

Abstract
Helicobacter pylori (H. pylori) induces chronic gastric inflammation, atrophic gastritis, intestinal metaplasia, and cancer. Although the risk of gastric cancer increases exponentially with the extent of atrophic gastritis, the precise mechanisms of gastric carcinogenesis have not been fully elucidated. H. pylori induces genetic and epigenetic changes in gastric epithelial cells through activating intracellular signaling pathways in a cagPAI-dependent manner. H. pylori eventually induces gastric cancer with chromosomal instability (CIN) or microsatellite instability (MSI), which are classified as two major subtypes of gastric cancer. Elucidation of the precise mechanisms of gastric carcinogenesis will also be important for cancer therapy.
1. Introduction (Figure 1)
Figure 1.

Summary of the three subtypes of gastric cancer that were associated with H. pylori.
Gastric cancer is the world's third leading cause of cancer-related death [1]. It is well known that the majority of gastric cancers are associated with Helicobacter pylori (H. pylori) infection [2]. Normal gastric mucosa, chronic superficial gastritis, atrophic gastritis, intestinal metaplasia, dysplasia, and adenocarcinoma are the chain of events with H. pylori. Atrophic gastritis and intestinal metaplasia exponentially increase the risk of developing gastric cancer (90-fold) [3]. Recent data from The Cancer Genome Atlas (TCGA) project led to the proposal of three subtypes of gastric cancer that were associated with H. pylori: (1) tumors with chromosomal instability (CIN), which display marked aneuploidy and focal amplification of receptor tyrosine kinases; (2) microsatellite unstable tumors (MSI), which have elevated rates of mutation, including mutations in genes encoding targetable oncogenic signaling proteins; and (3) genomically stable tumors (GS), which are enriched for the diffuse histological variant and fusions involving RHO-family GTPase-activating proteins or mutations of RHOA [4]. This review discusses pathogenesis and intracellular signaling pathways that are associated with H. pylori infection, which result in chronic inflammation, intestinal metaplasia, and gastric cancer.
2. H. pylori-Induced Chronic Inflammation and Intestinal Metaplasia (Figure 2)
Figure 2.
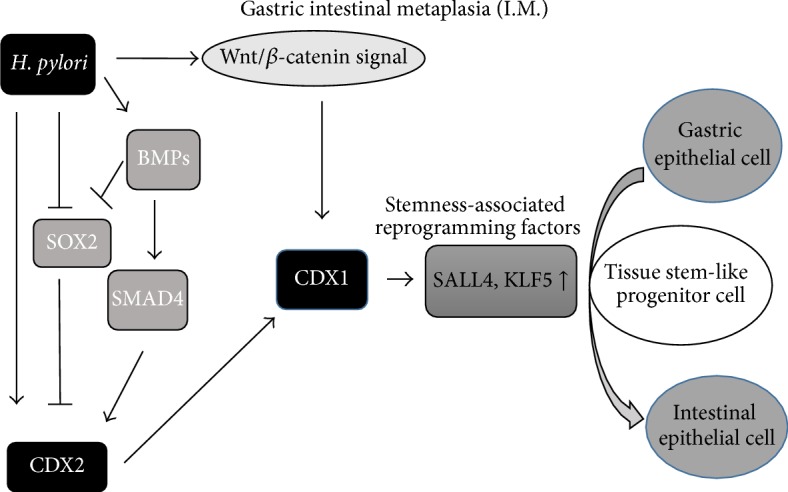
Putative mechanism for inducing intestinal metaplasia by H. pylori.
2.1. CDX1/2: Key Factors for Gastric Carcinogenesis and Intestinal Metaplasia
Gastric intestinal metaplasia (IM) is considered to be a preneoplastic lesion of the stomach consisting of the transdifferentiation of the gastric mucosa into an intestinal phenotype, both morphologically and functionally [5]. Two types of IM: complete type and incomplete type are defined [6]. Complete IM express only intestinal mucin marker (MUC2), whereas incomplete IM express both intestinal and gastric mucin marker (MUC5AC) in single cell level [7]. The Wnt target genes CDX1 and CDX2 are thought to play a pivotal role in establishing and maintaining intestinal metaplasia and carcinogenesis, due to the observation that the intestinal phenotype is induced in cdx1 or cdx2 transgenic mice and that the intestinal-type adenocarcinoma is induced in cdx2 transgenic mice [5]. Several mechanisms of how CDXs contribute to the development of intestinal metaplasia have been reported. In a mouse model expressing intestine-specific homeobox genes, CDX1 transgenic mice developed a complete form of intestinal metaplasia, representing absorptional epithelial cells, goblet cells, gastrointestinal endocrine cells, and Paneth cells, while the characteristics of gastric mucosa completely disappeared [8]. In addition, Fujii et al. reported recently that CDX1 induces stemness-associated reprogramming factors, KLF5 and SALL4, suggesting that CDXs directly contribute to the development of gastric intestinal metaplasia. CDX1-induced KLF5 and SALL4 converted gastric epithelial cells into tissue stem-like progenitor cells, which then transdifferentiated into intestinal epithelial cells. A requirement for transition of intestinal metaplasia into dedifferentiated stem/progenitor-like cells, which share properties in common with cancer stem cells, may underlie the predisposition of intestinal metaplasia to neoplastic transformation [9]. Taken together, CDX1-induced dedifferentiated stem/progenitor-like cells in incomplete type IM may be essential for development of a preneoplastic lesion and may explain the diversity of gastric cancer.
