

Abstract
Three-dimensional cryo-electron microscopy (cryo-EM) is an expanding structural biology technique that has recently undergone a quantum leap progression in its achievable resolution and its applicability to the study of challenging biological systems. Because crystallization is not required, only small amounts of sample are needed, and, because images can be classified in a computer, the technique has the potential to deal with compositional and conformational mixtures. Therefore, cryo-EM can be used to investigate complete and fully functional macromolecular complexes in different functional states, providing a richness of biological insight. In this review we underlie some of the principles behind the cryo-EM methodology of single particle analysis and discuss some recent results of its application to challenging systems of paramount biological importance. We place special emphasis on new methodological developments that are leading to an explosion of new studies, many of which are reaching resolutions that could only be dreamed of only a couple of years ago.
1 - INTRODUCTION AND HISTORICAL OVERVIEW OF 3D-EM RECONSTRUCTION
Characterizing the molecular mechanism of macromolecules is essential for a full understanding of the biochemical and cellular processes they carry out. Structural visualization is invaluable for such mechanistic understanding, especially when done for multiple functional states of the macromolecule of interest. The 20th century saw the development of powerful tools for macromolecular structure determination, most remarkably X-ray crystallography, which today stands as the most effective method to produce atomic models of proteins and nucleic acids. In spite of the countless successes of X-ray crystallography, some of the requirements of this technique impose limitations in its applicability. In particular, when samples prove hard to crystallize (as is often the case for integral membrane proteins), or the macromolecular complex cannot be produced in sufficient quantities/concentration to even attempt crystallization trials. Certain functionally relevant states may be hard to purify, and the sample may coexist in multiple conformational or compositional states under the range of accessible biochemical conditions. Some samples are inherently refractant to crystal packing, like most polymers. In certain cases, even when crystallization is achieved, the nature of the crystals (size of the unit cell, lack of order, etc) may make structural determination hard.
3D electron microscopy (3D-EM) is a potential alternative to X-ray crystallography that is quickly gaining popularity among structural biologists. In 3D-EM biological samples are directly visualized using transmission electron microcopy (TEM), which generates 2D images corresponding to a projection of the structure in the direction of the electron path (Fig. 1a). A 3D reconstruction is obtained by combining images corresponding to different views of the object under study (see below). Multiple views are naturally present in helical assemblies, such as in phage tails, helical viruses or cytoskeletal polymers. In such cases the helical parameters define the orientation of the different molecules in the array, and 3D “reconstruction” can be obtained using helical Fourier inversion methods (DeRosier and Klug, 1968). In certain cases, different views of the object are produced by tilting the sample stage, as it is the case of electron tomographic studies of unique structures that are imaged multiple times in different orientations, or in the case of 2D crystals, where different crystals are each imaged once, but in different orientations that are later combined. More generally in the study of purified macromolecular complexes, the sample is made of individual molecules that adopt random (or at least multiple) orientations on the EM grid and thus provide multiple views of the structure. In such cases, different strategies can be used to define the relative orientations of the projection images to produce a 3D reconstruction using computational tools referred to as “single particle” analysis. While helical Fourier methods and 2D crystallography pioneered the 3D-EM field, it is the general applicability of single particle analysis that is making this variety of EM studies predominant today in the pursuit of high-resolution macromolecular structure.
Figure 1. Basic concepts of cryo-EM structure determination.

(A) The projection-slice theorem states that the 2D projection of a 3D object in real-space (left column) is equivalent to taking a central 2D slice out of the 3D Fourier transform of that object (right column). The realspace projection direction (left; dashed red arrows) is perpendicular to the slice (right; red frame). (B–E) Many experimental 2D projections can be combined in a 3D reconstruction through an iterative process called “projection matching”. To determine the relative orientations of all experimental projections one first calculates reference projections of a 3D object in all directions (B). Then, one compares each experimental projection with all reference projections to find the best match of a given similarity measure (C). This orients all experimental projections relative to the 3D structure (D). The projection-slice theorem then implies that the 3D reconstruction can be calculated by positioning many 2D slices (the 2D Fourier transforms of all experimental projections) into the 3D transform (E) and calculating an inverse transform. Iterating steps (B–E) will gradually improve the orientations, and hence the resolution of the reconstruction.
To withstand the high vacuum in the electron microscope and to minimize the visible effects of radiation damage (problems not affecting many non-biological EM studies), biological samples can be either stained with a low concentration solution of heavy metals (typically uranium salts) and dried before being inserting into the scope, or ideally studied in a frozen hydrated state after vitrification (cryo-EM). The first method, negative staining, produces high contrast images, but is limited in resolution (to about 15 Å due to the grain size of the stain), may cause deformation of the most fragile samples during drying, and does not generally allow visualization of nucleic acids. This methodology has been used since the 1970’s and it is still common in the study of small macromolecules (< 250 kDA), and for ab initio reconstructions using geometrical principles that require tilting of the specimen (RCT and OTR) (Leschziner and Nogales, 2006; Radermacher, 1988), especially if the sample is expected to be heterogeneous. Although experimentally more demanding, cryo-EM of frozen hydrated samples does not require dehydration of the sample, can be used to visualize proteins as well as DNA and RNA, and has the potential to produce higher resolution structures.
From initial pioneering studies, a continuous evolution of the 3D-EM field has taken place over the last 50 years. But it is only recently that the data quality and the algorithms required for its analysis have come to a stage where applicability, resolution and throughput place 3D-EM in its path to become a main-stream structural biology technique. A number of pioneering studies stand as key landmarks in the evolution of 3D-EM (the list is by no means comprehensive).
1968 - DeRosier and Klug (DeRosier and Klug, 1968) publish the first 3D-EM reconstruction, using helical Fourier methods, of the bacteriophage T4 tail.
1979 - Taylor and Glaeser (Taylor and Glaeser, 1976) demonstrate, using electron diffraction, that high resolution information of a biological sample can be obtained using EM by overcoming the limitation of radiation damage using cryo-protection.
1987 - Dubochet and colleagues (Adrian et al., 1984; Dubochet et al., 1982) develop an easily applicable method for sample vitrification starting an era of cryo-EM visualization.
1990 - Henderson and colleagues (Henderson et al., 1990) produced the first atomic structure using electron crystallography of 2D crystals of bacteriorhodopsin.
1993 – Milligan, Rayment and colleagues (Rayment et al., 1993) used a “hybrid” approach and combine crystallographic atomic models of components and lower resolution 3D-EM structures to describe the interaction of myosin with actin filaments.
1995 - Unwin (Unwin, 1995) used time-resolved cryo-EM to produce the open state of the acetylcholine receptor following activation by acetylcholine and preceding deactivation.
1995 - Frank and colleagues (Frank et al., 1995) used single particle reconstruction methodology to propose a model of translation based on a 25 Å cryo-EM structure of the 70S ribosome.
1997 – Crowther and colleagues (Bottcher et al., 1997), in their study of Hepatitis B virus structure, obtained the first single particle study breaking the 10 Å barrier thus allowing visualization of alpha helices.
2005 – Walz, Harrison and colleagues (Gonen et al., 2005), using phase extension and highresolution electron diffraction, obtained the highest resolution structure to date by 3D-EM, 1.9 Å, in the study of aquaporin 0.
2010 – Zhou and colleagues (Liu et al., 2010) produced the first atomic structure of a virus (human adenovirus) by cryo-EM single particle reconstruction.
More recently, a new generation of electron detectors have drastically improved signal to noise ratios in the experimental data, while powerful new image processing algorithms have been developed for the correction of beam-induced motion and the classification of distinct structural states. Together, these developments are revolutionizing cryo-EM structure determination by single-particle analysis (Kuhlbrandt, 2014). In this review we cover the basic principles behind this technique, give recent examples of studies with unique biological insights not presently achievable by any other structural biology approach, and describe how these recent technological breakthroughs are leading to atomic resolution structures and characterization of complex functional mixtures by cryo-EM.
2 - PRINCIPLES IN SINGLE PARTICLE EM RECONSTRUCTION
As indicated above, TEM images correspond to 2D projections of the 3D particles (macromolecular objects) in the sample. When a macromolecular solution is prepared for study by TEM (by absorption, staining and drying in the case of negative stain preparation, or by blotting into a thin layer and quick freezing in cryo-EM), multiple copies of the same particle are generally present in many different orientations. How a collection of 2D projections from different directions can be combined into a 3D reconstruction can be understood from the “projection-slice theorem”. This theorem states that the Fourier transform of a 2D projection is a central slice through the 3D Fourier transform (passing through the origin) of the underlying structure, the projection direction being orthogonal to the slice (Figure 1A). Therefore, if one knows the directions of multiple 2D projections, one can position the corresponding 2D Fourier slices within the 3D transform and calculate the original 3D structure by computing the inverse Fourier transform.
