

Abstract
Background
Primary central nervous system post-transplant lymphoproliferative disorder (PCNS-PTLD) is a rare complication following solid organ transplantation (SOT). With increasing rates of SOT, PCNS-PTLD incidence is increasing.
Objective
Describe the characteristics of PCNS-PTLD patients requiring neurosurgical intervention.
Methods
From 2000-2011 ten patients with prior SOT underwent biopsy for evaluation of brain lesions and were diagnosed with PCNS-PTLD. Data collected included imaging characteristics, pathology, treatments administered, and survival outcomes.
Results
All patients had kidney transplantation, and 3 had concurrent pancreas transplantation. Median age at diagnosis was 49 years, with a median of 4.5 years from SOT to diagnosis (range 1.8-11.4 years). Presenting symptoms most often included focal neurologic deficits (n=6), although several patients had non-specific symptoms of headache and altered mental status. Brain lesions were generally multiple (n=7), supratentorial (n=8), and lobar or periventricular in distribution with ring-enhancement. Diagnosis was established by stereotactic (n=4) and open surgical biopsy (n=6). Treatments most frequently administered included reduction of immunosuppression (n=10), dexamethasone (n=10), rituximab (n=8), high-dose methotrexate (n=3), and whole-brain radiotherapy (n=6). Six patients remain alive without PCNS-PTLD relapse, including 4 patients who have sustained remissions beyond 2 years from diagnosis of PCNS-PTLD. Of 4 observed deaths, 1 was related to progressive PCNS-PTLD.
Conclusion
PCNS-PTLD must be considered in the differential diagnosis of any patient with prior SOT presenting with an intracranial lesion. Histologic diagnosis with brain biopsy is imperative given the risk for opportunistic infections that may have similar imaging findings and presentation. Prognosis is variable, although long-term survival has been reported.
Keywords: post-transplant lymphoproliferative disorder, immunosuppressed, Epstein-Barr virus, lymphoma, radiotherapy, rituximab
Introduction
Lymphomas account for approximately 1% of all primary brain tumors in the general population, but lymphoma is the most common primary neoplasm of the brain in allograft transplant recipients.1 Autopsy series of transplant patients have estimated the frequency of primary central nervous system post-transplant lymphoproliferative disorder (PCNS-PTLD) at 2 to 7%; however, this may be an over-estimate of the incidence of clinical disease.1-3 Due to the increasing number of successful organ transplantation procedures being performed, the incidence of post-transplant lymphoproliferative disorder (PTLD) is rising, and a broad range of medical and surgical specialties are increasingly being called upon to recognize this entity and respond with appropriate diagnostic and therapeutic approaches.
As PCNS-PTLD is a relatively uncommon complication post-transplantation, most literature related to PCNS-PTLD is in the form of isolated case reports and small case series. Here we present a single-institution case series of 10 patients. The report focuses on clinical presentation, imaging findings, pathological characteristics, and treatment modalities in patients with PCNS-PTLD.
Methods
PCNS-PTLD is defined as a lymphoproliferative disorder of the central nervous system (CNS) without evidence of systemic PTLD in patients who have previously undergone solid organ transplantation (SOT). This retrospective analysis was approved by the Institutional Review Board of the University of Wisconsin. Patients were selected by reviewing the pathologic diagnoses for all patients who underwent brain biopsy at the University of Wisconsin Hospital between 2000 through 2011, and identifying patients who received a diagnosis of PCNS-PTLD. Our pathologists reviewed all diagnoses. Magnetic resonance imaging (MRI) and computed tomography (CT) imaging were available for review in all patients. Imaging characteristics identified for each patient included lesion number, location, and presence or absence of contrast enhancement. All patients had undergone staging CT imaging to rule out systemic involvement by lymphoma. Supplemental staging evaluation with bone marrow biopsy, lumbar puncture, ophthalmologic evaluation, and full-spine MRI were not uniform. Clinical data regarding risk factors for PTLD were reviewed, including prior treatment with anti-thymocyte globulin (ATG) or muromonab-CD3 (OKT3) and prior episodes of allograft rejection. Data were also collected regarding clinical, radiographic, and pathologic features of PTLD as well as information relative to treatment and survival outcomes.
Illustrative cases
Case 1
History and examination
A 38 year-old Hispanic woman presented with a 2-week history of progressive left-sided facial weakness and numbness and left arm weakness. Her past medical history was significant for uncontrolled hypertension leading to renal failure and a living unrelated kidney transplant 21 months prior to the onset of symptoms. She had received induction immunosuppression with ATG pre-transplantation. Her immunosuppressive regimen post-transplantation included prednisone, tacrolimus, and mycophenolate, and she had not developed any events of organ rejection post-transplantation. Her physical exam at the time of presentation was significant for 4 of 5 left-sided strength testing of both upper and lower extremities. Cranial nerve testing was normal aside from some diminished sensation to light touch of the left hemi-face, although with symmetric movement of the facies.
She was admitted for evaluation of her neurologic complaints, and brain MRI showed multiple enhancing intracranial masses. She underwent an infectious work-up looking for opportunistic infections and embolic sources of infection, with negative results. Lumbar puncture cytology was negative. Her tacrolimus was empirically discontinued due to concerns for PTLD, with some initial improvement in her left-sided weakness. She underwent repeat brain MRI 10 days later when new symptoms of progressive left leg weakness and balance instability developed.
Brain MRI studies with and without gadolinium contrast were performed at initial presentation and repeated when her symptoms progressed to left leg weakness. Both studies demonstrated a total of 4 enhancing intraparenchymal lesions, 3 of which were located in the right thalamus and a fourth lesion within the right frontal lobe. Vasogenic edema was seen surrounding all lesions. The largest right thalamic lesion measured approximately 2 cm in size, with several smaller satellite lesions present (Figure 1). Leptomeningeal enhancement was seen within the bilateral parietal and occipital lobes, which had progressed on the follow-up MR examination (Figure 2). Full spine MRI and dilated ophthalmologic exam did not show any additional disease sites within the neuraxis. Staging CT imaging and bone marrow biopsy ruled out systemic involvement by PTLD.
Figure 1.
