

Abstract
Mitochondria are increasingly recognized as drivers of inflammatory responses. MAP kinase kinase 3 (MKK3), a dual-specificity protein kinase, is activated in inflammation and in turn activates p38 MAP kinase signaling. Here we show that MKK3 influences mitochondrial function and acts as a critical mediator of inflammation. MKK3 deficient (MKK3−/−) mice and bone marrow derived macrophages (BMDMs) secreted less cytokines than wild type (WT) after LPS exposure. There was improved mitochondrial function, as measured by basal oxygen consumption rate, mitochondrial membrane potential, and ATP production, in MKK3−/− BMDMs. After LPS exposure, MKK3−/− BMDM did not show significant increase in cellular reactive oxygen species (ROS) production as well as mitochondrial superoxide (MitoSOX) compared to WT. Activation of two important inflammatory mediators i.e. the nuclear translocation of NF-κB and caspase-1 activity (a key component of the inflammasome), were lower in MKK3−/− BMDMs. p-38 and JNK activation were lower in MKK3−/− BMDMs compared to WT after exposure to LPS. Knockdown of MKK3 by siRNA in wild type BMDMs improved mitochondrial membrane potential, reduced LPS induced caspase-1 activation and attenuated cytokine secretion. Our studies establish MKK3 as a regulator of mitochondrial function and inflammatory responses to LPS and suggest that MKK3 may be a therapeutic target in inflammatory disorders like sepsis.
Keywords: Mitochondria, Sepsis, Acute lung injury, Oxidants, NF-kB, Inflammasome, Caspase1, Inflammation
Introduction
Mitochondria are considered central hubs of inflammation [1] and new paradigm shifts show that mitochondria are capable of inducing multiple inflammatory pathways, reflecting their origins as primitive archaebacteria [2]. Mitochondrial reactive oxygen species (ROS) have been implicated in the activation of major inflammatory transcription factors such as NF-kB (Nuclear Factor kappa B) and AP-1 (Activator Protein -1) [3]. In addition, mitochondrial ROS, membrane potential, and the release of oxidized mitochondrial DNA activate another important inflammatory pathway, the inflammasome and caspase-1 [4]. Mitochondrial metabolism and innate immunity are intimately linked. Metabolic switching from OXPHOS (oxidative phosphorylation) to glycolytic for ATP generation likely determines the magnitude of an inflammatory response. For example, OXPHOS deficiency is associated with increased inflammatory responses in adipocytes [5]. In macrophages and dendritic cells, stimulation of the toll like receptor 4 (TLR4) by LPS inhibits mitochondrial activity and induces a change in the metabolism from oxidative phosphorylation to glycolysis, which is more inflammatory in nature [6, 7]. Thus it is speculated that maintenance of adequate OXPHOS metabolism will enable reduced inflammatory response to stimulants such as LPS, however the factors affecting mitochondrial function are not well known. Here we investigate the role of MKK3 in regulation of inflammation through the modulation of mitochondrial function.
Mitogen activated protein kinases (MAP kinases) are integral to intracellular signaling pathways mediating inflammation, cell survival/death, proliferation and differentiation in response to a wide variety of signals such as cytokines, growth factors, UV light, osmotic stress and lipopolysaccharide (LPS) [8]. MKK3 (MAP Kinase Kinase 3) is a major upstream kinase of the p38 MAP kinase and phosphorylates p38 in response to LPS. MKK3 and p38 MAP kinase are ancient and evolutionarily conserved components of metazoan defense against pathogen attack which predated the canonical Toll-like-receptors [9]. We have previously described the role of MKK3 in sepsis, a syndrome of excessive and inappropriate inflammatory response to infection. We also found that MKK3 deficiency leads to less lung inflammation and better survival in various models of sepsis such as cecal ligation and puncture (CLP) and LPS exposure [10]. Additionally, we found that MKK3 deficient endothelial cells have a larger pool of healthy mitochondria as shown by higher mitophagy, clearance of dysfunctional mitochondria, and increased mitochondrial biogenesis [11]. It has been reported that mitochondrial ROS regulates MKK3 and p38 but MKK3 regulation of mitochondrial function has not been reported, to the best of our knowledge [12]. Thus, we hypothesized that MKK3 deficiency will have specific effects on mitochondria and this in turn potently modulates inflammation.
We sought to examine the effect of MKK3 deletion on mitochondrial function and cytokine secretion after LPS exposure in bone marrow derived macrophages (BMDMs), which provide a good cell model to study inflammatory responses and mitochondrial function [13]. We show that MKK3 deletion leads to better mitochondrial function, lower production of ROS, and a reduction in NF-κB activation and caspase-1 activity, and lower cytokine release after LPS stimulation. Thus, we conclude that mitochondrial dysfunction contributes to LPS-induced inflammatory responses in BMDMs and that MKK3 plays an important role.
Materials and Methods
Animal Experiments
The MKK3−/− mice were originally provided by R. Davis (University of Massachusetts Medical School, Worcester, MA) and R. Flavell (Yale University, New Haven, CT) and have been backcrossed onto a C57BL/6 background for >15 generations. We have previously described how the MKK3−/− mice were generated [10]. MKK3−/− mice expressed normal levels of MKK6, MKK4, JNK, and p38 MAP kinases [14]. The mice were divided into 4 groups of 3 each, i.e. wild type control (C57BL/6 background), wild type LPS treated, MKK3−/− control, and MKK3−/− LPS treated. LPS (Escherichia coli 055:B5; Sigma-Aldrich) was given at single acute dose 40 mg/kg i.p. (LD100). Blood was collected after 3h of LPS treatment for serum cytokine analysis. All experiments were conducted in accordance with the NIH guidelines and approved by the institutional Animal Care and Use Committee.
