

Abstract
The most widely used approach for defining a genes’ function is to reduce or completely disrupt its normal expression. For over a decade, RNAi has ruled the lab, offering a magic bullet to disrupt gene expression in many organisms. However, new biotechnological tools - specifically CRISPR-based technologies - have become available and are squeezing out RNAi dominance in mammalian cell studies. These seemingly competing technologies leave research investigators with the question: ‘Which technology should I use in my experiment?’ This review offers a practical resource to compare and contrast these technologies, guiding the investigator when and where to use this fantastic array of powerful tools.
INTRODUCTION
The triumphal sequencing of the Human Genome Project (Lander et al., 2001) offered new insights into the fundamental inner workings of humans, promising a big step towards curing humankind of most diseases. More than 15 years after its completion, biologists are left with this unfulfilled promise and a vast amount of genomic sequence encoding tens of thousands of genes of unknown function (Consortium et al., 2007). Sequencing the genome was an incredible challenge but, in broader perspective, was only the first small step. The most difficult challenge lies ahead; deciphering the cryptic meaning of the 3.3 billion base pairs of DNA, by assigning functions to the tens of thousands of genes, and determining how they work together to make us human. This is the grand biological promise yet to be fulfilled, and with the recent development of new biotechnological tools, the biggest discoveries are yet to come.
The most direct way to decipher gene function is to disrupt normal gene expression and study the resulting phenotypes. Such loss-of-function experiments have been performed for more than 100 years, beginning with the work of Thomas Morgan who discovered that genes are on chromosomes and carry mutations responsible for phenotypes. With this knowledge, generations of scientists have worked hard to comprehensively mutate genes, using chemicals, radiation, and random virus integration to painstakingly map each phenotype to a specific mutant gene. This forward genetics approach has taken decades, as artisan techniques fraught with numerous technical challenges complicate the attempts to map random and seemingly minute lesions in a sea of genomic DNA. The sequencing of the human genome offered a map by which gene function could be deciphered, but lacked the means to selectively disrupt a specific gene in a nonrandom manner.
The discovery of RNA interference (RNAi) by Fire and Mello promised a magic bullet to target any gene, provided the investigator knows the DNA sequence (reverse genetics). The timing of the discovery could not have been more perfect, as the method was published shortly after the human genome sequence became freely available. The work of Fire and Mello astounded the scientific world by showing that simple injection of double-stranded RNA into C. elegans could potently silence any gene sequence and produce phenotypes that revealed gene function (Fire et al., 1998). As the mechanisms for RNAi were established, it soon found use in human cells to inhibit specific genes (Elbashir et al., 2001). Its ease of use made RNAi the method of choice for deciphering gene function. However, RNAi produces hypomorphic phenotypes, which do not always mirror the complete loss-of-function that often occurs with genetic mutation. This and other practical caveats encouraged scientists to develop new tools for reverse genetics.
Reverse genetics complete loss-of-function approaches became available with the discovery of zinc-finger nucleases (ZFNs) and later, transcription activator-like effector nucleases (TALENs) (Gaj et al., 2013). These approaches utilize customizable DNA binding domains (DBDs) that are engineered to recognize specific target DNA sequences. Fused to nucleases, DBDs can be used to introduce double-strand breaks (DSBs) and subsequent frame-shift mutations into genes, which can lead to their knockout (Sung et al., 2013).
A more recent addition to the genome editing toolbox is the CRISPR/Cas system. Its involvement in bacterial resistance against viral infections was initially described in Streptococcus thermophilus (Barrangou et al., 2007). Currently, the Type II CRISPR/Cas9 system from Streptococcus pyogenes is the most widely used CRISPR system and was successfully applied to edit human genomes (Cho et al., 2013; Cong et al., 2013; Jinek et al., 2013; Mali et al., 2013c). The full history of the discovery and development of the CRISPR/Cas system has been excellently reviewed previously (Doudna and Charpentier, 2014). Together, these powerful tools offer a new promise to rapidly and efficiently decipher any gene’s function.
With the increasing variety of molecular tools available for loss-of-function experiments, it can be difficult for researchers to select the most appropriate system. This review compares and contrasts these fantastic new tools and offers researchers a ‘practical guidebook’ for determining how to uncover gene function in cells. But, which one should you use?
RNA Interference (RNAi)
RNAi is currently the most extensively used reverse genetics approach to study gene function in mammalian cells. Its success can be attributed to an evolutionarily conserved endogenous pathway that regulates gene expression via small RNAs (Wilson and Doudna, 2013). The RNAi machinery can be ‘hijacked’ by introducing synthetic small RNAs into cells; this is commonly achieved using short-interfering RNAs (siRNAs) or short-hairpin RNAs (shRNAs) (Mohr et al., 2014). The result from either approach is similar, such that the introduced RNA is loaded into the RNA induced silencing complex (RISC), which in turn promotes the degradation of perfectly complementary target mRNA (Figure 1A) (Carthew and Sontheimer, 2009). Using this approach, target mRNA and subsequently target protein levels can be reduced postranscriptionally.
FIGURE 1. Mechanisms of action of RNAi and TALE/N.
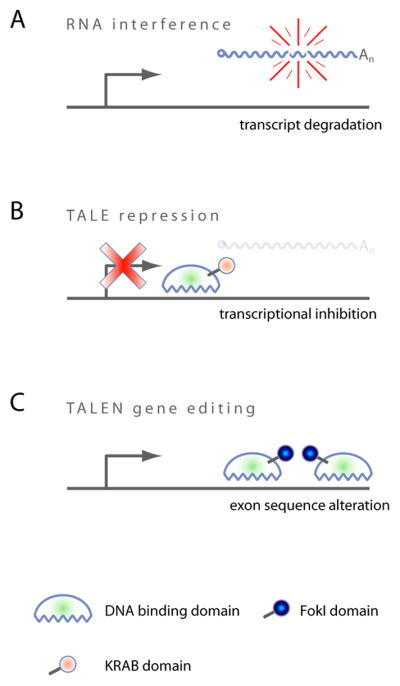
(A) RNA interference degrades transcripts post-transcriptionally in the cytoplasm. (B) TALE repression prevents transcription when targeted to the TSS. (C) Dimerized TALENs induce a site specific DSB which is repaired via error-prone NHEJ, potentially leading to frameshift mutations when exons are targeted.
However, given that the RNAi machinery appears to be mostly active in the cytoplasm, nuclear transcripts - for example long non-coding RNAs or lncRNAs - can be more difficult to effectively target (Derrien et al., 2012; Fatica and Bozzoni, 2014). Moreover, it was recently shown that the transcription of lncRNA itself can have functional consequences (Kornienko et al., 2013). Hence reducing lncRNAs at a post-transcriptional level via RNAi may not always reveal their full loss-of-function phenotype (Bassett et al., 2014).
A major advantage of RNAi is that the silencing machinery is present in practically every mammalian somatic cell. Hence no prior genetic manipulation of the target cell line is required, and a simple siRNA transfection can result in a loss-of-function phenotype.