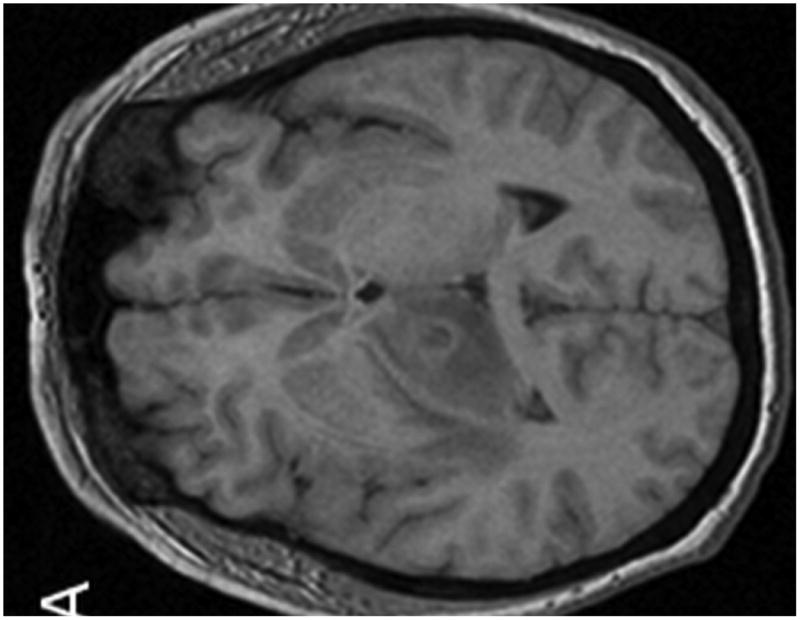
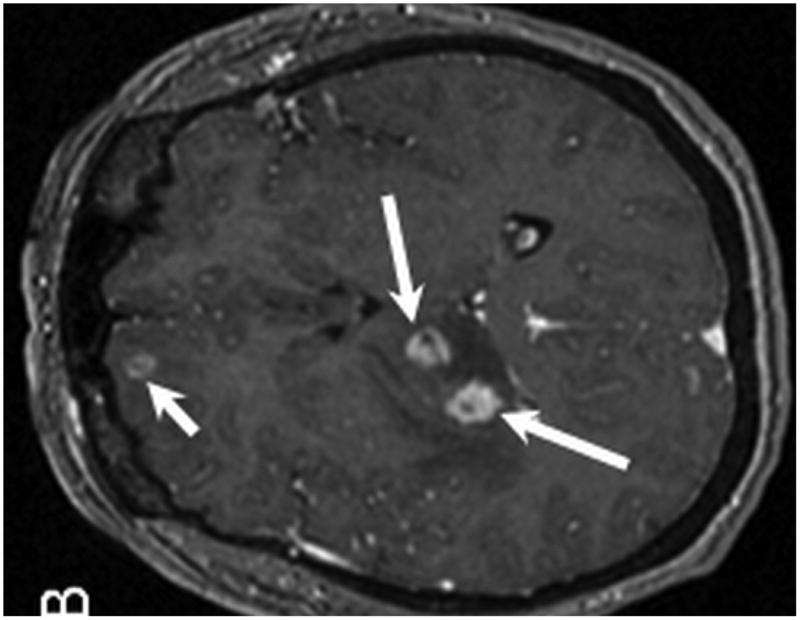
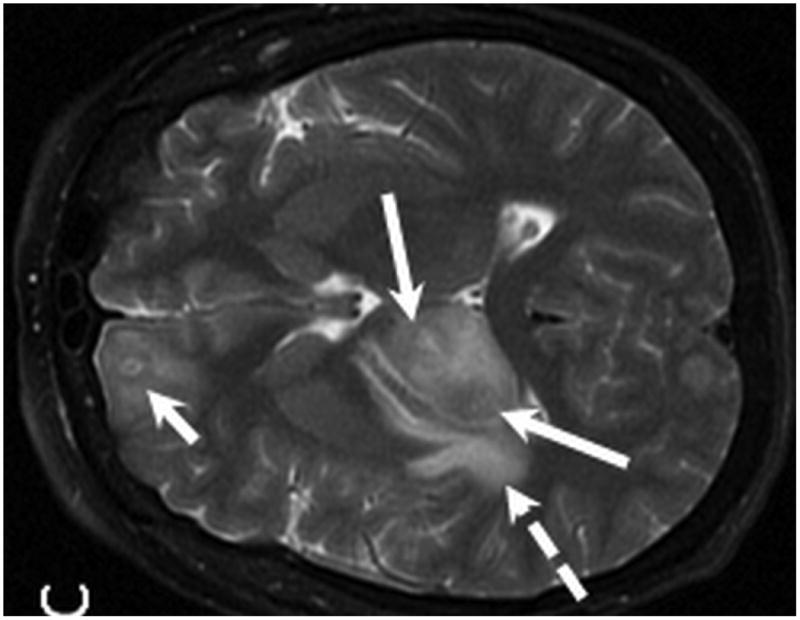
MR examination from Case 1 at the time of initial presentation. Axial T1 (A), axial T1 post contrast (B) and axial T2 weighted MR images of the brain demonstrate 3 of the patient's intraparenchymal masses. These lesions demonstrate peripheral enhancement (B) and are slightly hyperintense on T2 (C) . Two lesions are located within the right thalamus (B and C, long arrows) and one is identified within the right frontal lobe (B, C, short arrows). Note the T2 hyperintense vasogenic edema surrounding the lesions (C, dashed arrow).
Figure 2.
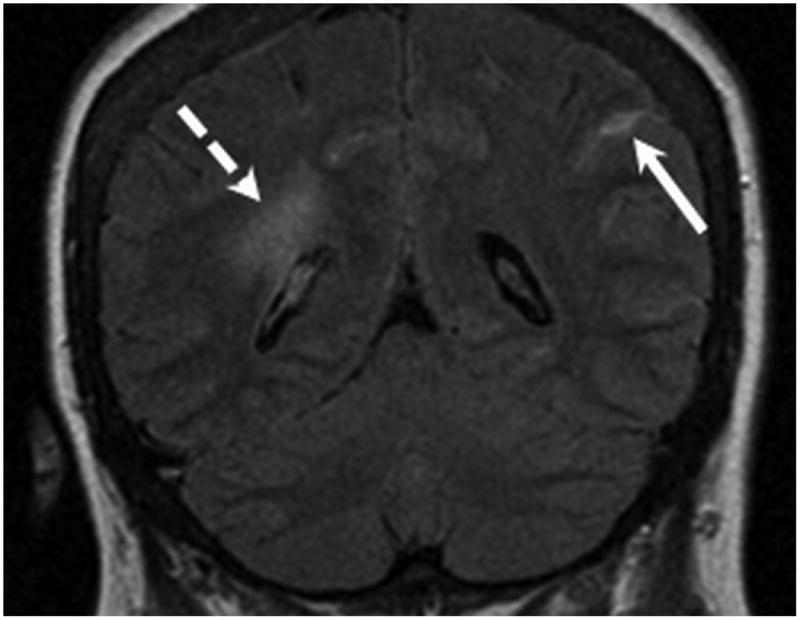
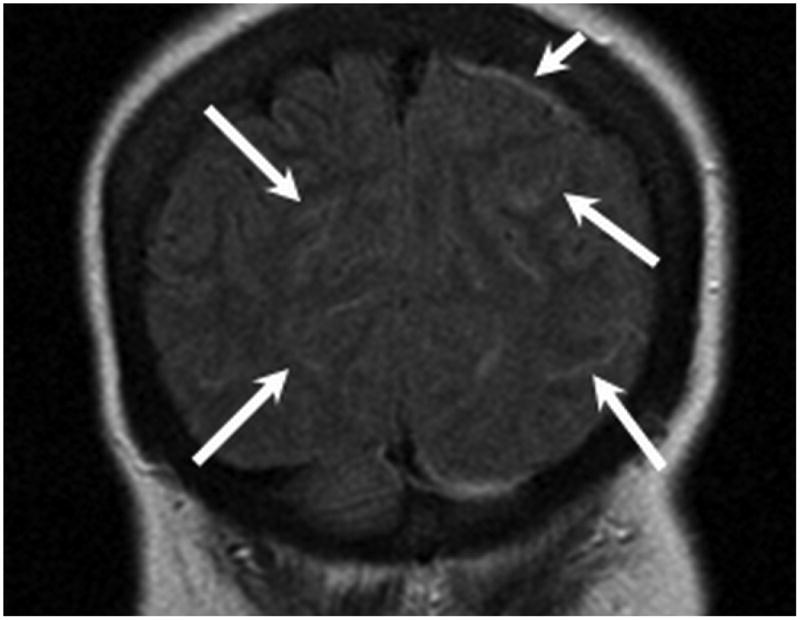
MR examination from Case 1 obtained 10 days following initial presentation. Coronal FLAIR MR images (A,B) were obtained following contrast which demonstrate abnormal signal within multiple bilateral parietal and occipital lobe sulci (long arrows) consistent with leptomeningeal involvement. Pachymeningeal involvement is also present adjacent to the left parietal lobe (B, short arrow). As in Figure 1, note the vasogenic edema within the peri-atrial white matter related to the thalamic lesions (B, dashed arrow).
A stereotactic-guided biopsy was performed via a right frontal approach on one of the enhancing right thalamic lesions. Pathological examination of the lesion revealed a monomorphic high-grade B-cell PTLD/lymphoma; immunohistochemistry analysis was positive for the mature B-cell marker CD20 and for EBV-encoded RNA (EBER). Repeat lumbar puncture after the diagnostic brain biopsy confirmed positive CSF cytology, with presence of 22 nucleated cells (>90% lymphocytes) and morphologic appearance of large atypical lymphocytes.