Bone marrow derived macrophage (BMDM) isolation and culture
BMDMs were isolated and differentiated using standard protocols. Primary macrophages were derived from bone marrow cells and were cultured for 7 days in RPMI1640 media containing 30% L929 cell conditioned medium, 10% fetal bovine serum and Penicillin/Streptomycin (Pen/Strep). L929 cell conditioned medium was prepared by growing L929 cells in RPMI1640 containing 10% fetal bovine serum and Pen/Strep for 10 days. BMDMs were harvested by treating with ice cold DPBS containing 5μM EDTA and plated as per experimental requirement.
Mitochondrial Membrane Potential (MMP)
BMDMs were seeded at a density 0.5×105 cells per well in a 96-well plate and exposed to LPS (0.1μg/ml) after 16h. Estimation of mitochondrial membrane potential (Δψm) was performed using JC-10 (Enzo Life Sciences), a membrane permeable fluorescent probe. JC-10 enters selectively into mitochondria and exists as two forms, monomeric or aggregate, depending upon Δψm. The JC-10 monomer form predominates in mitochondria with low Δψm and emits in the green wavelength (525–530 nm). The JC-10 aggregate form accumulates in mitochondria with high Δψm and emits in the orange wavelength (590 nm). The JC-10 aggregate/monomer ratio is proportional to MMP. The final concentration of JC-10 used was 5 μM in the media and incubated for 30 min before reading the fluorescence in a SpectraMax Gemini XS spectroflurometer (Molecular Devices).
ATP measurement
Total ATP was determined using the ATP Fluorometric assay kit (BioVision). BMDMs, 0.5×106 cells per well in six well cell culture plate, after 24h LPS treatment were lysed in 100 μL of ATP assay buffer and centrifuged. The supernatant (50 μL) was mixed with ATP Probe, ATP Converter, and Developer Mix and incubated for 30min at room temperature in dark. The fluorescence was read at Ex 535/Em 587nm using a spectroflurometer (Vmax, Molecular Devices). The values were calculated against the provided standard.
Measurement of mitochondrial bioenergetics
An XF96 Analyzer (Seahorse Biosciences, North Billerica MA) was used to measure bioenergetic function in intact BMDMs in real time. BMDMs were seeded into Seahorse Bioscience XF96 cell culture plates at the seeding density of 10,000 cells in 80μl media and allowed to adhere and grow for 24h in a 37°C humidified incubator with 5% CO2. Measurements of extracellular flux were made in unbuffered media. Extracellular acidification rate (ECAR) was calculated at baseline to compare the rate of glycolysis. Mitochondrial function was analysed by sequentially adding pharmacological inhibitors of oxidative phosphorylation. The resultant bioenergetic profile provides detailed information on individual components of mitochondrial bioenergetic components.
Measurement of cellular ROS and mitochondrial superoxide in BMDMs
The cellular level of ROS and mitochondrial superoxide were determined using CM-H2DCFDA and MitoSOX, respectively. BMDMs were cultured in 96-well plate at a density of 10,000 cells per well. After 16h the media was changed and CM-H2DCFDA (final concentration 1μM) and MitoSOX (final concentration 2μM) were added along with nuclear stain Hoechst (5μg/ml). After 60 min the cells were washed twice and suspended in DPBS and fluorescence was read at Ex 352/Em 461nm for Hoechst, Ex 492/Em 527nm for CM-H2DCFDA, and Ex 510/Em 580nm for MitoSOX. The cellular ROS and mitochondrial superoxide data were normalized to the nuclear stain and quantified.
Caspase-1 activity
BMDMs were seeded at 0.5×106 cells per well in 6-well cell culture plate. After 24h the media was changed and LPS (0.1μg/ml) treatment was done for 6h with the addition of ATP (5μM) for last 15 min. Cells were lysed with cell lysis buffer provided in the Caspase-1 Fluorometric Assay Kit (ab39412, Abcam). Caspase-1 activity was determined using 100μg cell lysate protein with the manufacturer’s protocol. The assay is based on spectrophotometric detection of the chromophore p-nitroanilide (p-NA) after cleavage from the labeled substrate YVAD-p-NA which absorbs at 405nm. Standard curve of pNA was prepared and data expressed as pNA released (μM)/mg protein.
NF-κB nuclear translocation immunostaining and image analysis
BMDMs were cultured on a coverslip in 6-well cell culture plate at about 70% confluency. Cells were treated with LPS (0.1μg/ml) for 45min and cells were washed with DPBS twice and fixed in 4% paraformaldehyde for 15 min. After fixation cells were washed with DPBS and permeabilised with 0.3% triton-X100. After blocking with DAKO protein-free blocking buffer for 1h, p-NF-κB (p65) antibody (Cat# 3031 Cell Signaling Tech., Inc.) was added to the blocking buffer and incubated overnight. After two DPBS washes, anti-rabbit AF-488 (Cat# A11008, Invitrogen) secondary antibody was incubated for 1h and washed with DPBS and mounted with mounting media containing DAPI. Images were taken by confocal microscopy and quantified using CellProfiler software, Broad Institute, MA. The amount of p-NF-κB staining within the nucleus was quantified and plotted.
Cytokine Analysis
Blood was collected by cardiac puncture in BD Microtainer serum separator tubes without additive. The blood samples were allowed to clot at room temperature for 30 minutes before centrifugation (2200g, 4°C, 10 min) and the serum was collected and stored at −80°C until analyzed. BMDM media (0.5×106 cells per well in 6 well cell culture plate) was collected after 24h LPS (0.1μg/ml) treatment and analyzed for cytokine levels using luminex fluorescent bead based technology (Bio-Rad). The cytokine standards were run along with samples to calculate the amounts in pg/ml.
