

Highlights
-
•
Translational regulation of viral and host mRNA is an important function of a subset of interferon-stimulated genes (ISGs).
-
•
These ISGs repress translation through binding to viral RNA, direct interaction with, or perturbation of the translation machinery components.
-
•
Several ISGs localize to cytoplasmic granules such as stress granules (SGs) and processing bodies (PBs), and interfere with the processing or function of microRNAs (miRNAs).
Keywords: translational regulation, microRNA processing, microRNA function
Abstract
Type I interferon (IFN) is one of the first lines of cellular defense against viral pathogens. As a result of IFN signaling, a wide array of IFN-stimulated gene (ISG) products is upregulated to target different stages of the viral life cycle. We review recent findings implicating a subset of ISGs in translational regulation of viral and host mRNAs. Translation inhibition is mediated either by binding to viral RNA or by disrupting physiological interactions or levels of the translation complex components. In addition, many of these ISGs localize to translationally silent cytoplasmic granules, such as stress granules and processing bodies, and intersect with the microRNA (miRNA)-mediated silencing pathway to regulate translation of cellular mRNAs.
ISGs block virus replication
In response to an infection, the host recognizes pathogen-associated molecular patterns (PAMPs) of invading microbes in the cell. Viral PAMPs are often nucleic acid-based, derived from their DNA, or from their RNA genomes. Several pattern recognition receptor families located in various cellular compartments work together to sense PAMPs leading to activation of the transcription factors IFN-regulatory factors 3 or 7 (IRF3/7) and nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB) (for a recent review, see [1]). The signaling events following PAMP recognition result in dimerization and translocation of IRF3/7 into the nucleus along with NFκB, leading to the transcription and expression of type I IFN and proinflammatory cytokines, which in turn get secreted by the cell. Autocrine or paracrine signaling in response to IFN induces downstream expression of an array of IFN-stimulated genes (ISGs), which function to establish an antiviral state [1].
ISGs act on different stages of the viral life cycle, from entry and replication to assembly and release. In order to productively infect the host and multiply, viruses usurp the host translation machinery to make viral proteins. Translational inhibition is a common mechanism utilized by ISGs to mediate antiviral effects [2]. Indeed, some of the best studied ISGs, protein kinase RNA-activated (PKR) and 2′-5′-oligoadenylate synthetase (OAS)/RNAseL function to block translation to limit virus replication (Box 1 ). This review focuses on the more recently described ISGs that regulate host or viral translation, localize to translationally silent granules, and interfere with miRNA-mediated silencing of host transcripts.
Box 1. Early discoveries on the mechanism of action of IFN.
Before the discovery of ISGs, it was known that treatment of animal cells with IFN confers upon them resistance to new virus infections. IFN is not directly antiviral; cellular transcription and protein synthesis were found to be required for IFN to work, suggesting that IFN signaling leads to the translation of an inhibitory protein(s). The inhibitory activity targets an early stage of the viral life cycle, specifically the translation of the viral mRNA 99, 100. Protein synthesis in lysates prepared from mouse L cells pretreated with IFN was blocked upon exposure to dsRNA 101, 102. It appeared that a dsRNA-dependent protein kinase(s) and an oligonucleotide inhibitor (pppA2′-5′A2′-5′A) were involved 103, 104, 105, 106, 107, 108, 109, 110, 111. It is now well appreciated that protein kinase RNA-activated (PKR) is a serine–threonine kinase and when activated by dsRNA becomes autophosphorylated and phosphorylates the α subunit of eIF2, leading to the inhibition of host and viral mRNA translation 112, 113, 114, 115, 116, 117, 118. In addition, activation of the 2′-5′-oligoadenylate synthetase (OAS) by dsRNA triggers the synthesis of 2′-5′A from ATP, which causes the dimerization and activation of a latent endoribonuclease (later referred to as RNAse L) 113, 119. RNAseL causes the degradation of viral or cellular RNA leading to translation inhibition 120, 121, 122.
Regulation of viral and host mRNA translation
Viruses are completely reliant on host cell translational machinery to produce the proteins encoded by their genes. In eukaryotic cells, translation is initiated (summarized in Figure 1 and recently reviewed in [3]) by binding of eukaryotic initiation factor (eIF) 4E to the m7G cap structure at the 5′ end of mRNAs. Meanwhile, poly(A)-binding protein (PABP) binds to the poly(A) tail at the 3′ end of mRNAs. Both eIF4E and PABP interact with the scaffold protein eIF4G, leading to mRNA circularization and recruitment of the 43S preinitiation complex, the minimal constituents of which include the eIF3 complex (13 subunits; a–m), the ternary complex eIF2-GTP-Met-tRNAi, and the 40S ribosomal subunit. The 43S complex then scans the 5′ untranslated region (UTR) of the mRNA until it reaches the translational start codon. Ribosome scanning is aided by the RNA helicase, eIF4A, which disrupts secondary and tertiary structures in the 5′ UTR. The 60S ribosomal subunit then joins the 40S ribosomal subunit to form the 80S ribosome, resulting in translation initiation and elongation; formation of polyribosomes (polysomes) where multiple ribosomes simultaneously translate the same mRNA can then take place. Because translation initiation is a complex and highly ordered process, most of the translational regulation in eukaryotic cells occurs at this step [3]. Several ISG products, such as zinc finger antiviral protein (ZAP), interferon-induced protein with tetratricopeptide repeats 1 (IFIT1), and schlafen 11 (SLFN11), have been shown to affect viral or global protein synthesis, and their modes of action are described in this section. The common strategies shared by these ISGs include direct binding to viral RNA, and interaction with or perturbation of the translation machinery components, preventing translation.
Figure 1.
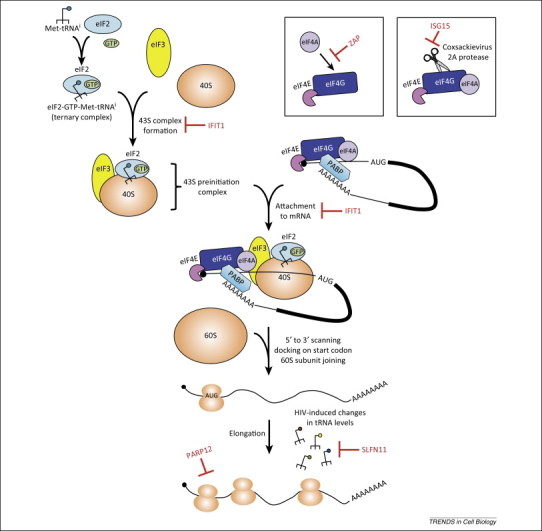
Interferon-stimulated genes (ISGs) repress translation by targeting host and viral factors important for regulation of translation initiation and elongation. Translation initiation involves binding of eukaryotic initiation factor (eIF) 4E to the m7G cap structure at the 5′ end of mRNAs (middle top). Both eIF4E and poly(A)-binding protein (PABP) interact with the scaffold protein eIF4G, leading to mRNA circularization. The 43S preinitiation complex, which consists of the eIF3 complex (13 subunits; a–m), the ternary complex eIF2-GTP-Met-tRNAi and the 40S ribosomal subunit, is then recruited (top left). The 43S complex scans the 5′ untranslated region (UTR) of the mRNA, which is unwound by the RNA helicase eIF4A until the initiating AUG is found. As a result, the 60S ribosomal subunit joins the 40S ribosomal subunit to form the 80S ribosome, resulting in translation initiation and elongation, and formation of polyribosomes (polysomes) on the mRNA (middle bottom). The ISGs that target some of the translation initiation and elongation steps are depicted in red.
