

Abstract
Clostridium cellulovorans is an anaerobic, cellulolytic bacterium, capable of effectively degrading and metabolizing various types of substrates, including cellulose, hemicellulose (xylan and galactomannan), and pectin. Among Clostridia, this ability to degrade and metabolize a wide range of hemicellulose and pectin substrates is a unique feature; however, the mechanisms are currently unknown. To clarify the mechanisms of hemicelluloses and pectin recognition and metabolism, we carried out a quantitative proteome analysis of C. cellulovorans cultured with these substrates. C. cellulovorans was cultured in the medium of glucose (control), xylan, galactomannan (Locus bean gum, LBG), or pectin for 36 h. Xylan and galactomannan were used to search for the common recognition mechanisms of hemicellulose, and pectin was used to search for unique recognition systems in C. cellulovorans. Using an isobaric tag method and liquid chromatograph/mass spectrometer equipped with a long monolithic silica capillary column, we identified 734 intracellular proteins from all substrates. We performed KEGG analyses and cluster analyses of the resulting proteins. In the KEGG analyses, we found common degradation mechanisms for hemicellulose and pectin. In the cluster analysis corresponding to the genome analysis, we detected substrate-specific clusters that include genes involved in substrate recognition, substrate degradation, and metabolism. Combining the results of the KEGG analyses and cluster analyses, we propose the mechanisms involved in the recognition and metabolism of hemicellulose and pectin in C. cellulovorans.
Electronic supplementary material
The online version of this article (doi:10.1186/s13568-015-0115-6) contains supplementary material, which is available to authorized users.
Keywords: Clostridium cellulovorans, Proteome analysis, Monolithic column, Substrate recognition, Hemicellulose, Pectin, Metabolism
Introduction
There has been enormous recent interest in biorefinery using non-edible biomass as a source for renewable fuels (Lynd et al. 1999). To achieve this aim, it is necessary to convert various biological substrates into the desired chemicals and fuels (Saha 2003). Terrestrial biomass consists of cellulose, hemicellulose, and pectin (Cosgrove 2005). Cellulose is a polymer of β-glucose that constitutes the main backbone of the plant cell wall, and composes of about 50% of plant cell walls. Hemicellulose forms a primary network with cellulose microfibrils in various conformations, and the hemicellulose binds with pectin. Polysaccharides belonging to hemicellulose can be extracted from plant cell walls with alkali solution. Common hemicellulose polysaccharides include xyloglucan, xylan, and mannan. Pectin is a family of polysaccharides that include homogalacturonan, rhamnogalacturonan-I, and rhamnogalacturonan-II. Polysaccharides belonging to pectin can be extracted with hot water or chelating agents. Homogalacturonan is a major component of pectin. It is believed that pectin crosslinking involves calcium and boron ions (Cosgrove 2005).
Of these three components of the plant cell walls, the degradation of cellulose has been best studied. For example, Clostridium thermocellum can metabolize cellulose and is used for the degradation of cellulose to glucose (Prawitwong et al. 2013). C. thermocellum belongs to the Family 4 (Cluster III) of Clostridia (Rainey and Stackebrandt 1993; Collins et al. 1994), and genetically engineered strains of C. cellulolyticum can metabolize cellulose and xylan among hemicellulose, and are used for producing isobutanol from cellulose (Higashide et al. 2011). However, available strains cannot utilize all forms of hemicellulose and pectin. For maximally efficient biorefinery, utilization of hemicellulose and pectin is also essential.
Clostridium cellulovorans is a gram-positive, mesophilic, and cellulosome-producing anaerobe of the Family 1 (Cluster I) of Clostridia that can degrade and directly metabolize various kinds of carbohydrates such as cellulose, hemicellulose (xylan and mannan), and pectin (Petitdemange et al. 1984; Tamaru et al. 2010). Furthermore, C. cellulovorans alters its production of secreted enzymes, depending on which substrates are available (Morisaka et al. 2012; Matsui et al. 2013; Esaka et al. 2015). Thus, C. cellulovorans can distinguish among varied substrates and adapt to survive in different environments.
In Clostridia, the systems by which cells recognize different substrates have been widely studied. For example, C. thermocellum was reported to recognize the availability of cellulose through the coordinated action of sigma and anti-sigma factors with cellulose binding domains (Nataf et al. 2010). C. cellulolyticum was reported to recognize cellulose availability with a carbon catabolite repression mechanism and xylan availability with a two-component system (TCS) (Celik et al. 2013; Xu et al. 2013). These findings imply that substrate recognition systems can show great variety between species. However, the substrate recognition systems of C. cellulovorans, which can degrade and directly metabolize a greater variety of substrates than other Clostridia studied, are so far unknown. This suggests that C. cellulovorans may be an attractive model organism for the study of substrate recognition systems.
In the present study, we carried out an “intracellular” proteome analysis using an LC–MS/MS system equipped with a long monolithic silica capillary column (500 cm) (Aoki et al. 2013; Morisaka et al. 2012) in order to elucidate substrate recognition systems of C. cellulovorans. As substrates, we used xylan, galactomannan (locust bean gum; LBG), and pectin. Studies on the recognition and metabolism of galactomannan and pectin have not been performed in cellulosome-producing Clostridia because the commonly used model organisms, C. cellulolyticum and C.thermocellum, are unable to metabolize these substrates. Based on our findings, we propose metabolic systems for each substrate and molecular models of substrate recognition system in C. cellulovorans. We further suggest that hemicellulose and pectin were degraded outside of the cell with constitutively produced enzymes, such as cellulosomal proteins. We could clarify that the TCS response regulator AraC was used in the substrate recognition system common to both xylan and galactomannan. Interestingly, for pectin, C. cellulovorans regulated the hydrolase/isomerase pathway and the lyase/5-dehydro-4-deoxy-gluconate pathway via the Crp transcriptional regulator and the IclR family transcriptional regulator, respectively.
Materials and methods
Cell culture and media
C. cellulovorans 743B (ATCC35296) was grown anaerobically as previously described (Sleat et al. 1984), differing only in the carbon source, which was replaced by 0.3% (w/v) glucose (Nacalai Tesque, Kyoto, Japan), 0.3% (w/v) xylan from beechwood (Sigma, MO, USA), 0.3% (w/v) pectin from apple (Sigma), or 0.3% (w/v) LBG from Caretonia silliqua seeds (Sigma).
Estimating the growth of the anaerobic bacteria
The growth curves of C. cellulovorans on each medium were determined by bacterial protein estimation, as previously described (Raman et al. 2009) with small modifications. Cells from 1 mL of cell culture were collected by centrifugation (13,000×g, 4°C, 10 min). The cell pellets were washed with 1 mL of phosphate-buffered saline (pH 7.4; PBS) and incubated with 800 μL of sodium deoxycholate (2%) for 20 min at 37°C. Two hundreds microliters of trichloroacetic acid (24%) was added to the suspension, which was then centrifuged (13,000×g, 4°C, 10 min). One hundred microliters of resolubilization solution (5% SDS, 2 N NaOH) was added to the suspension and vigorously mixed. The protein concentration was measured using a protein assay bicinchoninate kit (Nacalai Tesque), with bovine serum albumin used as a standard.
