

Summary
It has long been the dream of biologists to map gene expression at the single cell level. With such data one might track heterogeneous cell sub-populations, and infer regulatory relationships between genes and pathways. Recently, RNA sequencing has achieved single cell resolution. What is limiting is an effective way to routinely isolate and process large numbers of individual cells for quantitative in-depth sequencing. We have developed a high-throughput droplet-microfluidic approach for barcoding the RNA from thousands of individual cells for subsequent analysis by next-generation sequencing. The method shows a surprisingly low noise profile and is readily adaptable to other sequencing-based assays. We analyzed mouse embryonic stem cells, revealing in detail the population structure and the heterogeneous onset of differentiation after LIF withdrawal. The reproducibility of these high-throughput single cell data allowed us to deconstruct cell populations and infer gene expression relationships.
Introduction
Much of the physiology of metazoans is reflected in the temporal and spatial variation of gene expression among constituent cells. Some variation is stable and has helped us to define both adult cell types and many intermediate cell types in development (Hemberger et al. 2009). Other variation results from dynamic physiological events such as the cell cycle, changes in cell microenvironment, development, aging, and infection (Loewer and Lahav, 2011). Still other expression changes appear to be stochastic in nature (Paulsson, 2005; Swain et al. 2002), and may have important consequences (Losick and Desplan, 2008). To understand gene expression in development and physiology, biologists would ideally like to map changes in RNA levels, protein levels, and post-translational modifications in every cell. Analysis at the single cell level has until a decade ago principally been through in situ hybridization for RNA, immunostaining for proteins, or, more recently with fluorescent chimeric proteins. These methods allow only a few genes to be monitored in each experiment, however. More recently, pioneering work (e.g. (Chiang and Melton, 2003; Phillips and Eberwine, 1996)) has made possible global transcriptional profiling at the single cell level, though the number of cells is often limited. Although an RNA inventory at the single cell level does not offer a complete picture of the state of the cell, it can provide important insights into cellular heterogeneity and collective fluctuations in gene expression, as well as crucial information about the presence of distinct cell subpopulations in normal and diseased tissues. There is also hope that gene expression correlations within cell populations can be used to derive lineage structures (Qiu et al. 2011) and pathway structures de novo by reverse engineering (He et al. 2009).
Modern methods for RNA sequence analysis (RNA-Seq) can quantify the abundance of RNA molecules in a population of cells with great sensitivity. After considerable effort these methods have been harnessed to analyze RNA content in single cells. What is needed now are effective ways to isolate and process large numbers of individual cells for in-depth RNA sequencing, and to do so with quantitative precision. This requires cell isolation under uniform conditions, preferably with minimal cell loss, especially in the case of clinical samples. The requirements for the number of cells, the depth of coverage, and the accuracy of measurements will depend on experimental considerations, including factors such as the difficulty of obtaining material, the complexity of the cell population, and the extent to which cells are diversified in gene expression space. The depth of coverage necessary is hard to predict a priori, but the existence of rare cell types in populations of interest, such as occult tumor cells or tissue stem cell sub-populations (Simons and Clevers, 2011), combined with independent drivers of heterogeneity such as cell cycle and stochastic effects, suggests that analyzing large numbers of cells will be necessary.
The challenges of single cell RNA-Seq are easy to appreciate. Measurement accuracy is highly sensitive to the efficiency of its enzymatic steps; and the need for amplification from single cells risks introducing considerable errors. There are major obstacles to parallel processing of thousands of cells, and to handling small samples of cells efficiently so that nearly every cell is measured. Microfluidics has emerged as a promising technology for single-cell studies with the potential to address these challenges (Lecault et al. 2012; Wu et al. 2014). Microfluidic chips containing hundreds of valves can trap, lyse and assay biomolecules from single cells with higher precision and often with better efficiencies than micro-titer plates (Streets et al. 2014; Wu et al. 2014). For RNA sequencing of single cells, reduced reaction volumes improve the yields of cDNA and reduce technical variability (Islam et al. 2014; Wu et al. 2014). Yet the number of single cells that can be currently processed with microfluidic chips remains at ~70–90 cells per run, so analyzing large numbers of cells is difficult, and may take so much time that the cells are no longer viable. Moreover, capture efficiency of cells into microfluidic chambers is low, a potential issue for rare or clinical samples. An alternative is the use of microfluidic droplets suspended in carrier oil (Guo et al. 2012; Teh et al. 2008). Cells can be compartmentalized into droplets and assayed for different bio-molecules (Mazutis et al. 2013), their genes amplified (Eastburn et al. 2013) and droplets sorted at high-throughput rates (Agresti et al. 2010). Unlike conventional plates or valve-based microfluidics, droplets are intrinsically scalable: the number of reaction ‘chambers’ is not limited, and capture efficiencies are high since all cells in a sample volume can in principle be captured in droplets.
We exploited droplet microfluidics to develop a technique for indexing thousands of individual cells for RNA sequencing, which we term inDrop (indexing droplets) RNA sequencing. Another droplet-based RNA-seq technology is also described in this issue (McCarroll et al., 2015). Our method has a theoretical capacity to barcode tens of thousands of cells in a single run. Here we use hundreds to thousands of cells per run, since sequencing depth and cost becomes limiting for us at very high cell counts. We evaluated inDrop sequencing by profiling mouse embryonic stem (ES) cells before and after LIF withdrawal. A total of over 10,000 barcoded cells and controls were profiled, with ~3,000 ES and differentiating cells sequenced at greater depth for subsequent analysis. Our analysis identifies rare sub-populations expressing markers of distinct lineages that would be difficult to find by profiling a few hundred cells. We show that key pluripotency factors fluctuate in a correlated manner across the entire ES cell population, and we explore whether fluctuations might associate gene products with the pluripotent state. Upon differentiation, we observe dramatic changes in the correlation structure of gene expression, resulting from asynchronous inactivation of pluripotency factors, and the emergence of novel cell states. Altogether, our results showcase the potential of droplet methods to deconstruct large populations of cells and to infer gene expression relationships within a single experiment.
RESULTS
A microfluidic platform for droplet barcoding and analysis of single cells
The inDrop platform encapsulates cells into droplets with lysis buffer, reverse transcription (RT) reagents, and barcoded oligonucleotide primers (Fig. 1). mRNA released from each lysed cell remains trapped in the same droplet and is barcoded during synthesis of complementary DNA (cDNA). After barcoding, material from all cells is combined by breaking the droplets, and the cDNA library is sequenced using established methods (CEL-Seq) (Hashimshony et al. 2012; Jaitin et al. 2014). The major challenge is to ensure that each droplet carries primers encoding a different barcode. We synthesized a library of barcoded hydrogel microspheres (BHMs) that are co-encapsulated with cells (Figs. 2). Each BHM carries covalently coupled, photo-releasable primers encoding one of 147,456 barcodes, and the pool size could be increased in a straightforward manner. The current pool size allows randomly labeling 3,000 cells with 99% unique labeling (Extended Methods); many more cells can be processed by splitting a large emulsion into separate tubes.
Figure 1. A platform for DNA barcoding thousands of cells.

Cells are encapsulated into droplets with lysis buffer, reverse-transcription mix, and hydrogel microspheres carrying barcoded primers. After encapsulation primers are released. cDNA in each droplet is tagged with a barcode during reverse transcription. Droplets are then broken and material from all cells is linearly amplified before sequencing. UMI = unique molecular identifier.
Figure 2. Barcoding hydrogel microsphere synthesis.