In CDX2 transgenic mice, the expression of Shh, a morphogen associated with differentiation of the parietal cells of the stomach, was completely lost, both at the RNA level and when examined by immunohistochemistry [10]. Furthermore, the expression of Shh was decreased in the human intestinal metaplastic mucosa [11]. These phenomena support a key role for CDXs in the development of atrophic gastritis, intestinal metaplasia, and carcinogenesis.
In terms of signaling pathways, the Wnt and BMPs/SMAD4 pathways are both associated with the expression of CDX1/2. In addition to being a direct transcriptional target of the Wnt/β-catenin signaling pathway during mouse gut development, CDX1 is also induced by cag-positive H. pylori infection [9, 12]. BMPs/SMAD4 is known to be a fundamental pathway for the development of intestinal epithelium; it is upregulated upon H. pylori infection and thereafter induces the expression of the downstream target CDX2, as well as the downregulation of SOX2, an inhibitor of CDX2 [13–15]. CDX2 regulate MUC2 [16] by binding to enhancer sequences [17].
2.2. Genetic Alteration and Gene Expression in Intestinal Metaplasia
Gene alteration, such as aneuploidy of chromosomes [18], P53 mutations (38–45%) [19–22], P53 deletion (60%) [18], microsatellite instability (27%) [23], and mitochondrial microsatellite instability (33%) [24] were detected in IM. P53 mutations were mostly in incomplete type [20, 21]. Microsatellite instabilities were all in incomplete type [25]. Gene expression, such as MUC2 [6], LI-cadherin [26], KLF4 [27], intestinal trefoil factor (TFF3) [28], sucrose-isomaltase [29], villin [7], CD10 [30], and defensing [31], increased in IM. MUC2 is regulated by CDX2 [16, 32]. On the other hand, gene expression, such as Sonic hedgehog (Shh) [33], SOX2 [14], RUNX3 [34], and TFF1 and TFF2 [28], decreased in IM. Shh is particularly decreased in incomplete IM type [11].
Alteration of these gastric and intestinal phenotype markers was observed at the cellular level, as well as at the glandular level. In fact, neuroendocrine cells also showed intestinalization along with their exocrine counterparts. In animal models, incomplete type intestinal metaplasia appears first and then progresses to the complete type. In summary, intestinal metaplasia may be caused by the gradual intestinalization of stem/progenitor cells from the incomplete to the complete type [35].
3. H. pylori-Induced Genetic Changes
Several reports have suggested that H. pylori infection caused genetic alterations in gastric epithelial cells, mostly through the induction of reactive oxygen species (ROS) [36]. Matsumoto et al. reported that H. pylori induced aberrant expression of activation-induced cytidine deaminase (AID), known as an editor of DNA and RNA. AID was reported to cause mutations in the P53 and APC genes in gastric epithelial cells, relevant to the development of adenocarcinoma [37]. AID hypermutates immunoglobulin genes in B cell genome, contributing to variety acquisition of immunoglobulin. AID also target oncogenes, leading to B cell malignancy [38]. In addition, various cancers develop in AID transgenic mice, including gastric cancer [39]. In Matsumoto's report, H. pylori strongly induced AID expression in human gastric epithelial cells, through activation of the NF-κB pathway, and induced mutation of p53. As mutation of p53 was inhibited by blocking AID, p53 mutation induced by H. pylori mostly depends on AID. Since AID was upregulated via activation of the NF-κB pathway, proinflammatory cytokines—such as TNF-α or IL-1β—in gastric inflammation also reinforce the onset of AID as well as the direct stimulation of H. pylori in gastric epithelial cells [40].
4. H. pylori-Induced Epigenetic Changes
4.1. DNA Methylation Induced by H. pylori Infection
It has been reported that H. pylori could cause DNA methylation of many genes in gastric epithelial cells. Mongolian gerbils were infected with H. pylori and DNA methylation levels in the gastric mucosa were analyzed over time. Methylation levels were increased in the persistent infection group depending on the duration of infection [41–43]. Accordingly, H. pylori eradication led to a dramatic decrease in methylation levels [44, 45]. Since DNA methylation remained after infection with H. pylori and methylation could be inhibited with an immunosuppressive drug, it can be concluded that the inflammatory reaction induced by H. pylori infection, and not the presence of the bacterium itself, is more important in the process of DNA methylation [43]. H. pylori infection causes gastric mucosal inflammation responses, resulting in upregulation of IL-1β or Nos2, which in turn induce aberrant DNA methylation [46]. Several studies found that aberrant DNA methylation in gastric biopsies from H. pylori-positive patients correlated with a greater risk of developing gastric cancer [43, 47], suggesting that H. pylori-associated inflammation and subsequent induction of DNA methylation could have a potential role in gastric carcinogenesis. A large number of genes with different biological functions have been found to be methylated in gastric carcinogenesis. Among these, methylation of a DNA repair gene, MLH1, may play an important role in gastric carcinogenesis in MSI-positive gastric cancer, since MLH1 is silenced in this type of cancer.
4.2. H. pylori and Gastric CIMP
Aberrant DNA methylation in cancer encompasses global hypomethylation and regional hypermethylation, which are thought to be associated with genomic instability and inactivation of tumor-suppressor genes [48]. However, regional hypermethylation refers to the aberrant methylation of normally unmethylated sequences, most of which are clusters of CpG sites, denoted as CpG islands. The strong relationship between CIMP and MSI suggests that CIMP may be related to gene mutation. In fact, H. pylori infection significantly elevated the rate of CIMP positivity [49], suggesting that H. pylori caused aberrant DNA hypermethylation of specific genes, followed by induction of CIMP during gastric carcinogenesis.
5. Changes in Signaling Pathways Induced by H. pylori Infection
Numerous signaling pathways mediated by H. pylori are reportedly dependent on the cag pathogenicity island (cagPAI), especially the cagA gene. Elucidation of the signaling pathways activated by H. pylori infection may be important for the identification of targets for treatment.
