiation. J Neurosci. 2006;26:10967–10983. doi: 10.1523/JNEUROSCI.2572-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, Barres BA. Dicer1 and miR-219 are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65:597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery B, Agalliu D, Cahoy JD, Watkins TA, Dugas JC, Mulinyawe SB, Ibrahim A, Ligon KL, Rowitch DH, Barres BA. Myelin gene regulatory factor is a critical transcriptional regulator required for CNS myelination. Cell. 2009;138:172–185. doi: 10.1016/j.cell.2009.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Giuliani A, Pirondi S, D’Intino G, Giardino L, Aloe L, Levi-Montalcini R, Calza L. Thyroid hormone administration enhances remyelination in chronic demyelinating inflammatory disease. Proc Natl Acad Sci U S A. 2004;101:16363–16368. doi: 10.1073/pnas.0407262101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira AA, Nazario JC, Pereira MJ, Azevedo NL, Barradas PC. Effects of experimental hypothyroidism on myelin sheath structural organization. J Neurocytol. 2004;33:225–231. doi: 10.1023/b:neur.0000030697.78488.63. [DOI] [PubMed] [Google Scholar]
- Ferreira AA, Pereira MJ, Manhaes AC, Barradas PC. Ultrastructural identification of oligodendrocyte/myelin proteins in corpus callosum of hypothyroid animals. Int J Dev Neurosci. 2007;25:87–94. doi: 10.1016/j.ijdevneu.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Figueiredo BC, Almazan G, Ma Y, Tetzlaff W, Miller FD, Cuello AC. Gene expression in the developing cerebellum during perinatal hypo- and hyperthyroidism. Brain Res. 1993;17:258–268. doi: 10.1016/0169-328x(93)90010-m. [DOI] [PubMed] [Google Scholar]
- Franco PG, Silvestroff L, Soto EF, Pasquini JM. Thyroid hormones promote differentiation of oligodendrocyte progenitor cells and improve remyelination after cuprizone-induced demyelination. Exp Neurol. 2008 Aug;212(2):458–467. doi: 10.1016/j.expneurol.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Fruttiger M, Karlsson L, Hall AC, Abramsson A, Calver AR, Bostrom H, Willetts K, Bertold CH, Heath JK, Betsholtz C, Richardson WD. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development. 1999;126:457–467. doi: 10.1242/dev.126.3.457. [DOI] [PubMed] [Google Scholar]
- Furlow JD, Kanamori A. The transcription factor basic transcription element-binding protein 1 is a direct thyroid hormone response gene in the frog Xenopus laevis. Endocrinology. 2002;143:3295–3305. doi: 10.1210/en.2002-220126. [DOI] [PubMed] [Google Scholar]
- Gupta RK, Bhatia V, Poptani H, Gujral RB. Brain metabolite changes on in vivo proton magnetic resonance spectroscopy in children with congenital hypothyroidism. J Pediatr. 1995;126:389–392. doi: 10.1016/s0022-3476(95)70454-x. [DOI] [PubMed] [Google Scholar]
- Harsan LA, Steibel J, Zaremba A, Agin A, Sapin R, Poulet P, Guignard B, Parizel N, Grucker D, Boehm N, Miller RH, Ghandour MS. Recovery from chronic demyelination by thyroid hormone therapy: myelinogenesis induction and assessment by diffusion tensor magnetic resonance imaging. J Neurosci. 2008;28:14189–14201. doi: 10.1523/JNEUROSCI.4453-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey CB, Williams GR. Mechanism of thyroid hormone action. Thyroid. 2002;12:441–446. doi: 10.1089/105072502760143791. [DOI] [PubMed] [Google Scholar]
- Hinks GL, Franklin RJ. Distinctive patterns of PDGF-A, FGF-2, IGF-I, and TGF-beta1 gene expression during remyelination of experimentally-induced spinal cord demyelination. Mol Cell Neurosci. 1999;14:153–168. doi: 10.1006/mcne.1999.0771. [DOI] [PubMed] [Google Scholar]
- Hoopfer ED, Huang L, Denver RJ. Basic transcription element binding protein is a thyroid hormone-regulated transcription factor expressed during metamorphosis in Xenopus laevis. Dev Growth Differ. 2002;44:365–381. doi: 10.1046/j.1440-169x.2002.00650.x. [DOI] [PubMed] [Google Scholar]
- Ibarrola N, Rodriguez-Pena A. Hypothyroidism coordinately and transiently affects myelin protein gene expression in most rat brain regions during postnatal development. Brain Res. 1997;752:285–293. doi: 10.1016/s0006-8993(96)01480-1. [DOI] [PubMed] [Google Scholar]