MKK3 siRNA
For depletion of MKK3 in BMDMs, MEK-3 siRNA (m) (Santa Cruz, Cat# 35908) was used. Nonspecific siRNA scrambled duplex probes (sense, 5′-GCGCGCUUUGUAGGAUUCG-3′; antisense, 5′-CGAAUCCUACAAAGCGCGC-3′) were synthesized by Dharmacon Research Inc. as previously described. BMDMs (70% confluence) were transfected with the siRNAs (50nM) using the Mirus TransIT-TKO® reagent (Mirus, Madison, WI) according to the manufacturer’s manual for 48h. The experiments were performed after fresh media change.
Western Blotting
0.5×106 BMDMs per well in six well cell culture plate were treated with LPS (0.1μg/ml) for 60 min and washed twice with DPBS before making the lysate. Total cellular lysate was prepared using RIPA buffer (50 mM Tris–HCl, pH 7.4/150 mM NaCl/1% Triton X-100/1% deoxycholate/0.1% SDS/1% aprotinin) with protease and phosphatase inhibitors (Roche). 20μg protein was separated on precast mini-PROTEAN TGX gels, 4–20% (BioRad), and transferred to PVDF membrane (Millipore). The membrane was incubated for 1h at room temperature with 5% non-fat milk in tris buffered saline with 0.05% tween-20 (TBS-T) to block, and then incubated with primary antibodies. Secondary antibodies tagged with HRP were used as appropriate for primary antibody host. The blots were developed by chemiluminescent substrates (ECL, Amersham). The primary antibodies used were p38 (9212), p-p38 (9211), JNK (9252), p-JNK (9251), ERK (4372), p-ERK (4376), caspase-1 (2225), cleaved caspase-1 (4199) and p-NF-κB (3031) procured from Cell Signaling Technologies, Inc., Danvers, MA and HRP tagged β-actin antibody (sc-47778) was from Santa Cruz Biotech.
Statistical analysis
All the data are presented as the means ± S.E.M. The statistical analysis was performed using t-test analysis and p<0.05 was considered significant.
Results
Serum from MKK3−/− mice show attenuated inflammatory response against LPS injury
We compared the inflammatory response of MKK3−/− mice with wild type mice. The mice were treated with LPS and blood was collected after 3h to analyze serum cytokine levels. MKK3−/− mice had significantly lower serum levels of IL-1α, IL-1β, IL-5, IL-12 (p70), IL-17, Eotaxin, GM-CSF, IFN-γ, MIP-1α and MIP-1β compared to wild type mice. The other measured cytokines, IL-3, IL-4, IL-10, IL-12 (p40), and IL-13, were not significantly different but showed a similar trend. There were no differences in the baseline levels of any of the cytokines between the wild type and MKK3−/− mice (Figure 1).
Figure 1. Attenuated inflammatory responses in MKK3−/− mice against LPS induced injury.

After 3h of LPS (40mg/kg) intra peritoneal injection in mice, serum was collected to analyze levels of cytokines. MKK3−/− mice showed attenuated inflammatory response compared to wild type mice after LPS injury. The baseline cytokine levels in MKK3−/− mice were not different than WT mice. * represents significance between control and LPS treated within same genotype and # represents significance between the genotypes with control and LPS treatment groups, * or # represents significance at p<0.05 and ** or ## represents significance at p<0.01. Measurements were done from serum of 3 mice per group.
MKK3−/− BMDMs display reduced inflammatory response after LPS stimulation
To further study the inflammatory response at a cellular level we used BMDMs from MKK3−/− mice. BMDMs were exposed to LPS for 24h and media was collected to measure the cytokine levels. In both MKK3−/− and wild type BMDMs LPS stimulation lead to increased secretion of cytokines in the media. However, the BMDMs from MKK3−/− mice showed attenuated inflammatory response against LPS exposure which was more prominent than that found in the in vivo study. The levels of IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-6, IL-12 (p40), IL-12 (p70), IL-13, Eotaxin, G-CSF, GM-CSF, IFNγ, KC, MCP-1, RANTES, MIP-1β, and TNFα were significantly lower in MKK3−/− BMDM media compared to WT at baseline as well as after stimulation with LPS. The IL-9 and IL-17 levels were not different at baseline but after LPS stimulation MKK3−/− BMDMs had significantly lower levels compared to WT. The IL-10, an anti-inflammatory cytokine, level was significantly higher in MKK3−/− BMDMs at baseline and also after LPS stimulation compared to WT (Figure 2). The results indicate that MKK3 regulates cytokine response to LPS stimulus.
Figure 2. Reduced inflammatory responses in MKK3−/− BMDMs against LPS stimulation.

BMDM cell media was collected after 24h LPS (0.1μg/ml) treatment and cytokine levels were measured. MKK3−/− BMDMs showed lesser cytokine release after LPS treatment and also at baseline. * represents significance between control and LPS treated within same genotype and # represents significance between the genotypes with control and LPS treatment groups, * or # represents significance at p<0.05 and ** or ## represents significance at p<0.01. Data is representative of 3 independent experiments done in triplicates.