Sample preparation for quantitative proteome analysis
C. cellulovorans was grown in 50 mL cultures to late-logarithmic phase (36 h). Cells were concentrated by centrifugation (6,000×g, 4°C, 10 min), and the supernatant was discarded. Cell pellets were collected by centrifugation (13,000×g, 4°C, 10 min) and washed with 500 µL PBS, and centrifuged again. Cells were resuspended with 500 µL of lysis buffer [2% (w/v) 3-(3-cholamidopropyl)dimethylammonio-1-propanesulfonate, 10 mM dithiothreitol, 1% (v/v) protease inhibitor cocktail for bacterial cell lysis (Sigma), 7 M urea, and 2 M thiourea in 50 mM Tris–HCl (Nacalai Tesque)]. The cells were disrupted with a Bioruptor UCD-250T (Cosmo Bio, Tokyo, Japan) at 250 W, 15 s on-and-off cycles for 10 min, on ice. The solution was centrifuged (13,000×g, 4°C, 20 min), and the supernatant was collected and subjected to ultrafiltration with an Amicon Ultra-0.5 Centrifugal Filter Unit (10 kDa, Millipore, MA, USA) and buffer-exchanged with 200 mM triethyl ammonium bicarbonate (TEAB; Sigma). Proteins were reduced by adding tris (2-carboxyethyl) phosphine to 10 mM from a 200 mM stock, and the reaction was allowed to proceed for 60 min at 55°C. Following the incubation, 5 µL of iodoacetamide (375 mM) was added, and the reaction continued for 30 min at room temperature in the dark. Proteins were precipitated by adding 1 mL of ice-cold acetone and incubating the solution overnight at −20°C. The precipitated proteins were resuspended with 100 µL of 200 mM TEAB, 2 µg of sequencing grade modified trypsin (Promega, WI, USA) was added, and incubated overnight at 37°C. The four samples for proteome analysis (glucose, xylan, pectin, and galactomannan) were labeled using a tandem mass tag (TMT) 6-plex labeling kit (Thermo Fisher Scientific, MA, USA) with reporters at m/z = 126, 127, 129, and 130, respectively, in 41 μL acetonitrile. After 60 min at room temperature, 8 μL of 5% (w/v) hydroxylamine was added to each tube and mixed for 15 min. As an internal standard for quantification, a mixture of tryptic fragments from all substrates was combined with TMT-131 (reporter at m/z = 131). Aliquots were pooled and evaporated under vacuum, then dissolved in 60 μL of formic acid (0.1%) and subjected to LC–MS/MS analysis.
LC–MS/MS analysis
Proteome analysis was performed using an LC (Ultimate 3000; Thermo Fisher Scientific)–MS/MS (LTQ Orbitrap Velos Mass Spectrometer Thermo Fisher Scientific) system equipped with a long monolithic silica capillary column. Tryptic digests were separated by reverse-phase chromatography using a monolithic silica capillary column (500 cm long, 0.1 mm ID) (Morisaka et al. 2012), at a flow rate of 500 nL/min. A gradient was achieved by changing the ratio of the two eluents: eluent A, 0.1% (v/v) formic acid; eluent B, 80% acetonitrile containing 0.1% (v/v) formic acid. The gradient started with 5% B, increased to 45% B for 600 min, further increased to 95% B to wash the column for 140 min, returned to the initial condition, and was held for re-equilibration of the column. The separated analytes were detected using a mass spectrometer with a full scan range of 350–1,500 m/z (resolution 60,000), followed by 10 data-dependent higher-energy c-trap dissociation (HCD) MS/MS scans acquired for TMT reporter ions by using 40% normalized collision energy in HCD with 0.1 ms activation time in quantitative proteome analysis and with a full scan range of 350–1,500 m/z (resolution 60,000), followed by 10 data-dependent collision-induced dissociation (CID) MS/MS scans in a qualitative proteome analysis. An electrospray ionization (ESI) voltage was set at 2.3 kV. Triplicate analyses were performed for each sample in three independent experiments, and the collected data were reviewed for protein identification and quantification in a quantitative proteome analysis. Single analyses were performed for each sample of the three independent experiments in a qualitative proteome analysis. The collected data were reviewed for protein identification and quantification. Blank runs were inserted between runs of different samples.
Data analysis
Data analysis was performed using Proteome Discoverer 1.4 (Thermo Fisher Scientific). Protein identification was performed using the Mascot algorithm against the C. cellulovorans protein database (4,254 sequences) from NCBI (National Center for Biotechnology Information, http://www.ncbi.nlm.nih.gov/), with a precursor mass tolerance of 20 ppm and a fragment ion mass tolerance of 50 mmu in quantitative proteome analysis, and mass tolerance of 2.0 Da and a fragment ion mass tolerance of 0.8 Da in qualitative proteome analysis. Carbamidomethylation of cysteine was set as a fixed modification for quantitative proteome analysis, and carbamidomethylation of cysteine and a TMT 6-plex at the N-terminus and lysine were set as fixed modifications for qualitative proteome analysis. Protein quantification was performed using the reporter ions quantifier with the TMT 6-plex method. The data were then filtered with a cut-off criteria of q value ≤0.05, corresponding to a 5% false discovery rate (FDR) on a spectral level. Proteins with no missing values in three replicates were accepted as identified proteins for the protein quantification analysis. In the qualitative proteome analysis, proteins with scores >10 were accepted as identified proteins. Global median normalization was performed to normalize the quantity of each tryptic digest injected into the mass spectrometer. The heat map was generated using Cluster 3.0 (de Hoon et al. 2004), which can carry out hierarchical cluster analysis (HCA). Euclidean distance was used to measure the similarities of the protein profile patterns within the clustering analysis. To visualize the clustering results from Cluster 3.0, Java TreeView software was used (Saldanha 2004). The pathway map was constructed from Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/) (Kanehisa and Goto 2000), and identified proteins were adapted to this map.
Results
To identify substrate recognition systems of C. cellulovorans, we carried out a quantitative proteome analysis of cells grown anaerobically on media with the carbon sources-glucose, xylan, galactomannan (LBG), or pectin. Using a quantitative proteome approach based on isobaric tagging, we obtained protein profiles from cells grown on each carbon source. The workflow of the “intracellular” proteome analysis is illustrated in Figure 1.
Figure 1.
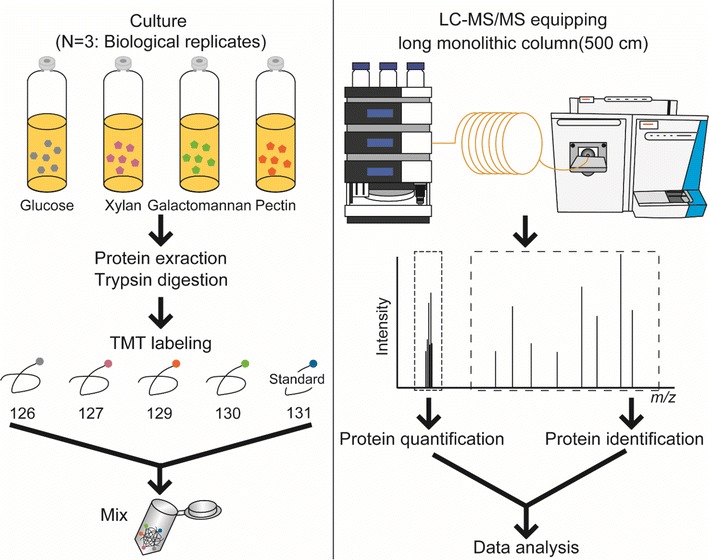
Experimental procedure of C. cellulovorans intracellular quantitative proteome analysis. Proteins prepared from cell lysates of C. cellulovorans grown in the presence of glucose, xylan, galactomannan (LBG), or pectin were individually reductive-alkylated and digested with trypsin. Tryptic fragments were labeled with tandem mass tags (TMTs). The labeled peptides were mixed and injected into the LC–MS/MS system with a long monolithic silica capillary column for mass measurement and collected data were used for protein quantification.