Specificity and off-target effects of RNAi
Since its discovery, off-target effects have been a growing concern when using RNAi as a tool to study gene function. It has been found that siRNAs can induce silencing of non-target mRNAs with limited sequence complementarity, often via interactions with the 3′UTR (Carthew and Sontheimer, 2009). These interactions occur when short regions of the mRNA 3′-UTR contain imperfect matches to the small RNA, triggering translational repression and/or degradation (Jackson et al., 2006; Sigoillot and King, 2011). In this way, depending on the sequence, one siRNA can potentially repress hundreds of transcripts (Sigoillot and King, 2011). These off-target effects appear to be dosage dependent (Wang et al., 2009) and phenotypes derived from them can be dominant over the intended on-target phenotypes (Franceschini et al., 2014).
In addition to sequence specific off-target effects, the artificial introduction of siRNAs or shRNAs into a target cell line can cause non-sequence specific off-target effects. As mentioned above, RNAi uses a natural pathway that regulates cellular gene expression levels, where the system is governed by endogenous microRNAs. Flooding the system with exogenous sequences can displace microRNAs from RISC. This can impair the functions of endogenous microRNAs, leading to alterations in gene transcript levels and consequently to off-target phenotypes (Khan et al., 2009). These observations fueled the effort to develop alternative reverse genetics approaches.
Transcription Activator-Like Effector (TALE)
A fundamentally different approach to modulate gene function became possible with the development of programmable DNA binding proteins: Transcription Activator-Like Effectors (TALEs). This approach makes use of synthetic transcription factor DNA binding domains (DBDs) that can be programmed to recognize specific DNA motifs. TALE DBDs contain 7–34 highly homologous direct repeats, each consisting of 33–35 amino acids. The key to specificity is contained in the two amino acid residues in positions 12 and 13 of each repeat, which are referred to as repeat variable diresidue (RVD) (Joung and Sander, 2013). Since the DNA:protein binding code of RVDs has been deciphered (Boch et al., 2009; Moscou and Bogdanove, 2009), it is possible to design TALEs that bind any desired target DNA sequence by engineering an appropriate DBD. Typically, the TALEs are designed to recognize 15 to 20 DNA base-pairs, balancing specificity with potential off targeting (Miller et al., 2011; Mussolino et al., 2011). This means that for every individual target site in the genome, it is necessary to design a different DBD, each consisting of ~500–700 amino acids, which can be a very lengthy process.
In the past years, the TALE’s programmability has been used to execute a variety of functions, simply by fusing various effector domains to the DBD.
TALE transcriptional repressor
Gene silencing by means of transcriptional repression can be achieved using KRAB-TALEs, where the transcriptional repressor Krüppel associated box (KRAB) domain (Margolin et al., 1994) is fused to DBDs that target the transcription start site (TSS) (Cong et al., 2012) (Figure 1B). A crucial difference of this approach when compared to RNAi is that TALE repression prevents the transcription of targeted genes in the nucleus, while RNAi degrades them post-transcriptionally in the cytoplasm. This allows, for instance, the study of functional effects that derive from transcription per se, as recently described for lncRNAs (Bassett et al., 2014). In another way though, TALE-repression mimics the hypomorphic effect of RNAi, such that gene function is reduced but not shut off permanently, as it is the case for TALENs.
TALE nuclease (TALEN)
TALE DBDs have also been successfully used to mutate genes in mammalian cells. In this strategy, nuclease effector domains such as FokI are fused to the TALE DBD, resulting in a Transcription Activator-Like Effector Nuclease or TALEN (Gaj et al., 2013). FokI is active only as a dimer; hence pairs of TALENs are constructed to position the FokI nuclease domains to adjacent genomic target sites, where they introduce DNA double strand breaks (DSBs) (Figure 1C). Although this strategy requires the construction of two TALENs per target site, great specificity is achieved since a DSB occurs only after correct positioning and dimerization of FokI (Christian et al., 2010). Once the DSB is introduced, DNA repair can be achieved via two different mechanisms: the high fidelity homologous recombination repair (HRR) or the error-prone non-homologous end joining (NHEJ) (Valerie and Povirk, 2003). Repair of double strand breaks via NHEJ frequently results in DNA target site deletions, insertions, or substitutions (Durai et al., 2005). In TALEN technology, this is desired and users typically target TALEN pairs to the most 5′ exons of genes, promoting early frame shift mutations or premature stop codons. Given the permanence of the genetic lesion, there is no need for maintained TALEN activity inside the target cell, once the mutation has been introduced. This is in contrast to the reversible nature of RNAi, where loss of the small RNA equates to the loss of any evoked phenotype.
In addition to the introduction of random reading frameshift mutations via NHEJ, TALENs can also be utilized to introduce non-random point mutations, targeted deletion or addition of large DNA fragments via the aforementioned HRR. In the past, genome editing in mammalian cells was primarily achieved via the introduction of donor DNA that contained homologous sequences to the target locus. However, this process works very inefficiently in mammalian cells (Vasquez et al., 2001). More recently it was found that the introduction of DNA DSBs increases the efficiency of gene editing via homologous recombination, in the presence of suitable donor DNA (Moehle et al., 2007). Since targeted DSBs can be easily introduced into DNA using TALENs, the combination of such DSBs and the transfection of a homologous donor DNA repair template has become a popular approach to precisely engineer mammalian genomes, as reviewed in detail by Gaj et al. (Gaj et al., 2013).
Specificity and off-target effects of TALEN technologies
Because of the requirement of FokI to dimerize in order to have nuclease activity, no single TALEN can ever introduce a double strand break into a target site. As a consequence, two TALENs have to off-target to adjacent sites in order to introduce an unspecific double strand break into the DNA. Consequently, the off-target activity observed from TALENs is typically low (Guilinger et al., 2014a; Hockemeyer et al., 2011; Mussolino et al., 2011). Unlike TALENs, TALE transcriptional repressors function as monomers and can hence mediate off-target effects autonomously.
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
Cas9 – an RNA guided DNA endonuclease
Similar to TALENs, the CRISPR associated protein 9 (Cas9) is also a programmable DNA nuclease. However, its mode of operation differs in several ways: First of all, Cas9 possesses innate nuclease activity. The enzyme is active as a monomer and catalyzes DSBs, (Hsu et al., 2014); hence, targeting a single Cas9 molecule to the early exons of genes generates random knockout phenotypes via DSBs and NHEJ repair, similar to targeting pairs of TALENs to the same site. As with TALENs, to achieve a complete knockout of that gene, every functional copy of a target gene needs to be disrupted. A maximum in the fraction of successfully edited cells is typically observed at around five to eight days post transduction (Wang et al., 2014; Zhou et al., 2014). Thus far, current protocols yield editing efficiencies of up to 50% when quantified via SURVEYOR assay (Hsu et al., 2013; Mali et al., 2013c). Even in the near haploid cell line KBM7, the maximum editing efficiency reported is 70% (Wang et al., 2014). Hence, to obtain homozygous knockout cell lines for a target gene of interest, it is usually necessary to screen several clonal lines for homozygous gene disruption (Ran et al., 2013b).
Another major difference between TALENs and Cas9 is the way both proteins recognize their genomic target sites. While TALENs encode the DNA target motif specificity in the amino acid sequence of their DBD, Cas9 requires a single chimeric guide RNA (sgRNA) to recognize its target site (Mali et al., 2013b) (Figure 2A). So, while for TALENs the amino acid sequence of their DBD has to be painstakingly re-designed for each target site, one invariant Cas9 nuclease can be targeted to a large variety of DNA motifs, simply by co-expression of a target site-specific sgRNA. It is this easy programmability that allowed the CRISPR/Cas9 platform to rapidly replace other genome editing platforms, including ZFNs and TALENs.