A) Microfluidic preparation of hydrogel microspheres containing a common DNA. Scale bars 100 μm. B) The common DNA primer: acrylic phosphoroamidite moiety (blue), photo-cleavable spacer (green), T7 RNA polymerase promoter sequence (red) and sequencing primer (blue). C,D) Method for combinatorial barcoding of the microspheres. E) The fully assembled primer: T7 promoter (red), sequencing primer (blue), barcodes (green), synthesis adaptor (dark brown), UMI (yellow) and poly-T primer (purple). See also Fig. S1.
To barcode the cells, we developed a microfluidic device with four inlets for the BHMs, cells, RT/lysis reagents and carrier oil; and one outlet port for droplet collection (Fig. 3). The device generates monodisperse droplets that can be varied in the range of 1–5 nL at a rate of ~10–100 drops per second, simultaneously mixing aliquots from the inlets (Fig. 3A-C; Movies S1, S2). The flow of deformable hydrogels inside the chip can be synchronized due to their close packing and regular release, allowing nearly 100% hydrogel droplet occupancy (Abate et al. 2009). Thus cells arriving into droplets will nearly always be co-encapsulated with barcoded primers. Due to the large cross-section of the microfluidics channel (60×80 μm2) there is no cell size bias in capture. In typical conditions, cells occupy only 10% of droplets, so two-cell events are rare (Fig. 3D), and cell aggregates are minimized by passing cells through a strainer or by FACS. Droplets must contain at least one cell and one gel to produce a barcoded library for sequencing; we observed that over 90% of these productive droplets contained exactly one cell and one gel (Fig. 3E). After cell and BHM encapsulation, primers are photo-released by ultraviolet exposure, a step critical for efficient RT (Figs. 1,3F).
Figure 3. A droplet barcoding device.

A) Microfluidic device design, see also Fig. S2. B,C) Snapshots of encapsulation (left) and collection (right) modules, see also Movies S1,S2. Arrows indicate cells (red), hydrogels (blue), and flow direction (black). Scale bars 100μm. D) Droplet occupancy over time. E) Cell and hydrogel co-encapsulation statistics showing a high 1:1 cell:hydrogel correspondence. F) BioAnalyzer traces showing dependence of library abundance on primer photo-release. H) Number of cells/controls as a function of collection volume.
With this system, we captured cells at a rate of 4,000–12,000/hour, or 2,000–3,000 cells barcoded for every 100μL of emulsion (Fig. 3G). As the cost of sequencing drops, higher scales may become routine.
Validation of random barcoding and droplet integrity
We tested droplet integrity by barcoding a ~50:50 mixture of mouse ES and human K562 erythroleukemia cells (Fig. 4A). In this test each barcode should associate entirely with either mouse or human transcripts; only two-cell events would lead to the appearance of barcodes with mixed profiles. Fig. 4A shows that indeed 96% of barcodes mapped to either the mouse or human transcriptome with more than 99% purity. This already low error rate (~4%) could be further reduced by dilution of the cell suspensions, or by sorting singlet droplets (Baret et al. 2009). However, the presence of rare two cell events does not obscure rare cell sub-populations, since even if 10% of cells are in doublets, then 90% of rare cells will be found as singlets. This is demonstrated later for ES cells, where we found a rare cell type representing <1% of the population.
Figure 4. Technical noise in droplet barcoding.

A) Droplet integrity control: mouse and human cells are co-encapsulated to allow unambiguous identification of barcodes shared across multiple cells; 4% of barcodes share mixed mouse/human reads. B) inDrops technical control schematic, and histogram of UMI-filtered mapped (UMIFM) reads per droplet. C) Unique gene symbols detected as a function of UMIFM reads per droplet. D) Mean UMIFM reads for spike-in molecules are linearly related to their input concentration, with a capture efficiency β=7.1%. E) Method sensitivity S as a function of input RNA abundance; red curve is the sensitivity limit of binomial sampling (S = 1 − e−βx). F) CV-mean plot of pure RNA after normalization. Data points correspond to individual gene symbols; solid curve is the binomial sampling noise limit. For abundant transcripts, droplet-to-droplet variability in method efficiency β sets a baseline CV (dashed curve: CVβ=5%), see also Fig. S3. G) Relationships between observed and biological values of gene CVs, Fano Factors and correlations, showing how low efficiency dampens Fano Factors (Eq. 2) and weakens correlations (Eq. 3).
We also tested that cell barcodes were randomly sampled from the intended pool of possible barcodes. A comparison of barcode identities across a total of 11,085 control droplets consistently showed excellent agreement with random sampling (Fig. S3A).
Baseline technical noise for inDrops
Two major sources of technical noise in single cell RNA-Seq are variability between cells in mRNA capture efficiency, and the intrinsic sampling noise resulting from capturing finite numbers of mRNA transcripts in each cell. The CEL-Seq protocol has been reported to have a capture efficiency of ~3% (Grun et al. 2014) or less (Jaitin et al. 2014), and a variability in capture efficiency of ~25% for pure RNA controls and ~50% for cells (coefficients of variation between samples) when performed in microtitre plates (Grun et al. 2014). Technical noise can also arise during library amplification, but this is mostly eliminated through the use of random unique molecular identifier (UMI) sequences, allowing bioinformatic removal of duplicated reads (Fu et al. 2011; Islam et al. 2014).
An ideal test of technical noise would compare two identical cells, but unfortunately there are no cells where one can assert that the abundance of transcripts would be equal. To test technical noise in our system, we analyzed a control sample of purified total RNA diluted to single cell concentration (10pg per droplet), mixed with ERCC RNA spike-in controls of known concentration (Baker et al. 2005) (Fig. 4B). We processed 953 droplets with an average of 30·103 (±21%) UMI-filtered mapped (UMIFM) reads per droplet (Fig. 4B), and low sequencing redundancy (averaging 2.3 reads/molecule; Fig. S3E). Each droplet gave 5–15·103 unique gene symbols (25,209 detected in total), correlating strongly with UMIFM counts (Fig. 4C). The method showed an excellent linear readout of the ERCC spike-in input concentration (Fig. 4D) down to concentrations of 0.5 molecules/droplet on average; below that limit, we tended to over-count transcripts, a bias seen previously (Grun et al. 2014; Hashimshony et al. 2012).
Another measure of method performance is its sensitivity, i.e. the likelihood of detecting an expressed gene. The sensitivity was almost entirely explained by binomial sampling statistics (Fig. 4E; Extended Methods), and thus depends on transcript abundance and the capture efficiency, measured from the ERCC spike-ins to be 7.1% (Fig. 4D). With this efficiency, sensitivity was 50% when 10 transcripts were present, and >95% when >45 transcripts were present (Fig. 4E). The sensitivity and capture efficiency are lower than those estimated for another single cell transcriptomics protocol (~20%) (Picelli et al. 2014), but are higher than those reported for CEL-Seq (3.4%) (Grun et al. 2014; Hashimshony et al. 2012). Moreover, the low sequencing redundancy suggests that deeper sequencing may further increase efficiency and thus sensitivity.