Growth confirmation
To determine the growth of C. cellulovorans on each of the substrates, we conducted bacterial protein estimation (Figure 2). As has previously been shown, C. cellulovorans can grow on xylan, pectin, or galactomannan as the sole carbon source (Sleat et al. 1984), although other cellulosome-producing Clostridia, such as C. thermocellum and C.cellulolyticum, cannot grow on pectin and galactomannan (Petitdemange et al. 1984; Prawitwong et al. 2013). Growth on glucose, xylan, and pectin was slower than that on galactomannan, but cells were collected from all cultures at similar growth phases. From the growth analysis, we selected a culture time of 36 h, as cells were in the late-logarithmic phase, which was appropriate for proteome analysis in terms of growth phase and protein concentration.
Figure 2.
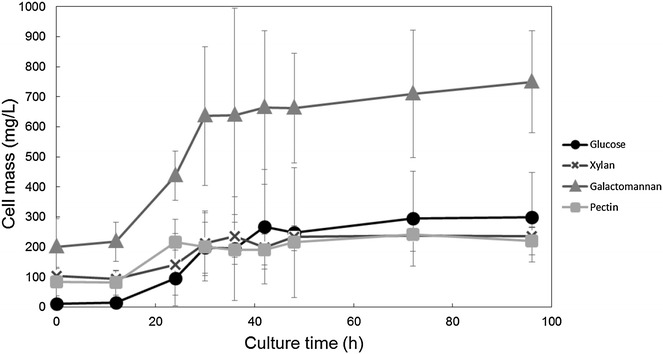
Confirmation of growth of C. cellulovorans cultured with four different substrates. Growth of C. cellulovorans was measured by estimation of protein in cell lysates to determine an appropriate culture time (Raman et al. 2009). At 36 h, C. cellulovorans appears to be in the late-logarithmic phase in all four substrates (glucose, xylan, galactomannan, pectin). These conditions were used for the proteome analysis. Error bars indicate SD (n = 3).
Qualitative and quantitative proteome analysis
LC–MS/MS equipped with a long monolithic silica capillary using a tandem mass tag (TMT) 6-plex isobaric tag column was employed for intracellular quantitative proteome analysis. C. cellulovorans has 4,254 protein-encoding genes in its genome (Tamaru et al. 2010). For protein identification, we constructed a protein database built from the genome of C. cellulovorans. In total, we could identify 734 proteins from all samples within our cutoff criteria (Additional file 1: Table S1). To correct for variations in the amount of TMT-labeled peptides infused into the mass spectrometer, we normalized all quantitative data to the median value of that analysis. For all data analysis, we used these normalized relative quantification values. We checked for reproducibility of quantitative protein profiles among three biological replicates for each substrate by HCA (Figure 3). Each array was clustered individually based on each biological replicate. Our results indicate that each sample was reproducibly quantified, and analytical and biological replication of results was ensured. Furthermore, we separately carried out a qualitative proteome analysis to compare the results of quantitative proteome analysis (Additional file 2: Table S2).
Figure 3.
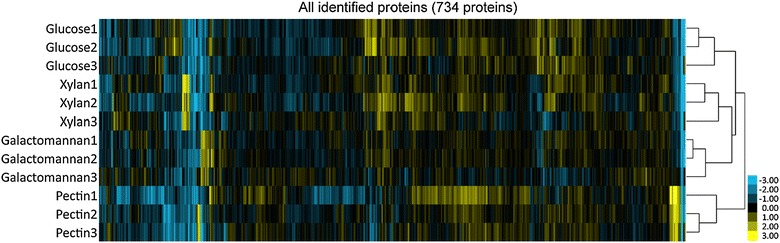
Hierarchical clustering analysis represented proteome profiles of C. cellulovorans with four different substrates. To standardize the data, the quantitative data were normalized using the median for each condition. The quantitative proteome data from three biological replicates of each substrate were used for the hierarchical clustering analysis. Each array was clustered in response to substrates, and each biological replicate was grouped together. This strategy ensures that the proteome analysis was biologically reproducible. Color bar indicates changes in protein abundance. Increased and decreased protein levels are shown in yellow and blue, respectively.
Substrate-specific proteins
To discover substrate-specific proteins (those that showed a significant change between different growth substrates), we carried out an empirical Bayes moderated t test (Smyth 2004). P values were adjusted with the Benjamini–Hochberg method to avoid the problem of multiple testing. The thresholds that we adopted were FDR-adjusted p value of <0.05 and fold-change of protein ratio >2.0 compared to glucose. Proteins for which the levels significantly changed were defined by comparison between glucose and each polysaccharide (xylan, galactomannan, and pectin) at these thresholds. All substrate-specific proteins detected are shown in Table 1. Using KEGG analysis and cluster analysis based on genome analysis, we focused mainly on metabolism-related and substrate recognition-related proteins for further analysis.
Table 1.
Substrate-specific proteins
| Specificity | Locus | Description in NCBI | Description in KEGG | Vs. Glucose | |
|---|---|---|---|---|---|
| Log2 (fold change) | FDR-adjusted p value | ||||
| Xylan | Clocel_0589 | Alpha-l-fucosidase | Alpha-l-fucosidase 2 | 2.67 | 2.07E−04 |
| Clocel_0590 | Xylose isomerase | Xylose isomerase | 3.35 | 1.43E−04 | |
| Clocel_0591 | Transaldolase | Transaldolase | 3.52 | 1.46E−06 | |
| Clocel_0592 | Xylulokinase | Xylulokinase | 2.85 | 7.29E−05 | |
| Clocel_1085 | Dinitrogenase iron–molybdenum cofactor biosynthesis protein | – | 2.32 | 4.24E−03 | |
| Clocel_1151 | Methyl-accepting chemotaxis sensory transducer | Methyl-accepting chemotaxis protein | 1.96 | 1.70E−02 | |
| Clocel_1430 | Glycoside hydrolase family protein | Alpha-d-xyloside xylohydrolase | 1.90 | 9.48E−03 | |
| Clocel_2573 | Hypothetical protein Clocel_2573 | Chemotaxis protein CheX | 1.60 | 3.95E−03 | |
| Clocel_2592 | Two component transcriptional regulator, AraC family | – | 3.72 | 1.49E−04 | |
| Clocel_2595 | Xylan 1,4-beta-xylosidase | Xylan 1,4-beta-xylosidase | 4.70 | 4.53E−06 | |
| Clocel_2596 | Sugar ABC transporter periplasmic protein | Ribose transport system substrate-binding protein | 4.62 | 1.06E−03 | |
| Clocel_2597 | Inner-membrane translocator | Ribose transport system permease protein | 4.01 | 7.36E−05 | |
| Clocel_2598 | ABC transporter | Ribose transport system ATP-binding protein | 4.34 | 1.89E−05 | |
| Clocel_2881 | PTS system lactose/cellobiose-specific transporter subunit IIB | PTS system, cellobiose-specific IIB component | 1.49 | 1.70E−02 | |
| Clocel_2940 | Putative phosphate transport regulator | Hypothetical protein | 1.44 | 2.45E−02 | |
| Clocel_3175 | phoH family protein | phoH-like ATPase | 1.64 | 3.84E−02 | |
| Clocel_3761 | ATP:guanido phosphotransferase | – | 1.31 | 1.37E−02 | |
| Clocel_3762 | UvrB/UvrC protein | – | 1.42 | 3.45E−02 | |
| Clocel_4277 | Aldo/keto reductase | – | 1.08 | 4.66E−02 | |
| Galactomannan | Clocel_0034 | Glycoside hydrolase family protein | Alpha-d-xyloside xylohydrolase | 1.73 | 1.04E−02 |
| Clocel_0391 | Glycosyltransferase | – | 1.83 | 1.96E−02 | |