FIGURE 2. Mechanisms of action of CRISPR-based technologies.
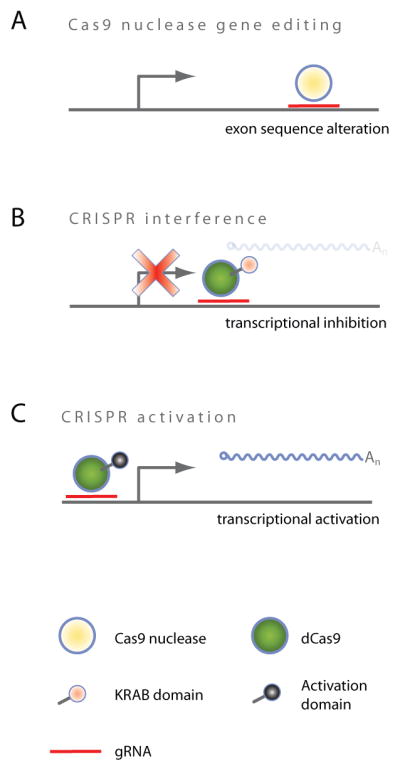
(A) Cas9 nuclease monomer is guided to its target site via sgRNA and induces site specific DSB which is repaired via error-prone NHEJ, potentially leading to frameshift mutations when exons are targeted. (B) CRISPR interference (CRISPRi) prevents transcription when targeted to the TSS. (C) CRISPR activation (CRISPRa) activates transcription when targeted to the TSS.
In the Type II CRISPR/Cas9 system from Streptococcus pyogenes, which is currently the most popular system used for genome editing in mammalian cells, the sgRNA is composed of two separately expressed RNAs, a CRISPR RNA (crRNA) and a trans-activating crRNA (tracrRNA), which are processed by the endogenous bacterial machinery to yield the mature gRNA (Chylinski et al., 2014; Deltcheva et al., 2011). Later it was shown that it is possible to express a single chimeric sgRNA, a fusion of crRNA and tracrRNA, which is able to act as a mature sgRNA in-vitro, without the necessity of further processing (Jinek et al., 2012). This chimeric sgRNA, similar to the fully processed native bacterial sgRNA, consists of a constant region that mediates interaction with Cas9 and a variable region that mediates interaction with the genomic target site. Chimeric sgRNAs currently used for genome engineering typically contain a 17–20 nt long variable region which is complementary to the genomic target sequence.
More recently, a modified chimeric sgRNA design was developed in order to enhance its expression and interaction with Cas9. For this purpose, a potential PolIII terminator was removed from the constant region of the sgRNA and a hairpin was extended by 5 bp (Chen et al., 2013) in order to restore the full S. pyogenes hairpin sequence identified by (Deltcheva et al., 2011).
Importantly, a major specificity determinant of Cas9 is the protospacer adjacent motif (PAM), a short region immediately 3′ of the target sequence (Sternberg et al., 2014). This PAM has the sequence NGG for Cas9 from Streptococcus pyogenes and varies for Cas proteins from different species (Horvath et al., 2008). Examples of orthogonal protospacer motifs include NGGNG (Horvath et al., 2008), NAAR (van der Ploeg, 2009), and NNAGAAW (Deveau et al., 2008). Although the dependency of the PAM motif limits Cas9 target space, it offers another level of specificity as discussed later, in the “Off targeting” section.
CRISPR interference (CRIPSRi)
The ease with which Cas9 can be programmed to bind a specific region of the genome prompted several groups to develop transcriptional repressors. Similar to TALE transcriptional repressors, an enzymatically inactive version of Cas9 (deadCas9 or dCas9) is fused to a repressor domain (e.g. KRAB), triggering heterochromatin mediated gene silencing at the TSS (Qi et al., 2013) (Figure 2B). This approach is termed CRISPRi (Gilbert et al., 2013) and leads to transcriptional reduction of the target RNA, similar to TALE transcriptional repression.
In comparison to RNAi, CRISPRi appears to produce a more consistent and robust knockdown given the same number of effector RNAs. For example, six out of eight sgRNAs were found to repress GFP levels by at least 75% (Gilbert et al., 2013) and a direct comparison to gene inhibition via RNAi shows significantly stronger loss-of-function phenotypes when using CRISPRi (Gilbert et al., 2014).
Like any technology, CRISPRi comes with its own caveats. Similar to RNAi, we still lack a solid understanding of the rules by which a given sgRNA may engage and be active on a given target site. We do know that the accessibility and location of the TSS is critical: Cas9 needs to physically access its target site. This process has been shown to be dependent on chromatin accessibility (Kuscu et al., 2014; Wu et al., 2014). Hence genomic regions with closed chromatin states may prevent binding and thus the function of Cas9.
In order to repress transcription with maximum efficiency, the KRAB domain should be targeted to a window of ~500 bp downstream from the TSS (Gilbert et al., 2014). Hence it is important to know the location of the TSS of targeted transcripts. Importantly, even in fully sequenced genomes, like the human genome, some TSSs may be experimentally hard to annotate, such as miRNA TSSs (Georgakilas et al., 2014). Moreover, many genes have alternative transcripts, some with TSSs very distant from each other. The usage of these TSSs can vary between cell types (Sandelin et al., 2007), so it is important to target the correct TSS for a given cell type. Moreover, different transcript variants expressed simultaneously from alternative TSSs can act redundantly (Davuluri et al., 2008). Consequently, if multiple transcript variants are expressed in a cell, blocking the transcription of one of them at a particular TSS may not be sufficient to produce a loss-of-function phenotype.
A different problem arises when distinct transcripts share the same TSS, as commonly is the case for intragenic non-coding RNAs (He et al., 2012). Targeting dCas9-KRAB to such TSSs could potentially block the transcription of all associated transcripts. Attributing the observed knockdown phenotype to one transcript or another thus requires additional experiments.
CRISPR activation (CRISPRa)
Gain-of-function studies in mammalian cells have traditionally been carried out by overexpressing transgenic open reading frames (ORFs or cDNAs) in target cells. This approach involves cloning an ORF behind a promoter and introducing it into target cells. More recently, CRISPR systems have been adapted to allow the expression of genes from their endogenous genomic locus, bypassing the need to clone and express the oftentimes-large ORFs into cells. To date, several dCas9-based approaches have successfully been used to activate transcription in mammalian cells (Cheng et al., 2013; Farzadfard et al., 2013; Maeder et al., 2013; Mali et al., 2013a; Perez-Pinera et al., 2013).
All approaches rely on targeting transcriptional activation domains to the TSS of genes; most commonly VP64 (Beerli et al., 1998) is fused to dCas9 (Figure 2C). Direct dCas9-VP64 fusion proteins, however, have been shown in numerous studies to efficiently activate transcription of endogenous genes only when they are co-expressed with multiple (3–5) sgRNAs targeting the same TSS (Cheng et al., 2013; Maeder et al., 2013; Mali et al., 2013a; Perez-Pinera et al., 2013). To overcome this limitation, several novel strategies were recently described (Chavez et al., 2015; Konermann et al., 2015; Tanenbaum et al., 2014). All approaches demonstrated efficient transcriptional activation of endogenous genes via expression of only one single sgRNA per cell.