In accuracy, the method showed very low levels of technical noise, assessed by comparing the coefficient of variation (CV = standard deviation/mean) of each gene across the cell population to its mean abundance. In a system limited only by sampling noise, all genes should obey CV=(mean)−1/2. Technical noise can lead to dispersion around this curve, and to a minimum “baseline” CV. After normalization, 99.5% of detected genes were consistent with the power law, with a baseline technical noise of <10% (N=25,209; p>0.01 χ2 test, no multiple hypothesis correction) (Fig. 4F). To our knowledge, this noise profile is among the cleanest obtained for single cell data to date, although the sampling noise is still high (see comparisons in Fig. S3H). Consistent with the low noise profile, the mean and CV values for genes measured in cells (see below) correlated well with results measured by single-molecule fluorescent in situ hybridization (Fig. S3 with data from (Grun et al. 2014); Pearson correlation R=0.92 for mean, and R=0.90 for CV).
Noise modeling of single cell data
Before analyzing cells, we developed a technical noise model of the effects of low sampling efficiency of transcripts, and of the effects of cell-to-cell variation (noise) in efficiency. Low efficiency and noise in efficiency affect both the observed cell-to-cell variability of gene expression, and the observed covariation of gene expression. We derived relationships between biological and observed quantities for the CVs of gene abundances across cells, gene Fano Factors (variance/mean), and pairwise correlations between genes (Fig. 4G and Theory section of Supplemental Information). The Fano Factor is commonly used to measure noisy gene expression, and yet is very sensitive to the efficiency β (Eq. 2): even without technical noise, only genes with a Fano Factor F ≳ 1/β will be noticeably variable in inDrops or other methods for single cell analysis. The addition of technical noise introduces a “baseline” CV (Brennecke et al. 2013; Grun et al. 2014), and spuriously amplifies true biological variation (Eq. 1). Low sampling efficiencies also dampen correlations between gene pairs in a predictable manner, setting an expectation to find relatively weak but nevertheless statistically significant correlations in our data (Eqs. 2–3). These results provide a basis for formally controlling for noise in single cell measurements.
Single cell profiling of mouse ES cells
Single cell transcriptomics can distinguish cell types of distinct lineages even with very low sequencing depths (Pollen et al. 2014). What is less clear is the type of information that can be determined from studying a relatively uniform population subject to stochastic fluctuations. To explore this, we chose to study mouse ES cells maintained in serum. These cells exhibit well-characterized fluctuations, but are still uniform compared to differentiated cell types and thus pose a challenge for single cell sequencing.
Previous studies have indicated that ES cells are heterogeneous in gene expression (Guo et al. 2010; Hayashi et al. 2008; MacArthur et al. 2012; Martinez Arias and Brickman, 2011; Ohnishi et al. 2014; Singer et al. 2014; Torres-Padilla and Chambers, 2014; Yan et al. 2013). Other studies, which sorted ES cells into populations expressing high or low levels of the pluripotency factors Nanog (Chambers et al. 2007; Kalmar et al. 2009), Rex1/Zfp42 (Singer et al. 2014; Toyooka et al. 2008) and Stella/Dppa3 (Hayashi et al. 2008), have suggested that ES cells fluctuate infrequently between two metastable epigenetic states corresponding to a pluripotent inner cell mass (ICM)-like state, and an epiblast-like state poised to differentiate. These pluripotency factors were found to correlate with the expression of the epigenetic modifier Dnmt3b and its regulator Prdm14, and with global differences in chromatin methylation (Singer et al. 2014; Yamaji et al. 2013). Evidence suggests that other sources of heterogeneity also exist in the ES cell population: fluctuations in the Primitive Endoderm (PrEn) marker Hex, for example, associate with a bias towards PrEn fate upon differentiation (Canham et al. 2010); fluctuations in Hes1 bias differentiation into Epiblast sub-lineages (Kobayashi et al. 2009); and rare expression of other markers (Zscan4, Eif1a and others) associate with a totipotent state with access to extra-embryonic fates (Macfarlan et al. 2012). Whether these multiple fate biases result from dynamic fluctuations of transcription factors or represent stable cell states is not known.
To test inDrop sequencing, we harvested different numbers of cells at different sequencing depths for each of the ES cell runs. We collected 935 ES cells for deep sequencing, and two further samples of 2,509 and 3,447 cells from a single dish as technical replicates. We further sampled 145, 302 and 2,160 cells after 2 days after LIF withdrawal; 683 cells after 4 days; and 169 and 799 cells after 7 days. The average number of reads per cell ranged up to 208·103, and the average UMIFM counts up to 29·103 (Table S1). Technical replicates showed very high reproducibility (Pearson correlation of CVs R>0.98, Fig. 5A inset); as did biological replicates (R=0.98), whereas differentiating cells showed distinct expression profiles (Fig. S4; R=0.94; 732 genes differentially expressed at more than 2-fold, see Table S2). The capture efficiency β, estimated from comparing UMIFM counts to smFISH results (Fig. S3), was slightly lower (4.5%) than for pure RNA.
Figure 5. inDrop sequencing reveals ES cell population structure.

A) CV-mean plot of the ES cell transcriptome. Pure RNA control (blue); genes significantly more variable than control (black). Solid and dashed curves are as in Fig. 4F [variability in cell size = 20%, see Theory Eq. (S4) in Supplemental Information]. Inset: gene CVs of two technical replicate cell populations (total n=5,956 cells), see also Fig. S4. B) Illustrative transcript counts showing low (Ttn), moderate (Trim28, Ly6a, Dppa5a) and high (Sparc, S100a6) expression variability; curve fits are Poisson (red) and Negative Binomial (blue) distributions. C) Above-Poisson (a.p.) noise, (CV2-1/mean) of pluripotency differentiation markers. D) Co-expression plots recapitulating known and novel gene expression relationships (see main text). E) The eigenvalue distribution of cell principal components (PC) reveals the number of non-trivial PCs detectable in the data (arrows), compared to eigenvalue distribution of randomized data (black) and to the Marcenko-Pastur distribution for a random matrix (red). F) The first four ES cell PCs and their coefficients, revealing three outlier populations. G) ES cell tSNE map revealing an axis of pluripotency-to-differentiation with fringe sub-populations at different points on the differentiation axis (see also Fig. S6). Top panel shows sub-populations visible in one projection. Lower panels show cells colored by abundance of specified gene sets (see Table S4).
Heterogeneous sub-populations of ES cell origin
For the 935 ES cells, we identified 2,044 significantly variable genes (Table S3, Fig. 5A,B) (10% FDR, statistical test in Extended Methods) expressed at a level of at least 5 UMIFM counts in at least one cell. The set of variable genes was enriched for annotations of metabolism and transcriptional regulation, and for targets of transcription factors associated with pluripotency (Sp1, Elk1, Nrf1, Myc, Max, Tcf3, Lef1), including transcription factors that directly interact with Pou5f1 and Sox2 promoter regions (Gao et al. 2013) (Gabpa, Jun, Yy1, Atf3) (Table S3, 10−120<p<10−10). Among the variable genes, we found pluripotency factors previously reported to fluctuate in ES cells (Nanog, Rex1/Zfp42, Dppa5a, Sox2, Esrrb) but, notably, the most highly variable genes included known markers of PrEn fate (Col4a1/2, Lama1/b1, Sox17, Sparc), markers of Epiblast fate (Krt8, Krt18, S100a6), and epigenetic regulators of the ES cell state (Dnmt3b). The vast majority of genes showed very low noise profiles, consistent with Poisson statistics (e.g. Ttn, Fig. 5B). We evaluated the above-Poisson noise, defined as η=CV2-1/μ (μ being the mean UMIFM count), for a select panel of genes (Fig. 5C) and found it to be in qualitative agreement with previous reports (Grun et al. 2014; Singer et al. 2014). Unlike the CV or the Fano Factor, η is expected to scale linearly with its true biological value even for low sampling efficiencies (Fig. 4G, Eq. (1)).