| Clocel_0684 | Thiazole biosynthesis protein ThiH | 2-Iminoacetate synthase | 1.57 | 4.69E−03 | |
| Clocel_2225 | Cobalamin (vitamin B12) biosynthesis CbiX protein | – | 1.39 | 4.52E−02 | |
| Clocel_2259 | pfkB domain-containing protein | 2-Dehydro-3-deoxygluconokinase | 1.31 | 1.04E−02 | |
| Clocel_2697 | Sialate O-acetylesterase | Sialate O-acetylesterase | 1.69 | 1.69E−02 | |
| Clocel_2800 | Alpha-galactosidase | Alpha-galactosidase | 2.99 | 5.49E−05 | |
| Clocel_2962 | Inosine-5′-monophosphate dehydrogenase | IMP dehydrogenase | 1.31 | 1.96E−02 | |
| Clocel_3175 | phoH family protein | phoH-like ATPase | 1.98 | 1.14E−02 | |
| Clocel_3194 | Mannose-6-phosphate isomerase | Mannose-6-phosphate isomerase | 3.77 | 6.86E−05 | |
| Clocel_3196 | Glycosidase-like protein | Beta-1,4-mannooligosaccharide/beta-1,4-mannosyl-N-acetylglucosamine phosphorylase | 2.65 | 5.29E−03 | |
| Clocel_3198 | N-acylglucosamine 2-epimerase | Mannobiose 2-epimerase | 3.19 | 2.10E−03 | |
| Clocel_3200 | Binding-protein-dependent transport system inner membrane protein | Multiple sugar transport system permease protein | 4.76 | 8.61E−04 | |
| Clocel_3201 | Extracellular solute-binding protein | Raffinose/stachyose/melibiose transport system substrate-binding protein | 4.23 | 5.49E−05 | |
| Clocel_3205 | Glycoside hydrolase family 2 | Beta-mannosidase | 2.55 | 1.51E−03 | |
| Clocel_3657 | Xylan 1,4-beta-xylosidase | Xylan 1,4-beta-xylosidase/alpha-N-arabinofuranosidase | 1.79 | 4.26E−02 | |
| Clocel_3857 | ABC transporter | Multiple sugar transport system ATP-binding protein | 2.15 | 1.03E−03 | |
| Clocel_4053 | LPXTG-motif cell wall anchor domain-containing protein | Sialate O-acetylesterase | 2.28 | 3.79E−02 | |
| Clocel_4087 | Aldose 1-epimerase | Aldose 1-epimerase | 1.45 | 2.68E−02 | |
| Clocel_4088 | Galactose-1-phosphate uridylyltransferase | UDP-glucose–hexose-1-phosphate uridylyltransferase | 3.38 | 1.83E−04 | |
| Clocel_4089 | UDP-glucose 4-epimerase | UDP-glucose 4-epimerase | 2.09 | 1.82E−03 | |
| Clocel_4277 | Aldo/keto reductase | – | 1.08 | 4.26E−02 | |
| Pectin | Clocel_0048 | Transcriptional regulator, AbrB family | Transcriptional pleiotropic regulator of transition state genes | 1.91 | 2.50E−03 |
| Clocel_0322 | tatD family hydrolase | tatD DNase family protein | 1.74 | 8.66E−03 | |
| Clocel_0513 | Extracellular solute-binding protein | Raffinose/stachyose/melibiose transport system substrate-binding protein | 2.12 | 2.50E−03 | |
| Clocel_0519 | Glycogen/starch/alpha-glucan phosphorylase | Starch phosphorylase | 1.88 | 1.37E−02 | |
| Clocel_1243 | Extracellular solute-binding protein | Multiple sugar transport system substrate-binding protein | 2.53 | 9.20E−05 | |
| Clocel_1892 | Acetate kinase | Acetate kinase | 1.31 | 6.43E−03 | |
| Clocel_2210 | Nicotinate-nucleotide–dimethyl-benzimidazole phosphoribosyltransferase | Nicotinate-nucleotide–dimethyl-benzimidazole phosphoribosyltransferase | 1.08 | 1.47E−02 | |
| Clocel_2214 | ATP:corrinoid adenosyltransferase BtuR/CobO/CobP |
Coblalamin adenosyltransferase | 1.37 | 4.25E−02 | |
| Clocel_2222 | Precorrin-3B C(17)-methyltransferase | Precorrin-3B C17-methyltransferase | 1.70 | 6.63E−03 | |
| Clocel_2225 | Cobalamin (vitamin B12) biosynthesis CbiX protein | – | 1.95 | 4.32E−03 | |
| Clocel_2227 | Precorrin-4 C(11)-methyltransferase | Precorrin-4/cobalt-precorrin-4 C11-methyltransferase | 2.11 | 7.92E−03 | |
| Clocel_2250 | Altronate dehydratase | Altronate hydrolase | 4.07 | 1.02E−05 | |
| Clocel_2251 | Mannitol dehydrogenase domain-containing protein | Tagaturonate reductase | 4.50 | 4.39E−06 | |
| Clocel_2253 | Crp family transcriptional regulator | – | 2.37 | 2.50E−03 | |
| Clocel_2254 | Glycoside hydrolase family protein | – | 4.36 | 4.39E−06 | |
| Clocel_2255 | Major facilitator superfamily protein | Oligogalacturonide transporter | 4.33 | 4.02E−05 | |
| Clocel_2256 | Glycosyl hydrolase family protein | Unsaturated rhamnogalacturonyl hydrolase | 4.19 | 1.49E−05 | |
| Clocel_2259 | pfkB domain-containing protein | 2-Dehydro-3-deoxygluconokinase | 3.75 | 1.49E−06 | |
| Clocel_2262 | Short-chain dehydrogenase/reductase SDR | Gluconate 5-dehydrogenase | 3.21 | 2.11E−02 | |
| Clocel_2263 | 4-Deoxy-l-threo-5-hexosulose-uronate ketol-isomerase | 4-Deoxy-l-threo-5-hexosulose-uronate ketol-isomerase | 3.15 | 4.92E−03 | |
| Clocel_2403 | Glucosamine/fructose-6-phosphate aminotransferase | Glucosamine–fructose-6-phosphate aminotransferase (isomerizing) | 1.45 | 4.57E−03 | |
| Clocel_2737 | Small GTP-binding protein | – | 1.10 | 2.98E−02 | |
| Clocel_3380 | LPXTG-motif cell wall anchor domain-containing protein | – | 4.03 | 1.27E−04 | |
| Clocel_3909 | Quorum-sensing autoinducer 2 (AI-2), LuxS | S-ribosylhomocysteine lyase | 1.19 | 2.34E−02 | |
| Clocel_4088 | Galactose-1-phosphate uridylyltransferase | UDP-glucose–hexose-1-phosphate uridylyltransferase | 1.62 | 1.73E−02 | |
| Clocel_4277 | Aldo/keto reductase | – | 1.31 | 8.08E−03 | |
To standardize data, obtained data were normalized with the median for all identified quantitative data. P values were adjusted with the Benjamini–Hochberg method to avoid the problem of multiple testing. For thresholds, we adopted an FDR-adjusted p value of <0.05 and fold change of protein ratio >2.0, compared to glucose.
Profiles of metabolism-related proteins
First, we focused on profiles of metabolism-related proteins, such as enzymes involved in substrate degradation and metabolism, and other characteristic metabolic pathway. C. cellulovorans is known to change production of carbohydrate-related enzymes secreted into media from exoproteome analyses (Esaka et al. 2015; Matsui et al. 2013) and alternation of production of different metabolic pathways depending on which substrates are available in culture is also predicted.
Degradation and metabolism of each substrate
We constructed a substrate degradation pathway from KEGG pathway maps, and presented the fold change of each protein (Figure 4). For xylan degradation- and metabolism-related proteins (Figure 4a), the levels of three proteins (Clocel_0590, 0592, and 2595) were significantly elevated in the presence of xylan (Table 1), but production of Clocel_2900 (Endo-1, 4-beta xylanase) was not specifically elevated (Additional file 1: Table S1).
Figure 4.