PARPs
Several members of the poly(ADP-ribose) polymerase (PARP) family are ISGs with antiviral activity (Box 2 ). Among them, the best-characterized antiviral protein is PARP13 or zinc finger antiviral protein (ZAP), which is encoded by the zinc finger CCCH-type, antiviral 1 (ZC3HAV1) gene. In the remainder of the review, we will refer to the protein as ZAP. ZAP is transcriptionally upregulated by type I IFN signaling and directly induced by phosphorylated IRF3 in virus-infected cells 4, 5. There are at least two splice variants of ZAP – ZAPL (PARP13.1) and ZAPS (PARP13.2) – where the long isoform encodes a PARP domain on the C terminus that is missing in the short isoform [6]. Although both isoforms are induced, ZAPS is upregulated more than ZAPL by virus and type I IFN 7, 8, 9. ZAP was first discovered as a potent antiviral factor against the retrovirus Moloney murine leukemia virus (MLV) in a cDNA library screen [10]. Since then, it has been shown to inhibit a broad range of RNA and DNA viruses, including other retroviruses, alphaviruses, filoviruses, and hepatitis B virus 8, 11, 12, 13, 14. It is not understood what determines the broad yet specific antiviral activity of ZAP. It binds viral RNA via its N-terminal zinc fingers, and ZAP-responsive sequence elements in MLV and Sindbis virus have been mapped [15]. ZAP recruits the exosome to target retroviral and specific host mRNAs for degradation 14, 16, 17, 18 but also acts to block viral genome translation [11]. ZAP dramatically reduces Sindbis virus production, and experiments utilizing temperature-sensitive Sindbis virus mutants that are unable to replicate the RNA genome at nonpermissive temperatures support a mechanism in which ZAP represses translation of the incoming viral genome [11]. ZAP also inhibits translation of luciferase reporters that carry the ZAP-responsive elements from HIV-1 and Sindbis virus [19]. ZAP binds to eIF4A and interferes with the interaction between eIF4A and eIF4G, and as a result blocks translation independently of mRNA degradation [19] (Figure 1).
Box 2. An overview of ADP-ribosylation.
Mono- and poly-ADP-ribosylation of proteins (Figure I ) are important post-translational modifications that are implicated in a variety of cellular processes, such as DNA repair, cell death, chromatin remodeling, and immune cell development 123, 124, 125, 126. Poly(ADP-ribose) polymerases (PARPs) transfer ADP-ribose from the co-substrate NAD+ onto specific amino acid residues of substrate proteins. Seventeen PARPs exist in humans, all with a conserved PARP domain, and nearly one-third of the PARP genes contain signatures of rapid evolution in primates [127]. Some of the PARPs carry the enzymatic activity to modify proteins with poly-ADP-ribose (pADPr) or mono-ADP-ribose (mADPr), whereas others are inactive, based primarily on the presence or absence of a catalytic triad motif of histidine, tyrosine, and glutamate (HYE) that is required for transferring the initial ADPr and/or multiple ADPr molecules [55]. Most of the PARP proteins can also auto-ADP-ribosylate themselves. Alternatively, poly-ADPr-glycohydrolase (PARG) removes the pADPr chain leaving the most proximal ADPr still attached to the target protein 128, 129. Searches for enzymes that reverse mono-ADP-ribosylated proteins back to their unmodified state have been elusive until recently when cellular macrodomains were shown to reverse mono-ADP-ribosylation 130, 131. The presence of macrodomain motifs in multiple diverse viruses such as alphaviruses, coronaviruses, rubella, and hepatitis E viruses suggests that the metabolism of this post-translational modification might be critical for viral antagonism of host responses. In this review, we will discuss some of the recent findings related to the novel roles of ISGs in the PARP family in stress granule formation and stress response-induced translational derepression of miRNA targets, involving PARP12, ZAPL/PARP13.1, and ZAPS/PARP13.2.
Figure I.
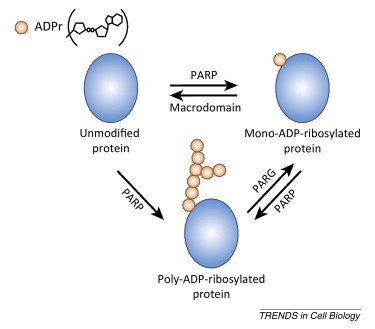
Enzymes responsible for addition and removal of ADP-ribose.
In addition to ZAP, other members of the PARP family are upregulated by IFN and have also been shown to inhibit alphaviruses. Murine PARP7, PARP10, and the long isoform of PARP12 (mPARP12L) block cellular translation and inhibit replication of Venezuelan equine encephalitis virus (VEEV), another member of the alphavirus genus 20, 21. mPARP12L also blocks infection of a variety of RNA viruses from other families, such as vesicular stomatitis virus (VSV), encephalomyocarditis virus (EMCV), and Rift Valley fever virus (RVFV) [20]. Similar to ZAP, mPARP12L affects protein translation, and the tethering of mPARP12L to a renilla luciferase reporter mRNA inhibits its translation [22]. Furthermore, mass spectrometry identified ribosomal proteins and proteins involved in translation as interacting partners of mPARP12L. mPARP12L interacts with ribosomes in the polysome-containing fractions at 4 hours post-infection (pi) with VEEV but facilitates disassembly of polysomes at later times of infection (12 h pi), which is dependent on its RNA-binding and PARP catalytic activities 21, 22 (Figure 1). mPARP12L mutants that are unable to transfer ADP-ribose to substrate proteins, including mPARP12L itself, fail to block translation; however, they are still able to inhibit replication of GFP-expressing VEEV. One possible explanation is that mPARP12L utilizes an unknown mechanism to block VEEV replication that is unrelated to poly-ADP-ribosylation and translational inhibition. The role of the PARP domain in mediating translational inhibition and the catalytic activity-independent antiviral function of mPARP12L warrant further studies.
IFIT1
IFIT proteins are localized in the cytoplasm and lack any obvious enzymatic domain or activity. They contain multiple tetratricopeptide repeats, which are important for protein–protein interactions. IFIT1 (also called p56 and ISG56) is among the better characterized members, and its expression is induced by dsRNA, IRF3, type I IFN, and a variety of viruses 23, 24. Similar to PARP proteins, the IFIT family targets viruses by translational repression [25]. Many cellular and viral mRNAs are methylated at the N-7 and 2′-O positions of the 5′ guanosine cap by nuclear and cytoplasmic methyltransferases, but the function of 2′-O methylation was unclear for many years. Recent studies found that 2′-O methylation of the 5′ cap of viral RNA serves as an immune evasion strategy for viruses that are otherwise recognized by IFIT1.
Human IFIT1 blocks West Nile virus (WNV), Japanese encephalitis virus (JEV), and coronavirus mutants that lack 2′-O-methyltransferase activity, and inhibition occurs by IFIT1 preferentially sequestering capped RNA lacking a 2′-O-methyl group and preventing eukaryotic translation initiation factors from binding to the RNA template 26, 27, 28, 29, 30 (Figure 1). Structural studies and binding assays also support a role for IFIT1 in recognizing single-stranded viral RNA bearing a 5′-triphosphate group, which results in translation inhibition, although binding appears to be of lower affinity than IFIT1 binding to 5′ capped RNA lacking 2′-O-methylation 28, 31, 32. Intriguingly, alphaviruses do not carry a 2′-O-methylated cap but instead have evolved a stable secondary structure in their 5′ UTRs to evade IFIT1 recognition and translational repression [33]. Taken together, IFIT1 is a critical innate immune effector that inhibits viruses whose RNAs lack a 2′-O-methylated cap.