- Jagannathan NR, Tandon N, Raghunathan P, Kochupillai N. Reversal of abnormalities of myelination by thyroxine therapy in congenital hypothyroidism: localized in vivo proton magnetic resonance spectroscopy (MRS) study. Brain Res. 1998;109:179–186. doi: 10.1016/s0165-3806(98)00081-9. [DOI] [PubMed] [Google Scholar]
- Leung WM, Nathaniel VE, Nathaniel EJ. A morphological and morphometric analysis of the optic nerve in the hypothyroid rat. Exp Neurol. 1992;117:51–58. doi: 10.1016/0014-4886(92)90110-c. [DOI] [PubMed] [Google Scholar]
- Malone MJ, Rosman NP, Szoke M, Davis D. Myelination of brain in experimental hypothyroidism. An electron-microscopic and biochemical study of purified myelin isolates. J Neurol Sci. 1975;26:1–11. doi: 10.1016/0022-510x(75)90108-2. [DOI] [PubMed] [Google Scholar]
- Marta CB, Adamo AM, Soto EF, Pasquini JM. Sustained neonatal hyperthyroidism in the rat affects myelination in the central nervous system. J Neurosci Res. 1998;53:251–259. doi: 10.1002/(SICI)1097-4547(19980715)53:2<251::AID-JNR14>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Martel J, Cayrou C, Puymirat J. Identification of new thyroid hormone-regulated genes in rat brain neuronal cultures. Neuroreport. 2002;13:1849–1851. doi: 10.1097/00001756-200210280-00003. [DOI] [PubMed] [Google Scholar]
- Matsushima GK, Morell P. The neurotoxicant, cuprizone, as a model to study demyelination and remyelination in the central nervous system. Brain Pathol. 2001;11:107–116. doi: 10.1111/j.1750-3639.2001.tb00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Tripathi RB. The life, death, and replacement of oligodendrocytes in the adult CNS. J Neurochem. 2008;107:1–19. doi: 10.1111/j.1471-4159.2008.05570.x. [DOI] [PubMed] [Google Scholar]
- Mitchell DL, DiMario JX. Bimodal, reciprocal regulation of fibroblast growth factor receptor 1 promoter activity by BTEB1/KLF9 during myogenesis. Mol Biol Cell. 2010;21:2780–2787. doi: 10.1091/mbc.E10-04-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore CS, Abdullah SL, Brown A, Arulpragasam A, Crocker SJ. How factors secreted from astrocytes impact myelin repair. J Neurosci Res. 2011;89:13–21. doi: 10.1002/jnr.22482. [DOI] [PubMed] [Google Scholar]
- Morita M, Kobayashi A, Yamashita T, Shimanuki T, Nakajima O, Takahashi S, Ikegami S, Inokuchi K, Yamashita K, Yamamoto M, Fujii-Kuriyama Y. Functional analysis of basic transcription element binding protein by gene targeting technology. Mol Cell Biol. 2003;23:2489–2500. doi: 10.1128/MCB.23.7.2489-2500.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussa GC, Mussa F, Bretto R, Zambelli MC, Silvestro L. Influence of thyroid in nervous system growth. Minerva Pediatr. 2001;53:325–353. [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Baron-Van Evercooren A. The role of SVZ-derived neural precursors in demyelinating diseases: from animal models to multiple sclerosis. J Neurol Sci. 2008;265:26–31. doi: 10.1016/j.jns.2007.09.032. [DOI] [PubMed] [Google Scholar]
- Noguchi T, Sugisaki T. Hypomyelination in the cerebrum of the congenitally hypothyroid mouse (hyt) J Neurochem. 1984;42:891–893. doi: 10.1111/j.1471-4159.1984.tb02768.x. [DOI] [PubMed] [Google Scholar]
- Oetting A, Yen PM. New insights into thyroid hormone action. Best Pract Res Clin Endocrinol Metab. 2007;21:193–208. doi: 10.1016/j.beem.2007.04.004. [DOI] [PubMed] [Google Scholar]
- Pei H, Yao Y, Yang Y, Liao K, Wu JR. Kruppel-like factor KLF9 regulates PPARgamma transactivation at the middle stage of adipogenesis. Cell Death Differ. 2011;18:315–327. doi: 10.1038/cdd.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pombo PM, Ibarrola N, Alonso MA, Rodriguez-Pena A. Thyroid hormone regulates the expression of the MAL proteolipid, a component of glycolipid-enriched membranes, in neonatal rat brain. J Neurosci Res. 1998;52:584–590. doi: 10.1002/(SICI)1097-4547(19980601)52:5<584::AID-JNR10>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Redwine JM, Armstrong RC. In vivo proliferation of oligodendrocyte progenitors expressing PDGFalphaR during early remyelination. J Neurobiol. 