After confirmation of the diagnosis of PTLD, tacrolimus and mycophenolate were discontinued. Dexamethasone 10 mg intravenously (IV) followed by 4 mg orally every 6 hours was initiated based on the associated vasogenic edema demonstrated on imaging. Given her young age and progressive symptoms, she was treated urgently with high-dose methotrexate 3.5 grams/m2 intravenous (IV) in combination with 4 weekly doses of rituximab 375 mg/m2 IV, with initial improvement in her left-sided weakness. After 3 cycles of high-dose methotrexate and a 4 week course of rituximab, she had significant regression in the multiple intraparenchymal brain lesions and clinically was experiencing minimal residual left-sided weakness. She then received external beam whole-brain radiation to a total dose of 37 Gy with an additional boost to the residual thalamic lesions. Post-radiotherapy, she received 2 courses of consolidative chemotherapy with temozolomide 150 mg/m2 orally days 1-5 of 28-day treatment cycles. Following chemoradiotherapy, she continued to have some mild residual left-sided weakness. Brain MRI confirmed residual T2 hyperintensity within the posterior right frontal lobe, parietal lobe, and right thalamus at the site of previously demonstrated PTLD lesions.
Serial imaging and follow-up for more than 3 years following the diagnosis of PTLD has demonstrated no evidence of recurrence. With reduction of immunosuppression to low-dose steroids alone, she has not developed any organ allograft rejection. Unfortunately, she developed worsening left hemi-body weakness with left upper extremity spasticity and hyperreflexia post-treatment, and brain imaging has shown progressive Wallerian degeneration most prominent at her sites of prior PTLD involvement (Figure 3).
Figure 3.
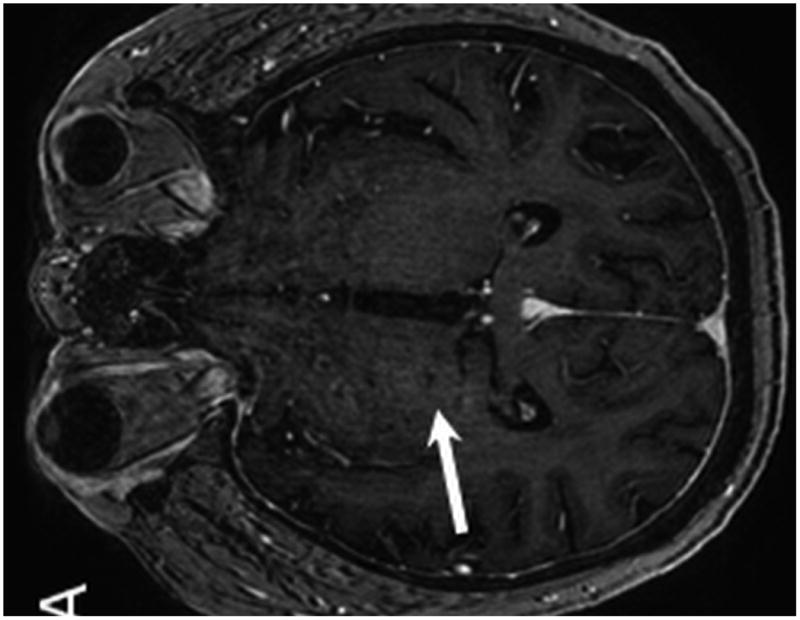
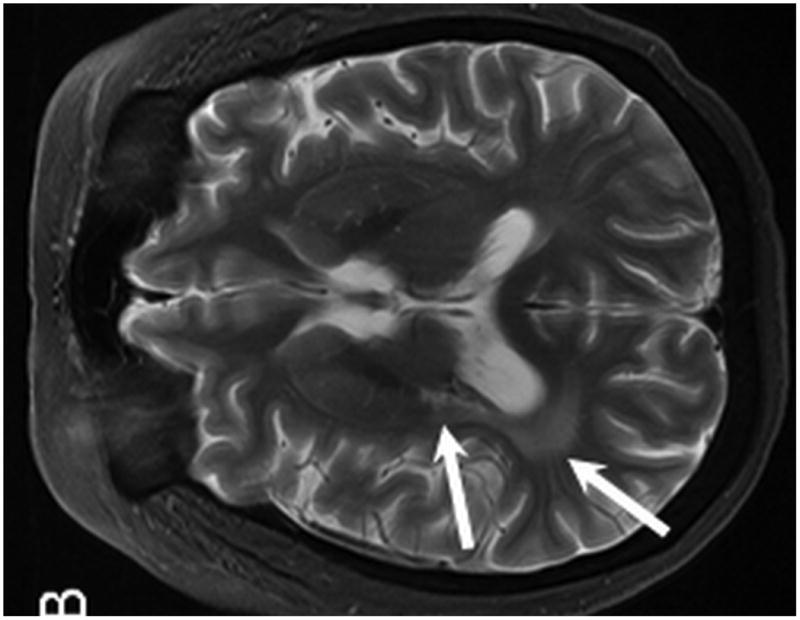
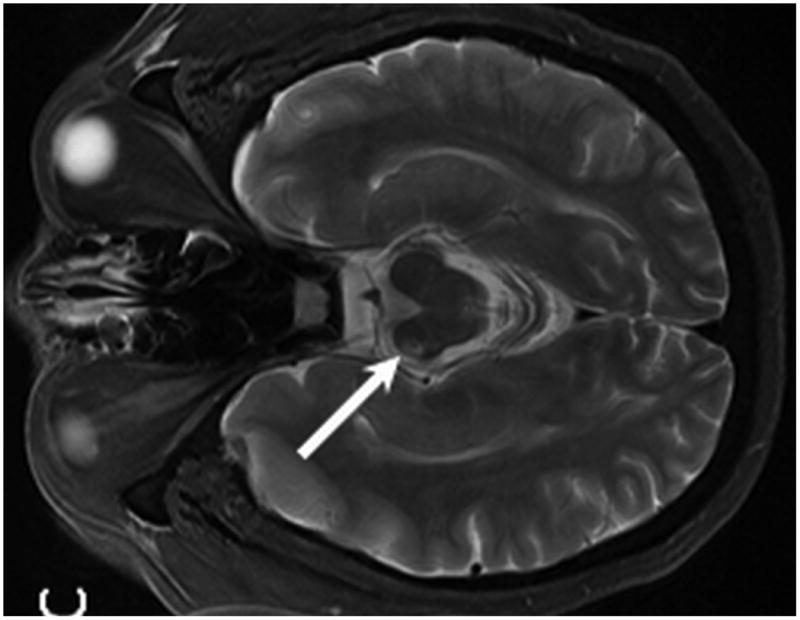
MR from Case 1 obtained 3.5 years following treatment. Axial T1 post contrast MR (A) shows no evidence of enhancement within the areas of prior PTLD within the right thalamus (A, arrow). Axial T2 W MR images (B, C) demonstrate interval development of abnormal T2 signal hyperintensity within the peri-atrial white matter and posterior limb of the right internal capsule consistent with post treatment leukoencephalopathy (B, arrows). Note the resultant Wallerian degeneration within the right cerebral peduncle (C, arrow).
Case 2
A 44 year-old Caucasian man presented with a 10-day history of nausea, vomiting and lethargy. His past medical history was significant for type 1 diabetes mellitus leading to kidney-pancreas transplantation approximately 5.8 years previously. His immunosuppressive regimen post-transplantation included prednisone, cyclosporine, and mycophenolate, and he had received additional courses of high-dose steroids, immune globulin, and rituximab for several events of prior acute allograft rejection. He was admitted to another hospital for evaluation of these complaints, and neurologic examination demonstrated no focal abnormalities. An initial brain MRI showed multiple intraparenchymal enhancing lesions. He was empirically treated for toxoplasmosis with sulfadiazine and pyrimethamine without clinical improvement, and with subsequent development of intermittent confusion and headaches. Serology and serum quantitative PCR analysis for toxoplasmosis were found to be negative, but analysis of serum quantitative titers of EBV by PCR analysis were elevated at 1340 copies/mL. PTLD was suspected and the patient was transferred to our facility for further treatment. On initial examination at our institution, the patient was alert and oriented with normal speech and symmetric strength; there were no focal neurologic abnormalities on detailed examination.