In addition to directly binding to the viral RNA, IFIT1 binds the eukaryotic translation initiation factor 3e (eIF3e) subunit in a yeast two-hybrid screen and inhibits translation both in vitro and in vivo by blocking eIF3 stabilization of the ternary complex eIF2-GTP-Met-tRNAi 34, 35 (Figure 1). IFIT1 was also identified as an eIF3-associated protein fractionating with hepatitis C virus (HCV) translation complexes in IFN-treated cells [36]. IFIT1 blocks translation driven by the HCV internal ribosome entry site (IRES) and the inhibition depends on eIF3 binding by IFIT1. IRESs are RNA elements that recruit the ribosome to internal sites in the mRNA in a cap-independent manner [3]. The HCV IRES interacts with eIF3 and 40S ribosomal subunit to initiate translation and therefore is prone to eIF3 inhibition by IFIT1. However, in another study that interrogated the effects of IFIT1 on different modes of translation, IFIT1 preferentially inhibits cap-dependent translation but not translation of the EMCV IRES [35]. Since the EMCV IRES is different than that of HCV and functions by a different mechanism, the former might have a different requirement for eIF3 and hence is more resistant to IFIT1 effects.
SLFN11
Many viral genes have a different codon usage than host genes; for example, high frequencies of adenosine (A) at wobble positions have been found in lentiviral genomes 37, 38, 39. It was shown that codons rarely used by the host cell are highly represented in the HIV-1 genome, and the virus induces changes in cellular transfer RNA (tRNA) levels to decode the rare codons and facilitate viral protein synthesis [39]. Human schlafen 11 (SLFN11) has been shown to block the production of wild type HIV-1 by inhibiting the translation of viral proteins [40]. Schlafen genes are a subset of ISGs that are found only in mammals and possess motifs resembling DNA/RNA helicase domains. Murine SFLN proteins are regulators of T cell development, and their expression is upregulated by lipopolysaccharide (LPS), poly(I:C), and type I IFN 40, 41, 42, 43. SLFN11 binds cellular tRNAs to counteract HIV-induced changes in tRNA levels and achieve viral translation inhibition [40] (Figure 1). The mechanism by which SLFN11 acts is not clear, but it provides an example of how ISGs can specifically block viral translation due to the rare codon usage of viruses.
ISG15
Some ISGs mediate antiviral activity by counteracting the effects that viruses have on host translation. IFN-stimulated gene of 15 kDa (ISG15) is a ubiquitin-like protein that is upregulated by type I IFN. It is covalently attached to lysine residues of target proteins, and has been found as a modification on newly translated proteins of both viral and host origin [44]. Replication of many viruses is blocked by ISG15 in vitro and/or in vivo, and for some of these viruses, ISG15 acts at the step of virus release [44]. Previously, Coxsackievirus B3 (CVB3) 2A protease was shown to cleave the host cell eukaryotic initiation factor eIF4G, leading to host cell translational shutoff in infected cells while allowing IRES-driven translation of the viral genome [45]. Interestingly, CVB3 2A protease is modified by ISG15, and the modified protease is less efficient at cleaving eIF4G in both HeLa cells and cardiomyocytes [46] (Figure 1). Indeed, inhibition of virus production and decreased CVB3-induced pathology were observed in wild type mice compared to ISG15 knockout animals [46]. The data suggest a previously unappreciated role for ISG15 modification in subversion of virus-induced translational shutoff. However, further studies are necessary to determine if this strategy applies to other viruses that cause host shutoff.
Localization to cytoplasmic granules and regulation of miRNA-mediated silencing
As cellular mRNA exits the nucleus, the translation initiation complex is recruited and assembled, leading to polysome formation and active translation of the mRNA. Polysomes can be dissembled by cues of translational silencing or deadenylation, and the mRNA is redirected to cytoplasmic RNA granules for regulated translation and/or decay (for recent reviews, see 47, 48). Upon exposure to stress-inducing stimuli, such as oxidation, hypoxia, and virus infection, translationally stalled mRNAs, 40S ribosomal subunits, and several RNA-binding proteins localize to distinct cytoplasmic foci called stress granules (SGs) (Figure 2 ). In most cases, phosphorylation of eIF2α, triggered by stress inducers including the chemical sodium arsenite, results in the assembly of SGs. Alternatively, acute energy starvation or the drug pateamine A can induce SG formation by an eIF2α phosphorylation-independent mechanism. SGs are believed to mediate antiviral processes and in response viruses have evolved strategies to interfere with SG formation, although in some cases SG formation acts in a proviral manner (for a recent review, see [49]). Further, cytoplasmic processing bodies (PBs) contain untranslated mRNAs and can serve as sites of mRNA degradation (Figure 2). There is evidence in support of dynamic interactions between SGs and PBs where mRNA species from dissociated polysomes get remodeled in SGs and some of them are subsequently targeted for degradation in PBs [48].
Figure 2.
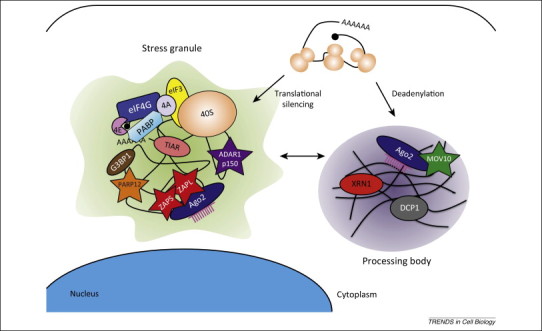
Interferon-stimulated genes (ISGs) localize to stress granules (SGs) and P bodies (PBs), translationally silent sites in the cytoplasm. Polysomes can be dissociated by signals of translational silencing or deadenylation, and the actively translating mRNA is redirected to SGs and PBs for repression or triage. Messenger ribonucleoproteins can also move from one type of granule to the other. ISGs that are localized to SGs upon stress induction and to PBs are shown as stars.
Argonaute (Ago) proteins localize to mammalian PBs and are effectors of the RNA-induced silencing complex (RISC) that is involved in miRNA-mediated translational regulation of cellular transcripts 50, 51, 52. The miRNA pathway (summarized in Figure 3 and recently reviewed in [53]) starts with the nuclear processing of longer primary miRNA (pri-miRNA) transcripts into pre-miRNA hairpins by the RNase III enzyme Drosha. The cytoplasmic RNase III enzyme Dicer then cleaves pre-miRNAs into miRNA duplexes where one strand is guided to its targets by Ago. In most cases, Ago-miRNA complexes engage mRNA targets with imperfect sequence complementarity leading to cleavage-independent repression of mRNA transcripts by translational silencing and to a lesser degree deadenylation and mRNA degradation. By contrast, a few miRNAs and small interfering RNAs (siRNAs) bind to target sites with extensive or perfect sequence complementarity, which results in mRNA cleavage.
Figure 3.
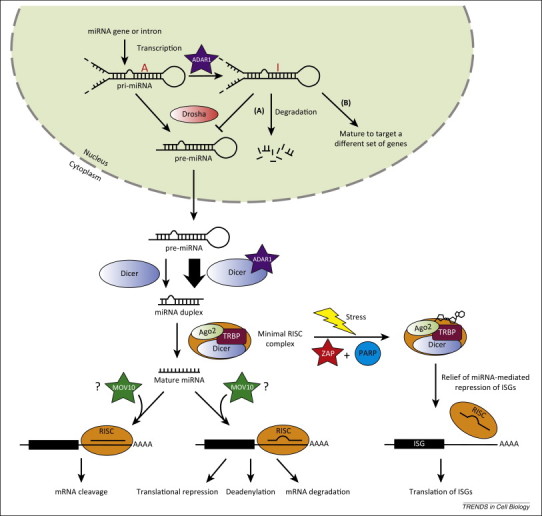
Interferon-stimulated genes (ISGs) affect microRNA (miRNA) processing, production, and actions. The mammalian RNAi pathway is depicted here. The RNase III enzyme Drosha first processes longer primary miRNA (pri-miRNA) transcripts into pre-miRNA hairpins in the nucleus. After nuclear export, pre-miRNAs are cleaved into miRNA duplexes by Dicer in the cytoplasm and loaded onto Argonaute 2 (Ago2). The minimal RNA-induced silencing complex (RISC) complex (Dicer-TRBP-Ago2) targets transcripts with extensive or perfect sequence complementarity to the miRNA leading to mRNA cleavage, whereas imperfect sequence complementarity between the target transcript and miRNA leads to translational silencing, deadenylation, and mRNA degradation. ISGs interacting with different components of the RNAi pathway are shown as stars. A-to-I editing by adenosine deaminase acting on RNA 1 (ADAR1) either leads to (A) decreased processing and increased degradation of pri-miRNA [97], or (B) silencing of a different set of gene targets [96]. The involvement of Moloney leukemia virus type 10 homolog (MOV10) in different repressive mechanisms of miRNA is controversial and therefore highlighted by question marks.