Similar to CRISPRi, the target site is crucial for CRISPRa to obtain maximal activation. The region upstream of the TSS (−400 to −50 nt) is typically considered to be the optimal target site for CRISPRa sgRNAs (Gilbert et al., 2014; Konermann et al., 2015). This is mutually exclusive with the CRISPRi target space downstream from the TSS (0 to +500 nt). The dependency of CRISPRa on the TSS of the targeted transcript means that challenges are similar to the aforementioned ones associated with CRISPRi. Most importantly, it is essential to know the location of the TSS to effectively activate expression and secondly, activation of multiple transcripts from one and the same TSS cannot be controlled individually.
RNA polymerase promoters for sgRNA expression
The vast majority of studies have used polymerase III promoters (U6, H1, 7SK, and tRNA promoters) to transcribe the sgRNA, given its natural ability to transcribe small RNAs at high levels (Kabadi et al., 2014). In some contexts, it may be desirable to express sgRNAs using polymerase II promoters. For example, tissue-specific expression could be achieved by the use of cell type specific promoters. However the use of polymerase II promoters complicates the ability to express functional sgRNAs, given the natural path of capped polyadenylated RNAs to shuttle through the ribosome as an mRNA. To overcome this issue, Nissim et al. developed several approaches that allow the expression of single and multiple sgRNAs from human RNA polymerase II promoters (Nissim et al., 2014), permitting CRISPRi and CRISPRa in transient transfection assays. This was accomplishable by flanking the sgRNA with either ribozyme or Cys4 RNA cleavage sites. Future studies should determine whether these approaches are transferable to single copy settings, such as genomically integrated sgRNA expression cassettes.
Specificity and off-target effects of CRISPR technologies
Specificity and target site recognition
A number of studies have examined Cas9 for off-targeting and found that the PAM plays an essential role in DNA target site recognition. A random collision model was proposed – in which Cas9 dissociates much more rapidly from the DNA when no PAM is present (Sternberg et al., 2014). Once Cas9 recognizes the PAM, it then interrogates the 5′-flanking DNA for sgRNA complementarity. According to this model, target site specificity is determined by the sequence adjacent to the PAM. In support of this model, Hsu et al. found that Cas9 can only cut target sites after recognizing the PAM (Hsu et al., 2013). They found that the proximal 8–14 nucleotides immediately upstream of the PAM were most important for target site cleavage. Other studies provided additional evidence for the existence of a seed region adjacent to the PAM (Cong et al., 2013; Fu et al., 2013). Both studies showed that mismatches in the seed region reduced Cas9 activity more than mismatches further away from the PAM.
Off-target effects
By means of dCas9-ChIP-seq, Wu at al. found that dCas9 binds to a large number of off-target sites (Wu et al., 2014). In fact, most dCas9-gRNA complexes were bound to sites other than the target site. Interestingly, all top ranking off-target motifs were 5′ of a PAM (NGG) and contained the correct 5 bp directly adjacent to the PAM. The degree of off-targeting appeared to be related to sgRNA levels. In a very similar study using dCas9-ChIP-seq, Kuscu et al. (Kuscu et al., 2014) came to the same conclusions, namely that the PAM is the strongest determinant for off-target binding followed by the nucleotides most proximal to the PAM. Both studies found that the frequency of DNA cleavage in the identified off-target binding sites was much lower than the frequency of binding.
To further investigate off-target cleavage in an unbiased manner, several groups have developed approaches to directly analyze unwanted DSBs produced by catalytically active Cas9 (Crosetto et al., 2013; Frock et al., 2015; Kim et al., 2015; Tsai et al., 2015). Using GUIDE-seq, Tsai et al. found that the number of off-target cutting events varies widely between different sgRNAs (from 0 to 150 off-target events per sgRNA) and does not correlate with GC content or genomic location of the target site. Moreover, these ChIP-seq independent approaches confirm that the process of DNA cleavage by Cas9 is distinct and separable from simple DNA binding.
While the large degree of non-specific binding of dCas9 appears to be a concern, it is important to remember that most dCas9 applications (such as CRISPRi or CRISPRa) are highly dependent on the genomic position of stable dCas9 binding (Gilbert et al., 2014). Inhibition as well as activation of gene expression appears to require dCas9-KRAB/-VP64 to be targeted to a narrow window around the TSS of genes (Gilbert et al., 2014). Hence, off-targeting of dCas9-KRAB/-VP64 to sites other than the TSS may not actually result in transcriptional repression or activation.
However, the KRAB domain’s ability to induce heterochromatin formation at any genomic locus could potentially lead to phenotypes resulting from displacement of regulatory factors from their target site. Data showing little to no anti-proliferative effects for thousands of negative control sgRNAs in a CRISPRi screen appears to support the hypothesis that off-target effects are not a major issue (Gilbert et al., 2014). This is in contrast to off-targeting of siRNA, which can inhibit and/or degrade mRNA from a large number of unspecific sites (Jackson et al., 2006) potentially leading to a large variety of phenotypes (Jackson and Linsley, 2010).
Increasing target site specificity of Cas9
Certain applications, such as for instance human gene therapy, require highly specific genome editing with minimized off-targeting. For this purpose, two separate Cas9 based systems have been developed to increase target site specificity and decrease off-target effects. The first approach involves the mutation of one of the two Cas9 nuclease domains, turning Cas9 from a nuclease into a nickase. Single DNA strand cuts, or nicks, are typically repaired with high fidelity (Ran et al., 2013a). Consequently, two Cas9 nickases need to target appropriate, adjacent sites in the genome in order to introduce DSBs, which are then repaired by the error-prone NHEJ. This approach has been shown to reduce off-targeting by 50–1,500-fold (Cho et al., 2014; Ran et al., 2013a).
A second approach very closely resembles TALENs and is based on the fusion of the FokI domain to a catalytically inactive Cas9. This approach makes use of the dependency of FokI on dimerization in order to cut the target DNA. It was shown that dCas9-FokI dimerization reduces off-target activity even further when compared to the Cas9 nickase approach (Guilinger et al., 2014b; Tsai et al., 2014). In addition to modifications of Cas9, truncations of the sgRNA to 17 or 18 nt have been shown to reduce off-targeting without reducing on-target efficiency (Fu et al., 2014).
Design rules and tools for si-, sh- and sgRNAs
To maximize on-target and minimize off-target effects, careful attention should be paid to siRNA, shRNA and sgRNA design. In this section we provide a very brief overview of the currently accepted design rules for each of those RNA reagents.
Design of siRNAs and shRNAs
Optimization of siRNA and shRNA design rules has been an active area of research for over a decade and various rules have been established (Fellmann and Lowe, 2014; Tafer, 2014). Typically, the duplex length ranges from 19–25 nucleotides, and sequences are designed to target regions of the ORF that are common among transcript variants. It has generally been recommended to avoid sequences with extremely high or low GC contents, miRNA seed matches, and internal complementarity. To minimize off-targeting, sequences having perfect or near-perfect complementarity to unintended target sites should be avoided (Birmingham et al., 2007). To assist researchers with si/shRNA sequence design, a large variety of computational tools have been developed. A listing of those web-based tools together with their URLs can be found here (Lagana et al., 2014). In addition, numerous commercially available resources exist that claim to have validated sequences and those enriched for potent activity.