5.1. NF-κB Pathway (Figure 3)
Figure 3.

H. pylori induced NF-κB signaling.
NF-κB is one of the major transcription factors that regulates inflammation and is constitutively activated in some gastric cancers [50]. H. pylori activates NF-κB in the gastric mucosa via cagPAI-dependent and cagPAI-independent pathways. H. pylori cag-positive strains deliver the certain protein into host cells via the cag PAI-encoded type IV secretion system (T4SS) [51–53]. The certain protein is thought to be injected into host epithelial cells where it interacts with TRAF6 and TAK1 to activate IKK. The IKK-complex contains two highly homologous kinase subunits, IKKα and IKKβ, in addition to the regulatory subunit NF-κB essential modulator (NEMO). The key factor required for activation of this pathway is still unknown. One possible explanation is that peptidoglycan injected into cells via the T4SS stimulates Nod1 activity leading to NF-κB activation [54]. During cag-independent activation of intracellular signaling, host immune cells are stimulated by lipopolysaccharide (LPS) produced by H. pylori via TLR pathways, followed by activation of the NF-κB pathway [55–58]. Among certain proteins injected into host cells, CagA is probably indispensable in the induction of an inflammatory reaction, as it has been reported that a CagA-knockout of H. pylori was unable to induce severe inflammation in Mongolian gerbils model [59, 60]. It has also been reported that overexpression of CagA induced NF-κB activation with subsequent IL-8 production [61].
NF-κB activation induces the release of proinflammatory cytokines, such as tumor necrosis factor- (TNF-) α, interleukin- (IL-) 1β, and IL-6 [62–65]. NF-κB also regulates other molecules that are involved in the chemokine response (IL-8, MCP-1), blockade of apoptosis (cIAPs, c-FLIP, A20, and BclX), angiogenesis (VEGF, IL-8), and invasion (MMP-2, MMP-9). All these factors may be related to carcinogenesis [66], and we have focused on inhibition of NF-κB as a potential avenue to inhibit cancer, by controlling the degree of gastritis caused by H. pylori infection [67].
5.2. The IL-6- (IL-11-) STAT3-CDX2 Pathway (Figure 4)
Figure 4.

Scheme of the IL-11/STAT3/CDX2 pathway.
It has been reported that the proinflammatory cytokine IL-6, which is upregulated upon H. pylori infection in the gastric mucosa, contributes to gastric tumorigenesis [68]. IL-6 binds to the α-subunit of its specific receptor, associates with gp130 homodimers at the cell membrane, and activates two main signaling pathways: SHP-2/ERK and JAK/STAT. In mouse models, gp130F/F mice spontaneously develop gastric inflammation and intestinal-type gastric tumors [69]. gp130 is IL-6 family receptor signaling subunit, and IL-6 family gp130 driver IL-11 drives hyperactivation of STAT3 contributing gastric phenotype. Gp130 F/F carries a knock-in mutation in gp130 [70]. The final step in the SHP-2/ERK pathway is gene regulation by the transcription factor AP-1, whereas in the JAK/STAT pathway, phosphorylated STAT3 dimers translocate to the nucleus and activate the transcription of target genes [71].
The H. pylori protein CagA recruits SHP2 to gp130; phosphorylated CagA shows enhanced SHP2 binding activity and ERK1/2 phosphorylation, whereas unphosphorylated CagA preferentially activates STAT3 [72]. SHP2/ERK signaling may lead to mucosal inflammation and carcinogenesis [73]. Phosphorylated STAT3 induces expression of genes that promote angiogenesis (e.g., VEGF), cell-cycle progression (e.g., cyclinD1), and cell survival (e.g., Bcl/xL, survivin) [69]. Mice in which gp130 is mutated and the STAT3 pathway is activated develop gastric cancer [74]. Taken together, these findings suggest that the IL-6/STAT3 pathway plays key roles in gastric carcinogenesis, not only via the IL6- (IL11-) STAT3-CDX2 pathway resulting in induction of intestinal metaplasia, but also via the unphosphorylated CagA/SHP2/ERK1/2 pathway, leading to induction of gastric epithelial proliferation and carcinogenesis.
5.3. c-Myc/p21/ERK-MAPK Pathway
It was reported that cagA coupled with PAR1 (MARK) phosphorylation resulted in disruption of tight junctions and the cellular polarity of epithelial cells [75]. CagA, which invaded epithelial cells, bound to PAR1 and stimulated the GEF (guanine nucleotide exchange factor)–H1–RhoA–ROCK (RhoA-associated kinase)–c-Myc–microRNA–p21 axis; this was caused by the liberation of cigulin from the GEF-H1-cingulin complex, followed by induction of miR-17 and miR-20a by c-Myc activation, resulting in inhibition of accumulation of p21. In other words, p21 causes cellular senescence by inhibiting activation of ERK-MAPK, and CagA causes abnormal cell proliferation by inhibiting p21, thus contributing to gastric carcinogenesis [76].
5.4. TLR Signaling
Toll-like receptors (TLRs) are a key family of microbial sensors of the host innate and adaptive immune systems [77]. For instance, genetic ablation of signaling adaptor MyD88 and the common TLR in mice alleviates intestinal tumorigenesis induced in ApcMin/+mice [78]. TLRs expressed in epithelial can promote noninflammatory epithelial responses including migration, cell survival, proliferation [79], and angiogenesis [80]. In the setting of H. pylori infection, gene expression of TLR2 and TLR4 is elevated in H. pylori-positive gastric patients [81], and TLR2 and TLR4 gene polymorphisms are associated with an increased risk of gastric cancer [82]. STAT3 directly upregulates epithelial expression of TLR2 in gastric tumors. Genetic and therapeutic targeting of TLR2 inhibited gastric tumorigenesis (but not inflammation), characterized by reduced proliferation and increased apoptosis of gastric epithelial cells [83]. Increased STAT3 pathway activation and TLR2 expression were also associated with poor survival in gastric cancer patients.