There are two major challenges to be overcome in single particle 3D reconstruction of biological samples. Firstly, in order to minimize radiation damage (specially of vitrified samples) it is necessary to image biological macromolecules with only a few electrons, typically 20–40 electrons/Å2. This results in extremely noisy images, with typical noise powers being more than 10 times larger than the signal. The way to gain signal is to average over many particles images, typically tens to hundreds of thousands. Secondly, the relative orientations of the particle images are unknown and need to be determined computationally. A generally applied method is to compare each experimental particle image with computationally generated projections (sometimes referred to as “reprojections”) of a 3D reference structure in all different directions (Figure 1B). In this “projection-matching” approach (Penczek et al., 1994) the initial reference may only need to “resemble” the true structure (image signal and asymmetry of the particle play a role on how closely the reference needs to be to the actual structure under study). Assuming good experimental coverage of all projection directions, the reconstruction obtained from particle images using the orientation angles best matching the reference reprojections will yield a better structure than the initial reference. Therefore, iterative application of this projection-matching algorithm should converge to the true structure of the particle under study, with resolution being determined by the amount of images used, as well as properties of the sample such as conformational and biochemical homogeneity (see later).
Initial reference structures for projection-matching refinement can either be generated from similar structures (e.g. low-resolution representations, partial complexes, homologs), or they can be determined de novo from EM images. In geometry-based ab initio approaches, which are generally considered robust and provide information on the handedness of the structure, the specimen is tilted with respect to the electron beam and two or more images of the same area of the specimen at different tilt angles are imaged. Different procedures exist to calculate a 3D reconstruction that exploit information coming from multiple visualizations of the same object in this controlled way, including random conical tilt (RCT) (Radermacher et al., 1987), orthogonal tilt reconstruction (OTR) (Leschziner and Nogales, 2006) and tomography (Lucic et al., 2005). Alternatively, one can exploit the observation that each pair of 2D projections shares a line (referred to as “common line”) in the 3D Fourier transform (a direct consequence of the projectionslice theorem) to analytically determine relative orientations between pairs of projections (or typically averages of identical views with increased signal-to-noise ratios) by maximizing the similarity of these “common lines.” The popular angular reconstitution method is an application of common lines principles implemented in real space (van Heel, 1987).
How well the initial reference resembles the true particle does matter. The projection-matching algorithm is a local optimizer, i.e. it will converge to the nearest local minimum. This means that if the initial model is not close enough to the true structure, the algorithm may converge onto an incorrect solution. Although modified projection-matching algorithms have been proposed that rely on stochastic steps to escape from local minima (Elmlund et al., 2010), those approaches are also not guaranteed to reach the correct structure. Consequently, any computer program could in principle remain stuck in a local minimum, and thereby generate an incorrect structure. An indication that the reconstruction is correct may be obtained if components of known crystallographic structure, not used as part of the reference-based angular assignment, can be unequivocally placed within the reconstruction using objective, quantitative docking. If no such structures exist, and the EM reconstruction does not reach a resolution of 9 Å, where α-helices become visible as rod-shaped densities, it may be difficult to distinguish correct reconstructions from incorrect ones. For this purpose a robust validation method was introduced that exploits the prior information of pairs of images recorded at different tilt angles (Henderson et al., 2011; Rosenthal and Henderson, 2003).
In addition to the high levels of noise and the unknown relative orientations of the individual particles discussed above, many cryo-EM samples present a third challenge for 3D reconstruction, that may, however, also represent a unique opportunity to gain insight into molecular mechanism. Very often, macromolecular complexes adopt multiple conformational or compositional states, and cryo-EM data sets are a mixture of 2D projections from multiple 3D structures. Analysis of such samples requires classification of the particle images into structurally homogeneous subsets, i.e. containing 2D projections of identical 3D structures. It is however difficult to distinguish projections of the same 3D structure in different directions from projections of different 3D structures. A commonly employed procedure is to use more than one reference structure for projection-matching. The first application of such strategy made use of prior knowledge (ribosome ratcheting) to generate correspondingly different initial references (Valle et al., 2002). Unfortunately, lack of preexisting knowledge about structural variability will limit the applicability of this method in a general case. More recently, unsupervised classification methods, i.e. methods that do not require prior knowledge about the structural variability in the sample, have been proposed (Scheres et al., 2007a). Among the most successful implementations of this methodology are those using maximum-likelihood approaches (discussed in more detail below). These methods have turned the “inconvenience” of sample mixtures into an opportunity to simultaneously determine multiple structural states, thus providing unique insights into the dynamics of the sample that are of direct relevance to our understanding of macromolecular function.
3 - DEFINING THE ARCHITECTURE OF COMPLEX ASSEMBLIES AT MEDIUM RESOLUTION: THE HUMAN TRANSCRIPTION PREINITIATION COMPLEX
The general applicability of single particle methods has made possible an ever expanding number of structural studies for the last two decades. But the resolution of most cryo-EM structures, with the exception of viruses or other symmetrical arrangements (e.g. GroEL, 20S proteosome), has until recently remained systematically limited below that minimally required to model atomic structures. Nevertheless, the application of hybrid methods has made it possible to combine 3D-EM structures with additional information to gain unique biological insight about the subunit architecture of large assemblies (Robinson et al., 2007). Beyond the docking of available atomic structures within a larger macromolecular assembly, additional information can help making sense of structures lagging in resolution. In cases where an expression and reconstitution system is in place, subunit or even domain localization can be obtained by direct visualization of terminally attached genetic tags such as MBP (Chen et al., 2008; Lander et al., 2012), or internally attached GFP (Ciferri et al., 2012). A recently popularized strategy is the use of proximity information generated from chemical crosslinking and mass spectrometry studies (Chen et al., 2010) in combination with 3D-EM reconstructions (Greber et al., 2014; Lasker et al., 2012). Alternatively, a method that has proven very effective for identifying the position of components within large assemblies, defining overall architecture and spatial relationships, has been to visualize partial complexes (where one or more subunits have been removed) (Schreiber et al., 2011) or to build up a large assembly one or more subunits at a time. A recent example of the latter concerns the architecture of the human transcription pre-initiation complex (He et al., 2013).
Initiation of eukaryotic transcription represents a major step in gene regulation, requiring the activity of a large number of protein complexes. The basal machinery includes RNA polymerase II (Pol II) and six general transcription factors (GTFs) (TFIIA, TFIIB, TFIID (or TBP), TFIIE, TFIIF, and TFIIH) that assemble into a ~2 MDa complex on core promoter DNA. This pre-initiation complex (PIC) is essential for transcription start site (TSS) selection, promoter melting, and Pol II promoter escape (Goodrich et al., 1996; Matsui et al., 1980; Roeder, 1996). In vitro studies of transcription initiation over the last 30 years have defined the sequential assembly of the PIC (Roeder, 1996). This information was used to build and structurally characterize TBP-based PICs of increasing size bound to promoter DNA. The effectiveness of this approach was initially tested by negatively stained single particle EM (which needs less sample, but precluded the visualization of DNA) and allowed to identify and localize each GTF within the context of the full assembly (Fig. 2A). The subsequent cryo-EM reconstructions lead to improved resolution and visualization of the DNA (Fig. 2B–C) (He et al., 2013). The starting point was a TBP-TFIIA-IIB-RNAPII complex on promoter DNA. Adding TFIIF resulted in a reconstruction where both extra density for protein and for DNA are visible, indicating that TFIIF stabilizes the core promoter DNA along the surface of RNAPII. Addition of TFIIE results in the topological trapping of the DNA on the RNAPII cleft (Fig. 2C). At 11 Å resolution, the reconstruction of the human PIC containing TBP-TFIIA-TFIIB-Pol IITFIIF and TFIIE starts to reveal the major and minor grooves of the promoter DNA (Fig. 2B). The crystal structures of TBP-TFIIA-DNA (Bleichenbacher et al., 2003), TBP-TFIIB-DNA (Tsai and Sigler, 2000) and yeast Pol II-TFIIB (Kostrewa et al., 2009) were unambiguously docked into the cryo-EM reconstruction as rigid bodies, as well as those of the RAP30 WD40 domain and the dimerization domain of RAP30 and RAP74 (Gaiser et al., 2000) (both within TFIIF) (Fig. 2C). The human PIC ultimately contained TBP-TFIIA-IIB-IIF-RNAPII-IIE-TFIIH on promoter DNA and showed that TFIIE positions TFIIH so that the active ATPase in transcription initiation, XPB, is down stream of the TSS (Fig. 2D).
Figure 2. Study of the human transcription PIC.