Brain MRI with and without gadolinium contrast revealed multiple intraparenchymal masses. The 2 largest enhancing lesions were identified within the right parietal lobe and left temporal lobe. The right parietal lobe mass exhibited peripheral enhancement and measured 1.6 × 1.3 cm with a significant amount of peritumoral vasogenic edema. This lesion had associated areas of hemosiderin staining on T2* consistent with petechial hemorrhage. The left temporal lobe mass was characterized by ill-defined enhancement and measured 1.7 × 1.6 cm with associated vasogenic edema (Figure 4). Additional lesions were identified within the left temporal lobe, left frontal lobe, corpus callosum/choroid plexus, left thalamus, and left caudate. Staging CT imaging did not reveal any sites of systemic PTLD involvement. A stereotactic-guided biopsy was performed on the large right parietal lesion. Pathological examination of the lesion revealed T-cell PTLD; immunohistochemistry analysis was positive for CD3 (pan T-cell marker) and EBER, and negative for the mature B-cell markers CD20 and CD79a.
Figure 4.
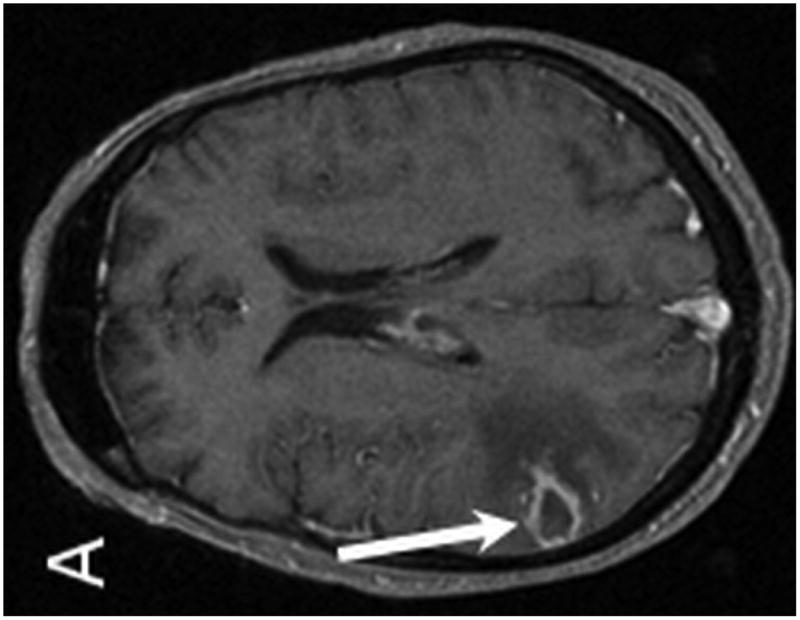
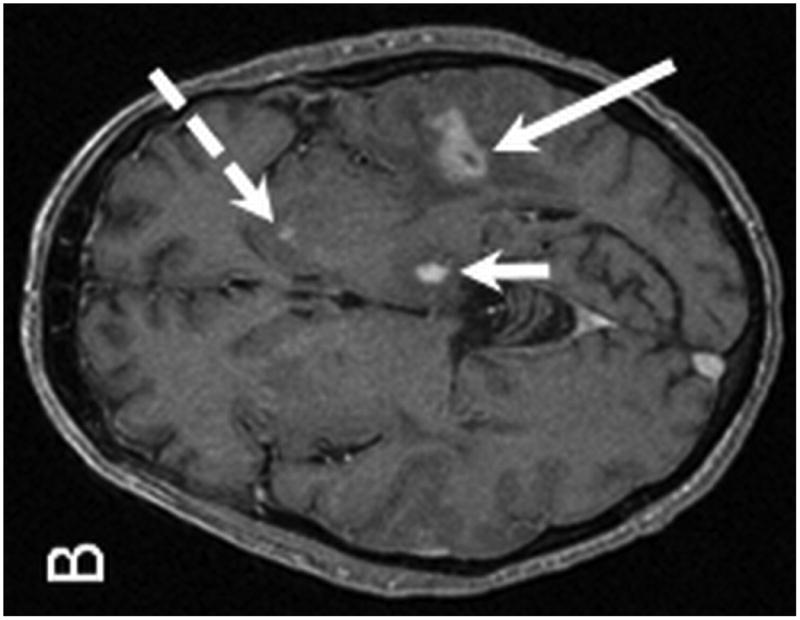
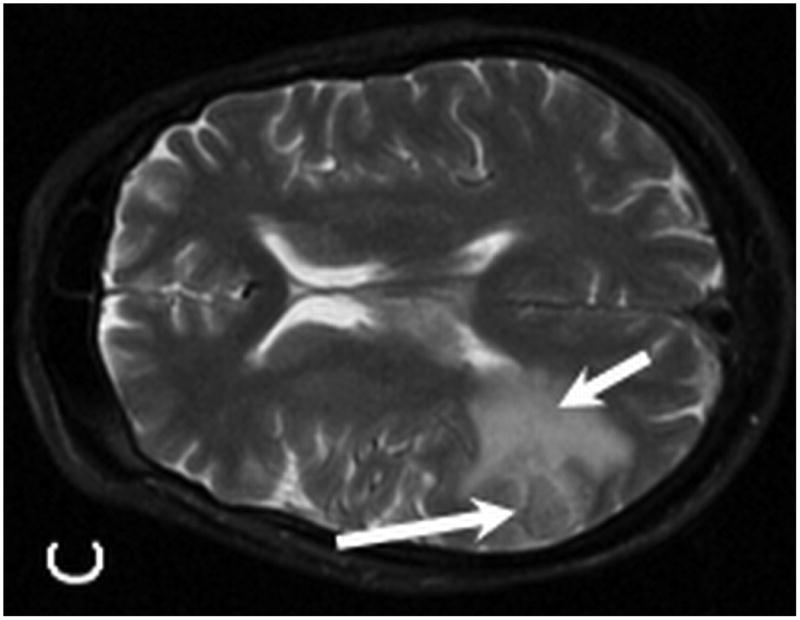
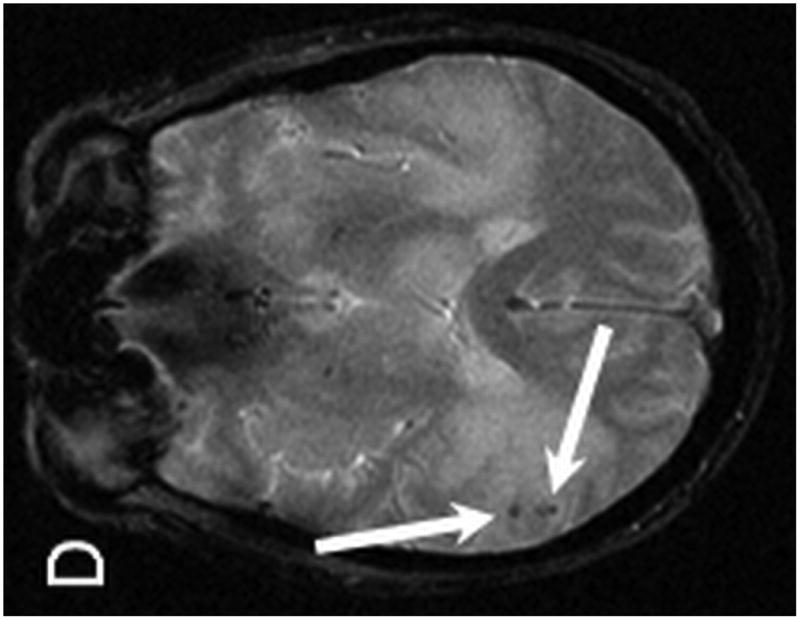
MR examination from Case 2 at the time of initial presentation. Axial T1 post contrast (A,B), axial T2 (C) and axial T2* (D) weighted MR images of the brain demonstrate four of the patient's enhancing intraparenchymal masses. The largest is located within the right parietal lobe and demonstrates peripheral enhancement (A, arrow). Additional smaller lesions are noted within the left temporal lobe (B, long arrow), left thalamus (B, short arrow) and left caudate (B, dashed arrow). The right dominant parietal lobe mass is slightly hyperintense on T2 (C, long arrow) with associated petechial hemorrhage noted on T2* (D, arrows). Note the extensive associated vasogenic edema surrounding the dominant lesion on T2 imaging (C, short arrow).