MKK3−/− BMDMs show higher basal respiration and ATP production
To understand the role of mitochondrial function in inflammatory response we measured the oxygen consumption rate (OCR) in MKK3−/− and WT BMDMs by Seahorse Biosciences Extracellular Flux Analyzer. We observed higher OCR in MKK3−/− BMDMs compared to WT (Figure 3A). We analyzed the basal respiration rate and ATP production, both of which were significantly higher in MKK3−/− than the WT (Figure 3C and D). The data shows higher oxygen demand in MKK3−/− with concomitantly higher production of ATP. The extracellular acidification rate (ECAR), a measure of glycolysis, was also higher in MKK3−/− BMDMs, which correlates with higher mitochondrial respiration (Figure 3B). These results indicate a higher metabolic rate in MKK3−/− BMDMs. After LPS stimulation we did not observe any changes in mitochondrial function compared to the baseline, in both WT and MKK3−/− BMDMs. There was a drop in the mitochondrial membrane potential (MMP) of WT BMDMs after LPS treatment but not in MKK3−/− BMDMs (Figure 3E). ATP content was higher in MKK3−/− BMDMs compared to WT and there was no change after LPS treatment (Figure 3F).
Figure 3. Improved mitochondrial bioenergetics in MKK3−/− BMDMs.

Mitochondrial bioenergetic profile of BMDMs was studied using the “Mito stress kit”, Seahorse Biosciences. MKK3−/− BMDMs showed higher Oxygen consumption rate (OCR) compared to wild type (WT) (A). There was corresponding increase in extracellular acidification rate (ECAR) in MKK3−/− BMDMs (B). Basal respiration and ATP production was calculated based on the OCR profile and we found that they were higher in MKK3−/− BMDMs (C and D). The MMP and ATP were measured in MKK3−/− and WT BMDMs after LPS treatment. MMP as well as ATP content was higher in MKK3−/− BMDMs compared to WT at baseline and after LPS treatment MMP dropped in WT but not in MKK3−/− BMDMs (E and F). ** represents significance at p<0.01. Data is representative of 3 experiments done in at least 4 replicates.
Lower cellular ROS and mitochondrial superoxide in MKK3−/− BMDMs
To compare the difference in oxidant levels in MKK3−/− and WT BMDMs we measured ROS after LPS exposure. WT BMDMs showed significant increase in ROS and MitoSOX levels but not the MKK3−/−. Even at baseline, MKK3−/− BMDMs had lower levels of total cellular ROS and MitoSOX (Figure 4).
Figure 4. Lower levels of reactive oxygen species (ROS) and mitochondrial superoxide in MKK3−/− BMDMs after LPS treatment.

BMDMs were treated with LPS (0.1 μg/ml) for 60 min and stained with CM-H2DCFDA and MitoSOX. Cells were fixed and imaged with confocal microscopy and quantified using CellProfiler software. MKK3−/− BMDMs have lower levels of both cellular ROS as well as mitochondrial superoxide after LPS exposure. * and ** represents significance at p<0.05 and 0.01, respectively. Data is representative of atleast 20 images.
MKK3−/− BMDMs show lower NF-κB nuclear translocation after LPS treatment
As nuclear translocation of NF-κB is a critical event in the activation of inflammatory gene response to inflammatory stimuli we studied it in our model system. BMDMs were treated with LPS and probed by immunocytochemistry for p-NF-κB along with nuclear staining. The slides were imaged by confocal microscopy and quantified by CellProfiler software. WT BMDMs after LPS treatment showed a significant increase in nuclear p-NF-κB but there was not a significant increase in MKK3−/− BMDMs (Figure 5). This suggests that the attenuated inflammatory response in MKK3−/− BMDMs after LPS stimulation is at least partly due to lower activation/nuclear translocation of p-NF-κB.
Figure 5. Lower NF-κB nuclear translocation in MKK3−/− BMDMs after LPS stimulation.

BMDMs were treated with LPS (0.1 μg/ml) for 45 min and immuno-stained with p-NF-κB antibody. Slides where imaged with confocal microscopy and quantified using CellProfiler software. There was an increase in nuclear p-NF-κB in WT but to a lesser extent in MKK3−/− BMDMs compared to respective controls. * represents significance at p<0.05. Data is representative of 3 experiments done in at least duplicates.
Lower caspase-1 activity in MKK3−/− BMDMs after LPS injury
Caspase-1 is a key component of the inflammasome, which drives the maturation of cytokines for extracellular secretion. We did not find significant activation of caspase-1 after LPS treatment in both WT and MKK3−/− BMDMs. When ATP was added for last 15 min of LPS treatment we observed a significant increase in caspase-1 activity in WT BMDMs but not in MKK3−/−(Figure 6A). Furthermore, MKK3−/− BMDM LPS+ATP treatment group had significantly lower cleaved caspase-1 than the WT as shown by western blot (Figure 6B). This data indicates that MKK3 also influences caspase-1 activation either directly or indirectly under inflammatory conditions.
Figure 6. Reduced caspase-1 activation in MKK3−/− BMDMs after LPS + ATP injury.

BMDMs were treated with LPS and ATP to check the activation of caspase-1. Caspase-1 activity was significantly increased in WT after LPS (0.1 μg/ml) + ATP (5uM) treatment but MKK3−/− BMDMs did not show any significant change compared to control (A). The levels of cleaved caspase-1 were lower in MKK3−/− BMDMs compared to WT after LPS + ATP treatment (B). * represents significance between control and LPS treated within same genotype and # represents significance between the genotypes within LPS treatment groups, and ** or ## represents significance at p<0.01. Data is representative of 3 experiments done in at least triplicates.