To test the idea that ES cells exhibit heterogeneity between a pluripotent ICM-like state and a more differentiated epiblast-like state, we contrasted the expression of candidate pluripotency and differentiation markers in single ES cells. Gene pair correlations (Fig. 5D) at first appear consistent with a discrete two-state view, since both the epiblast marker Krt8 and the PrEn marker Col4a1 were expressed only in cells low for Pou5f1 (shown) and other pluripotency markers (Fig. S6A). Also in agreement with previous studies (Toyooka et al. 2008), the differentiation-prone state was rare. The correlations also confirmed other known regulatory interactions in ES cells, for example Sox2, a known negative target of BMP signaling, was anti-correlated with the BMP target Id1. What was more surprising was the finding that multiple pluripotency factors (Nanog, Trim28, Esrrb, Sox2, Klf4, Zfp42) fluctuated in tandem across the bulk of the cell population, but not all pluripotency factors did so (Oct4/Pou5f1) (Fig. 5D and Fig. S6). These observations are not explained by a simple two-state model (Singer et al. 2014), since pluripotency factor levels are not determined only by differentiation state. Oct4/Pou5f1 instead correlated strongly with cyclin D3 (Fig. 5D and Fig. S5A), but not other cyclins, suggesting fluctuations of unknown origin.
What then is the structure of the ES cell population? We conducted a principal component analysis (PCA) of the ES cell population for the highly variable genes (Fig. 5E,F; sensitivity analysis in Fig. S5B; gene selection and normalization in Extended Methods). PCA reveals multiple non-trivial dimensions of heterogeneity (12 dimensions with 95% confidence) (Fig. 5E), which are not explained by independent fluctuations in each gene (Marčenko and Pastur, 1967; Plerou et al. 2002). Inspection of the first four principal components, and the principal genes contributing to these components (Fig. 5F, S5), revealed the presence of at least three small but distinct cell sub-populations: one rare population (6/935 cells) expressed very low levels of pluripotency markers and high levels of PrEn markers (Niakan et al. 2010); a second cell population (15/935 cells) expressed high levels of Krt8, Krt18, S100a6, Sfn and other markers of the epiblast lineage. The third population represented a seemingly uncharacterized state, marked by expression of heat shock proteins Hsp90, Hspa5 and other ER components such as the disulphide isomerase Pdia6. These sub-populations expressed low levels of pluripotency factors, suggesting they are biased toward differentiation or have already exited the pluripotent state. The latter population could also reflect stressed cells.
PCA analysis is a powerful tool for visualizing cell populations that can be fractionated with just two or three principal axes of gene expression. However, when more than three non-trivial principal components exist, PCA alone is not sufficient for dimensionality reduction of high-dimensional data. Using genes identified from PCA, we used t-distributed Stochastic Neighbor Embedding (t-SNE) (Amir el et al. 2013; Van der Maaten and Hinton, 2008) to further reduce dimensionality (Fig. 5G and Fig. S5C-L) (see Extended Methods). A continuum of states from high pluripotency to low pluripotency emerged, with several outlier populations at the population fringes. These included the three populations found by PCA, but also two additional fringe sub-populations characterized respectively by high expression of Prdm1/Blimp1 and Lin41/Trim71 (Fig. S5I-L). The first of these expressed moderate levels of the pluripotency factors, while the second expressed low levels. Thus, while we found evidence of ES cells occupying an epiblast-like state as previously reported, and indeed found evidence for collective fluctuations between ICM to epiblast-like states (Fig. 5G and Fig. S5), these fluctuations do not describe the full range of heterogeneity in the ES cell population.
Functional signatures in gene expression covariation
In complex mixtures of cells, correlations of gene expression patterns could arise from differences between mature cell lineages. In a population of a single cell type such as the ES cell population studied here, however, fluctuations in cell state might reveal functional dependencies among genes.
To test whether expression covariation might contain regulatory information, we explored the covariation partners of known pluripotency factors using a topological network analysis scheme, similar to approaches developed for comparing multiple bulk samples (Li and Horvath, 2007) (Fig. 6A; algorithm in Extended methods; sensitivity analysis of the method in Fig. S6A). This scheme identifies the set of genes most closely correlated with a given gene (or genes) of interest, and which also most closely correlate with each other. Given the sensitivity of correlations to sampling efficiency (Fig. 4G, Eq. (3)), we reasoned that a method based on correlation network topology would be more robust than using correlation magnitude. Remarkably, the network analysis strongly enriched for pluripotency factors: of the 20 nearest neighbors of Nanog, ten are documented pluripotency factors, three more are associated with pluripotency, and one (Slc2a3) is syntenic with Nanog (Scerbo et al. 2014). Only one gene (Rbpj) is dispensable for pluripotency (Oka et al. 1995). The analysis revealed a network of correlated pluripotency factors (Figs. S6B), with multiple pluripotency factors neighboring the same previously uncharacterized genes (Extended Methods and Fig. S6C). It is tempting to predict that at least some of these genes are also involved in maintaining the pluripotent state. For Sox2, the entire neighborhood consisted of factors directly or indirectly associated with pluripotency (Fig. 6C).
Figure 6. Regulatory information preserved in gene correlations.

A) A strategy for inferring robust gene associations from cell-to-cell variability with weak and/or highly connected gene correlations, see also Fig. S6. B-D) Gene neighborhoods of Nanog, Sox2, and Cyclin B. Grey boxes mark validated pluripotency factors; blue boxes mark factors previously associated with a pluripotent state. E,F) Correlations of 44 cell cycle-regulated transcripts in a somatic cell line (K562) and in mouse ES cells shows a loss of cell cycle dependent transcription in ES cells (gene names in Fig. S6). Genes are ordered by hierarchical clustering. Color scale applies to (E,F).
The same analysis may provide insight into other biological pathways, although pathways seemingly independent of ES cell biology had no meaningful topological network associations. This suggests that gene correlation networks in single cell data capture the fluctuations most specific to the biology of the cells being studied, but could be harnessed to study other pathways through weak experimental perturbations.
Cell cycle transcriptional oscillations in ES cells are weak compared to somatic cells
When the network analysis was applied to Cyclin B we found very few neighboring genes (Fig. 6C), raising the question of why single cell data does not reveal broader evidence of cell cycle-dependent transcription in ES cells. Previous studies have argued for an absence of ES cell cycle dependent transcription (White and Dalton, 2005). Cyclins (except cyclin B) are expressed uniformly throughout the cell cycle (Faast et al. 2004; Stead et al. 2002), and the activity of the E2F family of transcription factors, which normally oscillates in somatic cells, is also constitutive in ES cells (Stead et al. 2002). ES cells have a very short cell cycle of ~8–10 hours, with ~80% of cells in S phase (White and Dalton, 2005), and almost no G1 and G2 phases, so that cell cycle-dependent transcription could be difficult to detect.
We tested whether unperturbed ES cell data showed evidence of cell cycle transcriptional variation. As a control, we applied inDrops to human K562 erythroleukemia lymphoblasts (N=239 cells, average 27·103 UMIFM counts per cell), and focused on 44 transcripts previously categorized to a particular cell cycle phase (Whitfield et al. 2002). A hierarchical clustering of these genes ordered them across the K562 cell cycle, with anti-correlations between early and late cell cycle genes (Fig. 6E). When the same analysis was repeated for the ES cell population, we found correlations between the cell cycle genes were extremely weak, and only clustered a subset of G2/M genes (Fig. 6F). These results confirm that ES cells lack strong cell cycle oscillations in mRNA abundance, but they do show evidence of limited G2/M phase-specific transcription.