5.5. ROS/ASK1/JNK Pathway
ASK1 is reported to be one of the key players in the regulation of H. pylori-related cellular responses in gastric epithelial cells. ASK1 is involved in cellular responses induced by H. pylori, such as apoptosis and cytokine production. Furthermore, ASK1 and TAK1 have reciprocal interactions and differentially regulate the activation of downstream molecules, such as JNK, p38, and NF-κB [84].
JNK, which can be activated by H. pylori infection via both ASK1 and TAK1, plays an important role in gastric carcinogenesis. In human gastric cancer, the extent of activated JNK observed by immunostaining is approximately 30–40%, whereas activation was observed in almost all cases of H. pylori-infected gastric mucosa. In a mouse MNU gastric carcinogenic model, number and diameter of the tumor cells were significantly decreased in JNK knockout mice compared to WT mice [85]. The effect on cellular proliferation was also examined in vitro; it was found that cellular proliferation was inhibited by using a JNK inhibitor or an siRNA to knock down JNK expression. Constitutive activation of JNK is proposed to be due to a positive feedback loop: ASK/JNK/CyclinD1/Rb phosphorylation/ASK protein upregulation [86].
6. H. pylori-Induced Chronic Inflammation and Innate/Adaptive Immunity
As mentioned above, after infecting H. pylori on gastric epithelial cells, it can affect not only the proliferation of gastric epithelial cells, but also the activation of intracellular signaling, and that leads to perturbing the host's innate and adaptive immune system [87]. Among the inflammatory reactions induced by H. pylori infection, innate immune system, represented by infiltration of neutrophils and macrophages, plays key roles in production of proinflammatory cytokines/chemokines, which promote chronic inflammation [67]. On the other hand, adaptive immune system plays roles not only to produce proinflammatory cytokines and cytotoxic reaction to bacterium directory, but also in induction of anti-inflammatory cytokines, such as IL-10, to suppress the cytotoxic function of effector T cells, which enables the bacteria to evade immune system, resulting in chronic infection [88].
7. Conclusions and Future Perspectives
Since chronic inflammation can cause epithelial cell disturbance, the early eradication of H. pylori could provide a basic solution for prevention of gastric carcinogenesis caused by H. pylori. Accordingly, all patients in Japan with H. pylori-related gastritis are being recommended for eradication methods to decrease the risk of gastric cancer. However, as a considerable proportion of patients remain to be irreversible “field cancerization,” where the cancer-causing case does not cut off after H. pylori sanitization, precancerous intestinal metaplasia even after H. pylori eradication, it is difficult to identify those at high risk of gastric cancer. Regarding the treatment of advanced gastric cancer, HER2 has emerged as a successful molecular target, and the treatment of other RTK/RAS amplifications complies with the concept of oncogene addiction, dependency of cancer on one or a few genes for maintenance of the malignant phenotype. Elucidation of the mechanism of gastric carcinogenesis associated with H. pylori will aid the development of further targeted therapies, which will be accompanied by the advent of personalized cancer medicine, a field that is developing rapidly.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Ferlay J., Soerjomataram I., Dikshit R., et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. International Journal of Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Uemura N., Okamoto S., Yamamoto S., et al. Helicobacter pylori infection and the development of gastric cancer. The New England Journal of Medicine. 2001;345(11):784–789. doi: 10.1056/nejmoa001999. [DOI] [PubMed] [Google Scholar]
- 3.Sipponen P., Marshall B. J. Gastritis and gastric cancer: Western countries. Gastroenterology Clinics of North America. 2000;29(3):579–592. doi: 10.1016/s0889-8553(05)70131-x. [DOI] [PubMed] [Google Scholar]
- 4.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature. 2014;513(7517):202–209. doi: 10.1038/nature13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barros R., Freund J.-N., David L., Almeida R. Gastric intestinal metaplasia revisited: function and regulation of CDX2. Trends in Molecular Medicine. 2012;18(9):555–563. doi: 10.1016/j.molmed.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 6.Reis C. A., David L., Correa P., et al. Intestinal metaplasia of human stomach displays distinct patterns of mucin (MUC1, MUC2, MUC5AC, and MUC6) expression. Cancer Research. 1999;59(5):1003–1007. [PubMed] [Google Scholar]
- 7.Niwa T., Ikehara Y., Nakanishi H., et al. Mixed gastric- and intestinal-type metaplasia is formed by cells with dual intestinal and gastric differentiation. Journal of Histochemistry and Cytochemistry. 2005;53(1):75–85. doi: 10.1369/jhc.4A6443.2005. [DOI] [PubMed] [Google Scholar]