It has been shown that several ISGs localize to cytoplasmic granules (Figure 2) and, of these, the PARP proteins, Moloney leukemia virus type 10 homolog (MOV10), and adenosine deaminase acting on RNA 1 (ADAR1) are known to regulate miRNA-mediated silencing of target genes. Regulation is mediated by modifying or interacting with components of the RISC, or by editing miRNAs. In the instances described below, the immune response is altered as a result of translational derepession of antiviral gene transcripts or increased recognition of infected cells by immune subsets.
PARPs
Among the ISG members of the PARP family, human PARP12, PARP13.1 (ZAPL), and PARP13.2 (ZAPS) were found to localize to cytoplasmic SGs [54]. In response to arsenite-induced oxidative stress, PARP12 of both human and murine origin colocalizes in cytoplasmic puncta with well-characterized components of SGs, such as Ras GTPase-activating protein-binding protein 1 (G3BP1), the T-cell-restricted intracellular antigen 1-related protein (TIAR), and the eukaryotic translation initiation factor 3η (eIF3η) subunit 22, 54 (Figure 2). The ability to localize to SGs is dependent on mPARP12's N-terminal zinc fingers, suggesting a role for RNA binding in SG localization. Similar to PARP12, both ZAP isoforms localize to cytoplasmic SGs after sodium arsenite treatment (Figure 2), and in addition become poly-ADP-ribosylated, as does Ago2 [54]. ADP-ribose addition to ZAPS and ZAPL must be carried out by other cytoplasmic PARP proteins because ZAPL lacks auto-ADP-ribosylating activity and is predicted to be an inactive PARP enzyme, and ZAPS lacks the PARP domain completely 54, 55. There was some evidence suggesting that human PARP12 might be one of the SG-specific PARPs that ADP-ribosylates ZAP and/or Ago2 [54]. Interestingly, the short isoform of ZAP has a higher level of ADP-ribosylation than ZAPL upon pateamine A treatment, supporting distinct roles for ZAP isoforms in the cellular response to stress [54].
The known functional consequences of ADP-ribosylation are largely restricted to nuclear processes. Therefore, the finding that ADP-ribosylation of Ago2 increases upon stress is novel and implies important functions for this post-translational modification in cytoplasmic processes [54]. The resultant ADP-ribosylation of Ago2 after stress or overexpression of either ZAPS or ZAPL correlates with derepression of miRNA-mediated translational silencing of luciferase reporters that contain siRNA or miRNA binding sites in their 3′ UTRs 9, 54. Overexpression of human PARP12 also leads to relief of silencing of a luciferase reporter with miRNA binding sites, albeit less dramatically compared to ZAP overexpression [54]. Because many ISGs are anti-proliferative, their constitutive expression levels in the cells are presumably low and tightly regulated. It was postulated that due to an increased density of miRNA binding sites in the 3′ UTRs of ISGs compared to all cellular 3′ UTRs, the mRNAs of ISGs are selectively repressed under normal physiological conditions to maintain cellular homeostasis [9]. However, ISG mRNAs are derepressed during viral infection to allow upregulation of antiviral gene expression (Figure 3). In this case, ADP-ribose serves as the signaling molecule that switches off RISC activity in order to upregulate ISG expression and eliminate the virus. This derepression of miRNA activity on ISGs has so far been shown to contribute to inhibition of herpes simplex virus 1 and a mutant influenza A virus that induces a stronger antiviral response than the wild type virus [9]. ZAPS, ZAPL, and potentially PARP12 are important components of stress-induced relief of RISC activity, although the mechanism by which they act remains unclear.
As mentioned here, ZAP intersects with a variety of cellular processes potentially in an isoform-specific fashion and carries out complex functions that are seemingly contradictory and cannot be unified by a single mechanism. For example, it is not clear how ZAP can block translation of viral mRNA yet be involved in the derepression of miRNA-mediated silencing of cellular transcripts. It is possible that ZAP's binding to viral RNA, recruitment of exosome components, interaction with the translation machinery, and regulation of Ago2 all play a role in its antiviral activity although the relative contribution of these various functions to ZAP's inhibition of different viruses needs to be further investigated.
MOV10
MOV10 protein is a member of the superfamily 1 (SF-1) RNA helicase family [56] and contains seven highly conserved helicase motifs in its C-terminal region. MOV10, upregulated by type I IFN, has been shown to inhibit HCV, retroviruses, and both long terminal repeat (LTR) and non-LTR endogenous retroelements, but can facilitate the RNA-directed transcription of hepatitis delta virus 2, 57, 58, 59, 60, 61, 62. MOV10, identified in purified Ago1- and Ago2-containing complexes from human cells, localizes to cytoplasmic PBs (Figure 2) and is functionally required to mediate miRNA-guided mRNA silencing [63]. Interestingly, MOV10 is critical for regulation of local protein synthesis at synapses where cellular mRNAs normally suppressed by miRNAs enter the polysomes once MOV10 is targeted for degradation in response to synaptic stimulation [64].
One controversy in the RNAi field is what determines the different repressive mechanisms following miRNA and siRNA targeting. Identification of specific host factors or differential complex formation required for miRNA function may help elucidate the different outcomes of targeted transcripts. Using fractions isolated from cells a minimal RISC complex, consisting of Dicer, the HIV-1 TAR RNA binding protein (TRBP), and Ago2, cleaves target RNA with perfect complementarity; use of a pre-miRNA hairpin as the source of siRNA results in more efficient target cleavage than a mature miRNA duplex 65, 66. These studies provide evidence for the coupling of miRNA processing by Dicer and Ago2-mediated target cleavage. Alternatively, a larger fraction containing the minimal RISC complex components as well as MOV10, core proteins of the 60S ribosomal subunit, and eIF6, a ribosome inhibitory protein, resulted in translational repression and downregulation of target mRNAs [67] (Figure 3). It was proposed that eIF6 prevents ribosome association or recycling and recruits target RNAs to PBs, which are free of core translation components. In contrast to these studies, silencing experiments implicate MOV10 in both mRNA cleavage mediated by a perfectly matching miRNA and translation repression mediated by an imperfectly matching miRNA [68]. In support of the role of MOV10 in mRNA cleavage, APOBEC3G, another ISG that localizes to cytoplasmic PBs, inhibits miRNA function mediated by perfect complementarity sites by interfering with the interaction between Ago2 and MOV10, causing either abnormal assembly or maturation of the RISC complex 68, 69. Taken together, MOV10 interacts with Ago2 and is critical for the function of the RISC, but the relative contribution of MOV10 to different repressive mechanisms of miRNAs is still controversial and needs to be further explored.
ADAR1
Members of the ADAR family of enzymes convert A to inosine (I) in RNA species that carry double-stranded structures [70]. A-to-I RNA editing leads to nucleotide substitution as I is recognized by the cell as guanosine, and affects the coding of the protein that is ultimately made. Intriguingly, the majority of A-to-I editing targets in the cell are noncoding RNAs 71, 72, 73, 74, 75, 76. All members of the ADAR family (ADAR1, 2, and 3) contain a deaminase domain at their C-terminal end that is required for enzymatic activity and two to three double-stranded RNA-binding domains (dsRBDs). ADAR1 also contains Z-DNA-binding domains (ZBDs) in its amino-terminus. ADAR1 has been implicated in the editing of a variety of viral genomes, which either drives genetic diversity of the population and generates novel viral protein variants or limits viral replication and reduces infectivity 77, 78, 79, 80, 81, 82, 83, 84, 85, 86, 87, 88.