After confirmation of the diagnosis of PTLD, cyclosporine and mycophenolate were discontinued. Dexamethasone 10 mg IV followed by 4 mg orally every 6 hours was initiated based on the associated vasogenic edema demonstrated on imaging. The patient was started on valganciclovir due to circulating EBV in peripheral blood. He received external beam whole-brain radiation to a total dose of 30 Gy. At one month follow-up, he had complete resolution of his symptoms of headache, nausea, and confusion. Unfortunately, 5 months following the diagnosis of PTLD he had experienced allograft failure and was requiring hemodialysis and exogenous insulin, although had no radiographic evidence of PTLD relapse. He died from cardiovascular and infectious complications approximately 7 months following his diagnosis of PTLD.
Results
Participants
Ten patients were identified from our institution with biopsy-proven PCNS-PTLD (Table 1). The median age at diagnosis was 49 (range 38-67). Seven patients had undergone kidney transplantation, and 3 patients had undergone simultaneous kidney/pancreas transplantation. Among the 7 kidney transplant recipients, 1 underwent transplantation from a living related donor, 1 from a living unrelated donor, and 5 from cadaveric donors. Patients receiving allogeneic bone marrow transplantation were not actively excluded, but none of the patients receiving brain biopsies and subsequent diagnoses of PCNS-PTLD had undergone allogeneic bone marrow transplantation. Indications for SOT included hypertensive nephropathy (n=2), type I diabetes mellitus with diabetic nephropathy (n=4), focal segmental glomerulonephritis (n=2), lupus nephritis (n=1), and interstitial nephritis (n=1). None of the patients were known to have HIV disease, and had tested negative for HIV at the time of SOT. The median time from transplantation to diagnosis of intracranial PTLD was 4.5 years (range 1.8-11.4 years).
Table 1.
Demographics and Transplantation Data of 10 Patients with Primary CNS Post-Transplant Lymphoproliferative Disorder.
| Characteristic | |
|---|---|
| Sex | |
| Male | 4 (40%) |
| Female | 6 (60%) |
|
| |
| Age at diagnosis of PCNS-PTLD | Median 49 years (Range 38-67 years) |
|
| |
| Time from transplantation to PCNS-PTLD† | Median 4.5 years (Range 1.8-11.4 years) |
| <2 years | 3 (30%) |
| 2-5 years | 3 (30%) |
| ≥6 years | 4 (40%) |
|
| |
| Type of transplantation | |
| Kidney | 7 |
| Kidney/pancreas | 3 |
|
| |
| Indications for transplantation | |
| Type I diabetes | 4 |
| Hypertensive nephropathy | 2 |
| Focal segmental glomerulonephritis | 2 |
| Interstitial nephritis | 1 |
| Lupus Nephritis | 1 |
|
| |
| Presence of EBV viremia at diagnosis | |
| Undetectable | 1 |
| <500 copies/mL | 4 |
| 500-4000 copies/mL | 4 |
| >5000 copes/mL | 1 |
One patient had undergone repeat transplantation for recurrent focal segmental glomerulonephritis. Data indicate time from most recent transplant to diagnosis of PCNS-PTLD.
Abbreviations: PCNS-PTLD = Primary central nervous system post-transplant lymphoproliferative disorder.
Data on previous immunosuppressive therapies are shown in Table 2. Six patients had experienced previous episodes of organ allograft rejection with intensification of immunosuppression prior to the diagnosis of PCNS-PTLD.
Table 2.
Prior immunosuppressive therapy and EBV/CMV status of 10 patients with Primary CNS Post-Transplant Lymphoproliferative Disorder.
| N | |
|---|---|
| Prior rejection episodes of organ allograft | 9 (90%) |
| Steroid-refractory | 2 (20%) |
|
| |
| Prior exposure to immunosuppressive agents for treatment of organ rejection: | |
| OKT3 | 0 |
| ATG | 4† |
|
| |
| Immunosuppressive agents at time of PTLD diagnosis: | |
| Cyclosporine | 2 |
| Tacrolimus | 3 |
| Mycophenolate | 10 |
| Prednisone | 10 |
One patient had received ATG prophylactically as pre-transplant conditioning regimen.
Abbreviations: OKT3 = murine monoclonal antibody targeting CD3. ATG = anti-thymocyte globulin.
Main Results
Presenting symptoms
Presenting signs and symptoms were widely variable (Table 3). One patient was asymptomatic with incidental identification of PTLD lesions on brain imaging performed for evaluation of traumatic injury after a fall. One patient presented with headache, and 4 patients presented with confusion and mental status changes. Three patients presented with speech-related symptoms of aphasia and word finding difficulties. Two patients presented with facial droop. Focal/unilateral motor weakness was a presenting complaint in 4 patients. One patient presented with complaints of eye pain, with ophthalmologic evaluation revealing no ocular disease. Two patients experienced ataxia at diagnosis, and 2 patients presented with generalized seizures.
Table 3.
Presenting symptoms of patients with Primary CNS Post-Transplant Lymphoproliferative Disorder (n=10).
| Symptoms | N |
|---|---|
| Asymptomatic | 1 |
| Headache | 1 |
| Confusion/mental status changes | 4 |
| Speech difficulties (aphasia, word-finding difficulties) | 3 |
| Facial droop | 2 |
| Diplopia | 1 |
| Focal/unilateral motor weakness | 4 |
| Vertigo | 1 |
| Ataxia | 2 |
| Seizures | 2 |
MRI findings
MR imaging was performed in all patients and gadolinium contrast was administered to 9 patients. Contrast was not administered to 1 patient due to poor renal function. Lesions were multiple in 7 patients and solitary in 3 cases (Table 4). A total of 37 lesions were identified in 10 patients, which ranged in size from 2 to 36 millimeters. The majority of lesions were supratentorial, most often within a lobar and periventricular distribution. In the 9 patients who received contrast, all had masses that demonstrated some degree of enhancement. Eight of the cases had at least 1 lesion with ring enhancement and 1 patient with incomplete ring enhancement. Evidence of hemorrhage or hemosiderin staining was identified in 5 patients and was significant in 1 patient. Concomitant leptomeningeal involvement was present in 5 patients, which was significant in 2 cases.
Table 4.
Neuroimaging findings of 10 patients with Primary CNS Post-Transplant Lymphoproliferative Disorder.
| Findings | |
|---|---|
| Number of lesions | No, patients (n=10) |
| Solitary | 3 |
| Multiple | 7 |
| Lesion location | No. patients (n=10) |
| Supratentorial alone | 8 |
| Infratentorial alone | 1 |
| Both supra- and infratentorial | 1 |
| Meningeal disease | No. patients (n=5)† |
| Significant | 1 |
| Minor | 4 |
| Hemorrhage/hemosiderin‡ | No. patients (n=10) |
| Significant | 2 |
| Petechial | 3 |
| Ependymal contact‡ | No. patients (n=10) |
| Yes | 4 |
| No | 6 |
| Lesion enhancement (n=8)* | No. patients (n=10)† |
| Ring pattern | 9 |
| Incomplete ring | 1 |
| Specific lesion location | No. lesions (n=37) |
| Lobar | 15 |
| Basal ganglia | 3 |
| Periventicular | 11 |
| Corpus callosum/choroid plexus | 1 |
| Thalamic | 4 |
| Cerebellum/brain stem | 3 |
Only 9 patients received contrast, meningeal involvement and enhancement could not be assessed in the patient who did not receive contrast.