- 8.Mutoh H., Sakurai S., Satoh K., et al. Cdx1 induced intestinal metaplasia in the transgenic mouse stomach: comparative study with Cdx2 transgenic mice. Gut. 2004;53(10):1416–1423. doi: 10.1136/gut.2003.032482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fujii Y., Yoshihashi K., Suzuki H., et al. CDX1 confers intestinal phenotype on gastric epithelial cells via induction of stemness-associated reprogramming factors SALL4 and KLF5. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(50):20584–20589. doi: 10.1073/pnas.1208651109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mutoh H., Hayakawa H., Sashikawa M., Sakamoto H., Sugano K. Direct repression of Sonic Hedgehog expression in the stomach by Cdx2 leads to intestinal transformation. Biochemical Journal. 2010;427(3):423–434. doi: 10.1042/BJ20091177. [DOI] [PubMed] [Google Scholar]
- 11.Shiotani A., Iishi H., Uedo N., et al. Evidence that loss of sonic hedgehog is an indicator of Helicobater pylori-induced atrophic gastritis progressing to gastric cancer. American Journal of Gastroenterology. 2005;100(3):581–587. doi: 10.1111/j.1572-0241.2005.41001.x. [DOI] [PubMed] [Google Scholar]
- 12.Lickert H., Domon C., Huls G., et al. Wnt/β-catenin signaling regulates the expression of the homeobox gene Cdx1 in embryonic intestine. Development. 2000;127(17):3805–3813. doi: 10.1242/dev.127.17.3805. [DOI] [PubMed] [Google Scholar]
- 13.Camilo V., Barros R., Sousa S., et al. Helicobacter pylori and the BMP pathway regulate CDX2 and SOX2 expression in gastric cells. Carcinogenesis. 2012;33(10):1985–1992. doi: 10.1093/carcin/bgs233. [DOI] [PubMed] [Google Scholar]
- 14.Tsukamoto T., Inada K., Tanaka H., et al. Down-regulation of a gastric transcription factor, Sox2, and ectopic expression of intestinal homeobox genes, Cdx1 and Cdx2: inverse correlation during progression from gastric/intestinal-mixed to complete intestinal metaplasia. Journal of Cancer Research and Clinical Oncology. 2004;130(3):135–145. doi: 10.1007/s00432-003-0519-6. [DOI] [PubMed] [Google Scholar]
- 15.Bleuming S. A., Kodach L. L., Garcia Leon M. J., et al. Altered bone morphogenetic protein signalling in the Helicobacter pylori-infected stomach. Journal of Pathology. 2006;209(2):190–197. doi: 10.1002/path.1976. [DOI] [PubMed] [Google Scholar]
- 16.Mesquita P., Jonckheere N., Almeida R., et al. Human MUC2 mucin gene is transcriptionally regulated by Cdx homeodomain proteins in gastrointestinal carcinoma cell lines. The Journal of Biological Chemistry. 2003;278(51):51549–51556. doi: 10.1074/jbc.m309019200. [DOI] [PubMed] [Google Scholar]
- 17.Taylor J. K., Levy T., Suh E. R., Traber P. G. Activation of enhancer elements by the homeobox gene Cdx2 is cell line specific. Nucleic Acids Research. 1997;25(12):2293–2300. doi: 10.1093/nar/25.12.2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.César A. C. G., Borim A. A., Caetano A., Cury P. M., Silva A. E. Aneuploidies, deletion, and overexpression of TP53 gene in intestinal metaplasia of patients without gastric cancer. Cancer Genetics and Cytogenetics. 2004;153(2):127–132. doi: 10.1016/j.cancergencyto.2004.01.017. [DOI] [PubMed] [Google Scholar]
- 19.Shiao Y.-H., Rugge M., Correa P., Lehmann H. P., Scheer W. D. p53 Alteration in gastric precancerous lesions. American Journal of Pathology. 1994;144(3):511–517. [PMC free article] [PubMed] [Google Scholar]
- 20.Ochiai A., Yamauchi Y., Hirohashi S. p53 mutations in the non-neoplastic mucosa of the human stomach showing intestinal metaplasia. International Journal of Cancer. 1996;69(1):28–33. doi: 10.1002/(SICI)1097-0215(19960220)69:1<28::AID-IJC6>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 21.Gomyo Y., Osaki M., Kaibara N., Ito H. Numerical aberration and point mutation of p53 gene in human gastric intestinal metaplasia and well-differentiated adenocarcinoma: analysis by fluorescence in situ hybridization (FISH) and PCR-SSCP. International Journal of Cancer. 1996;66(5):594–599. doi: 10.1002/(sici)1097-0215(19960529)66:5lt;594::aid-ijc262;3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 22.Morgan C., Jenkins G. J. S., Ashton T., et al. Detection of p53 mutations in precancerous gastric tissue. British Journal of Cancer. 2003;89(7):1314–1319. doi: 10.1038/sj.bjc.6601302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kobayashi K., Okamoto T., Takayama S., Akiyama M., Ohno T., Yamada H. Genetic instability in intestinal metaplasia is a frequent event leading to well-differentiated early adenocarcinoma of the stomach. European Journal of Cancer. 2000;36(9):1113–1119. doi: 10.1016/S0959-8049(00)00066-6. [DOI] [PubMed] [Google Scholar]
- 24.Ling X.-L., Fang D.-C., Wang R.-Q., Yang S.-M., Fang L. Mitochondrial microsatellite instability in gastric cancer and its precancerous lesions. World Journal of Gastroenterology. 2004;10(6):800–803. doi: 10.3748/wjg.v10.i6.800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hamamoto T., Yokozaki H., Semba S., et al. Altered microsatellites in incomplete-type intestinal metaplasia adjacent to primary gastric cancers. Journal of Clinical Pathology. 1997;50(10):841–846. doi: 10.1136/jcp.50.10.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hinoi T., Lucas P. C., Kuick R., Hanash S., Cho K. R., Fearon E. R. CDX2 regulates liver intestine-cadherin expression in normal and malignant colon epithelium and intestinal metaplasia. Gastroenterology. 2002;123(5):1565–1577. doi: 10.1053/gast.2002.36598. [DOI] [PubMed] [Google Scholar]