Two major isoforms of ADAR1 (p110 and p150) exist in mammalian cells. The longer 150-kDa protein (p150) is IFN inducible and shuttles between the nucleus and cytoplasm whereas the amino-terminally truncated 110-kDa version (p110) is constitutively expressed and predominantly nuclear 89, 90, 91. However, recent studies demonstrated that both ADAR1 isoforms shuttle between the nucleus and cytoplasm 92, 93. ADAR1 p150 localizes to cytoplasmic SGs in HeLa cells upon oxidative or IFN-induced stress; this localization is dependent on the ZBDs found exclusively in p150 94, 95 (Figure 2).
Previous studies suggested conflicting roles for ADAR1 in both facilitating and interfering with miRNA processing through direct interaction with components of the RNAi pathway or A-to-I editing of miRNAs (Figure 3). Altered miRNA seeds due to A-to-I editing could lead to altered translation profiles. Both ADAR1 p110 and p150 physically interact with Dicer in an RNA-independent manner to increase the rate of cleavage of pre-miRNA by Dicer, and thus facilitate loading of miRNA onto the RISC [93]. As expected, miRNA expression is downregulated in ADAR1−/− mouse embryos compared to wild type, leading to increased protein levels of the predicted miRNA targets [93]. Furthermore, miR-376 cluster transcripts undergo extensive A-to-I editing in select human and mouse tissues as well as specific subregions of the brain, and the edited miRNAs target a different set of genes, suggesting a role for ADAR1 in tissue-specific regulation of gene expression [96]. Conversely, ADAR1 p110 can edit specific A residues in pri-miRNAs and negatively affect the miRNA pathway. Editing of one of the ADAR1 targets, pri-miR-142, leads to decreased processing by Drosha and its degradation by Tudor-SN, a RISC-specific ribonuclease that targets I-containing dsRNAs [97]. ADAR1 can also block virus infection through editing of miRNAs. Surprisingly, ADAR1 p110 but not the IFN-induced p150 is upregulated during cytomegalovirus (CMV) infection and edits miR-376a, the precursor of which is a known target of ADAR1 [96]. The edited mature miR-376a downregulates HLA-E expression, and as a result infected cells become more susceptible to elimination by natural killer cells, suggesting an indirect antiviral role for the constitutive form of ADAR1 [98].
Concluding remarks
The innate immune response has developed multiple strategies to attack viral intruders through the action of effector proteins induced by type I IFN. A common antiviral strategy shared among some of these ISGs is to cause dysregulation of viral or host translation through interaction with translation factors and components of the RNAi pathway, and facilitation of stress-induced post-translational modification and function of Ago2. However, several important questions remain to be addressed. Are SGs functional sites of antiviral signaling triggered by sensing of viral RNA and translational inhibition? For example, ZAP has been shown to interact with the cytosolic viral RNA sensor RIG-I to enhance IFN-β production and NFκB signaling [7]. Can viral RNA binding and sequestration by some of these SG-specific ISGs activate cytosolic sensors to relay innate immune signaling? Moreover, for some of the ISGs mentioned, for example, ZAP and ADAR1, one of their isoforms is constitutively expressed in the cell whereas the other is induced by IFN. It is possible that the constitutively expressed form is also upregulated by IFN, albeit to a lower level compared to the IFN-inducible form. To add to the complexity, there is evidence supporting differential upregulation of the two different ADAR1 isoforms by different viruses, blurring the line between constitutively expressed and IFN-inducible forms [98]. How do the different isoforms of ISGs coordinate their activity upon viral infection, and how does that affect the antiviral response? What are the mechanisms by which ADP-ribosylation (and other post-translational modifications) triggers changes in protein function? We have much to learn regarding the details of these translational control mechanisms. It will be interesting to ponder new information that is uncovered regarding the processes described here and how they might be exploited for manipulation of viral or host translation for novel antiviral or therapeutic strategies.
Acknowledgments
M.M.L. was supported by a Career Development Award from the Northeast Biodefense Center (U54-AI057158; Lipkin), M.R.M. was supported by grants R01AI057905, R21AI100188, and R03AI097089, and the Greenberg Medical Research Institute, and C.M.R. was supported by PHS grants U54-AI057158 (Lipkin) and R01 AI091707. We give tremendous thanks to W. Schneider and Y.P. Yu for help on shaping the review, and J. Luna, T. Scheel, W. Schneider, and Y.P. Yu for critical reading of the manuscript. We apologize to the many authors whose outstanding work we could not cover due to space limitations.
References
- 1.Schneider W.M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schoggins J.W. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 2011;472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jackson R.J. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ryman K.D. Sindbis virus translation is inhibited by a PKR/RNase L-independent effector induced by alpha/beta interferon priming of dendritic cells. J. Virol. 2005;79:1487–1499. doi: 10.1128/JVI.79.3.1487-1499.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang N. Viral induction of the zinc finger antiviral protein is IRF3-dependent but NF-kappaB-independent. J. Biol. Chem. 2010;285:6080–6090. doi: 10.1074/jbc.M109.054486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kerns J.A. Positive selection and increased antiviral activity associated with the PARP-containing isoform of human zinc-finger antiviral protein. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.0040021. Published online January 25, 2008. http://dx.doi.org/10.1371/journal.pgen.0040021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hayakawa S. ZAPS is a potent stimulator of signaling mediated by the RNA helicase RIG-I during antiviral responses. Nat. Immunol. 2011;12:37–44. doi: 10.1038/ni.1963. [DOI] [PubMed] [Google Scholar]
- 8.Mao R. Inhibition of hepatitis B virus replication by the host zinc finger antiviral protein. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003494. Published online July 11, 2013. http://dx.doi.org/10.1371/journal.ppat.1003494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seo G.J. Reciprocal inhibition between intracellular antiviral signaling and the RNAi machinery in mammalian cells. Cell Host Microbe. 2013;14:435–445. doi: 10.1016/j.chom.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao G. Inhibition of retroviral RNA production by ZAP, a CCCH-type zinc finger protein. Science. 2002;297:1703–1706. doi: 10.1126/science.1074276. [DOI] [PubMed] [Google Scholar]