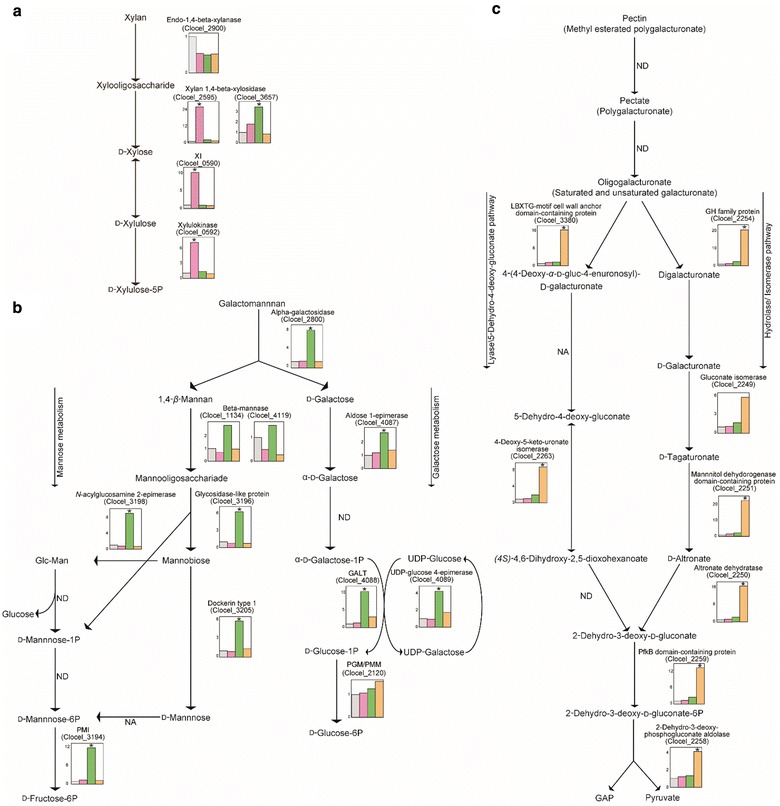
Degradation and metabolism pathways for each substrate, constructed from KEGG analysis. Each substrate degradation and metabolism pathway is shown: a xylan, b galactomannan (LBG), c pectin. For each protein, the fold change compared to glucose is shown. Asterisk denotes that protein levels are significantly elevated; as threshold, fold change >2.0, and FDR-adjusted p value <0.05 are adopted. ND protein not detected in this analysis. NA genes commonly assigned to the pathway, but not annotated. Fold change in glucose-grown cells is shown with a gray bar (=1); fold change in xylan-grown cells is shown with a pink bar; fold change of galactomannan-grown cells is shown with a green bar; fold change in pectin-grown cells is shown with an orange bar. XI xylose isomerase, PMI phosphomannose isomerase, GALT galactose-1-phosphate uridyltransferase, PGM/PMM phosphoglucomutase/phosphomannmutase alpha/beta/alpha domain I, 1P 1-phosphate, 6P 6-phosphate.
For galactomannan degradation- and metabolism-related proteins (Figure 4b), the levels of 9 proteins (Clocel_2259, 2800, 3194, 3196, 3198, 3205, 4087, 4088, and 4089) were significantly elevated in the presence of galactomannan (Table 1). By contrast, the levels of mannanase (Clocel_1134 and 4119) were not significantly elevated (Additional file 1: Table S1).
For pectin degradation- and metabolism-related proteins (Figure 4c), levels of 8 proteins (Clocel_2250, 2251, 2254, 2256, 2259, 2262, 2263, and 3380) were significantly elevated in the presence of pectin (Table 1). For the degradation and metabolism of pectin, both the “hydrolase/isomerase pathway” and the “lyase/5-dehydro-4-deoxy-gluconate pathway” were found (Richard and Hilditch 2009). Clocel_2254 is a member of the glycoside hydrolases (GH) 28 family (Cantarel et al. 2009), which is known to have endogalacturonase and exogalacturonase activities. Additionally, Clocel_3380 is a member of the polysaccharide lyase (PL) 9 family. BLAST analysis indicates that the protein has exogalacturonate lyase activity.
Genome and cluster analyses
To identify candidates related to substrate recognition systems, we performed genomic analysis based on C. cellulolyticum TCS-related gene clusters (Xu et al. 2013). For the thresholds of TCS-regulated cluster identification, we applied two criteria. First, a cluster must contain more than two components of a TCS pathway: an integral membrane histidine kinase (sensor histidine kinase), a transcriptional regulator (response regulator), and an extracellular solute-binding protein (sugar binding protein). Second, degradation-, metabolism-, or transport-related gene loci must be located within 2 of the TCS genes identified above. Using this threshold, we identified 14 candidate clusters related to TCS.
Combining the information of our genome analysis and the substrate-specific proteins, we identified xylan- and galactomannan-specific clusters that include TCS-related genes. We identified clusters corresponding to two substrates; the xylan-specific cluster included Clocel_2592, 2595, 2596, 2597, and 2598 (Table 1), and Clocel_2593 and Clocel_2594 (Additional file 2: Table S2). The galactomannan-specific cluster included Clocel_3194, 3196, 3198, 3200, 3201, and 3205 (Table 1), and Clocel_3195, 3197, 3199, 3202, 3203, 3204 (Figure 5; Additional file 2: Table S2). Each cluster includes three components of a TCS: a transcriptional regulator AraC, an integral membrane histidine kinase, and an extracellular solute-binding protein. We suggest that these genes are common components of a hemicellulose recognition system.
Figure 5.
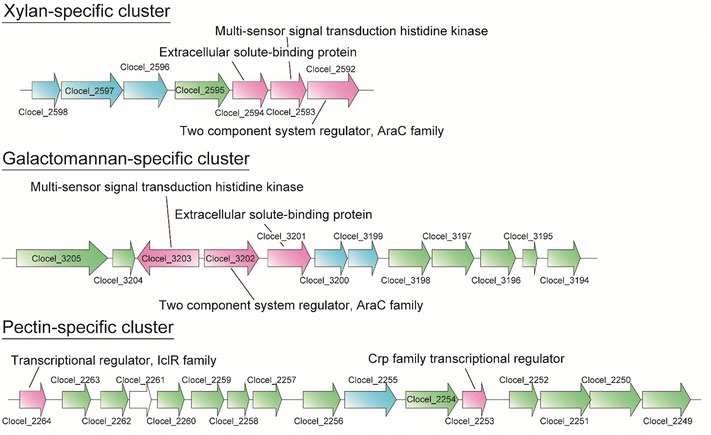
Candidates for gene clusters related to substrate recognition. We carried out genome analysis and identified candidates for gene clusters involved in TCS. Combining these results and substrate specific proteins, we found substrate-specific gene clusters related to recognition of each substrate. Green arrows indicate metabolism-related proteins, pink arrows indicate signal transduction-related proteins, and blue arrows indicate transport-related proteins. White arrows indicate pseudogenes.
From pectin, we found increased levels of proteins Clocel_2250, 2251, 2253, 2254, 2255, 2256, 2259, 2262, and 2263 (Table 1) and Clocel_2249, 2257, 2258, and 2260 (Additional file 2: Table S2). We suggest that this is a large cluster for the degradation and metabolism of pectin. For the degradation and metabolism of pectin, hydrolase/isomerase and lyase/5-dehydro-4-deoxy-gluconate pathways are known. The pectin-specific cluster which we identified contains genes related to both of these pathways, as well as for transporting pectin.