Identified in at least one of the lesions.
The enhancement characteristics are based on the dominant mass.
Key Results
Biopsy technique
The diagnosis of PCNS-PTLD was established with both open (n=6) and stereotactic (n=4) biopsy techniques. No patients required re-operation to obtain diagnostic material. There were no surgical wound infections and no patients required re-operation for hematoma or additional complications.
Brain biopsy, CSF, and peripheral blood findings
Diagnostic brain biopsies demonstrated EBER positivity in 9 cases, and EBER by in situ hybridization had not been performed in 1 case. Seven patients underwent lumbar punctures in a time proximate to their diagnoses of PCNS-PTLD. Protein levels were elevated in all 7 cerebrospinal fluid (CSF) samples, and 1 patient had concurrent evidence of elevated CSF glucose. In 4 of the 7 patients, the CSF demonstrated an excess of nucleated cells with a predominance of lymphocytes consistent with circulating lymphoma cells. Flow cytometry was not consistently performed on CSF samples to verify monoclonality of the lymphocytes identified on cytology.
Monomorphic diffuse large B cell was the most common histological subtype in this series, with 5 patients being affected. Of the remaining patients, 4 suffered from the polymorphic PTLD histological subtype and only one patient was diagnosed with the monomorphic peripheral T cell histological subtype. (Table 5). CD3 and CD20 immunohistochemistry staining was used to determine cell types.
Table 5.
Treatment and survival summary 10 patients with Primary CNS Post-Transplant Lymphoproliferative Disorder.
| Patient | Age | Time to PTLD diagnosis† (years) | Histological Subtype | Treatments | Overall survival (years)‡ | Survival status‡ | Organ failure‡ |
|---|---|---|---|---|---|---|---|
| 1 | 42 | 3.1 | Polymorphic | ROI, DEX HD-MTX (1 cycle) WBRT |
6.3 | Died (non-PTLD) | No |
| 2 | 67 | 11.4 | Monomorphic, Diffuse large B cell | ROI, DEX Rituximab WBRT |
4.9 | Died (non-PTLD) | No |
| 3 | 44 | 5.8 | Monomorphic, Peripheral T Cell | ROI, DEX Rituximab WBRT |
0.6 | Died (non-PTLD) | Yes |
| 4 | 47 | 2.7 | Monomorphic, Diffuse large B cell | ROI, DEX Rituximab WBRT |
4.3 | Alive | Yes |
| 5 | 38 | 1.8 | Polymorphic | ROI, DEX HD-MTX (3 cycles) Rituximab IT-MTX WBRT Temozolomide |
3.3 | Alive | No |
| 6 | 55 | 1.9 | Monomorphic, Diffuse large B cell | ROI, DEXHD-MTX (1 cycle) Rituximab IT-MTX WBRT Temozolomide |
2.1 | Alive | No |
| 7 | 56 | 6.3 | Polymorphic | ROI, DEX Rituximab |
1.4 | Alive | Yes |
| 8 | 43 | 9.7 | Monomorphic, Diffuse large B cell | ROI, DEX Rituximab |
0.2 | Died (PTLD) | No |
| 9 | 63 | 1.9 | Monomorphic, Diffuse large B cell | ROI, DEX WBRT Rituximab |
0.8 | Alive | No |
| 10 | 50 | 11.3 | Polymorphic | ROI, DEX | 0.3 | Alive | No |
Abbreviations: DEX = dexamethasone, HD-MTX = high-dose methotrexate 3.5 grams/m2 IV per cycle, IT-MTX = intrathecal methotrexate, ROI = reduction of immunosuppresion, WBRT = whole-brain radiotherapy.
Time from date of most recent SOT to date of PCNS-PTLD diagnosis.
Outcomes measured from date of PCNS-PTLD diagnosis.
Treatments
All patients were initially managed with reduction of immunosuppression concurrent with additional therapy (Table 5). Two patients were managed with minimization of immunosuppression alone. All patients receiving dexamethasone at the time of diagnosis for management of peritumoral edema. Eight patients received at least 1 course of rituximab. Whole-brain radiotherapy was administered to 7 patients. Three patients received high-dose methotrexate at a dose of 3.5 grams/m2, with 1 patient treated for 3 cycles and 2 patients treated with 1 cycle of therapy. Temozolomide 150 mg/m2 orally for 5 days of every 28-day treatment cycle was administered to 2 patients as consolidation therapy following whole-brain radiotherapy; these patients received 2 and 4 cycles of therapy.
Survival outcomes
Patient survival outcomes were followed through April 2012. The median duration of follow-up was 34.9 months (range 2.2-75.9 months). Four deaths occurred, with only 1 death related to progressive PCNS-PTLD. Two other deaths were related to systemic candidemia from an abdominal source and from thrombotic microangiopathy. One patient died from unknown causes, but on clinical assessment 5 months prior to the sudden death, the patient had normal graft function with no evidence of recurrent PCNS-PTLD. Patient survival outcomes with corresponding therapies administered are summarized in Table 5.
There were 4 longer-term survivors with remissions durable ≥2 years following a PCNS-PTLD diagnosis. Of these patients, 3 had received rituximab and whole-brain radiotherapy. Two of the longer-term survivors had also received pre-radiotherapy systemic high-dose methotrexate and consolidative temozolomide. For both of these patients, the planned treatment course of methotrexate had been terminated prematurely due to incomplete radiographic and clinical response.
Transplantation outcomes
Three patients experienced loss of organ allograft function following the diagnosis and treatment of PCNS-PTLD. All patients had been managed with reduction of immunosuppression for management of PCNS-PTLD, with loss of allograft function documented at 2, 6, and 23 months after the diagnosis of PCNS-PTLD. Of the 3 patients experiencing loss of allograft function, 2 remain alive at 33 and 52 months of follow-up, and 1 patient has died from non-PTLD related complications.
Discussion
PTLD represents a spectrum of disorders, ranging from early polyclonal expansion of lymphocyte populations to high-grade lymphomas. The majority of PCNS-PTLDs are B-cell non-Hodgkin lymphomas, although rare occurrences of T-cell PCNS-PTLD have been reported.4-6 Recipients of SOT have a 20- to 120-fold higher incidence of lymphomas/PTLD than the immunocompetent population7,8. With the exception of skin cancers, PTLD is the most commonly diagnosed malignancy in patients following SOT.9 Age and the type and intensity of immunosuppression are the most significant risk factors for development of PTLD. Higher degrees of immunosuppression required for small bowel, heart, and lung transplantation are associated with higher incidence rates of PTLD. For example, reported incidence rates for PTLD are as high as 20% following small bowel transplantation, but rates of 1-3% have been reported following kidney transplantation.10-13 The overall incidence of PTLD continues to rise as increasing number of SOTs are being successfully performed each year. The much higher number of kidney transplantations at our institution compared with other solid organ transplantations associated with higher intensity of immunosuppressive (i.e., small bowel transplants, cardiac transplants) and the excellent long-term survival outcomes of kidney transplantations are the probable explanation for the observation in this case series of all cases of PTLD diagnosed in patient receiving kidney or kidney/pancreas transplants.
In this report, late recurrences of EBV+ PTLD were common, with 40% of observed cases diagnosed ≥6 years post-transplant. There is emerging evidence that challenges the traditional paradigm of EBV+ PTLD typically occurring in the first year post-transplant. The large retrospective analysis by the International Primary CNS Lymphoma Collaborative Group reported a median time from transplant to diagnosis of PCNS-PTLD of 4.4 years10, which is similar to the observed median time of 4.5 years in this case series. In addition, the International Primary CNS Lymphoma Collaborative Group found that 7 of 34 patients (21%) had diagnoses of PTLD >10 year after transplantation, with only 1 patient demonstrating documented EBV negativity on diagnostic biopsy10. However, the distribution of later occurrences of PCNS-PTLD in our report is not completely explained. It is unclear if the prominent presentation of renal transplantation with typically lower degrees of immunosuppression relative to other solid organ transplants could be a contributing factor.