- 11.Bick M.J. Expression of the zinc-finger antiviral protein inhibits alphavirus replication. J. Virol. 2003;77:11555–11562. doi: 10.1128/JVI.77.21.11555-11562.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Muller S. Inhibition of filovirus replication by the zinc finger antiviral protein. J. Virol. 2007;81:2391–2400. doi: 10.1128/JVI.01601-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang X. Zinc-finger antiviral protein inhibits XMRV infection. PLoS ONE. 2012;7 doi: 10.1371/journal.pone.0039159. Published online June 15, 2012. http://dx.doi.org/10.1371/journal.pone.0039159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhu Y. Zinc-finger antiviral protein inhibits HIV-1 infection by selectively targeting multiply spliced viral mRNAs for degradation. Proc. Natl. Acad. Sci. U.S.A. 2011;108:15834–15839. doi: 10.1073/pnas.1101676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo X. The zinc finger antiviral protein directly binds to specific viral mRNAs through the CCCH zinc finger motifs. J. Virol. 2004;78:12781–12787. doi: 10.1128/JVI.78.23.12781-12787.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen G. p72 DEAD box RNA helicase is required for optimal function of the zinc-finger antiviral protein. Proc. Natl. Acad. Sci. U.S.A. 2008;105:4352–4357. doi: 10.1073/pnas.0712276105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo X. The zinc-finger antiviral protein recruits the RNA processing exosome to degrade the target mRNA. Proc. Natl. Acad. Sci. U.S.A. 2007;104:151–156. doi: 10.1073/pnas.0607063104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Todorova T. PARP13 regulates cellular mRNA post-transcriptionally and functions as a pro-apoptotic factor by destabilizing TRAILR4 transcript. Nat. Commun. 2014;5 doi: 10.1038/ncomms6362. Published online November 10, 2014. http://dx.doi.org/10.1038/ncomms6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu Y. Translational repression precedes and is required for ZAP-mediated mRNA decay. EMBO J. 2012;31:4236–4246. doi: 10.1038/emboj.2012.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atasheva S. New PARP gene with an anti-alphavirus function. J. Virol. 2012;86:8147–8160. doi: 10.1128/JVI.00733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Atasheva S. Interferon-stimulated poly(ADP-Ribose) polymerases are potent inhibitors of cellular translation and virus replication. J. Virol. 2014;88:2116–2130. doi: 10.1128/JVI.03443-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Welsby I. PARP12, an interferon-stimulated gene involved in the control of protein translation and inflammation. J. Biol. Chem. 2014;289:26642–26657. doi: 10.1074/jbc.M114.589515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo J. Induction of the human protein P56 by interferon, double-stranded RNA, or virus infection. Virology. 2000;267:209–219. doi: 10.1006/viro.1999.0135. [DOI] [PubMed] [Google Scholar]
- 24.Terenzi F. Distinct induction patterns and functions of two closely related interferon-inducible human genes, ISG54 and ISG56. J. Biol. Chem. 2006;281:34064–34071. doi: 10.1074/jbc.M605771200. [DOI] [PubMed] [Google Scholar]
- 25.Diamond M.S., Farzan M. The broad-spectrum antiviral functions of IFIT and IFITM proteins. Nat. Rev. Immunol. 2013;13:46–57. doi: 10.1038/nri3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Daffis S. 2′-O methylation of the viral mRNA cap evades host restriction by IFIT family members. Nature. 2010;468:452–456. doi: 10.1038/nature09489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habjan M. Sequestration by IFIT1 impairs translation of 2′O-unmethylated capped RNA. PLoS Pathog. 2013;9 doi: 10.1371/journal.ppat.1003663. Published online October 3, 2013. http://dx.doi.org/10.1371/journal.ppat.1003663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kimura T. Ifit1 inhibits Japanese encephalitis virus replication through binding to 5′ capped 2′-O unmethylated RNA. J. Virol. 2013;87:9997–10003. doi: 10.1128/JVI.00883-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Szretter K.J. 2′-O methylation of the viral mRNA cap by West Nile virus evades ifit1-dependent and -independent mechanisms of host restriction in vivo. PLoS Pathog. 2012;8 doi: 10.1371/journal.ppat.1002698. Published online May 10, 2012. http://dx.doi.org/10.1371/journal.ppat.1002698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zust R. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011;12:137–143. doi: 10.1038/ni.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbas Y.M. Structural basis for viral 5′-PPP-RNA recognition by human IFIT proteins. Nature. 2013;494:60–64. doi: 10.1038/nature11783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pichlmair A. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011;12:624–630. doi: 10.1038/ni.2048. [DOI] [PubMed] [Google Scholar]
- 33.Hyde J.L. A viral RNA structural element alters host recognition of nonself RNA. Science. 2014;343:783–787. doi: 10.1126/science.1248465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo J. A new pathway of translational regulation mediated by eukaryotic initiation factor 3. EMBO J. 2000;19:6891–6899. doi: 10.1093/emboj/19.24.6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hui D.J. Viral stress-inducible protein p56 inhibits translation by blocking the interaction of eIF3 with the ternary complex eIF2.GTP, Met-tRNAi. J. Biol. Chem. 2003;278:39477–39482. doi: 10.1074/jbc.M305038200. [DOI] [PubMed] [Google Scholar]
- 36.Wang C. Alpha interferon induces distinct translational control programs to suppress hepatitis C virus RNA replication. J. Virol. 2003;77:3898–3912. doi: 10.1128/JVI.77.7.3898-3912.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grantham P., Perrin P. AIDS virus and HTLV-I differ in codon choices. Nature. 1986;319:727–728. doi: 10.1038/319727b0. [DOI] [PubMed] [Google Scholar]
- 38.Meintjes P.L., Rodrigo A.G. Evolution of relative synonymous codon usage in human immunodeficiency virus type-1. J. Bioinform. Comput. Biol. 2005;3:157–168. doi: 10.1142/s0219720005000953. [DOI] [PubMed] [Google Scholar]
- 39.van Weringh A. HIV-1 modulates the tRNA pool to improve translation efficiency. Mol. Biol. Evol. 2011;28:1827–1834. doi: 10.1093/molbev/msr005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li M. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature. 2012;491:125–128. doi: 10.1038/nature11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Geserick P. Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int. Immunol. 2004;16:1535–1548. doi: 10.1093/intimm/dxh155. [DOI] [PubMed] [Google Scholar]
- 42.Schwarz D.A. Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity. 1998;9:657–668. doi: 10.1016/s1074-7613(00)80663-9. [DOI] [PubMed] [Google Scholar]
- 43.Sohn W.J. Novel transcriptional regulation of the schlafen-2 gene in macrophages in response to TLR-triggered stimulation. Mol. Immunol. 2007;44:3273–3282. doi: 10.1016/j.molimm.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 44.Morales D.J., Lenschow D.J. The antiviral activities of ISG15. J. Mol. Biol. 2013;425:4995–5008. doi: 10.1016/j.jmb.2013.09.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gradi A. Proteolysis of human eukaryotic translation initiation factor eIF4GII, but not eIF4GI, coincides with the shutoff of host protein synthesis after poliovirus infection. Proc. Natl. Acad. Sci. U.S.A. 1998;95:11089–11094. doi: 10.1073/pnas.95.19.11089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rahnefeld A. Ubiquitin-like protein ISG15 in host defense against heart failure in a mouse model of virus-induced cardiomyopathy. Circulation. 2014;130:1589–1600. doi: 10.1161/CIRCULATIONAHA.114.009847. http://dx.doi/10.1161/CIRCULATIONAHA.114.009847. Epub 2014 Aug 27. [DOI] [PubMed] [Google Scholar]
- 47.Anderson P., Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem. Sci. 2008;33:141–150. doi: 10.1016/j.tibs.2007.12.003. [DOI] [PubMed] [Google Scholar]
- 48.Buchan J.R., Parker R. Eukaryotic stress granules: the ins and outs of translation. Mol. Cell. 2009;36:932–941. doi: 10.1016/j.molcel.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Onomoto K. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014;35:420–428. doi: 10.1016/j.it.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu J. MicroRNA-dependent localization of targeted mRNAs to mammalian P-bodies. Nat. Cell Biol. 2005;7:719–723. doi: 10.1038/ncb1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pillai R.S. Inhibition of translational initiation by Let-7 MicroRNA in human cells. Science. 2005;309:1573–1576. doi: 10.1126/science.1115079. [DOI] [PubMed] [Google Scholar]
- 52.Sen G.L., Blau H.M. Argonaute 2/RISC resides in sites of mammalian mRNA decay known as cytoplasmic bodies. Nat. Cell Biol. 2005;7:633–636. doi: 10.1038/ncb1265. [DOI] [PubMed] [Google Scholar]
- 53.Yang J.S., Lai E.C. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol. Cell. 2011;43:892–903. doi: 10.1016/j.molcel.2011.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Leung A.K. Poly(ADP-ribose) regulates stress responses and microRNA activity in the cytoplasm. Mol. Cell. 2011;42:489–499. doi: 10.1016/j.molcel.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kleine H. Substrate-assisted catalysis by PARP10 limits its activity to mono-ADP-ribosylation. Mol. Cell. 2008;32:57–69. doi: 10.1016/j.molcel.2008.08.009. [DOI] [PubMed] [Google Scholar]