While siRNA/shRNA design rules have substantially improved over previous years, de novo RNAi design still exhibits variation in efficiency and specificity between small RNAs. Hence it is imperative to test every new reagent, to measure knockdown efficiency and to reproduce phenotypes with multiple si/shRNAs targeting the same gene.
Design of sgRNAs
In order to evoke gene knockout via early frameshift mutations, sgRNAs are frequently designed to target Cas9 nuclease to the N-terminal coding exons of protein encoding genes. For CRISPRi/a on the other hand, sgRNAs are currently designed to target a region around the TSS. Hence, for every CRISPR/Cas9 platform, the design of sgRNAs must fit the specific system. Nonetheless, common design rules for sgRNAs from different platforms have been empirically determined.
For all CRISPR systems described to date, the sgRNA should target a genomic DNA sequence that is adjacent to a PAM, without actually containing the PAM itself (Sternberg et al., 2014). Sequences flanking the PAM have been described to impact the success of sgRNA targeting. Specifically, CGGH is the recommended optimal PAM sequence for S. pyogenes (Doench et al., 2014). The typical length of an sgRNA is 20 nt, although slightly shorter sgRNAs (17–18 nt) have been found to reduce off-targeting without compromising efficiency (Fu et al., 2014). Similar to si/shRNA rules, extremely high or low GC contents and homopolymeric nucleotide stretches should be avoided (Doench et al., 2014; Gilbert et al., 2014). Importantly, targeting the sense or antisense strand appears to have no influence on efficiency of Cas9 nuclease (Doench et al., 2014) or CRISPRi/a (Gilbert et al., 2014). Given that sgRNAs appear to possess a seed region proximal to the PAM, sequences should be designed to avoid perfect or near-perfect matches to the seed sequence outside the target site (Fu et al., 2013; Wu et al., 2014). Local chromatin structure has been shown to strongly impact Cas9 binding to its target DNA site (Kuscu et al., 2014; Wu et al., 2014).
Several web-based software tools can assist researchers with the design of sgRNAs for genome editing, each of them taking into account empirically determined parameters for optimal sgRNA design, such as length and the GC contents of sgRNAs (Doench et al., 2014; Heigwer et al., 2014; Hsu et al., 2013). A list of those tools together with their URLs can be found here (Lagana et al., 2014). While rules for si/shRNA sequence design have been developed over many years, the field of sgRNA design is still developing and design rules will probably be improved with additional studies. Similar to si/shRNAs, it is currently recommended to test several sgRNA reagents and to confirm similar if not identical phenotypes in a given experiment with at least two different sgRNAs.
Choosing the right tool for the job
Do you want to generate a knockout or knockdown?
Each loss-of-function technology has its own advantages and limitations, so choosing the right tool for the job largely depends on the specific experimental design. As a guide, figure 3 gives an overview of the most common points to be considered before beginning an experiment; it highlights the appropriate tools that can be used in everyday scenarios.
FIGURE 3. User guide decision tree for help to choose the right tool.

(1) Known sequence. For your targeted organism, what type of known sequence information is available? Transcriptome sequence alone allows the targeting of mRNA (si/shRNAs) and DNA exons (TALENs and Cas9 nuclease). If your organisms’ genome has been sequenced and TSSs have been annotated, all technologies become available, since TALE-repression and CRISPRi/a target promoter regions. (2) Loss-of-function-phenotype. What type of loss-of-function phenotype do you want? While RNAi, TALE-repression, and CRISPRi induce reversible gene knockdown, TALEN and Cas9 nuclease induce a permanent knockout. (3) Time to phenotype. How long will it take from the initial planning of the experiment to finally having a loss-of-function phenotype in cells? Transfection of siRNAs remains the most direct and quickest route. For RNAi using shRNAs as well as CRISPRi/a and Cas9 nuclease, appropriate shRNA/sgRNA plasmids need to be cloned. Moreover, it may be necessary to validate the functionality of Cas9 in target cells. The most time consuming way to induce loss-of-function are TALENs, since each target site requires the design of a specific amino acid sequence of the TALEN DNA binding domain. (4) Type of phenotype. RNAi, TALE-repression and CRISPRi/a are best suited to generate hypomorphic phenotypes that mimic for instance the inhibition of a target by a drug. TALEN and Cas9 nuclease on the other hand can generate complete knockout phenotypes (null allele) via genome editing. (5) Cost of experiment. How much will the production of required reagents cost? shRNAs and sgRNAs are generated by simple cloning or they can be purchased. Because TALEN-based technologies require a more complicated synthesis, cloning them or purchasing them generally costs more. Performing RNAi via siRNAs requires the costly synthesis of short RNAs. (6) Ease of experiment. How much relative effort does it take until one arrives at the desired loss-of-function phenotype? RNAi via siRNAs necessitates only the purchase or synthesis of short RNAs, followed by a simple transfection into target cells. RNAi via shRNAs and CRISPR-based approaches additionally involve the cloning of shRNAs or sgRNAs. The design and cloning of TALEN-based technologies are relatively more complicated, making it less easy to use. (7) Off-targeting. How severe are the known off-target effects? So far, the described off-target effects for CRISPR as well as TALEN technology appear to be less of an issue compared to RNAi. While off-targeting of an si/shRNA to a given RNA can result in an undesired phenotype, it seems likely that TALEN and CRISPR off-targeting to the genomic DNA may not often produce an undesired phenotype.
It is probably fair to say that CRISPR/Cas9 has already leapfrogged TALEN technology, mostly because Cas9 offers an unprecedented simplicity to target a large variety of functional domains to various genomic sites. Hence in the following section we will focus on contrasting RNAi, CRISPRi and Cas9 nuclease for genome editing without specifically mentioning TALEN technology. Perhaps the most striking difference among these various approaches is that RNAi and CRISPRi induce transient knockdown of gene expression, whereas genome editing via Cas9 nuclease can induce permanent damage of the target DNA, leading to a null phenotype in some cases. Therefore, a major question is “do you want to generate a complete knockout or a hypomorphic knockdown?”
Gene knockout
For many applications, a full knockout may be desirable, and perhaps even required. For instance, in cases of slow protein degradation, reduced transcript levels - as caused by knockdown approaches - may not necessarily result in low protein levels. Moreover, if a gene product is not rate limiting, even strongly reduced protein levels may not produce a loss-of-function phenotype. A knockout on the other hand would sooner or later lead to full depletion of functional protein.
A significant advantage of Cas9 nuclease mediated genome editing compared to transcriptional repression via CRISPRi, is that it targets exonic regions rather than TSSs. Why is this important? First of all, exons are generally very well annotated and can be deduced from transcriptome sequencing data. More importantly though, genome editing can disrupt exons that are shared by all variants of a given gene, even if these are transcribed from separate TSSs. Like this, all transcript variants and even whole gene families can be knocked out using a single sgRNA against exons that are conserved between all family members.