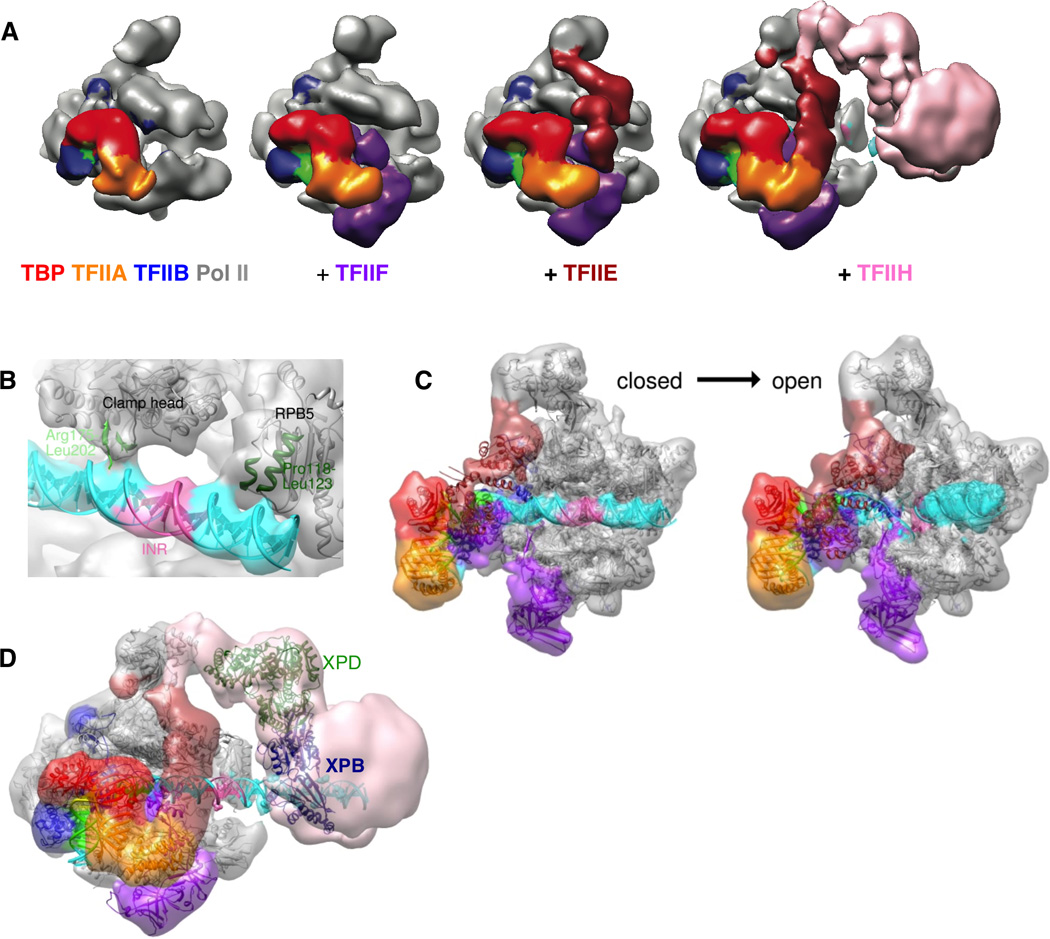
(A) Negative stain reconstructions of increasingly larger human PIC assemblies. (B) Detail of a PIC cryo-EM reconstruction showing Pol II contacts with duplex DNA in the closed PIC complex. (C) Comparison of the closed and open PIC cryo-EM structures. (D) Negative stain reconstruction of the TFIIH-containing PIC with available atomic models. Modified from He et al. (2013) Nature.
To gain information on the process of promoter opening, an artificial DNA template was used that served as a mimic of a transcription bubble (Fig. 2C). The relative movement of downstream DNA, together with the positioning of XPB, suggests how XPB acts as a DNA translocase to thread approximately 10bp of downstream double stranded DNA into the cleft, as previously proposed based on biochemical (Kim et al., 2000) and crosslinking data (Grunberg et al., 2012). A cryo-EM structure of the TFIIH-containing complex, together with higher resolution (see later for major recent technological breakthroughs), will shed further light into the human PIC, a biological system that had until recently defied structural characterization.
4 - GAINING FUNCTIONAL INSIGHT TRHOUGH THE CHARACTERIZATION OF MULTIPLE STATES: MICROTUBULE DYNAMICS
Among the most significant benefit of 3D-EM studies that emerges from the lack of crystallization requirement and the study of macromolecules in physiological conditions that are compatible with their biochemical functionality, is the description of multiple functional states in a mechanistic cycle. The different states can be controlled, separated or mimicked if a biochemical means is possible, and then studied one at a time (Goulet et al., 2014; Meyerson et al., 2014). Alternatively, they may coexist and be “computationally sorted” using the appropriate image analysis strategy (Agirrezabala et al., 2012; Clare et al., 2012). We will cover in some detail two recent examples, each one belonging to one of the two scenarios indicated above. In the first, microtubules in different states concerning nucleotide content were compared to gain information on how GTP hydrolysis leads to the dynamic behavior of these polymers (Alushin et al., 2014). The second, later in this review, concerns the use of Bayesian methodology to allow the “computational purification” of an intermediate in eukaryotic translation that otherwise exists as a rare state in a complex biochemical mix (Fernandez et al., 2013).
Microtubules (MTs) are polymers of αβ-tubulin that undergo dynamic instability (Mitchison and Kirschner, 1984), the stochastic switching between growth and shrinkage driven by GTP hydrolysis. The assembly capabilities of tubulin are determined by the exchangeable E-site nucleotide in β-tubulin. When this nucleotide is GTP, tubulin is competent for polymerization. Importantly, addition of a tubulin dimer to a growing microtubule end brings the catalytic residue in α-tubulin in contact with the E-site GTP of the adjacent dimer, coupling microtubule growth with GTP hydrolysis. The MT continues to grow only for as long as it contains GTP-tubulin subunits at its end. When this “GTP cap” is lost, it rapidly depolymerizes (Mitchison and Kirschner, 1984). The molecular mechanism by which GTP hydrolysis controls MT dynamics remains elusive. In order to understand dynamic instability it is necessary to define the effects of nucleotide state on tubulin conformation within the context of the microtubule lattice.
Recent cryo-EM reconstructions of dynamic MTs and MT made of tubulin bound to GMPCPP (a slowly-hydrolizable GTP analog), reaching better than 5 Å resolution (Fig. 3A), have been used in conjunction with Rosetta molecular modeling to generate pseudo-atomic structures that illustrate the changes in tubulin upon GTP hydrolysis within the MT (Fig. 3B) (Alushin et al., 2014). Comparison of the GMPCPP and GDP MT structures shows a compression of the interdimer interface around the E-site nucleotide (Fig. 3C–D). Changes within the β-subunit are mostly limited to the loops engaging the nucleotide. These changes and/or the absence of the gamma phosphate, are coupled to a significant movement of the T7 loop and helix H8 of the adjacent α subunit (Fig. 1D), which contains the presumed catalytic residue Glu254 (Lowe et al., 2001). In fact, all major structural changes occur in the alpha subunit, where the intermediate domain and C-terminal helices move with respect to the nucleotide-binding N-terminal domain (Fig. 3C). Interestingly, the differences in the GMPCPP- and GDP-bound tubulin structures within the MT are somehow reminiscent of those ascribed to the straight-to-bent transition during MT depolymerization (Gigant et al., 2000; Nogales and Wang, 2006), but are smaller in scale and appear to occur only in α-tubulin. This result is consistent with a model in which the energy of GTP hydrolysis is stored as internal strain that would subsequently relax during depolymerization (Caplow et al., 1994). This study demonstrated that microtubules are structurally tractable by cryo-EM at resolutions now approaching those obtained by X-ray crystallography, thus enabling the detailed study of nucleotide and drug effects, or the interaction of microtubule binding proteins, in the context of the native tubulin polymer. As the next sections indicate, recent methodology is likely to make possible yet higher resolution structures of MTs.
Figure 3. High-resolution cryo-EM structures of dynamic and stabilized microtubules.