MKK3 affects MAP kinase response to LPS
To study the effect of MKK3 on LPS-induced downstream signaling events we studied the MAPK (Mitogen Activated Protein Kinase) family. The MAPK family consists of the ERK1/2 (Extracellular Signal-Regulated Kinase), p38, and JNK (Jun N-terminal kinase). After LPS stimulation MKK3−/− BMDMs had lower induction of p-p38 and p-JNK compared to WT. There was no difference in p-ERK response in MKK3−/− and WT BMDMs (Figure 7). These results indicate that MKK3 affects activation of p38 and JNK, which in turn affects NF-κB phosphorylation.
Figure 7. MKK3−/− BMDMs show reduced activation of MAPK signaling after LPS stimulation.
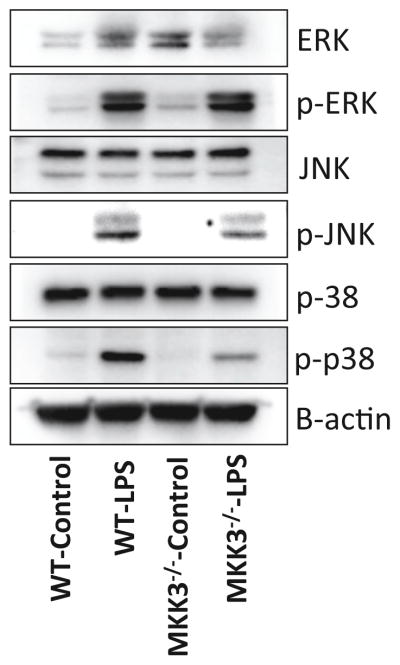
The MKK3−/− BMDMs after LPS (0.1 μg/ml) treatment showed lower levels of p-p38 and p-JNK compared to WT. There was no difference in p-ERK response after LPS treatment in both MKK3−/− and WT BMDMs. Data was normalized to b-actin content. Data is representative of 3 experiments done in at least triplicates with multiple western blotting runs.
MKK3 siRNA treatment in WT BMDMs confirms the MKK3−/− phenotype
To confirm our findings, we treated WT BMDMs with MKK3 siRNA and checked mitochondrial function and inflammatory responses. MKK3 siRNA treatment caused 70% decrease in MKK3 level as measured by PCR whereas there was no change in non-specific siRNA (NS siRNA)-treated BMDMs (Data not shown). MMP was higher in MKK3 siRNA-treated BMDMs compared to NS siRNA-treated BMDMs (Figure 8A). NF-κB nuclear translocation as well as caspase-1 activation in response to LPS was lower in MKK3 siRNA BMDMs compared to NS siRNA-treated BMDMs (Figure 8B and C). The baseline levels of IL-1α, IL-1β, IL-4, MCP-1, G-CSF, MIP-1α, MIP-1β, KC, and RANTES were lower in MKK3 siRNA-treated BMDMs. LPS exposure resulted in an increase of all the cytokines and chemokines measured but the response was significantly attenuated in MKK3 siRNA-treated BMDMs (Figure 8D). These results clearly implicate MKK3 in the inflammatory response of BMDMs after LPS stimulation.
Figure 8. MKK3 siRNA produces MKK3−/− phenotype in WT BMDMs.

WT BMDMs transfected with MKK3 siRNA show higher MMP compared to control i.e. non-specific siRNA (NS siRNA) transfected (A). NF-κB nuclear translocation was lower in MKK3 siRNA transfected BMDMs compared to control after LPS treatment (0.1 μg/ml) (B). Caspase-1 activation was lower in MKK3 siRNA transfected BMDMs after LPS (0.1 μg/ml) + ATP (5 μM) injury compared to control (C). Cytokine release was lower in LPS (0.1 μg/ml) exposed MKK3 siRNA treated BMDMs compared to NS siRNA treamtment (D). For MMP graph * represents significance at p<0.05. For Caspase-1 activity and NF-κB graph * represents significance among treatments within same genotype at p<0.05. For cytokine graphs * represents significance between control and LPS treated within same siRNA treatment and # represents significance between different siRNA treatment with control and LPS treatment groups, * or # represents significance at p<0.05 and ** or ## represents significance at p<0.01. Data is representative of 3 experiments done in at least triplicates.
Discussion
Sepsis is an acute systemic inflammatory condition [15]. This life-threatening condition leads to more than 200,000 deaths in US, making it the leading cause of mortality in ICU patients. The hallmark of sepsis is a dysregulated and overwhelming inflammatory response, characterized by massive cytokine release, oxidative stress, and mitochondrial dysfunction [16]. Several lines of evidence, genetic and functional, suggest that the mitochondrial function is the primary target during acute inflammation and that it is a central determinant of clinical outcome. It is proposed that inflammatory signaling leads to the phosphorylation of cytochrome oxidase (COX) and other mitochondrial targets followed by the suppression of mitochondrial function, reduced mitochondrial membrane potentials (MMP), and energy failure [17–19]. It has been shown that in animal models of severe inflammation ATP levels were significantly reduced compared with controls [20]. Similar results were reported in septic patients where reduced ATP levels were found in the “non-survivors” group compared to “survivors” group [21]. In addition organ failure in sepsis is linked to ATP depletion, as LPS induced lung injury is associated with reduced ATP [20, 22]. In a recently published study on gene expression profiling to identify genes associated with sepsis in human patients found that components of mitochondrial respiratory complex I (NDUFB8, NDUFC2, and NDUFS6) and ATP synthase (ATP5I) are down regulated in septic patients with a concomitant increase in the components of chemokine signaling pathway i.e. Forkhead box 3, Signal transducer and activator of transcription 3, and v-raf-1 murine leukemia viral oncogene homolog 1 [23]. In this context, we have earlier shown that MKK3−/− mice are resistant to septic injury [10]. Moreover the MKK3−/− cells and tissues showed higher mitochondrial biogenesis and mitophagy leading to a larger pool of healthy mitochondria [11]. In this study we show that MKK3 negatively affects mitochondrial function, which could be the driver of inflammatory outcome under septic conditions. The MKK3−/− BMDMs showed higher oxygen consumption at baseline and also had higher ATP production. The MMP and ATP levels were higher in MKK3−/− BMDMs, indicative of better mitochondrial function. This could be an important reason for the attenuated cytokine response against LPS-induced inflammation.