A true knockout - defined, as the complete disruption of a target gene’s function - should cause a homogenous knockout phenotype. However, it is important to keep in mind that genomic mutations introduced via NHEJ repair of DSBs are somewhat random (Valerie and Povirk, 2003). Therefore approximately one third of the introduced mutations via Cas9 nuclease would be expected to be in-frame and hence should not disrupt the ORF. That said, depending on the location within a protein, a few extra amino acids removed or added can alter activity, complicating the prediction of phenotypic outcome for each mutation and each gene. Hence, genetic heterogeneity could yield phenotypic heterogeneity.
An additional and often overlooked consideration is ploidy. Most common laboratory cancer cell lines exhibit changes in ploidy, with many lines carrying four or more copies of a chromosome (Paulsson et al., 2013; Rondon-Lagos et al., 2014). In these cases, true knockout phenotypes may only emerge once every functional copy of the target gene is mutated. In practice, this may be difficult - although not impossible - to accomplish. Depending on the experiment, it may be imperative to sequence the target loci to verify homozygous knockout when generating mutant clonal cell lines.
Gene knockdown
While not always capable of disrupting gene function completely, hypomorphic knockdowns can offer a number of advantages over knockouts. Since gene knockdown via RNAi or CRISPRi does not depend on frame shift mutations or ploidy, the aforementioned issues related to genetic heterogeneity are minimized. Moreover, gene knockdowns are reversible, a feature that has proven very useful in the past (Xue et al., 2007).
Furthermore, partial gene knockdown allows researchers to study the function of essential genes whose knockout would otherwise be cell-lethal. Additionally, applications such as drug target discovery could benefit from phenotypic hypomorphs, since an RNAi or CRISPRi knockdown - rather than a knockout - may mimic the effects of an inhibitive drug more closely. However, in some cases, a full knockout may represent an idealized loss-of-function phenotype for the most potent inhibitive drugs.
Another important issue is time: RNAi knockdown cell lines can be obtained within a matter of days – simply via siRNA transfection - and do not require time-consuming screening for homozygous knockout clones. This may be especially important in primary cells with limited proliferative capacity or limited cell health in culture models. Moreover, the selection of homozygous knockout clones may not faithfully represent the original composition cell populations. Hence, sub-populations that are underrepresented or for which the knockout was cell-lethal, may be excluded from subsequent analysis.
Similar to RNAi, CRISPRi appears to generate a homogenous phenotype for an entire population of cells, providing all cells are robustly expressing both the sgRNA and Cas9. As such, phenotypic heterogeneity is minimized compared to Cas9 nuclease mediated knockouts that depend on frameshift mutations or loss of specific DNA sequence elements.
RNAi – A phasing-out technology?
With the rapid development of CRISPR technology, a reasonable question to ask is – “Is the era of RNAi based tools over”? After all, RNAi often exhibits significant off-target effects and unpredictable knockdown efficiencies. In contrast, CRISPRi appears to be advantageous since knockdown efficiencies appear to be superior to RNAi. Although the field is still exploring the extent of CRISPR-centric off-targeting, some results suggest that CRISPRi barely produces off-target phenotypes (Gilbert et al., 2014). To answer the above question, one needs to consider the strengths and weaknesses of each tool.
As discussed above, RNAi seems to be the simplest system to create hypomorphic knockdowns. In contrast to CRISPRi, RNAi does not require additional genes to be introduced into cells prior to the actual knockdown experiment, since most cells come with the full RNAi silencing machinery. Hence the execution of RNAi is more rapid, saving investigators time and money.
Unlike CRISPRi, RNAi does not target TSSs. Therefore, RNAi can be used in species for which transcriptome but not genome sequencing data exists and whose TSSs have not yet been annotated via additional sequencing approaches like CAGE (Kodzius et al., 2006). Similar to genome editing via Cas9 nuclease, RNAi can target sequences that are conserved between transcript variants and gene family members. Hence one single si/shRNA can target all transcripts with conserved sequence - regardless of what TSS they originate from. This is in contrast to CRISPRi and could put RNAi ahead of CRISPRi based technologies.
Finally, RNAi targets RNA transcripts in the cells’ cytoplasm and not genomic DNA in the nucleus. Hence, accessibility does not appear to be impaired by chromatin states, as observed for CRISPR-based approaches (Kuscu et al., 2014; Wu et al., 2014). Taken together, despite the excitement surrounding CRISPR-based approaches, RNAi still retains a strong position in several experimental settings and should be expected to co-exist for some time.
Future perspectives
Clearly, the potential of CRISPR/Cas systems goes far beyond inducing loss-of-function and gain-of-function phenotypes. In principle, dCas9 can be fused to virtually any functional domain to target proteins to specific genomic sites, allowing locus-specific, modifications. For example, Cas9 could be used to modify epigenetic marks such as histone modifications, 5-methylcytosine or other functional modifications. As such it offers the opportunity to study the regulation of gene expression on an epigenetic level.
More recently, in-vitro assays have demonstrated the ability of Cas9 to act on single stranded RNA (O’Connell et al., 2014). This interesting discovery may make it possible to manipulate RNA molecules in much the same way as DNA. Future effort to apply Cas9-mediated RNA recognition in-vivo may allow applications like RNA cleavage (similar to RNAi), enhancement of translation or isolation of specific RNA:protein complexes under native conditions (O’Connell et al., 2014). Likewise, a large number of mysterious RNA modifications exist, and the ability of Cas9-fusion proteins to deliver specific modifications to RNA can offer a new tool to probe their function in RNA regulation.
Efforts to identify orthogonal Cas9 enzymes from different bacterial species are in full swing (Esvelt et al., 2013). Identification of such orthogonal Cas9 enzymes will allow us to target a larger variety of sequences due to the requirement of different PAMs. More importantly, it will make it possible to combine Cas9s with different functional domains, e.g. activating one gene while inhibiting another one. This modularity and the overall simplicity of combinatorial perturbation, places Cas9 in a very unique position to impact our understanding of complex regulatory circuits.
In sum, the versatility and programmability of Cas9 has made CRISPR the genome targeting and editing platform of choice for many applications. There is no doubt that we can expect additional exciting CRISPR applications to appear in the near future. Like with any new technology, there will be an active period of development, followed by an entrenchment of established methods. Numerous fields are already employing reagents and protocols to expedite discovery. And CRISPR continues a steady march toward the clinic, offering hope for patients, catalyzing ethical questions for humankind, and promising a new tool to one day intervene human disease.
TABLE 1.