Population dynamics of differentiating ES cells
Upon LIF withdrawal, ES cells differentiate by a poorly characterized process, leading to the formation of predominantly epiblast lineages. In our single cell analysis, following unguided differentiation by LIF withdrawal (Nishikawa et al., 1998), the differentiating ES cell population underwent significant changes in population structure, qualitatively seen by hierarchical clustering cells (Fig. 7A). As validation, and to dissect the changes in the cell population, we first inspected selected pluripotency factors and differentiation markers (Fig 7B,C and Table S2). As seen in bulk assays, the average expression of Zfp42 and Esrrb levels dropped rapidly; Pou5f1 and Sox2 dropped gradually; the epiblast marker Krt8 increased steadily; and Otx2, one of the earliest transcription factors initiating differentiation from the ICM to the epiblast state, transiently increased by day 2 and then decreased (Yang et al. 2014). The average gene expression was not however representative of individual cells: some cells failed to express epiblast markers and a fraction of these expressed pluripotency factors at undifferentiated levels even seven days after LIF withdrawal, (Fig 7C). This trend was supported by a PCA analysis of cells from all time points (Fig. 7D; see Extended Methods for gene selection and normalization), showing that after 7 days, 5% (N=799) of cells overlapped with the ES cell population. The greatest temporal heterogeneity was evident at four days post-LIF, with cells spread broadly along the first principal component between the ES cell and differentiating state. The PCA analysis also revealed a metabolic signature (GO annotation: Cellular Metabolic Process, p=1.4·10−8) consistent with the changes occurring upon differentiation (Yanes et al. 2010).
Figure 7. Heterogeneity in differentiating ES cells.

A) Changes in global population structure after LIF withdrawal seen by hierarchically clustering cell-cell correlations over highly variable genes. B,C) Average (B) and distribution (C) of gene expression after LIF withdrawal; violin plots in (C) indicate the fraction of cells expressing a given number of counts; points show top 5% of cells. D,E) First two PCs of 3,034 cells showing asynchrony in differentiation. F) Epiblast and PrEn cell fractions as a function of time. G) tSNE maps of differentiating ES cells, and of genes (right panel) reveal putative population markers (see also Fig. S7 and Table S4). H) Intrinsic dimensionality of gene expression variability in ES cells and following LIF withdrawal, showing a smaller fluctuation sub-space during differentiation. The pure RNA control lacks correlations and displays a maximal fluctuation sub-space.
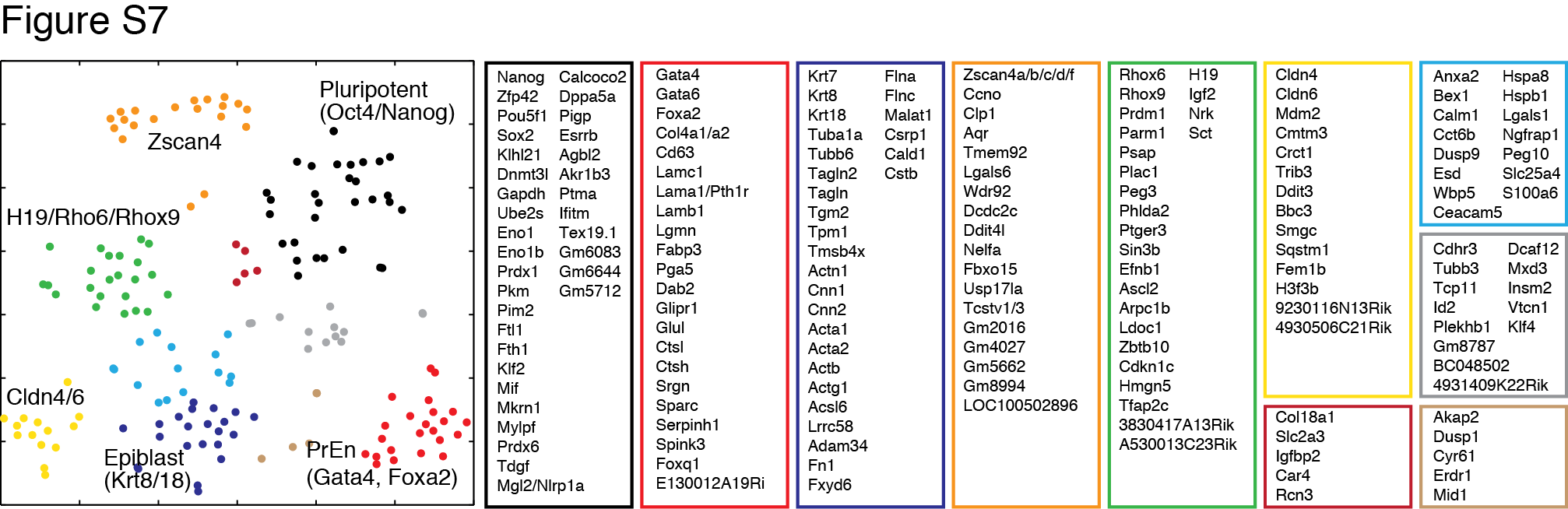
In addition to heterogeneity due to asynchrony, we visualized population structure by t-SNE and found distinct sub-populations, not all of which mapped to known cell types (Fig. 7G; sub-population markers tabulated in Table S4). tSNE of genes over the cells revealed clusters of genes marking distinct sub-populations (Fig. 7G right panel). At two and fours days post-LIF withdrawal, we identified cells expressing Zscan4 and Tcstv1/3, previously identified as rare totipotent cells expressing markers of the 2-cell stage (Macfarlan et al. 2012). At four and seven days, a population emerged expressing maternally imprinted genes (H19, Rhox6/9, Peg10, Cdkn1 and others), suggesting widespread DNA demethylation, possibly in early primordial germ cells. In addition, resident PrEn cells were seen at all time points (Figs. 7F,G), but failed to expand. In sum, the analysis exposes temporal heterogeneity in differentiation and distinct ES cell fates.
Refinement of gene expression upon differentiation
Our results allow testing suggestions that ES cells are characterized by promiscuous gene expression that becomes refined upon differentiation (Golan-Mashiach et al. 2005; Wardle and Smith, 2004). If so, differentiating cells should become confined to tighter domains in gene expression “space” than ES cells, as measured by the number of independent dimensions over which cells can be found. We evaluated the intrinsic dimensionality of the distribution of ES cells and differentiating cells in gene expression space using the method by (Kégl, 2002). Supporting the refinement hypothesis, we found that intrinsic dimensionality decreased after differentiation (Fig. 7H). Thus, ES gene expression fluctuations are weakly coupled compared to the more coherent differences following LIF withdrawal.
Discussion
We report here a platform for single cell capture, barcoding and transcriptome profiling, without physical limitations on the number of cells that can be processed. The method captures the majority of cells in a sample, has rapid collection times and has low technical noise. Such a method is suitable for small clinical samples including from tumors and tissue micro-biopsies, and opens up the possibility of routinely identifying cell types, even if rare, based on gene expression. This type of data is also valuable for identifying putative regulatory links between genes, by exploiting natural variation between individual cells. We gave simple examples of such inference, but this type of data lends itself to more formal reverse engineering.