- 27.Hinnebusch B. F., Siddique A., Henderson J. W., et al. Enterocyte differentiation marker intestinal alkaline phosphatase is a target gene of the gut-enriched Krüppel-like factor. The American Journal of Physiology—Gastrointestinal and Liver Physiology. 2004;286(1):G23–G30. doi: 10.1152/ajpgi.00203.2003. [DOI] [PubMed] [Google Scholar]
- 28.Taupin D., Pedersen J., Familari M., Cook G., Yeomans N., Giraud A. S. Augmented intestinal trefoil factor (TFF3) and loss of pS2 (TFF1) expression precedes metaplastic differentiation of gastric epithelium. Laboratory Investigation. 2001;81(3):397–408. doi: 10.1038/labinvest.3780247. [DOI] [PubMed] [Google Scholar]
- 29.Zweibaum A., Triadou N., Kedinger M., et al. Sucrase-isomaltase: a marker of foetal and malignant epithelial cells of the human colon. International Journal of Cancer. 1983;32(4):407–412. doi: 10.1002/ijc.2910320403. [DOI] [PubMed] [Google Scholar]
- 30.Shiroshita H., Watanabe H., Ajioka Y., Watanabe G., Nishikura K., Kitano S. Re-evaluation of mucin phenotypes of gastric minute well-differentiated-type adenocarcinomas using a series of HGM, MUC5AC, MUC6, M-GGMC, MUC2 and CD10 stains. Pathology International. 2004;54(5):311–321. doi: 10.1111/j.1440-1827.2004.01625.x. [DOI] [PubMed] [Google Scholar]
- 31.Tatematsu M., Tsukamoto T., Inada K. Stem cells and gastric cancer: role of gastric and intestinal mixed intestinal metaplasia. Cancer Science. 2003;94(2):135–141. doi: 10.1111/j.1349-7006.2003.tb01409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamamoto H., Bai Y.-Q., Yuasa Y. Homeodomain protein CDX2 regulates goblet-specific MUC2 gene expression. Biochemical and Biophysical Research Communications. 2003;300(4):813–818. doi: 10.1016/s0006-291x(02)02935-2. [DOI] [PubMed] [Google Scholar]
- 33.Faller G., Kirchner T. Immunological and morphogenic basis of gastric mucosa atrophy and metaplasia. Virchows Archiv. 2005;446(1):1–9. doi: 10.1007/s00428-004-1157-3. [DOI] [PubMed] [Google Scholar]
- 34.Li Q.-L., Ito K., Sakakura C., et al. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109(1):113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- 35.Tsukamoto T., Mizoshita T., Tatematsu M. Gastric-and-intestinal mixed-type intestinal metaplasia: aberrant expression of transcription factors and stem cell intestinalization. Gastric Cancer. 2006;9(3):156–166. doi: 10.1007/s10120-006-0375-6. [DOI] [PubMed] [Google Scholar]
- 36.Ohnishi S., Ma N., Thanan R., et al. DNA damage in inflammation-related carcinogenesis and cancer stem cells. Oxidative Medicine and Cellular Longevity. 2013;2013:9. doi: 10.1155/2013/387014.387014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto Y., Marusawa H., Kinoshita K., et al. Helicobacter pylori infection triggers aberrant expression of activation-induced cytidine deaminase in gastric epithelium. Nature Medicine. 2007;13(4):470–476. doi: 10.1038/nm1566. [DOI] [PubMed] [Google Scholar]
- 38.Yamane A., Resch W., Kuo N., et al. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nature Immunology. 2011;12(1):62–69. doi: 10.1038/ni.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Honjo T., Kinoshita K., Muramatsu M. Molecular mechanism of class switch recombination: linkage with somatic hypermutation. Annual Review of Immunology. 2002;20:165–196. doi: 10.1146/annurev.immunol.20.090501.112049. [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto T., Marusawa H., Endo Y., Ueda Y., Matsumoto Y., Chiba T. Expression of APOBEC2 is transcriptionally regulated by NF-κB in human hepatocytes. FEBS Letters. 2006;580(3):731–735. doi: 10.1016/j.febslet.2005.12.081. [DOI] [PubMed] [Google Scholar]
- 41.Maekita T., Nakazawa K., Mihara M., et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clinical Cancer Research. 2006;12(3, part 1):989–995. doi: 10.1158/1078-0432.ccr-05-2096. [DOI] [PubMed] [Google Scholar]
- 42.Nakajima T., Yamashita S., Maekita T., Niwa T., Nakazawa K., Ushijima T. The presence of a methylation fingerprint of Helicobacter pylori infection in human gastric mucosae. International Journal of Cancer. 2009;124(4):905–910. doi: 10.1002/ijc.24018. [DOI] [PubMed] [Google Scholar]
- 43.Niwa T., Tsukamoto T., Toyoda T., et al. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Research. 2010;70(4):1430–1440. doi: 10.1158/0008-5472.can-09-2755. [DOI] [PubMed] [Google Scholar]
- 44.Leung W. K., Man E. P. S., Yu J., et al. Effects of Helicobacter pylori eradication on methylation status of E-cadherin gene in noncancerous stomach. Clinical Cancer Research. 2006;12(10):3216–3221. doi: 10.1158/1078-0432.ccr-05-2442. [DOI] [PubMed] [Google Scholar]
- 45.Perri F., Cotugno R., Piepoli A., et al. Aberrant DNA methylation in non-neoplastic gastric mucosa of H. pylori infected patients and effect of eradication. American Journal of Gastroenterology. 2007;102(7):1361–1371. doi: 10.1111/j.1572-0241.2007.01284.x. [DOI] [PubMed] [Google Scholar]
- 46.Hur K., Niwa T., Toyoda T., et al. Insufficient role of cell proliferation in aberrant DNA methylation induction and involvement of specific types of inflammation. Carcinogenesis. 2011;32(1):35–41. doi: 10.1093/carcin/bgq219. [DOI] [PubMed] [Google Scholar]