ROS contribute to inflammatory responses by activating key signaling pathways including the redox sensitive transcription factor pathway of nuclear factor κB (NF-κB) and the inflammasome [24]. Mitochondria produce more than 90% of the body’s energy needs in the form of ATP, via oxidative phosphorylation. The majority of ROS production occurs within mitochondria during ATP generation. Also, being the major source of intracellular ROS, mitochondria are a major target of ROS-mediated damage. Mitochondrial ROS (mtROS) are important for cellular signaling but can also cause cell damage, and so are tightly regulated by endogenous antioxidant scavenging systems that limit damage [25]. Under conditions of stress or disease, ROS production is increased, antioxidant protection is decreased, mitochondrial damage occurs, and inflammation increases, creating a vicious cycle [26]. This self-sustaining and amplifying cycle between ROS generation, inflammatory responses, and mitochondrial impairment contributes to organ failure in patients with sepsis [19]. The MKK3−/− BMDMs showed lower levels of ROS and MitoSOX at baseline as well as after LPS treatment which could be due to either better ROS scavenging mechanisms or higher efficiency of respiratory complexes leading to reduced ROS production. Although ROS are clearly part of the septic sequence their specific role and importance relative to the energy crisis component has to be further examined. Based on our data it is apparent that MKK3 has dual effects on mitochondrial function and ROS generation as well as activation of MAP kinases, p-38 and JNK, affecting the inflammatory outcome.
Our study shows that MKK3 deficiency significantly reduced inflammatory responses by down regulating two major inflammatory pathways i.e. the NF-κB and the inflammasome. Inflammatory responses to stimuli can be seen as a two-step process. First, the pathogen associated molecular patterns (PAMPS) (including LPS, a prototypical PAMP) induce a transcriptional response with production of cytokine mRNA and proteins which is mediated by transcription factors such as NF-κB. The other step is, inflammasome activation, mediated by damage associated molecular patterns (DAMPS), which helps secretion of mature cytokines. Mitochondria are central to inflammatory responses by affecting both these steps [27]. It is known that the inflammatory transcription factors such as NF-κB are redox sensitive and are activated by mitochondrial ROS. We observed reduced activation of NF-κB after LPS exposure in MKK3−/− BMDMs which could be due to insufficient increase in ROS. Damaged mitochondria release DAMPS such as oxidized mitochondrial DNA into the cytoplasm that revs up the inflammatory response by activation of inflammasome [28]. The inflammasome is a caspase-1-activating multi-protein complex that promotes cleavage of many pro-cytokines to their biologically active forms [29] [30]. In addition, active caspase-1 cleaves other substrates like caspase-7 and sterol regulatory element-binding proteins (SREBP), therefore playing an important role in cell death and survival [31]. Our data suggests that improved mitochondrial function in MKK3−/− BMDMs prevents the activation of caspase-1, which is instrumental in inflammatory cytokine release after LPS exposure. There are reports supporting this argument where it has been shown that in the presence of activated NF-κB, mitochondrial ROS and oxidized mtDNA are important players in activation of inflammasome [32, 33].
Given the important role mitochondria have in generating inflammation, modulators of mitochondrial function could be potential anti-inflammatory agents. Several mitochondria-targeted therapies have been tested including treatment with mitochondrial substrates (e.g., carnitine, succinate, and MgCl2–ATP), cofactors (e.g., coenzyme Q and α-lipoic acid), antioxidants and ROS scavengers (e.g., MitoQ, SkQ, phenyl-tert-butylnitrone, N-acetylcysteine, and Tempol), and membrane stabilizers (e.g., cyclosporine A and melatonin), which restore mitochondrial function to some extent [34, 35]. However, interfering with inflammatory signaling along with mitochondrial function might result in better outcomes. For example, the synthetic cortisol homologue dexamethasone, which affects several signaling pathways including Wnt/β-cateninin, NFκB, MAPK/Erk, and PI3K signaling, was shown to partially rescue COX function 24 h after cecal ligation and puncture in the cortex and outer stripe of the outer medulla of mouse kidneys [36]. Based on our results it is apparent that MKK3 can be a potential target for reducing inflammation under septic conditions. Kinases are well known and druggable targets with many kinase inhibitors currently being developed [37]. Mitogen activated protein kinases (MAP kinases, MAPKs and p38), have been the focus of intense but ultimately unsuccessful search for inhibitors for anti-inflammatory and anti-sepsis applications. A limiting factor is that p38 inhibitors have significant side effects that restrict their clinical use [38]. MKK3 inhibition does not show any side effects associated with p38 inhibition because downstream signaling is tuned differently between the two kinases [39].
In conclusion, we demonstrate that MKK3 mediates LPS induced inflammatory responses through the improvement in mitochondrial function. Thus, MKK3 represents an attractive therapeutic target for the treatment of sepsis.
Acknowledgments
Sources of funding
PM is supported by American Heart Association grant, AHA 09FTF2090019, NIA R03 AG 042358-02 (GEMSSTAR), Yale Claude D. Pepper Older Americans Independence Center (P30 AG021342), and American Thoracic Society (ATS) Foundation Recognition award. PJL is supported by R01 HL090660, R01 HL071595 and FAMRI 82384.