We also identified a xylose-metabolic cluster containing proteins Clocel_0589, 0590, 0591, and 0592 (Table 1) that had increased levels specifically in the presence of xylan. A galactose-metabolic cluster containing proteins Clocel_4087, 4088, and 4089 (Table 1) and Clocel_4090 (Additional file 2: Table S2) had elevated levels specifically in the presence of galactomannan. The xylose-metabolic cluster contained two xylose metabolism-related proteins, Clocel_0590 and 0592 (Table 1; Figure 4a), transaldorase (Clocel_0591; Table 1), which is related to the pentose phosphate pathway, and alpha-fucosidase (Clocel_0589; Table 1). In the galactose-metabolic cluster, galactose metabolism-related genes are present (Figure 4b) and increased levels of proteins Clocel_4087, 4088, and 4089 (Table 1) and Clocel_4090 (Additional file 2: Table S2) were found.
Discussion
In total, we identified 734 proteins using our “intracellular” quantitative proteome analysis, and we were able to identify substrate-specific proteins using statistical analysis. Our method provided reproducibility, as evaluated by HCA (Figure 3). For substrate-specific proteins, we carried out genome analysis and cluster analysis following genome analysis on the basis of the results from our proteome analysis.
We first focused on metabolic pathways of each substrate in C. cellulovorans by KEGG analysis. For xylan metabolism, it is interesting to note that intracellular levels of Clocel_2595 (xylosidase) increased, but levels of Clocel_2900 (xylanase) were not enhanced. Although Clocel_2595 (xylosidase) and some xylanases were categorized as xylan-specific in “exoproteome” analysis (Matsui et al. 2013), C. cellulovorans cannot grow in medium containing xylose as the sole carbon source (Sleat et al. 1984). Combining our findings with the inability of C. cellulovorans to grow in xylose medium, we predict that C. cellulovorans metabolizes xylooligosaccharides intracellularly. We hypothesize the following pathway for xylan metabolism: C. cellulovorans degrades xylan to xylooligosaccharides extracellularly and transports them into the cytoplasm. Next, xylooligosaccharides are degraded to xylose in the cell, and xylose is metabolized and used by the pentose phosphate pathway. Indeed, we observed increased levels of Clocel_0591 (transaldolase), which is related to the pentose phosphate pathway, in xylan medium.
For galactomannnan metabolism, we focused on Clocel_3196 (glycosidase-like protein), which is a member of the GH130 family and is annotated as beta-1,4-mannooligosaccharide/beta-1,4-mannosyl-N-acetylglucosamine phosphorylase (Kawahara et al. 2012). Similarly annotated genes are part of mannan metabolic pathways in other bacteria (Senoura et al. 2011), and we observed increased levels of proteins in this pathway (Clocel_3198, 3205) in the presence of galactomannan. However, Clocel_3197, which is known to play an important role in the mannan metabolic pathway (Esaka et al. 2015), was only identified in our qualitative proteome analysis (Additional file 2: Table S2). This protein has been also detected with “exoproteome” analysis (Esaka et al. 2015). It is assumed that this protein acts both intracellularly and extracellularly through an unknown mechanism. In addition, we identified increased levels of alpha-galactosidase (Clocel_2800). In a previous study, this protein was identified in an “exoproteome” analysis, and it is predicted to degrade galactose side chains both intracellularly and extracellularly. Specifically, the extracellular collaboration between mannase and alpha galactosidase rapidly degrades galactomannan. Next, galactose side chains that were not degraded extracellularly are degraded by “intracellular” alpha galactosidase, and the resulting mannooligosaccharides and galactose are further metabolized.
We predicted a pectin metabolism pathway in C. cellulovorans by combining the results of the present study and the previously reported “exoproteome” analysis (Matsui et al. 2013). Extracellularly, pectin is degraded by pectin lyase (Clocel_0873, 1172, and 3380) to oligogalacturonate with both saturated and unsaturated ends. In parallel, pectin is also de-esterified to pectate (Clocel_0211 and 3114), resulting in simpler extracellular degradation pathways. The resulting metabolites are transported into the cell and metabolized with the hydrolase/isomerase and lyase/5-dehydro-4-deoxy-gluconate pathways. In Erwinia chrysathemi, which is a well-studied pathogenic bacterium, pectin is degraded using similar systems; i.e. pectin is degraded to oligogalacturonides outside of the cell. Next, oligogalacturonides are transported into cells and metabolized in the hydrolase/isomerase and lyase/5-dehydro-4-deoxy-gluconate pathways (Hugouvieux-Cotte-Pattat et al. 1996). While known for E. chrysathemi, this system has not been reported in other cellulosome-producing Clostridia. C. cellulovorans commonly degrades all sugar-substrates (polysaccharides) to oligosaccharides, not to monosaccharide, outside of the cell. Degradation of those oligosaccharides to monosaccharides and further metabolism take place intracellularly. Similar degradation systems have not been reported in Clostridia although C. cellulolyticum can metabolize xylose as a sole carbon source. Furthermore, in the presence of pectin, we found increased levels of an oligogalacturonides (esterified-oligogalacturonates) transporter (Clocel_2255). In the current analysis, we did not identify pectinesterase “intracellularly”, although it had previously been identified in an “exoproteome” analysis (Matsui et al. 2013). Together, these results suggest that oligogalacturonates are transported into C. cellulovorans, and degraded to monogalacturonates and further metabolized intracellularly. In addition to the degradation and metabolism of each substrate, levels of four proteins related to the synthesis of vitamin B12 were significantly elevated in cells cultured on pectin (Clocel_2214, 2222, 2225, and 2227). We are unable to find any pectin-specific proteins reported to have a requirement for vitamin B12 (Sañudo-Wilhelmy et al. 2014). For the degradation and metabolism of pectin, C. cellulovorans uses pectin lyase (Clocel_2259) (EC 4.2.1.7), which showed elevated levels in our analysis. We speculate that this enzyme may require vitamin B12.
Within the pectin clusters, a Crp family transcriptional regulator (Clocel_2253) was identified upstream of the hydrolase/isomerase pathway, and levels of this regulator were significantly increased. Crp family transcriptional regulators are known to regulate gluconate-related genes in Escherichia coli (Peekhaus and Conway 1998), and we propose that this protein similarly regulates the hydrolase/isomerase pathway in C. cellulovorans. Upstream of Clocel_2263, an IclR family transcriptional regulator encoded genes (Clocel_2264) was annotated (Figure 5). In our quantitative proteome analysis, we did not identify this protein, but in the qualitative proteome analysis, this protein was identified and displayed higher peptide spectrum matches (PSMs) in pectin than in xylan and galactomannan (Additional file 2: Table S2). The KdgR protein regulates the degradation of pectin in Erwinia chrysanthemi, and is in the IclR family transcriptional regulator group (Molina-Henares et al. 2006). BLAST analysis indicates that Clocel_2264 has similarity with the KdgR protein (e = 5E-48). Based on these findings, we proposed that IclR family transcriptional regulators control the expression of the lyase/5-dehydro-4-deoxy-gluconate pathway.
Based on the metabolism-related protein profiles and cluster analysis performed in the present study, we propose molecular models for substrate recognition of C. cellulovorans in hemicellulose (xylan and galactomannan) and pectin (Figure 6). For hemicellulose, polysaccharides (xylan and galactomannan) are first degraded to oligosaccharides (xylooligosaccharides and mannooligosaccharides) in the extracellular environment, and these oligosaccharides are recognized by extracellular solute binding proteins (Clocel_2594 in Additional file 2: Table S2; Clocel_3201 in Table 1). Oligosaccharide-binding proteins then transduce signals to integral membrane histidine kinases (Clocel_2593 and 3203 in Additional file 2: Table S2). Next, intracellular transcriptional regulators (Clocel_2592 in Table 1; Clocel_3202 in Additional file 2: Table S2) are phosphorylated by the activation of integral membrane histidine kinases. Finally, genes related to the degradation and metabolism of hemicellulose are upregulated, allowing C. cellulovorans to metabolize substrates more efficiently (Figure 6a).
Figure 6.