- 56.Fairman-Williams M.E. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010;20:313–324. doi: 10.1016/j.sbi.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arjan-Odedra S. Endogenous MOV10 inhibits the retrotransposition of endogenous retroelements but not the replication of exogenous retroviruses. Retrovirology. 2012;9:53. doi: 10.1186/1742-4690-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Burdick R. P body-associated protein Mov10 inhibits HIV-1 replication at multiple stages. J. Virol. 2010;84:10241–10253. doi: 10.1128/JVI.00585-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haussecker D. Capped small RNAs and MOV10 in human hepatitis delta virus replication. Nat. Struct. Mol. Biol. 2008;15:714–721. doi: 10.1038/nsmb.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li X. The MOV10 helicase inhibits LINE-1 mobility. J. Biol. Chem. 2013;288:21148–21160. doi: 10.1074/jbc.M113.465856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu C. Moloney leukemia virus type 10 inhibits reverse transcription and retrotransposition of intracisternal a particles. J. Virol. 2012;86:10517–10523. doi: 10.1128/JVI.00868-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang X. Moloney leukemia virus 10 (MOV10) protein inhibits retrovirus replication. J. Biol. Chem. 2010;285:14346–14355. doi: 10.1074/jbc.M110.109314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Meister G. Identification of novel argonaute-associated proteins. Curr. Biol. 2005;15:2149–2155. doi: 10.1016/j.cub.2005.10.048. [DOI] [PubMed] [Google Scholar]
- 64.Banerjee S. A coordinated local translational control point at the synapse involving relief from silencing and MOV10 degradation. Neuron. 2009;64:871–884. doi: 10.1016/j.neuron.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 65.Chendrimada T.P. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–744. doi: 10.1038/nature03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gregory R.I. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–640. doi: 10.1016/j.cell.2005.10.022. [DOI] [PubMed] [Google Scholar]
- 67.Chendrimada T.P. MicroRNA silencing through RISC recruitment of eIF6. Nature. 2007;447:823–828. doi: 10.1038/nature05841. [DOI] [PubMed] [Google Scholar]
- 68.Liu C. APOBEC3G inhibits microRNA-mediated repression of translation by interfering with the interaction between Argonaute-2 and MOV10. J. Biol. Chem. 2012;287:29373–29383. doi: 10.1074/jbc.M112.354001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wichroski M.J. Human retroviral host restriction factors APOBEC3G and APOBEC3F localize to mRNA processing bodies. PLoS Pathog. 2006;2 doi: 10.1371/journal.ppat.0020041. Published online May 12, 2006. http://dx.doi.org/10.1371/journal.ppat.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.George C.X. Adenosine deaminases acting on RNA, RNA editing, and interferon action. J. Interferon Cytokine Res. 2011;31:99–117. doi: 10.1089/jir.2010.0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Athanasiadis A. Widespread A-to-I RNA editing of Alu-containing mRNAs in the human transcriptome. PLoS Biol. 2004;2:e391. doi: 10.1371/journal.pbio.0020391. http://dx.doi.org/10.1371/journal.pbio.0020391. Epub 2004 Nov 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kim D.D. Widespread RNA editing of embedded alu elements in the human transcriptome. Genome Res. 2004;14:1719–1725. doi: 10.1101/gr.2855504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Levanon E.Y. Systematic identification of abundant A-to-I editing sites in the human transcriptome. Nat. Biotechnol. 2004;22:1001–1005. doi: 10.1038/nbt996. [DOI] [PubMed] [Google Scholar]
- 74.Ramaswami G. Accurate identification of human Alu and non-Alu RNA editing sites. Nat. Methods. 2012;9:579–581. doi: 10.1038/nmeth.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sakurai M. Inosine cyanoethylation identifies A-to-I RNA editing sites in the human transcriptome. Nat. Chem. Biol. 2010;6:733–740. doi: 10.1038/nchembio.434. [DOI] [PubMed] [Google Scholar]
- 76.Wulff B.E. Elucidating the inosinome: global approaches to adenosine-to-inosine RNA editing. Nat. Rev. Genet. 2011;12:81–85. doi: 10.1038/nrg2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baczko K. Clonal expansion of hypermutated measles virus in a SSPE brain. Virology. 1993;197:188–195. doi: 10.1006/viro.1993.1579. [DOI] [PubMed] [Google Scholar]
- 78.Carpenter J.A. Evidence for ADAR-induced hypermutation of the Drosophila sigma virus (Rhabdoviridae) BMC Genet. 2009;10 doi: 10.1186/1471-2156-10-75. Published online November 26, 2009. http://dx.doi.org/10.1186/1471-2156-10-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cattaneo R. Biased hypermutation and other genetic changes in defective measles viruses in human brain infections. Cell. 1988;55:255–265. doi: 10.1016/0092-8674(88)90048-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ko N.L. Hyperediting of human T-cell leukemia virus type 2 and simian T-cell leukemia virus type 3 by the dsRNA adenosine deaminase ADAR-1. J. Gen. Virol. 2012;93:2646–2651. doi: 10.1099/vir.0.045146-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumar M., Carmichael G.G. Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3542–3547. doi: 10.1073/pnas.94.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Y. Circulating type 1 vaccine-derived poliovirus may evolve under the pressure of adenosine deaminases acting on RNA. J. Matern. Fetal Neonatal Med. 2014 doi: 10.3109/14767058.2014.979147. Published online November 14, 2014. http://dx.doi.org/10.3109/14767058.2014.979147. [DOI] [PubMed] [Google Scholar]
- 83.Luo G.X. A specific base transition occurs on replicating hepatitis delta virus RNA. J. Virol. 1990;64:1021–1027. doi: 10.1128/jvi.64.3.1021-1027.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rima B.K. Stability of the parainfluenza virus 5 genome revealed by deep sequencing of strains isolated from different hosts and following passage in cell culture. J. Virol. 2014;88:3826–3836. doi: 10.1128/JVI.03351-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Suspene R. Double-stranded RNA adenosine deaminase ADAR-1-induced hypermutated genomes among inactivated seasonal influenza and live attenuated measles virus vaccines. J. Virol. 2011;85:2458–2462. doi: 10.1128/JVI.02138-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taylor D.R. New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1. J. Virol. 2005;79:6291–6298. doi: 10.1128/JVI.79.10.6291-6298.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tenoever B.R. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science. 2007;315:1274–1278. doi: 10.1126/science.1136567. [DOI] [PubMed] [Google Scholar]