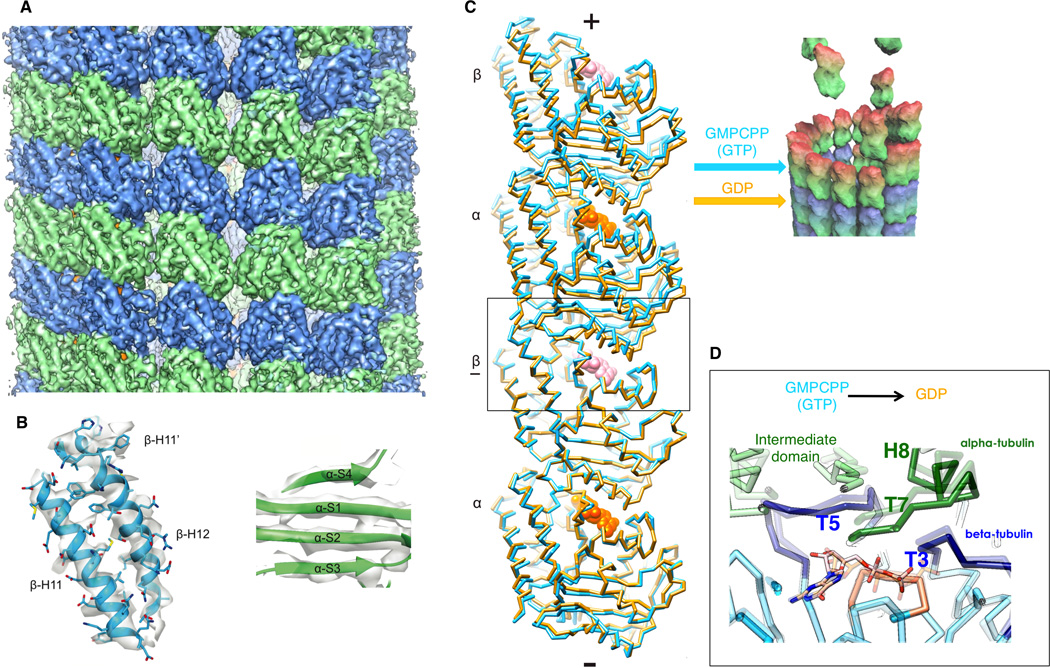
(A) Cryo-EM map of the GMPCPP MT (4.7 Å resolution). α-tubulin, green, β-tubulin, blue. (B) β- tubulin C-terminal helices (left) and beta strands in the α-tubulin intermediate domain (right) from the Rosetta GMPCPP MT model. (C) Cα traces of two longitudinally associated tubulin dimers from the GMPCPP (gold) and GDP (light purple) Rosetta models, superimposed on the underlined β tubulin, showing that hydrolysis results in a compression of the E-site at the interdimer interface (box). View is tangential to the microtubule lumen. The cartoon of a MT GTP cap illustrates how the two states described could coexists at a MT end. (D) Interdimer interface by the E-site nucleotide showing the conformational changes from GMPCPP (semitransparent) to GDP (solid).
5 – RECENT TECHNICAL BREAKTHROUGHS
Instrumentation and image processing developments have both been critical in the evolution of the 3D-EM field. An obvious example critical for the success of the cryo-EM methodology was the advent of commercial cold stages that allowed imaging of samples maintained at liquid nitrogen temperature without sacrificing resolution. Image quality also improved with the microscope technology, such as the field emission gun (FEG), intermediate voltages or improved illumination systems, while throughput went up with the automation of data collection (Suloway et al., 2005). Even as important, if not more so in the last two decades, has been the development of robust algorithms for single particle analysis (Frank et al., 1996; Grigorieff, 2007; Scheres et al., 2008; Tang et al., 2007; van Heel et al., 1996). Only about two years ago, state of the art studies of the ribosome, a test bed sample for many new approaches attempting to break resolution barriers for samples lacking symmetry, were leading to structures at about 5–7 Å resolution (Armache et al., 2010; Seidelt et al., 2009). Such studies required the averaging of hundreds of thousands, if not millions, of particle images, and therefore data sets were collected over multiple, long sessions at the microscope. Furthermore, a significant percentage of the micrographs in a typical cryo-EM data collection session was often thrown away altogether due to beam-induced motion, a phenomenon still poorly understood that results in “blurring” of the image (loss of high resolution information) (Henderson and Glaeser, 1985). In spite of experimental efforts at reducing beam-induced motion, more notably spot-scan-imaging (Downing, 1991), this image degradation process has been a major limiting factor in the cryo-EM field. Another major shortcoming has been the poor performance of digital cameras, especially at the preferred electron energy for imaging biological samples (300 keV).
During the last two years the cryo-EM field has undergone what some are now describing as a “revolution” (Kuhlbrandt, 2014). This positive transformation relates to the advent of the first commercial direct electron detection devices (DDDs) after years of development at the MRC-LMB by Henderson and Faruqui, and at LBNL and UCSD by Denes, Xuong and coworkers. The higher signal-to-noise ratio of these detectors with respect to traditional film or the scintillator-based CCD cameras, was an obvious gain in a field limited by low signal. But DDDs came with an additional bonus. Grigorieff and colleagues were the first to demonstrate, using rotavirus particles, that the high contrast together with the fast read out of the DDD allowed splitting the total dose (typically 20 Å/e2 over a couple of seconds) into short frames (e.g. 20, with typically 1 e/Å2 dose each) where the blurring due to beam induced movement is minimized, and where frame alignment is carried out computationally after data collection (Brilot et al., 2012; Campbell et al., 2012). Because particle images with higher signal-to-noise ratios can now be aligned more accurately, the reconstruction improves in two different ways. On one hand, one needs to average fewer particles because they are less noisy; on the other hand, each particle contributes to higher resolutions as it is aligned more accurately. Moreover, higher resolution references lead to even more accurate alignments, thus resulting in a strong synergistic effect.
The availability of better images from DDDs has coincided with the development of better image processing algorithms. Many different software packages have now been used in conjunction with DDD data sets to calculate (near-)atomic resolution reconstructions (Bischoff et al., 2014; Koh et al., 2014; Wang et al., 2014). In what follows, we will concentrate on maximum likelihood image processing approaches, which are quickly gaining in popularity.
The development of maximum-likelihood approaches in cryo-EM parallels that seen for macromolecular X-ray crystallography since the 1990s. Both X-ray and cryo-EM structure determination represent “incomplete data problems”, where part of the data needed to determine the structure is considered to be missing. The missing data in X-ray diffraction experiments are the phases of the X-ray reflections; in cryo-EM the missing data are the relative orientations of the individual particles. In the 1980s Gerard Bricogne proposed a Bayesian framework for the crystallographic “phase problem” (Bricogne, 1988). Whereas conventional least-squares approaches to this problem attempted to estimate an optimal value for the phase of each reflection, the Bayesian approach integrates over probability distributions of all possible values. Nowadays, the great majority of macromolecular X-ray structures are determined using maximum-likelihood methods that are based on this statistical framework.
A similar approach was first introduced by Fred Sigworth for the “orientation problem” of single-particle analysis in the simplified case of aligning a set of 2D images against a single 2D reference structure (Sigworth, 1998). In this instance, the relative orientations (in-plane rotations and translations) of all particles images can be considered as the missing data. Again, whereas conventional least-squares approaches assign a single, best orientation to each particle image, the maximum likelihood approach integrates over the probability distribution of all possible orientations. An extension of this idea to image classification, by considering also the assignment to a user-defined number of classes as missing data, proved to be particularly powerful to separate mixtures into structurally homogeneous subsets, both in 2D (Scheres et al., 2005) and in 3D (Scheres et al., 2007a). Application of 3D maximum likelihood (ML3D) classification to a data set of E.coli ribosomes with sub-stoichiometric binding of elongation factor G (EF-G) provided the first indication that different structural states could be separated from a mixture of 2D projection images without depending on prior knowledge about the structural variability in the data (Scheres et al., 2007). Nowadays, multiple maximum-likelihood classification approaches have been implemented in a variety of software packages (de la Rosa-Trevin et al., 2013; Lyumkis et al., 2013; Scheres, 2012a). Apart from separating different conformational or compositional states, 3D classification is also important to reach atomic-resolution structures in general. It appears that a significant number of particle images in a typical cryo-EM data set contribute negatively to the reconstruction process. A possible reason may be poor preservation of those particles during the blotting/freezing procedures. Thereby, 3D classification algorithms play a crucial role in separating such particles from those that do contain useful high-resolution information.
Recently, a variant of the maximum-likelihood approach was introduced in the form of an (empirical) Bayesian approach (Scheres, 2012b). The difference with earlier maximum-likelihood approaches and the Bayesian approach lies in a regularization term that expresses prior knowledge about the 3D reconstruction in the refinement process. By imposing smoothness on the reconstructed maps, the Bayesian approach intrinsically inhibits the accumulation of noise in the reconstruction. A formulation of the reconstruction problem in Fourier space allows for an improved description of correlated experimental noise (Scheres et al., 2007b), and the correction of CTF effects on both phases and amplitudes. Because important parameters about the power of the noise and the signal are inferred from the data, and because overfitting is monitored by the independent refinement of two separate halves of the data (Scheres and Chen, 2012), the Bayesian approach has the potential to yield clean, high-resolution reconstructions with a minimum of user intervention. Its implementation in the RELION software package has provided the community with a powerful tool for objective, high-resolution structure determination and the handling of structurally heterogeneous data sets (Scheres, 2012b).