We thank Richard A. Flavell, Yale University, and Roger J. Davis, University of Massachusetts Medical School, for their kind gift of the MKK3-deficient mice and Gerald S. Shadel and A. Phillip West, Yale University for their assistance with the Seahorse experiments.
Footnotes
Author Disclosure Statement
No conflicts of interest, financial or otherwise, are declared by the author(s).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lee I, Huttemann M. Energy crisis: The role of oxidative phosphorylation in acute inflammation and sepsis. Biochimica et biophysica acta. 2014;1842:1579–1586. doi: 10.1016/j.bbadis.2014.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huttemann M, Helling S, Sanderson TH, Sinkler C, Samavati L, Mahapatra G, Varughese A, Lu G, Liu J, Ramzan R, Vogt S, Grossman LI, Doan JW, Marcus K, Lee I. Regulation of mitochondrial respiration and apoptosis through cell signaling: cytochrome c oxidase and cytochrome c in ischemia/reperfusion injury and inflammation. Biochimica et biophysica acta. 2012;1817:598–609. doi: 10.1016/j.bbabio.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Debelec-Butuner B, Alapinar C, Varisli L, Erbaykent-Tepedelen B, Hamid SM, Gonen-Korkmaz C, Korkmaz KS. Inflammation-mediated abrogation of androgen signaling: an in vitro model of prostate cell inflammation. Molecular carcinogenesis. 2014;53:85–97. doi: 10.1002/mc.21948. [DOI] [PubMed] [Google Scholar]
- 4.Andrades ME, Ritter C, Dal-Pizzol F. The role of free radicals in sepsis development. Frontiers in bioscience. 2009;1:277–287. doi: 10.2741/E27. [DOI] [PubMed] [Google Scholar]
- 5.Ryu MJ, Kim SJ, Kim YK, Choi MJ, Tadi S, Lee MH, Lee SE, Chung HK, Jung SB, Kim HJ, Jo YS, Kim KS, Lee SH, Kim JM, Kweon GR, Park KC, Lee JU, Kong YY, Lee CH, Chung J, Shong M. Crif1 deficiency reduces adipose OXPHOS capacity and triggers inflammation and insulin resistance in mice. PLoS genetics. 2013;9:e1003356. doi: 10.1371/journal.pgen.1003356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, Bosca L. Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. Journal of immunology. 2010;185:605–614. doi: 10.4049/jimmunol.0901698. [DOI] [PubMed] [Google Scholar]
- 7.Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, Cross JR, Jung E, Thompson CB, Jones RG, Pearce EJ. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–4749. doi: 10.1182/blood-2009-10-249540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoue T, Boyle DL, Corr M, Hammaker D, Davis RJ, Flavell RA, Firestein GS. Mitogen-activated protein kinase kinase 3 is a pivotal pathway regulating p38 activation in inflammatory arthritis. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5484–5489. doi: 10.1073/pnas.0509188103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim DH, Ausubel FM. Evolutionary perspectives on innate immunity from the study of Caenorhabditis elegans. Current opinion in immunology. 2005;17:4–10. doi: 10.1016/j.coi.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 10.Mannam P, Zhang X, Shan P, Zhang Y, Shinn AS, Zhang Y, Lee PJ. Endothelial MKK3 is a critical mediator of lethal murine endotoxemia and acute lung injury. Journal of immunology. 2013;190:1264–1275. doi: 10.4049/jimmunol.1202012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, Merkel J, Kang MJ, Dela Cruz CS, Ahasic AM, Pisani MA, Trentalange M, West AP, Shadel GS, Elias JA, Lee PJ. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. American journal of physiology. Lung cellular and molecular physiology. 2014;306:L604–619. doi: 10.1152/ajplung.00272.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Emerling BM, Platanias LC, Black E, Nebreda AR, Davis RJ, Chandel NS. Mitochondrial reactive oxygen species activation of p38 mitogen-activated protein kinase is required for hypoxia signaling. Molecular and cellular biology. 2005;25:4853–4862. doi: 10.1128/MCB.25.12.4853-4862.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Emre Y, Hurtaud C, Nubel T, Criscuolo F, Ricquier D, Cassard-Doulcier AM. Mitochondria contribute to LPS-induced MAPK activation via uncoupling protein UCP2 in macrophages. The Biochemical journal. 2007;402:271–278. doi: 10.1042/BJ20061430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brancho D, Tanaka N, Jaeschke A, Ventura JJ, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes & development. 2003;17:1969–1978. doi: 10.1101/gad.1107303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sriskandan S, Altmann DM. The immunology of sepsis. The Journal of pathology. 2008;214:211–223. doi: 10.1002/path.2274. [DOI] [PubMed] [Google Scholar]
- 16.Herzum I, Renz H. Inflammatory markers in SIRS, sepsis and septic shock. Current medicinal chemistry. 2008;15:581–587. doi: 10.2174/092986708783769704. [DOI] [PubMed] [Google Scholar]
- 17.Waxman AB, Kolliputi N. IL-6 protects against hyperoxia-induced mitochondrial damage via Bcl-2-induced Bak interactions with mitofusins. American journal of respiratory cell and molecular biology. 2009;41:385–396. doi: 10.1165/rcmb.2008-0302OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kolliputi N, Waxman AB. IL-6 cytoprotection in hyperoxic acute lung injury occurs via PI3K/Akt-mediated Bax phosphorylation. American journal of physiology. Lung cellular and molecular physiology. 2009;297:L6–16. doi: 10.1152/ajplung.90381.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rocha M, Herance R, Rovira S, Hernandez-Mijares A, Victor VM. Mitochondrial dysfunction and antioxidant therapy in sepsis. Infectious disorders drug targets. 2012;12:161–178. doi: 10.2174/187152612800100189. [DOI] [PubMed] [Google Scholar]