- 88.Zahn R.C. A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus. J. Virol. 2007;81:457–464. doi: 10.1128/JVI.00067-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Desterro J.M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003;116:1805–1818. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- 90.Eckmann C.R. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol. Biol. Cell. 2001;12:1911–1924. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Strehblow A. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol. Biol. Cell. 2002;13:3822–3835. doi: 10.1091/mbc.E02-03-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fritz J. RNA-regulated interaction of transportin-1 and exportin-5 with the double-stranded RNA-binding domain regulates nucleocytoplasmic shuttling of ADAR1. Mol. Cell Biol. 2009;29:1487–1497. doi: 10.1128/MCB.01519-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ota H. ADAR1 forms a complex with dicer to promote microRNA processing and RNA-induced gene silencing. Cell. 2013;153:575–589. doi: 10.1016/j.cell.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ng S.K. Proteins that contain a functional Z-DNA-binding domain localize to cytoplasmic stress granules. Nucleic Acids Res. 2013;41:9786–9799. doi: 10.1093/nar/gkt750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Weissbach R., Scadden A.D. Tudor-SN and ADAR1 are components of cytoplasmic stress granules. RNA. 2012;18:462–471. doi: 10.1261/rna.027656.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kawahara Y. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yang W. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat. Struct. Mol. Biol. 2006;13:13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nachmani D. MicroRNA editing facilitates immune elimination of HCMV infected cells. PLoS Pathog. 2014;10 doi: 10.1371/journal.ppat.1003963. Published online February 27, 2014. http://dx.doi.org/10.1371/journal.ppat.1003963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Joklik W.K., Merigan T.C. Concerning the mechanism of action of interferon. Proc. Natl. Acad. Sci. U.S.A. 1966;56:558–565. doi: 10.1073/pnas.56.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Metz D.H., Esteban M. Interferon inhibits viral protein synthesis in L cells infected with vaccinia virus. Nature. 1972;238:385–388. doi: 10.1038/238385a0. [DOI] [PubMed] [Google Scholar]
- 101.Content J. Blocks in elongation and initiation of protein synthesis induced by interferon treatment in mouse L cells. Eur. J. Biochem. 1975;54:1–10. doi: 10.1111/j.1432-1033.1975.tb04106.x. [DOI] [PubMed] [Google Scholar]
- 102.Kerr I.M. Increased sensitivity of cell-free protein synthesis to double-stranded RNA after interferon treatment. Nature. 1974;250:57–59. doi: 10.1038/250057a0. [DOI] [PubMed] [Google Scholar]
- 103.Ball L.A., White C.N. Oligonucleotide inhibitor of protein synthesis made in extracts of interferon-treated chick embryo cells: comparison with the mouse low molecular weight inhibitor. Proc. Natl. Acad. Sci. U.S.A. 1978;75:1167–1171. doi: 10.1073/pnas.75.3.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Cooper J.A., Farrell P.J. Extracts of interferon-treated cells can inhibit reticulocyte lysate protein synthesis. Biochem. Biophys. Res. Commun. 1977;77:124–131. doi: 10.1016/s0006-291x(77)80173-3. [DOI] [PubMed] [Google Scholar]
- 105.Hovanessian A.G. Synthesis of low molecular weight inhibitor of protein synthesis with enzyme from interferon-treated cells. Nature. 1977;268:537–540. doi: 10.1038/268537a0. [DOI] [PubMed] [Google Scholar]
- 106.Kerr I.M., Brown R.E. pppA2′p5′A2′p5′A: an inhibitor of protein synthesis synthesized with an enzyme fraction from interferon-treated cells. Proc. Natl. Acad. Sci. U.S.A. 1978;75:256–260. doi: 10.1073/pnas.75.1.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Kerr I.M. Nature of inhibitor of cell-free protein synthesis formed in response to interferon and double-stranded RNA. Nature. 1977;268:540–542. doi: 10.1038/268540a0. [DOI] [PubMed] [Google Scholar]
- 108.Lebleu B. Interferon, double-stranded RNA, and protein phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 1976;73:3107–3111. doi: 10.1073/pnas.73.9.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Roberts W.K. Interferon-induced inhibition of protein synthesis in L-cell extracts: an ATP-dependent step in the activation of an inhibitor by double-stranded RNA. Proc. Natl. Acad. Sci. U.S.A. 1976;73:3136–3140. doi: 10.1073/pnas.73.9.3136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Roberts W.K. Interferon-mediated protein kinase and low-molecular-weight inhibitor of protein synthesis. Nature. 1976;264:477–480. doi: 10.1038/264477a0. [DOI] [PubMed] [Google Scholar]
- 111.Zilberstein A. Specific phosphorylation in vitro of a protein associated with ribosomes of interferon-treated mouse L cells. FEBS Lett. 1976;68:119–124. doi: 10.1016/0014-5793(76)80418-8. [DOI] [PubMed] [Google Scholar]
- 112.Farrell P.J. Phosphorylation of initiation factor elF-2 and the control of reticulocyte protein synthesis. Cell. 1977;11:187–200. doi: 10.1016/0092-8674(77)90330-0. [DOI] [PubMed] [Google Scholar]
- 113.Hovanessian A.G., Kerr I.M. The (2′-5′) oligoadenylate (pppA2′-5′A2′-5′A) synthetase and protein kinase(s) from interferon-treated cells. Eur. J. Biochem. 1979;93:515–526. doi: 10.1111/j.1432-1033.1979.tb12850.x. [DOI] [PubMed] [Google Scholar]
- 114.Kramer G. Specificity of the protein kinase activity associated with the hemin-controlled repressor of rabbit reticulocyte. Proc. Natl. Acad. Sci. U.S.A. 1976;73:3078–3082. doi: 10.1073/pnas.73.9.3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Levin D., London I.M. Regulation of protein synthesis: activation by double-stranded RNA of a protein kinase that phosphorylates eukaryotic initiation factor 2. Proc. Natl. Acad. Sci. U.S.A. 1978;75:1121–1125. doi: 10.1073/pnas.75.3.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Levin D. Regulation of protein synthesis in reticulocyte lysates: phosphorylation of methionyl-tRNAf binding factor by protein kinase activity of translational inhibitor isolated from hemedeficient lysates. Proc. Natl. Acad. Sci. U.S.A. 1976;73:3112–3116. doi: 10.1073/pnas.73.9.3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Samuel C.E. Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc. Natl. Acad. Sci. U.S.A. 1979;76:600–604. doi: 10.1073/pnas.76.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Williams B.R. Signal integration via PKR. Sci STKE. 2001 doi: 10.1126/stke.2001.89.re2. Published online July 3, 2001. http://dx.doi.org/10.1126/stke.2001.89.re2. [DOI] [PubMed] [Google Scholar]
- 119.Ratner L. Interferon, double-stranded RNA and RNA degradation. Fractionation of the endonucleaseINT system into two macromolecular components; role of a small molecule in nuclease activation. Biochem. Biophys. Res. Commun. 1978;81:947–954. doi: 10.1016/0006-291x(78)91443-2. [DOI] [PubMed] [Google Scholar]
- 120.Baglioni C. Interferon action may be mediated by activation of a nuclease by pppA2′p5′A2′p5′A. Nature. 1978;273:684–687. doi: 10.1038/273684a0. [DOI] [PubMed] [Google Scholar]
- 121.Clemens M.J., Williams B.R. Inhibition of cell-free protein synthesis by pppA2′p5′A2′p5′A: a novel oligonucleotide synthesized by interferon-treated L cell extracts. Cell. 1978;13:565–572. doi: 10.1016/0092-8674(78)90329-x. [DOI] [PubMed] [Google Scholar]
- 122.Silverman R.H. Control of the ppp(a2′p)nA system in HeLa cells. Effects of interferon and virus infection. Eur. J. Biochem. 1982;124:131–138. doi: 10.1111/j.1432-1033.1982.tb05915.x. [DOI] [PubMed] [Google Scholar]
- 123.Poirier G.G. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc. Natl. Acad. Sci. U.S.A. 1982;79:3423–3427. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rosado M.M. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013;139:428–437. doi: 10.1111/imm.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schreiber V. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J. Biol. Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 126.Virag L. Poly(ADP-ribose) signaling in cell death. Mol. Aspects Med. 2013;34:1153–1167. doi: 10.1016/j.mam.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 127.Daugherty M.D. Rapid evolution of PARP genes suggests a broad role for ADP-ribosylation in host-virus conflicts. PLoS Genet. 2014;10 doi: 10.1371/journal.pgen.1004403. Published online May 29, 2014. http://dx.doi.org/10.1371/journal.pgen.1004403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dunstan M.S. Structure and mechanism of a canonical poly(ADP-ribose) glycohydrolase. Nat. Commun. 2012;3 doi: 10.1038/ncomms1889. Published online June 6, 2012. http://dx.doi.org/10.1038/ncomms1889. [DOI] [PubMed] [Google Scholar]
- 129.Slade D. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Jankevicius G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013;20:508–514. doi: 10.1038/nsmb.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Rosenthal F. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013;20:502–507. doi: 10.1038/nsmb.2521. [DOI] [PubMed] [Google Scholar]