- 47.Nakajima T., Enomoto S., Yamashita S., et al. Persistence of a component of DNA methylation in gastric mucosae after Helicobacter pylori eradication. Journal of Gastroenterology. 2010;45(1):37–44. doi: 10.1007/s00535-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 48.Park S.-Y., Yoo E. J., Cho N.-Y., Kim N., Kang G. H. Comparison of CpG island hypermethylation and repetitive DNA hypomethylation in premalignant stages of gastric cancer, stratified for Helicobacter pylori infection. Journal of Pathology. 2009;219(4):410–416. doi: 10.1002/path.2596. [DOI] [PubMed] [Google Scholar]
- 49.Zong L., Seto Y. CpG island methylator phenotype, Helicobacter pylori, Epstein-Barr virus, and microsatellite instability and prognosis in gastric cancer: a systematic review and meta-analysis. PLoS ONE. 2014;9(1) doi: 10.1371/journal.pone.0086097.e86097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee B. L., Lee H. S., Jung J., et al. Nuclear factor-kappaB activation correlates with better prognosis and Akt activation in human gastric cancer. Clinical Cancer Research. 2005;11(7):2518–2525. doi: 10.1158/1078-0432.CCR-04-1282. [DOI] [PubMed] [Google Scholar]
- 51.Tomb J. F., White O., Kerlavage A. R., et al. The complete genome sequence of the gastric pathogen Helicobacter pylori . Nature. 1997;388(6642):539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 52.Covacci A., Telford J. L., Del Giudice G., Parsonnet J., Rappuoli R. Helicobacter pylori virulence and genetic geography. Science. 1999;284(5418):1328–1333. doi: 10.1126/science.284.5418.1328. [DOI] [PubMed] [Google Scholar]
- 53.Odenbreit S., Püls J., Sedlmaier B., Gerland E., Fischer W., Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287(5457):1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 54.Viala J., Chaput C., Boneca I. G., et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature Immunology. 2004;5(11):1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- 55.Hirata Y., Ohmae T., Shibata W., et al. MyD88 and TNF receptor-associated factor 6 are critical signal transducers in Helicobacter pylori-infected human epithelial cells. Journal of Immunology. 2006;176(6):3796–3803. doi: 10.4049/jimmunol.176.6.3796. [DOI] [PubMed] [Google Scholar]
- 56.Maeda S., Yoshida H., Ogura K., et al. H. pylori activates NF-κB through a signaling pathway involving IκB kinases, NF-κB-inducing kinase, TRAF2, and TRAF6 in gastric cancer cells. Gastroenterology. 2000;119(1):97–108. doi: 10.1053/gast.2000.8540. [DOI] [PubMed] [Google Scholar]
- 57.Lamb A., Chen L.-F. Role of the Helicobacter pylori-Induced inflammatory response in the development of gastric cancer. Journal of Cellular Biochemistry. 2013;114(3):491–497. doi: 10.1002/jcb.24389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lamb A., Chen L.-F. The many roads traveled by Helicobacter pylori to NFκB activation. Gut Microbes. 2010;1(2):109–113. doi: 10.4161/gmic.1.2.11587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shibata W., Hirata Y., Maeda S., et al. CagA protein secreted by the intact type IV secretion system leads to gastric epithelial inflammation in the Mongolian gerbil model. Journal of Pathology. 2006;210(3):306–314. doi: 10.1002/path.2040. [DOI] [PubMed] [Google Scholar]
- 60.Rieder G., Merchant J. L., Haas R. Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in mongolian gerbils. Gastroenterology. 2005;128(5):1229–1242. doi: 10.1053/j.gastro.2005.02.064. [DOI] [PubMed] [Google Scholar]
- 61.Brandt S., Kwok T., Hartig R., König W., Backert S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(26):9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Noach L. A., Bosma N. B., Jansen J., Hoek F. J., van Deventer S. J. H., Tytgat G. N. J. Mucosal tumor necrosis factor-α interleukin-1β, and interleukin-8 production in patients with Helicobacter pylori infection. Scandinavian Journal of Gastroenterology. 1994;29(5):425–429. doi: 10.3109/00365529409096833. [DOI] [PubMed] [Google Scholar]
- 63.Fan X.-G., Chua A., Fan X.-J., Keeling P. W. N. Increased gastric production of interleukin-8 and tumour necrosis factor in patients with Helicobacter pylori infection. Journal of Clinical Pathology. 1995;48(2):133–136. doi: 10.1136/jcp.48.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Basso D., Scrigner M., Toma A., et al. Helicobacter pylori infection enhances mucosal interleukin-1 beta, interleukin-6, and the soluble receptor of interleukin-2. International Journal of Clinical and Laboratory Research. 1996;26(3):207–210. doi: 10.1007/bf02592984. [DOI] [PubMed] [Google Scholar]
- 65.Yamaoka Y., Kita M., Kodama T., Sawai N., Kashima K., Imanishi J. Induction of various cytokines and development of severe mucosal inflammation by cagA gene positive Helicobacter pylori strains. Gut. 1997;41(4):442–451. doi: 10.1136/gut.41.4.442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Maeda S., Omata M. Inflammation and cancer: role of nuclear factor-kappaB activation. Cancer Science. 2008;99(5):836–842. doi: 10.1111/j.1349-7006.2008.00763.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yanai A., Maeda S., Shibata W., et al. Activation of IκB kinase β and NF-κB is essential for Helicobacter pylori-induced chronic gastritis in Mongolian gerbils. Infection and Immunity. 2008;76(2):781–787. doi: 10.1128/iai.01046-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kinoshita H., Hirata Y., Nakagawa H., et al. Interleukin-6 mediates epithelial-stromal interactions and promotes gastric tumorigenesis. PLoS ONE. 2013;8(4) doi: 10.1371/journal.pone.0060914.e60914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tebbutt N. C., Giraud A. S., Inglese M., et al. Reciprocal regulation of gastrointestinal homeostasis by SHP2 and STAT-mediated trefoil gene activation in gp130 mutant mice. Nature Medicine. 2002;8(10):1089–1097. doi: 10.1038/nm763. [DOI] [PubMed] [Google Scholar]