With several commercially available direct-electron detectors using different engineered implementations, and with the availability of new image processing algorithms, the field was set for a quantum leap. In 2013, using the new detector technology and a modification of the Bayesian image processing approach to correct for beam-induced motion in each particle, it was shown that near-atomic resolution structures (once again of the ribosome) could be obtained, and that they only required a small fraction of the particle images required previously (tens of thousands instead of millions) (Bai et al., 2013). Soon after, in another landmark paper, using a whole-image beam-induced motion correction algorithm and many more particles, an even higher resolution was obtained for the 20S proteasome (another test sample for cryo-EM), which allowed for the first time to build an atomic model in a cryo-EM map from a particle with relatively lowsymmetry (Li et al., 2013). Since then, a number of studies have demonstrated that samples beyond viruses and ribosomes can be 3D reconstructed by cryo-EM to better than 4Å resolution, thus allowing for ab initio atomic modeling (Allegretti et al., 2014). Examples now include systems that were traditionally considered too small for cryo-EM reconstruction, like the TRPV1 channel, a tetramer of less than 500 kDA (Liao et al., 2013), beta-galactosidase (another tetramer of less than 500 kDa) (Bartesaghi et al., 2014), and gamma-secretase, an asymmetric complex of four membrane-proteins (with 170 kDa of protein mass) that is described in more detail below (Lu et al., 2014). Moreover, combined with the newly developed classification algorithms, biologically relevant states can now be identified and structurally characterized from a mixture of many other different states, as also described below for various eukaryotic translation complexes.
6. ATOMIC STRUCTURES FROM STRUCTURALLY HETEROGENEOUS SAMPLES: EUKARYOTIC TRANSLATION COMPLEXES
Ribosomes have long been a pet sample for the development and implementation of single particle cryo-EM reconstruction methods. Although these developments have resulted in an abundance of biologically insightful translation complexes, the structural characterization of ribosomes at atomic resolution had traditionally been the realm of X-ray crystallography. Ribosome purifications from thermophilic bacteria proved particularly well suited for crystallization. However, progress with eukaryotic ribosomes has been much slower. Today, hundreds of prokaryotic ribosome crystal structures have been described (Schmeing and Ramakrishnan, 2009), but only a few crystal structures of eukaryotic ribosome complexes are available (Ben-Shem et al., 2011; Klinge et al., 2011; Rabl et al., 2011). Although many of the early developments in single-particle analysis were driven by work on prokaryotic ribosomes, by 2010 cryo-EM studies of eukaryotic ribosomes were abundant and reconstructions were reaching 5–6 Å (Armache et al., 2010; Seidelt et al., 2009). Still, at these resolutions it was not possible to build atomic models de novo from the reconstructed density maps.
The first ribosome study to exploit the technical breakthroughs discussed above used a prototype of a back-thinned Falcon-II direct electron detector that was modified to intercept 16 movie frames during 1-second exposures (Bai et al., 2013). To account for both beam-induced rotations and translations, the Bayesian approach in the RELION program was extended to align running averages of several movie frames against the 3D reference. This approach revealed similar patterns of beam-induced motions as observed before by Grigorieff and co-workers (Brilot et al., 2012), where within a single field of view, particles would move in many different directions. Beam-induced rotations were observed to be in the range of 1–2 degrees; beam-induced translations were in the order of 3–5 Å. After correction for these motions, a reconstruction from only 35,000 particles yielded an overall resolution of 4.5 Å, while the best parts of the map showed clear side chain densities for many of the larger amino acids.
Subsequent application of the same techniques, but using higher magnification, led to atomic resolution reconstructions for the large subunit of the yeast (Amunts et al., 2014) and human (Brown et al., 2014; Greber et al., 2014) mitochondrial ribosomes, the mammalian cytoplasmic ribosome in complex with the Sec61 translocon (Voorhees et al., 2014), the yeast ribosome in complex with cricket paralysis virus internal ribosome entry site (CrPV IRES) (Fernandez et al., 2014), and the cytoplasmic ribosome of the Plasmodium falciparum parasite in complex with the anti-protozoan drug emetine (Wong et al., 2014). In all these studies, beam-induced motion correction resulted in near 3 Å resolution maps from approximately 50 to 100 thousand particles. 3D classification played an essential role in obtaining high-quality maps, as all these samples contained mixtures of different structural states. A major source of conformational variability in ribosomes is the relative rotation of the small ribosomal subunit with respect to the large one. This ratchet-like motion, essential for ribosome function, needs to be accounted for in image classification. In addition to this general issue, the yeast mitochondrial ribosomes samples were contaminated with 30% of cytoplasmic ribosomes, which stuck to the cytoplasmic side of the purified mitochondrial membranes. Because these different ribosomes could be separated in the computer by 3D classification of the particle images, further biochemical purification, which was hindered by limited amounts of material, could be circumvented (Figure 4A). For both the Sec61 and the CrPV IRES studies, 3D classification identified multiple functional states, providing further biological insights into the functioning of these complexes from a single cryo-EM experiment. A striking example of 3D classification was obtained in a study of the yeast ribosome in complex with eukaryotic initiation factor 5b (eIF5B) (Fernandez et al., 2013). In this case, only 4% of the particles had the initiation factor bound in the targeted state. Still, 3D classification was able to identify this minority class, and beam-induced motion correction led to a 6.5 Å map from only 5,000 particles. The observation that very small classes may indeed be identified suggests that it may be possible to obtain many atomic resolution maps of a ribosomal sample from a single cryo- EM experiment, provided that the initial data set comprises millions of particles. Once a homogeneous subpopulation of at least fifty thousand ribosomes has been identified, 3D reconstruction with beam-induced motion correction typically yields near 3 Å resolution maps. Because the phase information is preserved in cryo-EM images, these maps are often more interpretable than X-ray maps at the same nominal resolution, allowing de novo building of an atomic model (Figure 4B). This capability was of paramount importance for the mitochondrial ribosome, as these ribosomes have markedly diverged from their prokaryotic counterparts during 1.8 billion years of separated evolution. For the large subunit of the yeast mitochondrial ribosome, homologous structures were not available for approximately half of the proteins, and an atomic model for 600 kDa of polypeptide had to be built de novo in the cryo-EM map (Brown et al., 2015).
Figure 4. High-resolution cryo-EM structures from heterogeneous samples.

(A) To some extent, the classical approach in structural biology to study biochemically purified, homogeneous samples may be bypassed by cryo-EM image processing, where images of a mixture may be separated in the computer using powerful classification algorithms to obtain high-resolution structures for multiple components in the mixture. (B) Provided enough particles may be identified for each component, atomic-resolution maps may be generated for each of them. (C) Even small-molecule compounds may be built inside the high-resolution maps, in this case the eukaryotic translation inhibitor emetine is shown bound to the cytoplasmic ribosome from the P. falciparum parasite.
Within a time span of one and a half years, the new cryo-EM techniques have produced atomic models for the large subunits of the mitochondrial ribosome from yeast and human, as well as the cytoplasmic ribosomes from mammals, P. falciparum, and yeast (the latter in complex with eIF5B and CrPV IRES). X-ray crystallographic studies of the same samples could have easily taken many years, and even if successful, the final resolution would probably not be much better. Does that mean X-ray crystallography studies on ribosomes are now a thing of the past? Probably not entirely. Although the P. falciparum ribosome map clearly resolved the emetine compound (figure 4C), in silico design of novel therapeutics would probably still benefit from higher resolution than the 3.2 Å in this study. Also, screening many different compounds by repeating the same cryo-EM experiment would require considerable efforts in both data collection and processing. Therefore, as long as the problem of crystallization can be overcome, both throughput and resolution may still be better using X-ray diffraction (Garreau de Loubresse et al., 2014).
7. NEAR-ATOMIC RESOLUTION STRUCTURES FOR SMALLER COMPLEXES: HUMAN GAMMA-SECRETASE
While the ribosome, with a molecular mass of several megadalton and a high RNA-content, constitutes a very favorable sample for cryo-EM, smaller complexes present a bigger challenge. The signal to noise ratio in the images drops with particle size, and accurate alignment and classification of smaller particles becomes a severe bottleneck. Gamma-secretase is a membrane-embedded protease complex that is formed by four different proteins (presenilin, Pen- 2, Aph-1, and Nicastrin), with a molecular mass of 230 kDa (with 60 kDa corresponding to glycosylation on Nicastrin). This complex cleaves many trans-membrane proteins that are involved in various signaling pathways, such as the Notch receptor and the amyloid precursor protein (APP) (see (De Strooper et al., 2012) for a recent review). Abnormal Notch signaling is linked to cancer, while aberrant cleavage of APP leads to accumulation of β-amyloid plaques in the brain and to Alzheimer’s disease. Difficulties in producing large enough quantities of homogeneous and stable, detergent-solubilized complexes have so far precluded crystallization, in spite of decades of efforts by many labs (Li et al., 2009). EM characterization of this sample has also proven challenging. By 2013, five different groups had produced five completely different reconstructions for the same complex (Lazarov et al., 2006; Li et al., 2014; Ogura et al., 2006; Osenkowski et al., 2009; Renzi et al., 2011), reflecting the difficulty of 3D reconstruction of small particles with the previously available technology, and underscoring the need for robust validation techniques (Rosenthal and Henderson, 2003).