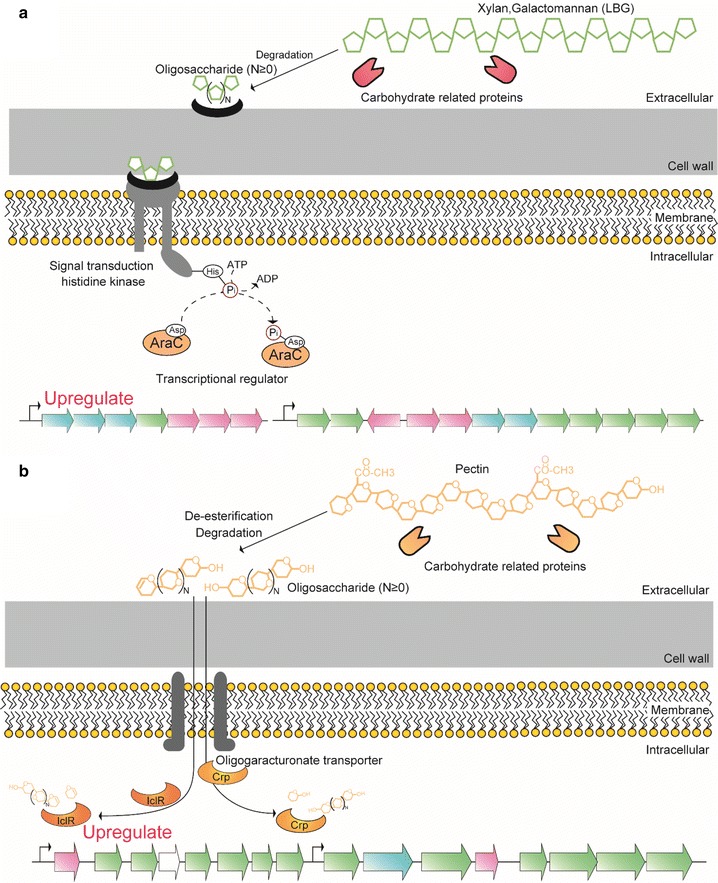
Proposed substrate recognition systems of hemicellulose and pectin in C. cellulovorans. For hemicellulose, polysaccharides are degraded to derived oligosaccharides outside of the cell, and extracellular solute-binding proteins bind these substrates. Solute-binding proteins induce signal-to-signal transduction integral membrane histidine kinases. Next the activated kinases phosphorylate transcriptional regulator AraC, and target genes (shown in Figure 5) are upregulated (a). For pectin, polysaccharides are de-esterified by pectinesterase and degraded by pectin lyase. Next, derived oligosaccharides are transported into cell, and these or other metabolites bind to a transcriptional regulator. Then, genes belonging to the target cluster (shown in Figure 5) are upregulated (b).
For pectin, two regulators (Clocel_2253 in Table 1; Clocel_2264 in Additional file 2: Table S2) work in substrate recognition. We predict that oligosaccharides derived from pectin or other pectin metabolites bind to these regulators allowing oligosaccharides to be transported into the cell (Figure 6b). This is a similar system to the pectin-recognition system in E. chrysathemi (Molina-Henares et al. 2006), but E. chrysathemi does not have two regulators operating hydrolase/isomerase and lyase/5-dehydro-4-deoxy-gluconate pathways.
The xylan recognition system of C. cellulovorans is very similar to that of C. cellulolyticum, but based on previous research (Xu et al. 2013), the composition of clusters is clearly different. In the C. cellulolyticum cluster, metabolism-related enzymes were not included. We therefore predict that these genes may not be regulated by TCS (Xu et al. 2013). However, the C. cellulovorans cluster includes metabolism- and transport-related genes. Thus, we speculate that the recognition system for xylan in C. cellulovorans regulates both the production of degradation- and transport-related enzymes and metabolism-related enzymes.
Substrate recognition systems in C. cellulovorans are one of the survival strategies. In other bacteria, monosaccharides such as glucose and xylose are mainly transported, but this bacterium recognizes oligosaccharides of hemicellulose and pectin, and can optimize degradation-, metabolism-, and transport-related proteins’ profiles according to the substrate faster than other bacteria. This feature maybe suitable for competing with other anaerobic bacteria.
In conclusion, we performed “intracellular” quantitative proteome analysis of C. cellulovorans with glucose, xylan, galactomannan, and pectin and identified 734 proteins with LC–MS/MS equipped long monolithic silica capillary columns. We focused on substrate-recognition-, degradation-, and metabolism-related proteins, and carried out cluster analysis based on genome analysis together with the intracellular proteome analysis and the previous “exoproteome” analysis (Matsui et al. 2013; Esaka et al. 2015). Based on our results, we propose a mechanism for the recognition of hemicellulose and pectin in C. cellulovorans. For hemicellulose recognition, C. cellulovorans uses TCS and regulates all substrate-related genes with a single regulator. In the pectin recognition system, C. cellulovorans regulates hydrolase/isomerase and lyase/5-dehydro-4-deoxy-gluconate pathways with two regulators, Clocel_2253 and Clocel_2264, respectively. Judging from the presence of an oligogalacturonate transporter in the pectin regulatory clusters, the regulation of pectin metabolism-related genes appears to be controlled by the oligogalacturonates derived from pectin, or other metabolic products. This system has not been described for other cellulosome-producing Clostridia. Due to the multiplicity of regulator proteins involving substrate recognition, we suggest that C. cellulovorans has more distinct promoter regions that respond to each substrate. This information should be helpful for the elucidation of the environmental sensing strategy of C. cellulovorans.
Authors’ contributions
SA and KE generated the strains used. SA, KE and HM performed most of the mass measurement. SA, KE and HM performed most of the statistical analyses. SA, KE, HM, KK and MU designed the study and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This research was supported by JST, CREST. This work was also partially supported by the Program for Promotion of Basic and Applied Researches for Innovations in Bio-oriented Industry (BRAIN).
Compliance with ethical guidelines
Competing interest The authors declare that they have no competing interest.
Abbreviations
- ATCC
American type culture collection
- CID
collision induced dissociation
- EC
enzyme commission number
- ESI
electrospray ionization
- FDR
false discovery rate
- GH
glycoside hydrolases
- HCA
hierarchical cluster analysis
- HCD
higher-energy c-trap dissociation
- HPLC
high performance liquid chromatography
- ID
internal diameter
- KEGG
Kyoto encyclopedia of genes and genomes
- LBG
locust bean gum
- LC–MS
liquid chromatography–mass spectrometry
- NCBI
National Center for Biotechnology Information
- PBS
phosphate buffered saline
- PL
polysaccharide lyase
- SDS
sodium dodecyl sulfate
- TCS
two component system
- TEAB
triethyl ammonium bicarbonate
- TMT
tandem mass tag
Additional files
The 734 proteins identified in quantitative proteome analysis. Proteome analytes were injected to LC–MS/MS system. Obtained data were used for protein identification with Proteome Discoverer software 1.4. Three independent biological experiments were carried out, and proteins with no missing values were accepted as identified protein. As a result, 734 proteins were successfully identified.
All proteins identified in the qualitative proteome analysis. Supplemental qualitative proteome analysis was carried out to strengthen the quantitative proteome analysis. Three independent biological experiments were carried out, and proteins with MASCOT scores >10 were accepted as identified protein. Score: MASCOT score; PSMs: Peptides spectrum matches.
Contributor Information
Shunsuke Aburaya, Email: aburaya.shunsuke.37r@st.kyoto-u.ac.jp.
Kohei Esaka, Email: k.esaka17@gmail.com.
Hironobu Morisaka, Email: morisaka@kais.kyoto-u.ac.jp.
Kouichi Kuroda, Email: k_kuro@kais.kyoto-u.ac.jp.
Mitsuyoshi Ueda, Email: miueda@kais.kyoto-u.ac.jp.