- 70.Howlett M., Menheniott T. R., Judd L. M., Giraud A. S. Cytokine signalling via gp130 in gastric cancer. Biochimica et Biophysica Acta. 2009;1793(11):1623–1633. doi: 10.1016/j.bbamcr.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 71.Kanda N., Seno H., Konda Y., et al. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 2004;23(28):4921–4929. doi: 10.1038/sj.onc.1207606. [DOI] [PubMed] [Google Scholar]
- 72.Lee I. O., Kim J. H., Choi Y. J., et al. Helicobacter pylori CagA phosphorylation status determines the gp130-activated SHP2/ERK and JAK/STAT signal transduction pathways in gastric epithelial cells. The Journal of Biological Chemistry. 2010;285(21):16042–16050. doi: 10.1074/jbc.m110.111054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang T. C., Goldenring J. R. Inflammation intersection: gp130 balances gut irritation and stomach cancer. Nature Medicine. 2002;8(10):1080–1082. doi: 10.1038/nm1002-1080. [DOI] [PubMed] [Google Scholar]
- 74.Ernst M., Najdovska M., Grail D., et al. STAT3 and STAT1 mediate IL-11-dependent and inflammation-associated gastric tumorigenesis in gp130 receptor mutant mice. Journal of Clinical Investigation. 2008;118(5):1727–1738. doi: 10.1172/JCI34944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Saadat I., Higashi H., Obuse C., et al. Helicobacter pylori CagA targets PAR1/MARK kinase to disrupt epithelial cell polarity. Nature. 2007;447(7142):330–333. doi: 10.1038/nature05765. [DOI] [PubMed] [Google Scholar]
- 76.Saito Y., Murata-Kamiya N., Hirayama T., Ohba Y., Hatakeyama M. Conversion of Helicobacter pylori CagA from senescence inducer to oncogenic driver through polarity-dependent regulation of p21. Journal of Experimental Medicine. 2010;207(10):2157–2174. doi: 10.1084/jem.20100602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nature Immunology. 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 78.Bollrath J., Greten F. R. IKK/NF‐κB and STAT3 pathways: central signalling hubs in inflammation-mediated tumour promotion and metastasis. The EMBO Reports. 2009;10(12):1314–1319. doi: 10.1038/embor.2009.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shaykhiev R., Behr J., Bals R. Microbial patterns signaling via toll-like receptors 2 and 5 contribute to epithelial repair, growth and survival. PLoS ONE. 2008;3(1) doi: 10.1371/journal.pone.0001393.e1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.West X. Z., Malinin N. L., Merkulova A. A., et al. Oxidative stress induces angiogenesis by activating TLR2 with novel endogenous ligands. Nature. 2010;467(7318):972–976. doi: 10.1038/nature09421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Uno K., Kato K., Atsumi T., et al. Toll-like receptor (TLR) 2 induced through TLR4 signaling initiated by Helicobacter pylori cooperatively amplifies iNOS induction in gastric epithelial cells. American Journal of Physiology—Gastrointestinal and Liver Physiology. 2007;293(5):G1004–G1012. doi: 10.1152/ajpgi.00096.2007. [DOI] [PubMed] [Google Scholar]
- 82.Hold G. L., Rabkin C. S., Chow W.-H., et al. A functional polymorphism of toll-like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology. 2007;132(3):905–912. doi: 10.1053/j.gastro.2006.12.026. [DOI] [PubMed] [Google Scholar]
- 83.Tye H., Kennedy C. L., Najdovska M., et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012;22(4):466–478. doi: 10.1016/j.ccr.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 84.Hayakawa Y., Hirata Y., Kinoshita H., et al. Differential roles of ASK1 and TAK1 in Helicobacter pylori-induced cellular responses. Infection and Immunity. 2013;81(12):4551–4560. doi: 10.1128/iai.00914-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Shibata W., Maeda S., Hikiba Y., et al. c-Jun NH2-terminal kinase 1 is a critical regulator for the development of gastric cancer in mice. Cancer Research. 2008;68(13):5031–5039. doi: 10.1158/0008-5472.can-07-6332. [DOI] [PubMed] [Google Scholar]
- 86.Hayakawa Y., Hirata Y., Nakagawa H., et al. Apoptosis signal-regulating kinase 1 and cyclin D1 compose a positive feedback loop contributing to tumor growth in gastric cancer. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(2):780–785. doi: 10.1073/pnas.1011418108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Salama N. R., Hartung M. L., Müller A. Life in the human stomach: persistence strategies of the bacterial pathogen Helicobacter pylori . Nature Reviews Microbiology. 2013;11(6):385–399. doi: 10.1038/nrmicro3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ismail H. F., Fick P., Zhang J., Lynch R. G., Berg D. J. Depletion of neutrophils in IL-10-/- mice delays clearance of gastric Helicobacter infection and decreases the Th1 immune response to Helicobacter . Journal of Immunology. 2003;170(7):3782–3789. doi: 10.4049/jimmunol.170.7.3782. [DOI] [PubMed] [Google Scholar]