The use of a K2 Summit direct electron detector in single-electron counting mode to image a gamma-secretase sample that was transiently co-expressed in mammalian cells and subsequently stabilized in amphipols, yielded sufficient signal to identify individual gammasecretase complexes. However, the movie frame alignment procedure in RELION that was developed for ribosomes no longer worked for the much noisier gamma-secretase images, and a new procedure was developed that allowed tracking beam-induced movements for smaller particles (Scheres, 2014). This procedure yielded a reconstruction with an overall resolution of 4.5 Å from 144,000 particles (Lu et al., 2014). At this resolution, individual β-strands and many bulky side chains were visible in the map, but smaller side chains and most loops were unresolved (Fig. 5). Also, the density for the trans-membrane domain was worse than for the soluble Nicastrin domain, and the map in this region showed insufficient detail to unambiguously assign which of the four proteins gave rise to which of 19 trans-membrane helices. As such, the exact location of the active site, which resides in two of the trans-membrane helices of presenilin, could not be identified. Therefore, a higher resolution reconstructed of gamma-secretase is still eagerly awaited. Better imaging, a larger data set that will allow for more extensive classification, and further stabilization of the complex by ligand binding may be feasible routes towards this exciting goal.
Figure 5. Near-atomic resolution cryo-EM structure of human gamma-secretase.

(A) Overall view of the complex with the trans-membrane domain (TMD), which is made up of the four different proteins, in blue, and the extra-cellular domain (ECD) of Nicastrin in green. (B) Representative density for the soluble domain showing a region of the map with separated betastrands. (C) View inside the TMD perpendicular to the membrane showing the horse-shoe like arrangement of the trans-membrane helices with a thick and a thin end. The lack of good sidechain density in this region of the map prohibited the assignment of each helix to the four different proteins.
8. CONCLUSIONS AND OUTLOOK
Single particle cryo-EM has emerged as a powerful alternative to X-ray crystallography that is broadly applicable. Recent technical breakthroughs, both in instrumentation and image processing software, have dramatically increased the achievable resolution, the throughput, and the capacity of cryo-EM to describe conformational and compositional mixtures. As a consequence, a flood of biological insight is being gained on systems traditionally refractant to structural characterization, including membrane proteins, large assemblies that can only be produced in small amounts (e.g. general transcriptional machinery) or states that are rare and part of a complex mixture (e.g. translation intermediates).
Still, significant technical issues remain. The new detectors are still less efficient than what should be possible in theory. Fragile complexes may fall apart during cryo-EM grid preparation, due to physical forces on the sample during blotting or due to interactions with the hydrophobic air-water interface. Continuous forms of structural variability are not adequately dealt with in existing image classification algorithms. And, complexes smaller than 200 kDa remain challenging because of low signal-to-noise ratios in the images. Therefore, continuing developments of sample preparation (Russo and Passmore, 2014a, b) and image processing techniques, as well as the development of even better detectors and the commercialization of phase plates, which will allow imaging smaller particles (see (Chang et al., 2010; Danev et al., 2014) for two recent, commercialized implementations), may likely lead to even further improvements in the near future.
Finally, the fact that recent successes are attracting many new researchers to the cryo-EM field, does not come without challenges. Among them are: (1) the elevated costs of purchasing and maintaining expensive EM equipment by many institutes; (2) the enormous computational load that DDD movie processing and Bayesian refinements of all these new data bring; and (3) the requirement of easy-to-use tools for structure validation. The first two points call for a new model of shared instruments and access to supercomputer resources that funding agencies should keep in mind with the expansion of the cryo-EM community. The third point has special relevance in the context of this influx of new users of cryo-EM, who may not be fully aware of the potential pitfalls of the methodology if not used adequately.
While we look forward to an increasing number of successful applications of single particle cryo-EM, it is important to remain aware that not all reconstructions, even those using the most up to date instruments and software, can be expected to reach atomic resolution easily. For many biological macromolecules the inability to grow crystals is related to conformational freedom, which also makes cryo-EM structure determination to atomic resolution challenging. This is particularly the case for structures rich in flexible elements that may sample a continuum of states. While lack of high resolution may be disappointing, a structural description of this conformational landscape may have unique value for such samples. Ultimately, it is the biological insight one gains from it, rather than the single number of resolution, that determines the value of a 3D-EM reconstruction.
Acknowledgements
This work was funded by NIGMS (GM051487 and GM63072 to E.N), and by the UK Medical Research Council (MC_UP_A025_1013) and the Miller Institute for Basic Research in Science to S.H.W.S. E.N. is a Howard Hughes Medical Institute Investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adrian M, Dubochet J, Lepault J, McDowall AW. Cryo-electron microscopy of viruses. Nature. 1984;308:32–36. doi: 10.1038/308032a0. [DOI] [PubMed] [Google Scholar]
- Agirrezabala X, Liao HY, Schreiner E, Fu J, Ortiz-Meoz RF, Schulten K, Green R, Frank J. Structural characterization of mRNA-tRNA translocation intermediates. Proc Natl Acad Sci U S A. 2012;109:6094–6099. doi: 10.1073/pnas.1201288109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allegretti M, Mills DJ, McMullan G, Kuhlbrandt W, Vonck J. Atomic model of the F420-reducing [NiFe] hydrogenase by electron cryo-microscopy using a direct electron detector. eLife. 2014;3:e01963. doi: 10.7554/eLife.01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alushin GM, Lander GC, Kellogg EH, Zhang R, Baker D, Nogales E. High-resolution microtubule structures reveal the structural transitions in alphabeta-tubulin upon GTP hydrolysis. Cell. 2014;157:1117–1129. doi: 10.1016/j.cell.2014.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A, Brown A, Bai XC, Llacer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343:1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armache JP, Jarasch A, Anger AM, Villa E, Becker T, Bhushan S, Jossinet F, Habeck M, Dindar G, Franckenberg S, et al. Cryo-EM structure and rRNA model of a translating eukaryotic 80S ribosome at 5.5-A resolution. Proc Natl Acad Sci U S A. 2010;107:19748–19753. doi: 10.1073/pnas.1009999107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayaz P, Ye X, Huddleston P, Brautigam CA, Rice LM. A TOG:alphabeta-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Science. 2012;337:857–860. doi: 10.1126/science.1221698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai XC, Fernandez IS, McMullan G, Scheres SH. Ribosome structures to near-atomic resolution from thirty thousand cryo-EM particles. eLife. 2013;2:e00461. doi: 10.7554/eLife.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartesaghi A, Matthies D, Banerjee S, Merk A, Subramaniam S. Structure of beta-galactosidase at 3.2-A resolution obtained by cryo-electron microscopy. Proc Natl Acad Sci U S A. 2014;111:11709–11714. doi: 10.1073/pnas.1402809111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. The structure of the eukaryotic ribosome at 3.0 A resolution. Science. 2011;334:1524–1529. doi: 10.1126/science.1212642. [DOI] [PubMed] [Google Scholar]
- Bischoff L, Berninghausen O, Beckmann R. Molecular basis for the ribosome functioning as an L-tryptophan sensor. Cell reports. 2014;9:469–475. doi: 10.1016/j.celrep.2014.09.011. [DOI] [PubMed] [Google Scholar]
- Bleichenbacher M, Tan S, Richmond TJ. Novel interactions between the components of human and yeast TFIIA/TBP/DNA complexes. J Mol Biol. 2003;332:783–793. doi: 10.1016/s0022-2836(03)00887-8. [DOI] [PubMed] [Google Scholar]
- Bottcher B, Wynne SA, Crowther RA. Determination of the fold of the core protein of hepatitis B virus by electron cryomicroscopy. Nature. 1997;385:88–91. doi: 10.1038/386088a0. [DOI] [PubMed] [Google Scholar]
- Bricogne G. A Bayesian statistical theory of the phase problem. I. A multichannel maximum-entropy formalism for constructing generalized joint probability distributions of structure factors. Avcta Cryst. 1988;A44:517–545. [Google Scholar]