Isolated CNS involvement of PTLD is uncommon. Aside from individual case reports, there have been fewer than 100 patients with PCNS-PTLD described in case series since the first report of PCNS-PTLD in 1970.14-18 Presenting signs and symptoms of PCNS-PTLD are typical of those observed with other intracranial mass lesions including confusion, seizures, hemiparesis, aphasia, or symptoms of increased intracranial pressure such as headaches and depressed level of consciousness.19 Rare presentations of PCNS-PTLD such as fatal hemorrhage or inflammatory demyelinating symptoms have also been reported.5,20 The presenting signs and symptoms observed in this case series are typical of previous reports of PCNS-PTLD, with most patients in this report presenting with focal neurologic deficits and/or symptoms of elevated intracranial pressure.
Clinical evaluation of PTLD involving the CNS should include complete staging CT imaging or PET imaging to exclude additional systemic disease sites. Given the risk for diffuse neuraxial involvement, additional staging evaluation should include complete imaging of the spine (preferably with MRI) and dilated ophthalmologic exam. CSF analysis has also been variably reported to aid in diagnosis of PTLD, but in the majority of cases, tissue-based diagnosis is necessary.2
Neuroimaging plays an integral role in the diagnosis and management of transplant patients presenting with neurologic symptoms. The imaging findings of some of these patients were addressed in previous publications. MRI is the preferred imaging modality to evaluate transplant patients because of its greater sensitivity and broader depth of tissue contrast. PCNS-PTLD has several characteristic imaging features. Multifocal supratentorial involvement predominates in essentially all case series of PCNSL-PTLD with some also suggesting a periventricular region predilection.6,10 Over 50% of patients in our series also had concurrent meningeal involvement, which has also been reported in other series.20 The majority of PCNS-PTLD demonstrate some degree of contrast enhancement, with reported rates of ring-enhancement observed in 30-70% of PCNS-PTLD cases.6,10 The peripheral enhancement pattern seen in patients with PTLD is likely related to necrosis within the tumor, although steroid therapy may result in an observed relative decrease in contrast enhancement.20-21
This histologic diagnosis of PCNS-PTLD requires biopsy confirmation, typically by stereotactic biopsy or open biopsy. Histologic biopsy is critical to rule out the possibility of opportunistic infection mimicking PCNS-PTLD (i.e., ring-enhancing lesions seen with toxoplasmosis). Traditionally, complete or near complete surgical resection of brain lesions has been discouraged in cases of suspected PCNS-PTLD. Incomplete surgical resection of primary CNS lymphoma was shown in one large retrospective analysis to be an adverse prognostic indicator compared with diagnostic biopsy alone.7 Some earlier treatment approaches with localized radiotherapy for primary CNS lymphomas demonstrated high rates of relapse outside of the radiotherapy field, suggesting occult dissemination of lymphoma throughout the brain, which may minimize the role of an intensive local tumor control strategy with surgical resection in management of CNS lymphomas and PCNS-PTLD.19 However, a more recent report by the German Primary CNS Lymphoma Study Group reported a significant improvement in both progression-free and overall survival in patients undergoing subtotal or gross total resections compared with patients undergoing diagnostic biopsies. These data are particularly compelling in support of the benefit of surgical resection based on the prospective nature of the phase III randomized trial enrolling more than 500 patients.22 Given the inherently radiotherapy- and chemotherapy-sensitive nature of lymphomas and the risk for diffuse occult neuraxis involvement, strategies such as chemotherapy and whole-brain radiotherapy to more broadly treat the neuraxis are currently preferred options. However, the data reported by the German CNS Lymphoma Study Group raise the question as to whether more intensive local control with surgical resection may be an equally important component of treatment for PCNS-PTLD in addition to systemic therapy and radiotherapy.
Treatment options for PCNS-PTLD include reduction of immunosuppression, whole-brain radiotherapy, and systemic chemotherapy and/or monoclonal antibody therapy. Reduction of immunosuppression alone has shown benefit in management of PTLD, particularly in early systemic polyclonal PTLD lesions.2,5-6 In cases of PCNS-PTLD where patients frequently have neurologic deficits as presenting signs of disease, rapid disease control with anti-neoplastic therapy (i.e., chemotherapy or radiotherapy) is recommended concurrent with reduction of immunosupression. The largest retrospective case series of 34 patients with PCNS-PTLD reported by the International Primary CNS Lymphoma Collaborative Group documented improved survival outcomes among patients treated with radiotherapy-based regimens (median overall survival 26.4 months) and rituximab-based regimens (median overall survival 13.5 months).10 Only 2 patients in this case series were managed with reduction in immunosuppression alone, although 1 of the patients was alive with a durable remission after 11 years.10 Although there is no clear standard therapy approach for PCNS-PTLD, most of the existing data suggest that reduction of immunosuppression alone is insufficient treatment for PCNS-PTLD in the majority of cases.
High-dose methotrexate-based chemotherapy has been associated with the most durable outcomes for treatment of primary CNS lymphomas in immunocompetent individuals.8,14-17,23 However, incorporation of high-dose methotrexate into therapy for PCNS-PTLD is limited by its associated risk for nephrotoxicity, which is particularly problematic in renal transplantation patients who already have graft function at risk following simultaneous reduction in immunosuppression. In addition, patients who have received whole brain radiotherapy after high-dose methotrexate have been observed to experience higher rates of leukoencephalopathy,8,14-15,24-25 Only 3 of the 8 patients in our case series received high-dose methotrexate based chemotherapy, which is similar to the reported use of high-dose methotrexate in other recently reported case series of PCNS-PTLD.6,10 One of the patients in our series receiving high-dose methotrexate followed by radiotherapy has experienced significant neurotoxicity (Case 1).
Whole brain radiotherapy and rituximab are the most frequently reported therapies used for treatment of PCNS-PTLD. Advantages of radiotherapy include the known radiosensitivity of lymphoma/PTLD at modest doses of 3000-4500 cGy, the high rates of complete response to whole-brain radiotherapy, and the lack of systemic toxicity and risk to allograft function.15-17 Two recent case series reported very high response rates exceeding 80% for patients receiving radiotherapy with or without chemotherapy.6,10 One report observed improved survival of 36 months compared with 7 months for patients receiving radiotherapy-containing therapies versus other non-radiotherapy treatment approaches.6
The anti-CD20 monoclonal antibody rituximab has also been frequently incorporated into treatment regimens for PTLD, based on the favorable toxicity profile of this agent and known activity in systemic PTLD.18,26 For example, a report from the International Primary CNS Lymphoma Collaborative Group observed that of 8 patients treated with rituximab (and without systemic chemotherapy or radiotherapy), all achieved an objective response with 7 patients alive after a median duration of 23 months of follow-up.10 Novel approaches of rituximab administration with intrathecal dosing have been explored, although are not yet incorporated as standard treatment options in PCNS-PTLD.27-31
An emerging immunotherapy for treatment of PTLD is administration of autologous or allogeneic EBV-specific cytotoxic T-lymphocytes (CTL) to enhance the inherent suppression of the CTL response post-transplant. As EBV induces proliferation and transformation of B lymphocytes, suppressing EBV-infected cells may results in regression of EBV+ PTLD. Previous reports have demonstrated feasibility and activity of autologous EBV-specific CTLs in patients with solid organ transplants at high-risk for PTLD or active PTLD disease,32-34 but the time and expense required to generate cells lines for individual administration severely restricts its availability for clinical use. A report from the United Kingdom reported promising results with allogeneic EBV-specific CTL infusions in 33 pediatric and adult patients with EBV+ PTLD. A response rate of 65% was observed at 5 weeks, with a 6 month response rate of 52%.35 Interestingly, this report included 4 patients with PCNS-PTLD, with 3 patients achieving a partial response at 5 weeks and 2 of the patients demonstrating an improvement in response to a complete response by 6 months.35 The allogeneic EBV-specific CTLs administered in this phase II multicenter trial were provided through a dedicated blood bank, however such banks are currently not available for general clinical use.35
Prognosis for patients with PCNS-PTLD is widely variable depending on patient age, extent of disease at diagnosis, underlying co-morbidities, risk for loss of allograft function, and therapy administered. Some more recent reports of larger case series have suggested improving outcomes for patients with PCNS-PTLD, with median survivals of 26-47 months with use of modern treatment approaches.6,10 Earlier recognition and diagnosis of this uncommon complication post-transplantation may allow for improved therapy options and improved outcomes.