We have developed the droplet platform initially for whole-transcriptome RNA sequencing; however the technology is highly flexible and should be readily adaptable to other applications requiring barcoding of RNA/DNA molecules. Our initial implementation of the method made use of a very simple droplet microfluidic chip, consisting of just a single flow-focusing junction. Future versions of the platform might take further advantage of droplet technology for multi-step reactions, or select target cells by sorting droplets on-chip (Guo et al. 2012).
The method in its current form still suffers some limitations. The major technical drawback we encountered was the mRNA capture efficiency of ~7%, which has only recently become robustly quantifiable using UMI-based filtering (Fu et al. 2011; Islam et al. 2014). Although higher than for several previously published methods, the efficiency is nonetheless too low to allow reliable detection in every cell of genes with transcript abundances lower than 20–50 transcripts. The method is therefore most reliable for profiling medium to highly abundant components of cells, missing some key transcriptional regulators, although we were able to detect almost all mouse transcription factors (1,350 out of 1,405) in a subset of cells, with the key ES cell transcription factors (Pou5f1, Sox2, Zfp42 and 44 other transcription factors) detected in over 90% of all cells. This is a general problem affecting single cell RNA sequencing, which will require improved cell lytic approaches or optimized enzymatic reactions in library preparation. A second drawback of the method is the random barcoding strategy, which does not allow individual cell identities (marked by shape, size, lineage or location) to be associated with a given barcode.
Despite these limitations, the current method can provide important data addressing many biological problems. This is illustrated by the challenging problem of ES cell heterogeneity and its dynamics during early differentiation. ES cells are not divided into large sub-populations of distinct cell types, and therefore analysis of their heterogeneity requires a sensitive method. Our analysis showed that, in the presence of serum and LIF, fluctuations in Oct4/Pou5f1 are decoupled from other pluripotency factors. We also found sub-populations of Epiblast and PrEn lineages, and other less well characterized ES cell sub-populations. This heterogeneity may reflect reversible fluctuations, or cells undergoing irreversible differentiation. The unbiased identification of small cell sub-populations requires the scale enabled by droplet methods.
Experimental Procedures
Microfluidic operation
The microfluidic device (80μm deep) was manufactured by soft lithography following standard protocols (Extended Methods). During operation, cells, RT/lysis mix and collection tubes were kept on ice. Flow rates were 100 μL/hr for cell suspension, 100 μL/hr for RT/lysis mix, 10–20 μL/hr for BHMs and 90 μL/hr for carrier oil to produce 4 nL drops. BHMs were washed 3x and concentrated by centrifugation 2x at 5krcf, then loaded directly into tubing for injection into the device. Cells were loaded at 50k–100k/mL in 16%v/v Optiprep (Sigma), and maintained in suspension using a micro-stir bar placed in the syringe. The carrier oil was HFE-7500 fluorinated fluid (3M) with 0.75% (w/w) EA surfactant (RAN Biotechnologies). See Extended Methods for BHM synthesis, buffer compositions, equipment, and detailed microfluidic protocols.
Library preparation
After cell encapsulation primers were released by 8 min UV exposure (365 nm at ~10 mW/cm2, UVP B-100 lamp) while on ice. The emulsion was incubated at 50°C for 2 hours, then 15 min at 70°C, then on ice. The emulsion was split into aliquots of 100–3500 cells, and demulsified by adding 0.2× 20% (v/v) perfluorooctanol, 80% (v/v) HFE-7500 and brief centrifugation. Broken droplets were stored at −20C and processed as per CEL-SEQ protocol, see Extended Methods.
Tissue culture
IB10 ES cells are a line derived from the mouse 129/Ola strain (subcloned from E14), kindly provided by Dr. Eva Thomas. Cells were maintained on flasks pre-coated with gelatin at density ~3×105 cells/mL. ES media contained phenol red free DMEM (Gibco), 15%v/v fetal bovine serum (Gibco), 2 mM L-glutamine, 1xMEM non-essential amino acids (Gibco), 1%v/v penicillin-streptomycin antibiotics, 110μM β-mercaptoethanol, 100μM sodium pyruvate. ESC base media was supplemented with 1000 U/mL Leukemia Inhibitory Factor (LIF). See Extended Methods for dissociation protocol and K562 cell culture.
Data analysis
See Extended Methods for custom bioinformatics, count normalization, method sensitivity, identification of highly variable genes, PCA and tSNE, and network neighborhood analysis.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We thank Mira Guo for guidance in hydrogel synthesis; Diego Jaitin for guidance on the CEL-SEQ/MARS-SEQ protocol; Clarissa Scholes and Angela DePace for feedback and for creating Figs 1,3; Rebecca Ward for help in editing. This study was supported by NIH SCAP Grant R21DK098818. AMK holds a Career Award at the Scientific Interface from the Burroughs-Wellcome Fund; LM holds a Marie Curie International Outgoing Fellowship (300121); AV is supported by the HSCI Medical Scientist Training Fellowship and the Harvard Presidential Scholars Fund. LP is supported by NIH Grant 5R01HD073104-03. Work in the MWK lab was supported by NIH (R01 GM026875, R01 GM103785, R01 HD073104), and in the DAW lab by NSF (DMR-1310266), the Harvard Materials Research Science and Engineering Center (DMR-1420570), DARPA (HR0011-11-C-0093), and NIH (P01HL120839). AMK, LM, IA, DAW and MWK have submitted patent applications (US62/065,348, US62/066,188, US62/072,944) for the work described.
Footnotes
Accession codes
Gene Expression Omnibus: raw sequence data and processed UMIFM counts are available under accession code GSE65525.