- Brilot AF, Chen JZ, Cheng A, Pan J, Harrison SC, Potter CS, Carragher B, Henderson R, Grigorieff N. Beam-induced motion of vitrified specimen on holey carbon film. J Struct Biol. 2012;177:630–637. doi: 10.1016/j.jsb.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SH, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014 doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Long F, Nicholls RA, Toots J, Emsley P, Murshudov G. Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Crystallogr. D. Biol. Crystallogr. 2015;71:136–153. doi: 10.1107/S1399004714021683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MG, Cheng A, Brilot AF, Moeller A, Lyumkis D, Veesler D, Pan J, Harrison SC, Potter CS, Carragher B, et al. Movies of ice-embedded particles enhance resolution in electron cryo-microscopy. Structure. 2012;20:1823–1828. doi: 10.1016/j.str.2012.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplow M, Ruhlen RL, Shanks J. The free energy of hydrolysis of a microtubule-bound nucleotide triphosphate is near zero: all of the free energy for hydrolysis is stored in the microtubule lattice. J. Cell Biol. 1994;127:779–788. doi: 10.1083/jcb.127.3.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WH, Chiu MT, Chen CY, Yen CF, Lin YC, Weng YP, Chang JC, Wu YM, Cheng H, Fu J, et al. Zernike phase plate cryoelectron microscopy facilitates single particle analysis of unstained asymmetric protein complexes. Structure. 2010;18:17–27. doi: 10.1016/j.str.2009.12.001. [DOI] [PubMed] [Google Scholar]
- Chen Z, Speck C, Wendel P, Tang C, Stillman B, Li H. The architecture of the DNA replication origin recognition complex in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 2008;105:10326–10331. doi: 10.1073/pnas.0803829105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, et al. Architecture of the RNA polymerase IITFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. Molecular architecture of human polycomb repressive complex 2. eLife. 2012;1:e00005. doi: 10.7554/eLife.00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clare DK, Vasishtan D, Stagg S, Quispe J, Farr GW, Topf M, Horwich AL, Saibil HR. ATP-triggered conformational changes delineate substrate-binding and - folding mechanics of the GroEL chaperonin. Cell. 2012;149:113–123. doi: 10.1016/j.cell.2012.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danev R, Buijsse B, Khoshouei M, Plitzko JM, Baumeister W. Volta potential phase plate for in-focus phase contrast transmission electron microscopy. Proc Natl Acad Sci U S A. 2014;111:15635–15640. doi: 10.1073/pnas.1418377111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Rosa-Trevin JM, Oton J, Marabini R, Zaldivar A, Vargas J, Carazo JM, Sorzano CO. Xmipp 3.0: an improved software suite for image processing in electron microscopy. J Struct Biol. 2013;184:321–328. doi: 10.1016/j.jsb.2013.09.015. [DOI] [PubMed] [Google Scholar]
- DeRosier DJ, Klug A. Reconstruction of three dimensional structures from electron micrographs. Nature. 1968;217:130–134. doi: 10.1038/217130a0. [DOI] [PubMed] [Google Scholar]
- Downing KH. Spot-scan imaging in transmission electron microscopy. Science. 1991;251:53–59. doi: 10.1126/science.1846047. [DOI] [PubMed] [Google Scholar]
- Dubochet J, Lepault J, Freeman R, Berriman JA, Homo JC. Electron microscopy of frozen water and aqueous solutions. J. Microsc. 1982;128:219–237. [Google Scholar]
- Elmlund D, Davis R, Elmlund H. Ab initio structure determination from electron microscopic images of single molecules coexisting in different functional states. Structure. 2010;18:777–786. doi: 10.1016/j.str.2010.06.001. [DOI] [PubMed] [Google Scholar]
- Fan L, Fuss JO, Cheng QJ, Arvai AS, Hammel M, Roberts VA, Cooper PK, Tainer JA. XPD helicase structures and activities: insights into the cancer and aging phenotypes from XPD mutations. Cell. 2008;133:789–800. doi: 10.1016/j.cell.2008.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez IS, Bai XC, Hussain T, Kelley AC, Lorsch JR, Ramakrishnan V, Scheres SH. Molecular architecture of a eukaryotic translational initiation complex. Science. 2013;342:1240585. doi: 10.1126/science.1240585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez IS, Bai XC, Murshudov G, Scheres SH, Ramakrishnan V. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell. 2014;157:823–831. doi: 10.1016/j.cell.2014.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Frank J, Zhu J, Penczek P, Li Y, Srivastava S, Verschoor A, Radermacher M, Grassucci R, Lata RK, Agrawal RK. A model of protein synthesis based on cryoelectron microscopy of the E. coli ribosome. Nature. 1995;376:441–444. doi: 10.1038/376441a0. [DOI] [PubMed] [Google Scholar]
- Gaiser F, Tan S, Richmond TJ. Novel dimerization fold of RAP30/RAP74 in human TFIIF at 1.7 A resolution. J Mol Biol. 2000;302:1119–1127. doi: 10.1006/jmbi.2000.4110. [DOI] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 2014;513:517–522. doi: 10.1038/nature13737. [DOI] [PubMed] [Google Scholar]
- Gigant B, Curmi PA, Martin-Barbey C, Charbaut E, Lachkar S, Lebeau L, Siavoshian S, Sobel A, Knossow M. The 4 angstrom X-ray structure of a tubulin : stathmin-like domain complex. Cell. 2000;102:809–816. doi: 10.1016/s0092-8674(00)00069-6. [DOI] [PubMed] [Google Scholar]
- Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich JA, Cutler G, Tjian R. Contacts in context: promoter specificity and macromolecular interactions in transcription. Cell. 1996;84:825–830. doi: 10.1016/s0092-8674(00)81061-2. [DOI] [PubMed] [Google Scholar]
- Goulet A, Major J, Jun Y, Gross SP, Rosenfeld SS, Moores CA. Comprehensive structural model of the mechanochemical cycle of a mitotic motor highlights molecular adaptations in the kinesin family. Proc Natl Acad Sci U S A. 2014;111:1837–1842. doi: 10.1073/pnas.1319848111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505:515–519. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- Grigorieff N. FREALIGN: high-resolution refinement of single particle structures. J Struct Biol. 2007;157:117–125. doi: 10.1016/j.jsb.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Grunberg S, Warfield L, Hahn S. Architecture of the RNA polymerase II preinitiation complex and mechanism of ATP-dependent promoter opening. Nat Struct Mol Biol. 2012;19:788–796. doi: 10.1038/nsmb.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Fang J, Taatjes DJ, Nogales E. Structural visualization of key steps in human transcription initiation. Nature. 2013;495:481–486. doi: 10.1038/nature11991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Baldwin JM, Ceska TA, Zemlin F, Beckman E, Downing KH. Model for the structure of bacteriorhodopsin based on high-resolution eletron cryomicroscopy. J. Mol. Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- Henderson R, Chen S, Chen JZ, Grigorieff N, Passmore LA, Ciccarelli L, Rubinstein JL, Crowther RA, Stewart PL, Rosenthal PB. Tilt-pair analysis of images from a range of different specimens in single-particle electron cryomicroscopy. J Mol Biol. 2011;413:1028–1046. doi: 10.1016/j.jmb.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson R, Glaeser RM. Quantitative analysis of image contrast in electron micrographs of beam-sensitive crystals. Ultramicroscopy. 1985;16:139–150. [Google Scholar]
- Kim TK, Ebright RH, Reinberg D. Mechanism of ATP-dependent promoter melting by transcription factor IIH. Science. 2000;288:1418–1422. doi: 10.1126/science.288.5470.1418. [DOI] [PubMed] [Google Scholar]
- Klinge S, Voigts-Hoffmann F, Leibundgut M, Arpagaus S, Ban N. Crystal structure of the eukaryotic 60S ribosomal subunit in complex with initiation factor 6. Science. 2011;334:941–948. doi: 10.1126/science.1211204. [DOI] [PubMed] [Google Scholar]
- Koh CS, Brilot AF, Grigorieff N, Korostelev AA. Taura syndrome virus IRES initiates translation by binding its tRNA-mRNA-like structural element in the ribosomal decoding center. Proc Natl Acad Sci U S A. 2014;111:9139–9144. doi: 10.1073/pnas.1406335111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostrewa D, Zeller ME, Armache KJ, Seizl M, Leike K, Thomm M, Cramer P. RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature. 2009;462:323–330. doi: 10.1038/nature08548. [DOI] [PubMed] [Google Scholar]
- Kuhlbrandt W. Biochemistry. The resolution revolution. Science. 2014;343:1443–1444. doi: 10.1126/science.1251652. [DOI] [PubMed] [Google Scholar]
- Lander GC, Estrin E, Matyskiela ME, Bashore C, Nogales E, Martin A. Complete subunit architecture of the proteasome regulatory particle. Nature. 2012;482:186–191. doi: 10.1038/nature10774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasker K, Forster F, Bohn S, Walzthoeni T, Villa E, Unverdorben P, Beck F, Aebersold R, Sali A, Baumeister W. Molecular architecture of the 26S proteasome holocomplex determined by an integrative approach. Proc Natl Acad Sci USA. 2012;109:1380–1387. doi: 10.1073/pnas.1120559109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leschziner AE, Nogales E. The orthogonal tilt reconstruction method: An approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol. 2006;153:284–299. doi: 10.1016/j.jsb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, Gubbens S, Agard DA, Cheng Y. Electron counting and beam-induced motion correction enable near-atomicresolution single-particle cryo-EM. Nat Methods. 2013;10:584–590. doi: 10.1038/nmeth.2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao M, Cao E, Julius D, Cheng Y. Structure of the TRPV1 ion channel determined by electron cryo-microscopy. Nature. 2013;504:107–112. doi: 10.1038/nature12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Jin L, Koh SB, Atanasov I, Schein S, Wu L, Zhou ZH. Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science. 2010;329:1038–1043. doi: 10.1126/science.1187433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe J, Li H, Downing KH, Nogales E. Refined structure of alpha betatubulin at 3.5 A resolution. J Mol Biol. 2001;313:1045–1057. doi: 10.1006/jmbi.2001.5077. [DOI] [PubMed] [Google Scholar]