References
- Aoki W, Ueda T, Tatsukami Y, Kitahara N, Morisaka H, Kuroda K, et al. Time-course proteomic profile of Candida albicans during adaptation to a fetal serum. Pathog Dis. 2013;67(1):67–75. doi: 10.1111/2049-632X.12003. [DOI] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. The carbohydrate-active enzymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009;37(suppl 1):D233–D238. doi: 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celik H, Blouzard J-C, Voigt B, Becher D, Trotter V, Fierobe H-P, et al. A two-component system (XydS/R) controls the expression of genes encoding CBM6-containing proteins in response to straw in Clostridium cellulolyticum. PLoS One. 2013;8(2):e56063. doi: 10.1371/journal.pone.0056063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins M, Lawson P, Willems A, Cordoba J, Fernandez-Garayzabal J, Garcia P, et al. The phylogeny of the genus Clostridium: proposal of five new genera and eleven new species combinations. Int J Syst Bacteriol. 1994;44(4):812–826. doi: 10.1099/00207713-44-4-812. [DOI] [PubMed] [Google Scholar]
- Cosgrove DJ. Growth of the plant cell wall. Nat Rev Mol Cell Biol. 2005;6(11):850–861. doi: 10.1038/nrm1746. [DOI] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20(9):1453–1454. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- Esaka K, Aburaya S, Morisaka H, Kuroda K, Ueda M. Exoproteome analysis of Clostridium cellulovorans in natural soft-biomass degradation. AMB Express. 2015;5(1):1–8. doi: 10.1186/s13568-014-0089-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashide W, Li Y, Yang Y, Liao JC. Metabolic engineering of Clostridium cellulolyticum for production of isobutanol from cellulose. Appl Environ Microbiol. 2011;77(8):2727–2733. doi: 10.1128/AEM.02454-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugouvieux-Cotte-Pattat N, Condemine G, Nasser W, Reverchon S. Regulation of pectinolysis in Erwinia chrysanthemi. Annu Rev Microbiol. 1996;50:213–257. doi: 10.1146/annurev.micro.50.1.213. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara R, Saburi W, Odaka R, Taguchi H, Ito S, Mori H, et al. Metabolic mechanism of mannan in a ruminal bacterium, Ruminococcus albus, involving two mannoside phosphorylases and cellobiose 2-epimerase: discovery of a new carbohydrate phosphorylase, β-1,4-mannooligosaccharide phosphorylase. J Biol Chem. 2012;287(50):42389–42399. doi: 10.1074/jbc.M112.390336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynd LR, Wyman CE, Gerngross TU. Biocommodity engineering. Biotechnol Prog. 1999;15(5):777–793. doi: 10.1021/bp990109e. [DOI] [PubMed] [Google Scholar]
- Matsui K, Bae J, Esaka K, Morisaka H, Kuroda K, Ueda M. Exoproteome profiles of Clostridium cellulovorans grown on various carbon sources. Appl Environ Microbiol. 2013;79(21):6576–6584. doi: 10.1128/AEM.02137-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina-Henares AJ, Krell T, Guazzaroni ME, Segura A, Ramos JL. Members of the IclR family of bacterial transcriptional regulators function as activators and/or repressors. FEMS Microbiol Rev. 2006;30(2):157–186. doi: 10.1111/j.1574-6976.2005.00008.x. [DOI] [PubMed] [Google Scholar]
- Morisaka H, Matsui K, Tatsukami Y, Kuroda K, Miyake H, Tamaru Y, et al. Profile of native cellulosomal proteins of Clostridium cellulovorans adapted to various carbon sources. AMB Express. 2012;2(1):1–5. doi: 10.1186/2191-0855-2-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nataf Y, Bahari L, Kahel-Raifer H, Borovok I, Lamed R, Bayer EA, et al. Clostridium thermocellum cellulosomal genes are regulated by extracytoplasmic polysaccharides via alternative sigma factors. Proc Natl Acad Sci USA. 2010;107(43):18646–18651. doi: 10.1073/pnas.1012175107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peekhaus N, Conway T. Positive and negative transcriptional regulation of the Escherichia coli gluconate regulon gene gntT by GntR and the cyclic AMP (cAMP)-cAMP receptor protein complex. J Bacteriol. 1998;180(7):1777–1785. doi: 10.1128/jb.180.7.1777-1785.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitdemange E, Caillet F, Giallo J, Gaudin C. Clostridium cellulolyticum sp. nov., a cellulolytic, mesophilic: species from decayed grass. Int J Syst Bacteriol. 1984;34(2):155–159. doi: 10.1099/00207713-34-2-155. [DOI] [Google Scholar]
- Prawitwong P, Waeonukul R, Tachaapaikoon C, Pason P, Ratanakhanokchai K, Deng L, et al. Direct glucose production from lignocellulose using Clostridium thermocellum cultures supplemented with a thermostable β-glucosidase. Biotechnol Biofuels. 2013;6(1):184. doi: 10.1186/1754-6834-6-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rainey FA, Stackebrandt E. 16S rDNA analysis reveals phylogenetic diversity among the polysaccharolytic clostridia. FEMS Microbiol Lett. 1993;113(2):125–128. doi: 10.1111/j.1574-6968.1993.tb06501.x. [DOI] [PubMed] [Google Scholar]
- Raman B, Pan C, Hurst GB, Rodriguez M, Jr, McKeown CK, Lankford PK, et al. Impact of pretreated switchgrass and biomass carbohydrates on Clostridium thermocellum ATCC 27405 cellulosome composition: a quantitative proteomic analysis. PLoS One. 2009;4(4):e5271. doi: 10.1371/journal.pone.0005271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Hilditch S. d-Galacturonic acid catabolism in microorganisms and its biotechnological relevance. Appl Microbiol Biotechnol. 2009;82(4):597–604. doi: 10.1007/s00253-009-1870-6. [DOI] [PubMed] [Google Scholar]
- Saha BC. Hemicellulose bioconversion. J Ind Microbiol Biotechnol. 2003;30(5):279–291. doi: 10.1007/s10295-003-0049-x. [DOI] [PubMed] [Google Scholar]
- Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20(17):3246–3248. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- Sañudo-Wilhelmy SA, Gómez-Consarnau L, Suffridge C, Webb EA. The role of B vitamins in marine biogeochemistry. Ann Rev Mar Sci. 2014;6:339–367. doi: 10.1146/annurev-marine-120710-100912. [DOI] [PubMed] [Google Scholar]
- Senoura T, Ito S, Taguchi H, Higa M, Hamada S, Matsui H, et al. New microbial mannan catabolic pathway that involves a novel mannosylglucose phosphorylase. Biochem Biophys Res Commun. 2011;408(4):701–706. doi: 10.1016/j.bbrc.2011.04.095. [DOI] [PubMed] [Google Scholar]
- Sleat R, Mah RA, Robinson R. Isolation and characterization of an anaerobic, cellulolytic bacterium, Clostridium cellulovorans sp. nov. Appl Environ Microbiol. 1984;48(1):88–93. doi: 10.1128/aem.48.1.88-93.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004) Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3(1) [DOI] [PubMed]
- Tamaru Y, Miyake H, Kuroda K, Nakanishi A, Kawade Y, Yamamoto K, et al. Genome sequence of the cellulosome-producing mesophilic organism Clostridium cellulovorans 743B. J Bacteriol. 2010;192(3):901–902. doi: 10.1128/JB.01450-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Huang R, Teng L, Wang D, Hemme CL, Borovok I, et al. Structure and regulation of the cellulose degradome in Clostridium cellulolyticum. Biotechnol Biofuels. 2013;6(1):73. doi: 10.1186/1754-6834-6-73. [DOI] [PMC free article] [PubMed] [Google Scholar]