Author contributions
AMK, LM, IA and MWK conceived the method; LM developed the microfluidic device; AMK, LM, IA performed experiments; VL supervised ES cell culture; AMK and AV wrote the UMI filtering pipeline; NT and AMK developed and performed statistical noise analysis; AMK and LP developed and performed cell population and dimensionality analysis; AMK, LM and MWK wrote the manuscript. DAW and MWK supervised the study. All authors read and commented on the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abate AR, Chen CH, Agresti JJ, Weitz DA. Beating Poisson encapsulation statistics using close-packed ordering. Lab on a chip. 2009;9:2628–2631. doi: 10.1039/b909386a. [DOI] [PubMed] [Google Scholar]
- Agresti JJ, Antipov E, Abate AR, Ahn K, Rowat AC, Baret JC, Marquez M, Klibanov AM, Griffiths AD, Weitz DA. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proceedings of the National Academy of Sciences. 2010;107:4004–4009. doi: 10.1073/pnas.0910781107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir el AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SC, Bauer SR, Beyer RP, Brenton JD, Bromley B, Burrill J, Causton H, Conley MP, Elespuru R, Fero M, et al. The External RNA Controls Consortium: a progress report. Nature methods. 2005;2:731–734. doi: 10.1038/nmeth1005-731. [DOI] [PubMed] [Google Scholar]
- Baret JC, Miller OJ, Taly V, Ryckelynck M, El-Harrak A, Frenz L, Rick C, Samuels ML, Hutchison JB, Agresti JJ, et al. Fluorescence-activated droplet sorting (FADS): efficient microfluidic cell sorting based on enzymatic activity. Lab on a chip. 2009;9:1850–1858. doi: 10.1039/b902504a. [DOI] [PubMed] [Google Scholar]
- Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. Accounting for technical noise in single-cell RNA-seq experiments. Nature methods. 2013;10:1093–1095. doi: 10.1038/nmeth.2645. [DOI] [PubMed] [Google Scholar]
- Canham MA, Sharov AA, Ko MS, Brickman JM. Functional heterogeneity of embryonic stem cells revealed through translational amplification of an early endodermal transcript. PLoS biology. 2010;8:e1000379. doi: 10.1371/journal.pbio.1000379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers I, Silva J, Colby D, Nichols J, Nijmeijer B, Robertson M, Vrana J, Jones K, Grotewold L, Smith A. Nanog safeguards pluripotency and mediates germline development. Nature. 2007;450:1230–1234. doi: 10.1038/nature06403. [DOI] [PubMed] [Google Scholar]
- Chiang MK, Melton DA. Single-Cell Transcript Analysis of Pancreas Development. Developmental Cell. 2003;4:383–393. doi: 10.1016/s1534-5807(03)00035-2. [DOI] [PubMed] [Google Scholar]
- Cinelli P, Casanova EA, Uhlig S, Lochmatter P, Matsuda T, Yokota T, Rulicke T, Ledermann B, Burki K. Expression profiling in transgenic FVB/N embryonic stem cells overexpressing STAT3. BMC developmental biology. 2008;8:57. doi: 10.1186/1471-213X-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejosez M, Krumenacker JS, Zitur LJ, Passeri M, Chu LF, Songyang Z, Thomson JA, Zwaka TP. Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell. 2008;133:1162–1174. doi: 10.1016/j.cell.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastburn DJ, Sciambi A, Abate AR. Ultrahigh-throughput Mammalian single-cell reverse-transcriptase polymerase chain reaction in microfluidic drops. Analytical chemistry. 2013;85:8016–8021. doi: 10.1021/ac402057q. [DOI] [PubMed] [Google Scholar]
- Faast R, White J, Cartwright P, Crocker L, Sarcevic B, Dalton S. Cdk6-cyclin D3 activity in murine ES cells is resistant to inhibition by p16(INK4a) Oncogene. 2004;23:491–502. doi: 10.1038/sj.onc.1207133. [DOI] [PubMed] [Google Scholar]
- Fu GK, Hu J, Wang PH, Fodor SP. Counting individual DNA molecules by the stochastic attachment of diverse labels. Proc Natl Acad Sci U S A. 2011;108:9026–9031. doi: 10.1073/pnas.1017621108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F, Wei Z, An W, Wang K, Lu W. The interactomes of POU5F1 and SOX2 enhancers in human embryonic stem cells. Sci Rep. 2013;3 doi: 10.1038/srep01588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan-Mashiach M, Dazard JE, Gerecht-Nir S, Amariglio N, Fisher T, Jacob-Hirsch J, Bielorai B, Osenberg S, Barad O, Getz G, et al. Design principle of gene expression used by human stem cells: implication for pluripotency. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:147–149. doi: 10.1096/fj.04-2417fje. [DOI] [PubMed] [Google Scholar]
- Grun D, Kester L, van Oudenaarden A. Validation of noise models for single-cell transcriptomics. Nature methods. 2014;11:637–640. doi: 10.1038/nmeth.2930. [DOI] [PubMed] [Google Scholar]
- Guo G, Huss M, Tong GQ, Wang C, Li Sun L, Clarke ND, Robson P. Resolution of cell fate decisions revealed by single-cell gene expression analysis from zygote to blastocyst. Dev Cell. 2010;18:675–685. doi: 10.1016/j.devcel.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Guo MT, Rotem A, Heyman JA, Weitz DA. Droplet microfluidics for high-throughput biological assays. Lab on a chip. 2012;12:2146–2155. doi: 10.1039/c2lc21147e. [DOI] [PubMed] [Google Scholar]
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell reports. 2012;2:666–673. doi: 10.1016/j.celrep.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Lopes SM, Tang F, Surani MA. Dynamic equilibrium and heterogeneity of mouse pluripotent stem cells with distinct functional and epigenetic states. Cell Stem Cell. 2008;3:391–401. doi: 10.1016/j.stem.2008.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Balling R, Zeng AP. Reverse engineering and verification of gene networks: Principles, assumptions, and limitations of present methods and future perspectives. Journal of Biotechnology. 2009;144:190–203. doi: 10.1016/j.jbiotec.2009.07.013. [DOI] [PubMed] [Google Scholar]
- Hemberger M, Dean W, Reik W. Epigenetic dynamics of stem cells and cell lineage commitment: digging Waddington’s canal. Nature reviews Molecular cell biology. 2009;10:526–537. doi: 10.1038/nrm2727. [DOI] [PubMed] [Google Scholar]
- Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lonnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nature methods. 2014;11:163–166. doi: 10.1038/nmeth.2772. [DOI] [PubMed] [Google Scholar]
- Jaitin DA, Kenigsberg E, Keren-Shaul H, Elefant N, Paul F, Zaretsky I, Mildner A, Cohen N, Jung S, Tanay A, et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science. 2014;343:776–779. doi: 10.1126/science.1247651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmar T, Lim C, Hayward P, Munoz-Descalzo S, Nichols J, Garcia-Ojalvo J, Martinez Arias A. Regulated fluctuations in nanog expression mediate cell fate decisions in embryonic stem cells. PLoS biology. 2009;7:e1000149. doi: 10.1371/journal.pbio.1000149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kégl B. Intrinsic dimension estimation using packing numbers. Paper presented at: Advances in neural information processing systems.2002. [Google Scholar]
- Kobayashi T, Mizuno H, Imayoshi I, Furusawa C, Shirahige K, Kageyama R. The cyclic gene Hes1 contributes to diverse differentiation responses of embryonic stem cells. Genes Dev. 2009;23:1870–1875. doi: 10.1101/gad.1823109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecault V, White AK, Singhal A, Hansen CL. Microfluidic single cell analysis: from promise to practice. Current opinion in chemical biology. 2012;16:381–390. doi: 10.1016/j.cbpa.2012.03.022. [DOI] [PubMed] [Google Scholar]
- Li A, Horvath S. Network neighborhood analysis with the multi-node topological overlap measure. Bioinformatics. 2007;23:222–231. doi: 10.1093/bioinformatics/btl581. [DOI] [PubMed] [Google Scholar]
- Lian WX, Yin RH, Kong XZ, Zhang T, Huang XH, Zheng WW, Yang Y, Zhan YQ, Xu WX, Yu M, et al. THAP11, a novel binding protein of PCBP1, negatively regulates CD44 alternative splicing and cell invasion in a human hepatoma cell line. FEBS Letters. 2012;586:1431–1438. doi: 10.1016/j.febslet.2012.04.016. [DOI] [PubMed] [Google Scholar]