- Lu P, Bai XC, Ma D, Xie T, Yan C, Sun L, Yang G, Zhao Y, Zhou R, Scheres SH, et al. Three-dimensional structure of human gamma-secretase. Nature. 2014;512:166–170. doi: 10.1038/nature13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucic V, Forster F, Baumeister W. Structural studies by electron tomography: from cells to molecules. Annu Rev Biochem. 2005;74:833–865. doi: 10.1146/annurev.biochem.73.011303.074112. [DOI] [PubMed] [Google Scholar]
- Lyumkis D, Brilot AF, Theobald DL, Grigorieff N. Likelihood-based classification of cryo-EM images using FREALIGN. J Struct Biol. 2013;183:377–388. doi: 10.1016/j.jsb.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow E-M, Mandelkow E, Milligan RA. Microtubules dynamics and microtubules caps: a time-resolved cryo-electron microscopy study. J. Cell Biol. 1991;114:977–991. doi: 10.1083/jcb.114.5.977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Segall J, Weil PA, Roeder RG. Multiple factors required for accurate initiation of transcription by purified RNA polymerase II. J Biol Chem. 1980;255:11992–11996. [PubMed] [Google Scholar]
- Meyerson JR, Kumar J, Chittori S, Rao P, Pierson J, Bartesaghi A, Mayer ML, Subramaniam S. Structural mechanism of glutamate receptor activation and desensitization. Nature. 2014 doi: 10.1038/nature13603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Nettles JH, Li H, Cornett B, Krahn JM, Snyder JP, Downing KH. The binding mode of epothilone A on alpha,beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wang HW. Structural mechanisms underlying nucleotide-dependent self-assembly of tubulin and its relatives. Curr Opin Struct Biol. 2006 doi: 10.1016/j.sbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Nogales E, Wolf SG, Downing KH. Structure of the ab tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- Penczek PA, Grassucci RA, Frank J. The ribosome at improved resolution: new techniques for merging and orientation refinement in 3D cryo-electron microscopy of biological particles. Ultramicroscopy. 1994;53:251–270. doi: 10.1016/0304-3991(94)90038-8. [DOI] [PubMed] [Google Scholar]
- Prota AE, Bargsten K, Zurwerra D, Field JJ, Diaz JF, Altmann KH, Steinmetz MO. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science. 2013;339:587–590. doi: 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiation factor 1. Science. 2011;331:730–736. doi: 10.1126/science.1198308. [DOI] [PubMed] [Google Scholar]
- Radermacher M. Three-dimensional reconstruction of single particles from random and nonrandom tilt series. J. Elect. Micros. Tech. 1988;9:359–394. doi: 10.1002/jemt.1060090405. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J. Micros. 1987;146:113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- Rayment I, Holden HM, Whittaker M, Yohn CB, Lorenz M, Holmes KC, Milligan RA. Structure of the actin-myosin complex and its implications for muscle contraction [see comments] Science. 1993;261:58–65. doi: 10.1126/science.8316858. [DOI] [PubMed] [Google Scholar]
- Robinson C, Sali A, Baumeister W. The molecular sociology of the cell. Nature. 2007;450:973–982. doi: 10.1038/nature06523. [DOI] [PubMed] [Google Scholar]
- Roeder RG. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem Sci. 1996;21:327–335. [PubMed] [Google Scholar]
- Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single-particle electron cryomicroscopy. J Mol Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- Russo CJ, Passmore LA. Controlling protein adsorption on graphene for cryo- EM using low-energy hydrogen plasmas. Nat Methods. 2014a;11:649–652. doi: 10.1038/nmeth.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo CJ, Passmore LA. Electron microscopy: Ultrastable gold substrates for electron cryomicroscopy. Science. 2014b;346:1377–1380. doi: 10.1126/science.1259530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. A Bayesian view on cryo-EM structure determination. J Mol Biol. 2012a;415:406–418. doi: 10.1016/j.jmb.2011.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J Struct Biol. 2012b;180:519–530. doi: 10.1016/j.jsb.2012.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Methods. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Gao H, Valle M, Herman GT, Eggermont PP, Frank J, Carazo JM. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat Methods. 2007a;4:27–29. doi: 10.1038/nmeth992. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Nunez-Ramirez R, Gomez-Llorente Y, San Martin C, Eggermont PP, Carazo JM. Modeling experimental image formation for likelihood-based classification of electron microscopy data. Structure. 2007b;15:1167–1177. doi: 10.1016/j.str.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Nunez-Ramirez R, Sorzano CO, Carazo JM, Marabini R. Image processing for electron microscopy single-particle analysis using XMIPP. Nat Protoc. 2008;3:977–990. doi: 10.1038/nprot.2008.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH, Valle M, Nunez R, Sorzano CO, Marabini R, Herman GT, Carazo JM. Maximum-likelihood multi-reference refinement for electron microscopy images. J Mol Biol. 2005;348:139–149. doi: 10.1016/j.jmb.2005.02.031. [DOI] [PubMed] [Google Scholar]
- Schmeing TM, Ramakrishnan V. What recent ribosome structures have revealed about the mechanism of translation. Nature. 2009;461:1234–1242. doi: 10.1038/nature08403. [DOI] [PubMed] [Google Scholar]
- Schreiber A, Stengel F, Zhang Z, Enchev RI, Kong EH, Morris EP, Robinson CV, da Fonseca PC, Barford D. Structural basis for the subunit assembly of the anaphase-promoting complex. Nature. 2011;470:227–232. doi: 10.1038/nature09756. [DOI] [PubMed] [Google Scholar]
- Seidelt B, Innis CA, Wilson DN, Gartmann M, Armache JP, Villa E, Trabuco LG, Becker T, Mielke T, Schulten K, et al. Structural insight into nascent polypeptide chain-mediated translational stalling. Science. 2009;326:1412–1415. doi: 10.1126/science.1177662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ. A maximum-likelihood approach to single-particle image refinement. J. Struct. Biol. 1998;122:328–339. doi: 10.1006/jsbi.1998.4014. [DOI] [PubMed] [Google Scholar]
- Suloway C, Pulokas J, Fellmann D, Cheng A, Guerra F, Quispe J, Stagg S, Potter CS, Carragher B. Automated molecular microscopy: the new Leginon system. J Struct Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, Ludtke SJ. EMAN2: an extensible image processing suite for electron microscopy. J. Struct. Biol. 2007;157:38–46. doi: 10.1016/j.jsb.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Taylor KA, Glaeser RM. Electron microscopy of frozen hydrated biological specimens. J. Ultrast. Res. 1976;55:448–456. doi: 10.1016/s0022-5320(76)80099-8. [DOI] [PubMed] [Google Scholar]
- Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006;41:105–178. doi: 10.1080/10409230600648736. [DOI] [PubMed] [Google Scholar]
- Tsai FTF, Sigler PB. Structural basis of preinitiation complex assembly on human Pol II promoters. EMBO J. 2000;19:25–36. doi: 10.1093/emboj/19.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unwin N. Acetylcholine receptor channel imaged in the open state. Nature. 1995;373:37–43. doi: 10.1038/373037a0. [DOI] [PubMed] [Google Scholar]
- Valle M, Sengupta J, Swami NK, Grassucci RA, Burkhardt N, Nierhaus KH, Agrawal RK, Frank J. Cryo-EM reveals an active role for aminoacyl-tRNA in the accommodation process. EMBO J. 2002;21:3557–3567. doi: 10.1093/emboj/cdf326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.van Heel M. Angular reconstitution: A posteriory assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–124. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- 101.van Heel M, Harauz G, Orlova EV, Schmidt R, Schatz M. A new generation of the IMAGIC image processing system. J Struct Biol. 1996;116:17–24. doi: 10.1006/jsbi.1996.0004. [DOI] [PubMed] [Google Scholar]
- 102.Voorhees RM, Fernandez IS, Scheres SH, Hegde RS. Structure of the mammalian ribosome-Sec61 complex to 3.4 A resolution. Cell. 2014;157:1632–1643. doi: 10.1016/j.cell.2014.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wang Z, Hryc CF, Bammes B, Afonine PV, Jakana J, Chen DH, Liu X, Baker ML, Kao C, Ludtke SJ, et al. An atomic model of brome mosaic virus using direct electron detection and real-space optimization. Nature communications. 2014;5:4808. doi: 10.1038/ncomms5808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wong W, Bai XC, Brown A, Fernandez IS, Hanssen E, Condron M, Tan YH, Baum J, Scheres SH. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife. 2014;3 doi: 10.7554/eLife.03080. [DOI] [PMC free article] [PubMed] [Google Scholar]