1998;37:413–428. doi: 10.1002/(sici)1097-4695(19981115)37:3<413::aid-neu7>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Reynolds R, Dawson M, Papadopoulos D, Polito A, Di Bello IC, Pham-Dinh D, Levine J. The response of NG2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J Neurocytol. 2002;31:523–536. doi: 10.1023/a:1025747832215. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pena A, Ibarrola N, Iniguez MA, Munoz A, Bernal J. Neonatal hypothyroidism affects the timely expression of myelin-associated glycoprotein in the rat brain. J Clin Invest. 1993;91:812–818. doi: 10.1172/JCI116301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scobie KN, Hall BJ, Wilke SA, Klemenhagen KC, Fujii-Kuriyama Y, Ghosh A, Hen R, Sahay A. Kruppel-like factor 9 is necessary for late-phase neuronal maturation in the developing dentate gyrus and during adult hippocampal neurogenesis. J Neurosci. 2009;29:9875–9887. doi: 10.1523/JNEUROSCI.2260-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmen RC, Eason RR, McQuown JR, Linz AL, Kang TJ, Chatman L, Jr, Till SR, Fujii-Kuriyama Y, Simmen FA, Oh SP. Subfertility, uterine hypoplasia, and partial progesterone resistance in mice lacking the Kruppel-like factor 9/basic transcription element-binding protein-1 (Bteb1) gene. J Biol Chem. 2004;279:29286–29294. doi: 10.1074/jbc.M403139200. [DOI] [PubMed] [Google Scholar]
- Simmen FA, Xiao R, Velarde MC, Nicholson RD, Bowman MT, Fujii-Kuriyama Y, Oh SP, Simmen RC. Dysregulation of intestinal crypt cell proliferation and villus cell migration in mice lacking Kruppel-like factor 9. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1757–G1769. doi: 10.1152/ajpgi.00013.2007. [DOI] [PubMed] [Google Scholar]
- Simmen RC, Pabona JM, Velarde MC, Simmons C, Rahal O, Simmen FA. The emerging role of Kruppel-like factors in endocrine-responsive cancers of female reproductive tissues. J Endocrinol. 2010;204:223–231. doi: 10.1677/JOE-09-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skripuletz T, Lindner M, Kotsiari A, Garde N, Fokuhl J, Linsmeier F, Trebst C, Stangel M. Cortical demyelination is prominent in the murine cuprizone model and is strain-dependent. Am J Pathol. 2008;172:1053–1061. doi: 10.2353/ajpath.2008.070850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolt CC, Rehberg S, Ader M, Lommes P, Riethmacher D, Schachner M, Bartsch U, Wegner M. Terminal differentiation of myelin-forming oligodendrocytes depends on the transcription factor Sox10. Genes Dev. 2002;16:165–170. doi: 10.1101/gad.215802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temple S, Raff MC. Clonal analysis of oligodendrocyte development in culture: evidence for a developmental clock that counts cell divisions. Cell. 1986;44:773–779. doi: 10.1016/0092-8674(86)90843-3. [DOI] [PubMed] [Google Scholar]
- Tokumoto YM, Tang DG, Raff MC. Two molecularly distinct intracellular pathways to oligodendrocyte differentiation: role of a p53 family protein. EMBO J. 2001;20:5261–5268. doi: 10.1093/emboj/20.18.5261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velarde MC, Geng Y, Eason RR, Simmen FA, Simmen RC. Null mutation of Kruppel-like factor9/basic transcription element binding protein-1 alters periimplantation uterine development in mice. Biol Reprod. 2005;73:472–481. doi: 10.1095/biolreprod.105.041855. [DOI] [PubMed] [Google Scholar]
- Walters SN, Morell P. Effects of altered thyroid states on myelinogenesis. J Neurochem. 1981;36:1792–1801. doi: 10.1111/j.1471-4159.1981.tb00433.x. [DOI] [PubMed] [Google Scholar]
- Ying M, Sang Y, Li Y, Guerrero-Cazares H, Quinones-Hinojosa A, Vescovi AL, Eberhart CG, Xia S, Laterra J. Kruppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29:20–31. doi: 10.1002/stem.561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng Z, Velarde MC, Simmen FA, Simmen RC. Delayed parturition and altered myometrial progesterone receptor isoform A expression in mice null for Kruppel-like factor 9. Biol Reprod. 2008;78:1029–1037. doi: 10.1095/biolreprod.107.065821. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Anderson DJ. The bHLH transcription factors OLIG2 and OLIG1 couple neuronal and glial subtype specification. Cell. 2002;109:61–73. doi: 10.1016/s0092-8674(02)00677-3. [DOI] [PubMed] [Google Scholar]