- Loewer A, Lahav G. We are all individuals: causes and consequences of non-genetic heterogeneity in mammalian cells. Current opinion in genetics & development. 2011;21:753–758. doi: 10.1016/j.gde.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losick R, Desplan C. Stochasticity and cell fate. 2008:65. doi: 10.1126/science.1147888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur BD, Sevilla A, Lenz M, Muller FJ, Schuldt BM, Schuppert AA, Ridden SJ, Stumpf PS, Fidalgo M, Ma’ayan A, et al. Nanog-dependent feedback loops regulate murine embryonic stem cell heterogeneity. Nat Cell Biol. 2012;14:1139–1147. doi: 10.1038/ncb2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature. 2012;487:57–63. doi: 10.1038/nature11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marčenko VA, Pastur LA. DISTRIBUTION OF EIGENVALUES FOR SOME SETS OF RANDOM MATRICES. Mathematics of the USSR-Sbornik. 1967;1:457. [Google Scholar]
- Martinez Arias A, Brickman JM. Gene expression heterogeneities in embryonic stem cell populations: origin and function. Curr Opin Cell Biol. 2011;23:650–656. doi: 10.1016/j.ceb.2011.09.007. [DOI] [PubMed] [Google Scholar]
- Mazutis L, Gilbert J, Ung WL, Weitz DA, Griffiths AD, Heyman JA. Single-cell analysis and sorting using droplet-based microfluidics. Nat Protoc. 2013;8:870–891. doi: 10.1038/nprot.2013.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niakan KK, Ji H, Maehr R, Vokes SA, Rodolfa KT, Sherwood RI, Yamaki M, Dimos JT, Chen AE, Melton DA, et al. Sox17 promotes differentiation in mouse embryonic stem cells by directly regulating extraembryonic gene expression and indirectly antagonizing self-renewal. Genes Dev. 2010;24:312–326. doi: 10.1101/gad.1833510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa SI, Nishikawa S, Hirashima M, Matsuyoshi N, Kodama H. Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages. Development. 1998;125:1747–1757. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
- Ohnishi Y, Huber W, Tsumura A, Kang M, Xenopoulos P, Kurimoto K, Oles AK, Arauzo-Bravo MJ, Saitou M, Hadjantonakis AK, et al. Cell-to-cell expression variability followed by signal reinforcement progressively segregates early mouse lineages. Nat Cell Biol. 2014;16:27–37. doi: 10.1038/ncb2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka C, Nakano T, Wakeham A, de la Pompa JL, Mori C, Sakai T, Okazaki S, Kawaichi M, Shiota K, Mak TW, et al. Disruption of the mouse RBP-J kappa gene results in early embryonic death. Development. 1995;121:3291–3301. doi: 10.1242/dev.121.10.3291. [DOI] [PubMed] [Google Scholar]
- Paulsson J. Models of stochastic gene expression. Physics of Life Reviews. 2005;2:157–175. [Google Scholar]
- Phillips J, Eberwine JH. Antisense RNA Amplification: A Linear Amplification Method for Analyzing the mRNA Population from Single Living Cells. Methods. 1996;10:283–288. doi: 10.1006/meth.1996.0104. [DOI] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Bjorklund AK, Winberg G, Sagasser S, Sandberg R. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc. 2014;9:171–181. doi: 10.1038/nprot.2014.006. [DOI] [PubMed] [Google Scholar]
- Plerou V, Gopikrishnan P, Rosenow B, Amaral LAN, Guhr T, Stanley HE. Random matrix approach to cross correlations in financial data. Physical Review E. 2002;65:066126. doi: 10.1103/PhysRevE.65.066126. [DOI] [PubMed] [Google Scholar]
- Pollen AA, Nowakowski TJ, Shuga J, Wang X, Leyrat AA, Lui JH, Li N, Szpankowski L, Fowler B, Chen P, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014 doi: 10.1038/nbt.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu P, Simonds EF, Bendall SC, Gibbs KD, Jr, Bruggner RV, Linderman MD, Sachs K, Nolan GP, Plevritis SK. Extracting a cellular hierarchy from high-dimensional cytometry data with SPADE. Nat Biotech. 2011;29:886–891. doi: 10.1038/nbt.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scerbo P, Markov GV, Vivien C, Kodjabachian L, Demeneix B, Coen L, Girardot F. On the origin and evolutionary history of NANOG. PloS one. 2014;9:e85104. doi: 10.1371/journal.pone.0085104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons BD, Clevers H. Strategies for homeostatic stem cell self-renewal in adult tissues. Cell. 2011;145:851–862. doi: 10.1016/j.cell.2011.05.033. [DOI] [PubMed] [Google Scholar]
- Singer ZS, Yong J, Tischler J, Hackett JA, Altinok A, Surani MA, Cai L, Elowitz MB. Dynamic heterogeneity and DNA methylation in embryonic stem cells. Molecular cell. 2014;55:319–331. doi: 10.1016/j.molcel.2014.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320–8333. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- Streets AM, Zhang X, Cao C, Pang Y, Wu X, Xiong L, Yang L, Fu Y, Zhao L, Tang F, et al. Microfluidic single-cell whole-transcriptome sequencing. Proc Natl Acad Sci U S A. 2014;111:7048–7053. doi: 10.1073/pnas.1402030111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain PS, Elowitz MB, Siggia ED. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc Natl Acad Sci U S A. 2002;99:12795–12800. doi: 10.1073/pnas.162041399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh SY, Lin R, Hung LH, Lee AP. Droplet microfluidics. Lab on a chip. 2008;8:198–220. doi: 10.1039/b715524g. [DOI] [PubMed] [Google Scholar]
- Torres-Padilla ME, Chambers I. Transcription factor heterogeneity in pluripotent stem cells: a stochastic advantage. Development. 2014;141:2173–2181. doi: 10.1242/dev.102624. [DOI] [PubMed] [Google Scholar]
- Toyooka Y, Shimosato D, Murakami K, Takahashi K, Niwa H. Identification and characterization of subpopulations in undifferentiated ES cell culture. Development. 2008;135:909–918. doi: 10.1242/dev.017400. [DOI] [PubMed] [Google Scholar]
- Van der Maaten L, Hinton G. Visualizing data using t-SNE. Journal of Machine Learning Research. 2008;9:85. [Google Scholar]
- Wang J, Alexander P, Wu L, Hammer R, Cleaver O, McKnight SL. Dependence of mouse embryonic stem cells on threonine catabolism. Science. 2009;325:435–439. doi: 10.1126/science.1173288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle FC, Smith JC. Refinement of gene expression patterns in the early Xenopus embryo. Development. 2004;131:4687–4696. doi: 10.1242/dev.01340. [DOI] [PubMed] [Google Scholar]
- White J, Dalton S. Cell cycle control of embryonic stem cells. Stem cell reviews. 2005;1:131–138. doi: 10.1385/SCR:1:2:131. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Molecular biology of the cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu AR, Neff NF, Kalisky T, Dalerba P, Treutlein B, Rothenberg ME, Mburu FM, Mantalas GL, Sim S, Clarke MF, et al. Quantitative assessment of single-cell RNA-sequencing methods. Nat Meth. 2014;11:41–46. doi: 10.1038/nmeth.2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, Nakato R, Yamada Y, Shirahige K, Saitou M. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell. 2013;12:368–382. doi: 10.1016/j.stem.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Yan L, Yang M, Guo H, Yang L, Wu J, Li R, Liu P, Lian Y, Zheng X, Yan J, et al. Single-cell RNA-Seq profiling of human preimplantation embryos and embryonic stem cells. Nat Struct Mol Biol. 2013;20:1131–1139. doi: 10.1038/nsmb.2660. [DOI] [PubMed] [Google Scholar]
- Yanes O, Clark J, Wong DM, Patti GJ, Sánchez-Ruiz A, Benton HP, Trauger SA, Desponts C, Ding S, Siuzdak G. Metabolic oxidation regulates embryonic stem cell differentiation. Nat Chem Biol. 2010;6:411–417. doi: 10.1038/nchembio.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SH, Kalkan T, Morissroe C, Marks H, Stunnenberg H, Smith A, Sharrocks Andrew D. Otx2 and Oct4 Drive Early Enhancer Activation during Embryonic Stem Cell Transition from Naive Pluripotency. Cell reports. 2014;7:1968–1981. doi: 10.1016/j.celrep.2014.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.