Comparisons between RNAi, TALE, and CRISPR centric technologies
| RNAi | TALE repression | TALEN | Cas9 nuclease | CRISPRi | CRISPRa | |
|---|---|---|---|---|---|---|
| Loss-of-function mechanism | Post-transcriptional RNA degradation | Repression of transcription | Frame shift DNA mutation | Frame shift DNA mutation | Repression of transcription | Activation of transcription |
| Result | Reversible knockdown | Reversible knockdown | Permanent knockout | Permanent knockout | Reversible knockdown | Reversible activation |
| Transgenes | si/shRNA | TALE-KRAB | TALEN | Cas9 nuclease gRNA |
dCas9-KRAB gRNA |
dCas9-VP64 gRNA |
| Guiding sequence | si/shRNA | DBD | DBD | gRNA | gRNA | gRNA |
| Required sequence information | Transcriptome | Annotated TSS | Transcriptome | Transcriptome | Annotated TSS | Annotated TSS |
| Off-target space | Transcriptome | Window around TSS | Genome, Requires FokI dimerization | Genome, Cuts as monomer | Window around TSS | Window around TSS |
| Transcript variants | All variants via conserved region | Only variants from the same TSS | All variants via conserved region | All variants via conserved region | Only variants from the same TSS | Only variants from the same TSS |
Acknowledgments
We thank James A. Blau, Luke A. Gilbert and Rodrigo Mora for critically proof reading the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–1712. doi: 10.1126/science.1138140. [DOI] [PubMed] [Google Scholar]
- Bassett AR, Akhtar A, Barlow DP, Bird AP, Brockdorff N, Duboule D, Ephrussi A, Ferguson-Smith AC, Gingeras TR, Haerty W, et al. Considerations when investigating lncRNA function in vivo. eLife. 2014;3:e03058. doi: 10.7554/eLife.03058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli RR, Segal DJ, Dreier B, Barbas CF., 3rd Toward controlling gene expression at will: specific regulation of the erbB-2/HER-2 promoter by using polydactyl zinc finger proteins constructed from modular building blocks. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:14628–14633. doi: 10.1073/pnas.95.25.14628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birmingham A, Anderson E, Sullivan K, Reynolds A, Boese Q, Leake D, Karpilow J, Khvorova A. A protocol for designing siRNAs with high functionality and specificity. Nature protocols. 2007;2:2068–2078. doi: 10.1038/nprot.2007.278. [DOI] [PubMed] [Google Scholar]
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, Lahaye T, Nickstadt A, Bonas U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez A, Scheiman J, Vora S, Pruitt BW, Tuttle M, EPRI, Lin S, Kiani S, Guzman CD, Wiegand DJ, et al. Highly efficient Cas9-mediated transcriptional programming. Nature methods. 2015;12:326–328. doi: 10.1038/nmeth.3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li GW, Park J, Blackburn EH, Weissman JS, Qi LS, et al. Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell. 2013;155:1479–1491. doi: 10.1016/j.cell.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell research. 2013;23:1163–1171. doi: 10.1038/cr.2013.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nature biotechnology. 2013;31:230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome research. 2014;24:132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian M, Cermak T, Doyle EL, Schmidt C, Zhang F, Hummel A, Bogdanove AJ, Voytas DF. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186:757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chylinski K, Makarova KS, Charpentier E, Koonin EV. Classification and evolution of type II CRISPR-Cas systems. Nucleic acids research. 2014;42:6091–6105. doi: 10.1093/nar/gku241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Zhou R, Kuo YC, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nature communications. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, et al. Nucleotide-resolution DNA doublestrand break mapping by next-generation sequencing. Nature methods. 2013;10:361–365. doi: 10.1038/nmeth.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davuluri RV, Suzuki Y, Sugano S, Plass C, Huang TH. The functional consequences of alternative promoter use in mammalian genomes. Trends in genetics : TIG. 2008;24:167–177. doi: 10.1016/j.tig.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Deltcheva E, Chylinski K, Sharma CM, Gonzales K, Chao Y, Pirzada ZA, Eckert MR, Vogel J, Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG, et al. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome research. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonte J, Fremaux C, Boyaval P, Romero DA, Horvath P, Moineau S. Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. Journal of bacteriology. 2008;190:1390–1400. doi: 10.1128/JB.01412-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature biotechnology. 2014;32:1262–1267. doi: 10.1038/nbt.3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, Charpentier E. Genome editing. The new frontier of genome engineering with CRISPR-Cas9. Science. 2014;346:1258096. doi: 10.1126/science.1258096. [DOI] [PubMed] [Google Scholar]
- Durai S, Mani M, Kandavelou K, Wu J, Porteus MH, Chandrasegaran S. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic acids research. 2005;33:5978–5990. doi: 10.1093/nar/gki912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nature methods. 2013;10:1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farzadfard F, Perli SD, Lu TK. Tunable and multifunctional eukaryotic transcription factors based on CRISPR/Cas. ACS synthetic biology. 2013;2:604–613. doi: 10.1021/sb400081r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatica A, Bozzoni I. Long non-coding RNAs: new players in cell differentiation and development. Nature reviews Genetics. 2014;15:7–21. doi: 10.1038/nrg3606. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Lowe SW. Stable RNA interference rules for silencing. Nature cell biology. 2014;16:10–18. doi: 10.1038/ncb2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A, Xu S, Montgomery MK, Kostas SA, Driver SE, Mello CC. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- Franceschini A, Meier R, Casanova A, Kreibich S, Daga N, Andritschke D, Dilling S, Ramo P, Emmenlauer M, Kaufmann A, et al. Specific inhibition of diverse pathogens in human cells by synthetic microRNA-like oligonucleotides inferred from RNAi screens. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:4548–4553. doi: 10.1073/pnas.1402353111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frock RL, Hu J, Meyers RM, Ho YJ, Kii E, Alt FW. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nature biotechnology. 2015;33:179–186. doi: 10.1038/nbt.3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Foden JA, Khayter C, Maeder ML, Reyon D, Joung JK, Sander JD. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nature biotechnology. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPRCas nuclease specificity using truncated guide RNAs. Nature biotechnology. 2014;32:279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj T, Gersbach CA, Barbas CF., 3rd ZFN, TALEN, and CRISPR/Casbased methods for genome engineering. Trends in biotechnology. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgakilas G, Vlachos IS, Paraskevopoulou MD, Yang P, Zhang Y, Economides AN, Hatzigeorgiou AG. microTSS: accurate microRNA transcription start site identification reveals a significant number of divergent pri-miRNAs. Nature communications. 2014;5:5700. doi: 10.1038/ncomms6700. [DOI] [PubMed] [Google Scholar]
- Gilbert LA, Horlbeck MA, Adamson B, Villalta JE, Chen Y, Whitehead EH, Guimaraes C, Panning B, Ploegh HL, Bassik MC, et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell. 2014 doi: 10.1016/j.cell.2014.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert LA, Larson MH, Morsut L, Liu Z, Brar GA, Torres SE, Stern-Ginossar N, Brandman O, Whitehead EH, Doudna JA, et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/j.cell.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger JP, Pattanayak V, Reyon D, Tsai SQ, Sander JD, Joung JK, Liu DR. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nature methods. 2014a;11:429–435. doi: 10.1038/nmeth.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nature biotechnology. 2014b;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Li Z, Chen P, Huang H, Hurst LD, Chen J. Young intragenic miRNAs are less coexpressed with host genes than old ones: implications of miRNAhost gene coevolution. Nucleic acids research. 2012;40:4002–4012. doi: 10.1093/nar/gkr1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigwer F, Kerr G, Boutros M. E-CRISP: fast CRISPR target site identification. Nature methods. 2014;11:122–123. doi: 10.1038/nmeth.2812. [DOI] [PubMed] [Google Scholar]