- 20.Brealey D, Karyampudi S, Jacques TS, Novelli M, Stidwill R, Taylor V, Smolenski RT, Singer M. Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. American journal of physiology. Regulatory, integrative and comparative physiology. 2004;286:R491–497. doi: 10.1152/ajpregu.00432.2003. [DOI] [PubMed] [Google Scholar]
- 21.Lawrence KL, White PH, Morris GP, Jennemann J, Phelan DL, Hotchkiss RS, Kollef MH. CD4+ lymphocyte adenosine triphosphate determination in sepsis: a cohort study. Critical care. 2010;14:R110. doi: 10.1186/cc9059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vanasco V, Magnani ND, Cimolai MC, Valdez LB, Evelson P, Boveris A, Alvarez S. Endotoxemia impairs heart mitochondrial function by decreasing electron transfer, ATP synthesis and ATP content without affecting membrane potential. Journal of bioenergetics and biomembranes. 2012;44:243–252. doi: 10.1007/s10863-012-9426-3. [DOI] [PubMed] [Google Scholar]
- 23.Tang BM, McLean AS, Dawes IW, Huang SJ, Lin RC. The use of gene-expression profiling to identify candidate genes in human sepsis. American journal of respiratory and critical care medicine. 2007;176:676–684. doi: 10.1164/rccm.200612-1819OC. [DOI] [PubMed] [Google Scholar]
- 24.Gorbunov NV, Garrison BR, McDaniel DP, Zhai M, Liao PJ, Nurmemet D, Kiang JG. Adaptive redox response of mesenchymal stromal cells to stimulation with lipopolysaccharide inflammagen: mechanisms of remodeling of tissue barriers in sepsis. Oxidative medicine and cellular longevity. 2013;2013:186795. doi: 10.1155/2013/186795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayir H, Kagan VE. Bench-to-bedside review: Mitochondrial injury, oxidative stress and apoptosis--there is nothing more practical than a good theory. Critical care. 2008;12:206. doi: 10.1186/cc6779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bereiter-Hahn J. Do we age because we have mitochondria? Protoplasma. 2014;251:3–23. doi: 10.1007/s00709-013-0515-x. [DOI] [PubMed] [Google Scholar]
- 27.Savage CD, Lopez-Castejon G, Denes A, Brough D. NLRP3-Inflammasome Activating DAMPs Stimulate an Inflammatory Response in Glia in the Absence of Priming Which Contributes to Brain Inflammation after Injury. Frontiers in immunology. 2012;3:288. doi: 10.3389/fimmu.2012.00288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, Ruggiero EA, Crawford DR. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. Journal of Alzheimer’s disease: JAD. 2012;30:617–627. doi: 10.3233/JAD-2012-120145. [DOI] [PubMed] [Google Scholar]
- 29.Verma D, Lerm M, Blomgran Julinder R, Eriksson P, Soderkvist P, Sarndahl E. Gene polymorphisms in the NALP3 inflammasome are associated with interleukin-1 production and severe inflammation: relation to common inflammatory diseases? Arthritis and rheumatism. 2008;58:888–894. doi: 10.1002/art.23286. [DOI] [PubMed] [Google Scholar]
- 30.Abais JM, Zhang C, Xia M, Liu Q, Gehr TW, Boini KM, Li PL. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxidants & redox signaling. 2013;18:1537–1548. doi: 10.1089/ars.2012.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martinon F. Detection of immune danger signals by NALP3. Journal of leukocyte biology. 2008;83:507–511. doi: 10.1189/jlb.0607362. [DOI] [PubMed] [Google Scholar]
- 32.Zhang Z, Xu X, Ma J, Wu J, Wang Y, Zhou R, Han J. Gene deletion of Gabarap enhances Nlrp3 inflammasome-dependent inflammatory responses. Journal of immunology. 2013;190:3517–3524. doi: 10.4049/jimmunol.1202628. [DOI] [PubMed] [Google Scholar]
- 33.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature immunology. 2011;12:222–230. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikh SM. Therapeutic targeting of the mitochondrial dysfunction in septic acute kidney injury. Current opinion in critical care. 2013;19:554–559. doi: 10.1097/MCC.0000000000000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galley HF. Bench-to-bedside review: Targeting antioxidants to mitochondria in sepsis. Critical care. 2010;14:230. doi: 10.1186/cc9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi HM, Jo SK, Kim SH, Lee JW, Cho E, Hyun YY, Cha JJ, Kang YS, Cha DR, Cho WY, Kim HK. Glucocorticoids attenuate septic acute kidney injury. Biochemical and biophysical research communications. 2013;435:678–684. doi: 10.1016/j.bbrc.2013.05.042. [DOI] [PubMed] [Google Scholar]
- 37.Klebl BM. Chemical kinomics - a target gene family approach in chemical biology. Drug discovery today. Technologies. 2004;1:25–34. doi: 10.1016/j.ddtec.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 38.Fisk M, Gajendragadkar PR, Maki-Petaja KM, Wilkinson IB, Cheriyan J. Therapeutic potential of p38 MAP kinase inhibition in the management of cardiovascular disease. American journal of cardiovascular drugs: drugs, devices, and other interventions. 2014;14:155–165. doi: 10.1007/s40256-014-0063-6. [DOI] [PubMed] [Google Scholar]
- 39.Liao Y, Wang X, Huang M, Tam JP, Liu DX. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology. 2011;420:106–116. doi: 10.1016/j.virol.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]