- Hockemeyer D, Wang H, Kiani S, Lai CS, Gao Q, Cassady JP, Cost GJ, Zhang L, Santiago Y, Miller JC, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature biotechnology. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath P, Romero DA, Coute-Monvoisin AC, Richards M, Deveau H, Moineau S, Boyaval P, Fremaux C, Barrangou R. Diversity, activity, and evolution of CRISPR loci in Streptococcus thermophilus. Journal of bacteriology. 2008;190:1401–1412. doi: 10.1128/JB.01415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. Development and applications of CRISPR-Cas9 for genome engineering. Cell. 2014;157:1262–1278. doi: 10.1016/j.cell.2014.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nature biotechnology. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Burchard J, Schelter J, Chau BN, Cleary M, Lim L, Linsley PS. Widespread siRNA “off-target” transcript silencing mediated by seed region sequence complementarity. Rna. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nature reviews Drug discovery. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nature reviews Molecular cell biology. 2013;14:49–55. doi: 10.1038/nrm3486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic acids research. 2014;42:e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan AA, Betel D, Miller ML, Sander C, Leslie CS, Marks DS. Transfection of small RNAs globally perturbs gene regulation by endogenous microRNAs. Nature biotechnology. 2009;27:549–555. doi: 10.1038/nbt.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Bae S, Park J, Kim E, Kim S, Yu HR, Hwang J, Kim JI, Kim JS. Digenome-seq: genome-wide profiling of CRISPR-Cas9 off-target effects in human cells. Nature methods. 2015;12:237–243. doi: 10.1038/nmeth.3284. [DOI] [PubMed] [Google Scholar]
- Kodzius R, Kojima M, Nishiyori H, Nakamura M, Fukuda S, Tagami M, Sasaki D, Imamura K, Kai C, Harbers M, et al. CAGE: cap analysis of gene expression. Nature methods. 2006;3:211–222. doi: 10.1038/nmeth0306-211. [DOI] [PubMed] [Google Scholar]
- Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, Hsu PD, Habib N, Gootenberg JS, Nishimasu H, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–588. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornienko AE, Guenzl PM, Barlow DP, Pauler FM. Gene regulation by the act of long non-coding RNA transcription. BMC biology. 2013;11:59. doi: 10.1186/1741-7007-11-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuscu C, Arslan S, Singh R, Thorpe J, Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nature biotechnology. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- Lagana A, Shasha D, Croce CM. Synthetic RNAs for Gene Regulation: Design Principles and Computational Tools. Frontiers in bioengineering and biotechnology. 2014;2:65. doi: 10.3389/fbioe.2014.00065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Maeder ML, Linder SJ, Cascio VM, Fu Y, Ho QH, Joung JK. CRISPR RNA-guided activation of endogenous human genes. Nature methods. 2013;10:977–979. doi: 10.1038/nmeth.2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Aach J, Stranges PB, Esvelt KM, Moosburner M, Kosuri S, Yang L, Church GM. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature biotechnology. 2013a;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nature methods. 2013b;10:957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. RNA-guided human genome engineering via Cas9. Science. 2013c;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin JF, Friedman JR, Meyer WK, Vissing H, Thiesen HJ, Rauscher FJ., 3rd Kruppel-associated boxes are potent transcriptional repression domains. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4509–4513. doi: 10.1073/pnas.91.10.4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JC, Tan S, Qiao G, Barlow KA, Wang J, Xia DF, Meng X, Paschon DE, Leung E, Hinkley SJ, et al. A TALE nuclease architecture for efficient genome editing. Nature biotechnology. 2011;29:143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, Urnov FD, Holmes MC. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohr SE, Smith JA, Shamu CE, Neumuller RA, Perrimon N. RNAi screening comes of age: improved techniques and complementary approaches. Nature reviews Molecular cell biology. 2014;15:591–600. doi: 10.1038/nrm3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic acids research. 2011;39:9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissim L, Perli SD, Fridkin A, Perez-Pinera P, Lu TK. Multiplexed and programmable regulation of gene networks with an integrated RNA and CRISPR/Cas toolkit in human cells. Molecular cell. 2014;54:698–710. doi: 10.1016/j.molcel.2014.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell MR, Oakes BL, Sternberg SH, East-Seletsky A, Kaplan M, Doudna JA. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature. 2014 doi: 10.1038/nature13769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsson K, Harrison CJ, Andersen MK, Chilton L, Nordgren A, Moorman AV, Johansson B. Distinct patterns of gained chromosomes in high hyperdiploid acute lymphoblastic leukemia with t(1;19)(q23;p13), t(9;22)(q34;q22) or MLL rearrangements. Leukemia. 2013;27:974–977. doi: 10.1038/leu.2012.263. [DOI] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nature methods. 2013;10:973–976. doi: 10.1038/nmeth.2600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, Lim WA. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013a;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nature protocols. 2013b;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondon-Lagos M, Verdun Di Cantogno L, Marchio C, Rangel N, Payan-Gomez C, Gugliotta P, Botta C, Bussolati G, Ramirez-Clavijo SR, Pasini B, et al. Differences and homologies of chromosomal alterations within and between breast cancer cell lines: a clustering analysis. Molecular cytogenetics. 2014;7:8. doi: 10.1186/1755-8166-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandelin A, Carninci P, Lenhard B, Ponjavic J, Hayashizaki Y, Hume DA. Mammalian RNA polymerase II core promoters: insights from genome-wide studies. Nature reviews Genetics. 2007;8:424–436. doi: 10.1038/nrg2026. [DOI] [PubMed] [Google Scholar]
- Sigoillot FD, King RW. Vigilance and validation: Keys to success in RNAi screening. ACS chemical biology. 2011;6:47–60. doi: 10.1021/cb100358f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg SH, Redding S, Jinek M, Greene EC, Doudna JA. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung YH, Baek IJ, Kim DH, Jeon J, Lee J, Lee K, Jeong D, Kim JS, Lee HW. Knockout mice created by TALEN-mediated gene targeting. Nature biotechnology. 2013;31:23–24. doi: 10.1038/nbt.2477. [DOI] [PubMed] [Google Scholar]
- Tafer H. Bioinformatics of siRNA design. Methods in molecular biology. 2014;1097:477–490. doi: 10.1007/978-1-62703-709-9_22. [DOI] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell. 2014 doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Wyvekens N, Khayter C, Foden JA, Thapar V, Reyon D, Goodwin MJ, Aryee MJ, Joung JK. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nature biotechnology. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai SQ, Zheng Z, Nguyen NT, Liebers M, Topkar VV, Thapar V, Wyvekens N, Khayter C, Iafrate AJ, Le LP, et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature biotechnology. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22:5792–5812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- van der Ploeg JR. Analysis of CRISPR in Streptococcus mutans suggests frequent occurrence of acquired immunity against infection by M102-like bacteriophages. Microbiology. 2009;155:1966–1976. doi: 10.1099/mic.0.027508-0. [DOI] [PubMed] [Google Scholar]
- Vasquez KM, Marburger K, Intody Z, Wilson JH. Manipulating the mammalian genome by homologous recombination. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:8403–8410. doi: 10.1073/pnas.111009698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Wang X, Varma RK, Beauchamp L, Magdaleno S, Sendera TJ. Selection of hyperfunctional siRNAs with improved potency and specificity. Nucleic acids research. 2009;37:e152. doi: 10.1093/nar/gkp864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson RC, Doudna JA. Molecular mechanisms of RNA interference. Annual review of biophysics. 2013;42:217–239. doi: 10.1146/annurev-biophys-083012-130404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nature biotechnology. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhu S, Cai C, Yuan P, Li C, Huang Y, Wei W. Highthroughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509:487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]