Limitations
Staging evaluation for the patient population was non-uniform, as previously described. Similarly, clinical and radiographic surveillance post-treatment was non-uniform. However, the primary outcome investigated was survival, which is unlikely to be significantly impacted by the variability in initial work-up and surveillance. All of the reported patients had received renal cell transplantation, which could limit the extrapolation of the data to other types of non-renal transplantations.
Generalizability
These results represent a patient population typical of many medical centers providing solid organ transplantation and post-transplantation medical care. The focus on presenting symptoms and imaging findings are particularly relevant given the strong generalizability of these data. In addition, the described medical therapies and radiotherapy are standard therapy approaches available widely at any medical practice providing care for malignancies.
Conclusions
PCNS-PTLD must be considered in the differential diagnosis of any immunosupressed patient following SOT presenting with an intracranial lesion. Presenting symptoms can include focal neurologic complaints or symptoms suggestive of elevated intracranial pressure. Imaging studies may show findings that mimic opportunistic infection (i.e., multifocal, ring-enhancing), further emphasizing the need for diagnostic histologic biopsy. There is no general consensus on treatment approaches for PCNS-PTLD, although treatment options most often include immunosuppression reduction, monoclonal antibody therapy (i.e., rituximab), radiation therapy, or chemotherapy. Prognosis is widely variable, although long-term survival has been reported with PCNS-PTLD. Further increases in incidence rates are expected with all subtypes of PTLD as successful organ transplantation continues to expand, further highlighting the importance of recognizing the distinct clinical entity of PCNS-PTLD.
Acknowledgments
This work was supported by the National Institutes of Health [P30 CA14520].
Footnotes
Disclosures: The authors report no conflict of interest regarding the materials or methods used in this study or the findings specified in this paper.
References
- 1.Penn I, Porat G. Central nervous system lymphomas in organ allograft recipients. Transplantation. 1995 Jan 27;59(2):240–244. [PubMed] [Google Scholar]
- 2.Castellano-Sanchez AA, Li S, Qian J, Lagoo A, Weir E, Brat DJ. Primary central nervous system posttransplant lymphoproliferative disorders. Am J Clin Pathol. 2004 Feb;121(2):246–253. doi: 10.1309/N82C-TQ1J-0XEV-EFQB. [DOI] [PubMed] [Google Scholar]
- 3.Gallardo D, Ferra C, Berlanga JJ, et al. Neurologic complications after allogeneic bone marrow transplantation. Bone Marrow Transplant. 1996 Dec;18(6):1135–1139. [PubMed] [Google Scholar]
- 4.Jamali FR, Otrock ZK, Soweid AM, et al. An overview of the pathogenesis and natural history of post-transplant T-cell lymphoma (corrected and republished article originally printed in Leukemia & Lymphoma, June 2007; 48(6): 1237 - 1241) Leuk Lymphoma. 2007 Sep;48(9):1780–1784. doi: 10.1080/10428190701608657. [DOI] [PubMed] [Google Scholar]
- 5.Kim IY, Jung S, Jung TY, Kang SS, Choi C. Primary central nervous system lymphoma presenting as an acute massive intracerebral hemorrhage: case report with immunohistochemical study. Surg Neurol. 2008 Sep;70(3):308–311. doi: 10.1016/j.surneu.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 6.Snanoudj R, Durrbach A, Leblond V, et al. Primary brain lymphomas after kidney transplantation: presentation and outcome. Transplantation. 2003 Sep 27;76(6):930–937. doi: 10.1097/01.TP.0000079253.06061.52. [DOI] [PubMed] [Google Scholar]
- 7.Bataille B, Delwail V, Menet E, et al. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg. 2000 Feb;92(2):261–266. doi: 10.3171/jns.2000.92.2.0261. [DOI] [PubMed] [Google Scholar]
- 8.DeAngelis LM, Seiferheld W, Schold SC, Fisher B, Schultz CJ. Combination chemotherapy and radiotherapy for primary central nervous system lymphoma: Radiation Therapy Oncology Group Study 93-10. J Clin Oncol. 2002 Dec 15;20(24):4643–4648. doi: 10.1200/JCO.2002.11.013. [DOI] [PubMed] [Google Scholar]
- 9.LaCasce AS. Post-transplant lymphoproliferative disorders. Oncologist. 2006 Jun;11(6):674–680. doi: 10.1634/theoncologist.11-6-674. [DOI] [PubMed] [Google Scholar]
- 10.Cavaliere R, Petroni G, Lopes MB, Schiff D. Primary central nervous system post-transplantation lymphoproliferative disorder: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Cancer. 2010 Feb 15;116(4):863–870. doi: 10.1002/cncr.24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottschalk S, Rooney CM, Heslop HE. Post-transplant lymphoproliferative disorders. Annu Rev Med. 2005;56:29–44. doi: 10.1146/annurev.med.56.082103.104727. [DOI] [PubMed] [Google Scholar]
- 12.Libertiny G, Watson CJ, Gray DW, Welsh KI, Morris PJ. Rising incidence of post-transplant lymphoproliferative disease in kidney transplant recipients. Br J Surg. 2001 Oct;88(10):1330–1334. doi: 10.1046/j.0007-1323.2001.01924.x. [DOI] [PubMed] [Google Scholar]
- 13.Taylor AL, Marcus R, Bradley JA. Post-transplant lymphoproliferative disorders (PTLD) after solid organ transplantation. Crit Rev Oncol Hematol. 2005 Oct;56(1):155–167. doi: 10.1016/j.critrevonc.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 14.Abrey LE, DeAngelis LM, Yahalom J. Long-term survival in primary CNS lymphoma. J Clin Oncol. 1998 Mar;16(3):859–863. doi: 10.1200/JCO.1998.16.3.859. [DOI] [PubMed] [Google Scholar]
- 15.Abrey LE, Yahalom J, DeAngelis LM. Treatment for primary CNS lymphoma: the next step. J Clin Oncol. 2000 Sep;18(17):3144–3150. doi: 10.1200/JCO.2000.18.17.3144. [DOI] [PubMed] [Google Scholar]
- 16.DeAngelis LM, Yahalom J, Thaler HT, Kher U. Combined modality therapy for primary CNS lymphoma. J Clin Oncol. 1992 Apr;10(4):635–643. doi: 10.1200/JCO.1992.10.4.635. [DOI] [PubMed] [Google Scholar]
- 17.Glass J, Gruber ML, Cher L, Hochberg FH. Preirradiation methotrexate chemotherapy of primary central nervous system lymphoma: long-term outcome. J Neurosurg. 1994 Aug;81(2):188–195. doi: 10.3171/jns.1994.81.2.0188. [DOI] [PubMed] [Google Scholar]
- 18.Gonzalez-Barca E, Domingo-Domenech E, Capote FJ, et al. Prospective phase II trial of extended treatment with rituximab in patients with B-cell post-transplant lymphoproliferative disease. Haematologica. 2007 Nov;92(11):1489–1494. doi: 10.3324/haematol.11360. [DOI] [PubMed] [Google Scholar]
- 19.Shibamoto Y, Hayabuchi N, Hiratsuka J, et al. Is whole-brain irradiation necessary for primary central nervous system lymphoma? Patterns of recurrence after partial-brain irradiation. Cancer. 2003 Jan 1;97(1):128–133. doi: 10.1002/cncr.11035. [DOI] [PubMed] [Google Scholar]
- 20.Phan TG, O'Neill BP, Kurtin PJ. Posttransplant primary CNS lymphoma. Neuro Oncol. 2000 Oct;2(4):229–238. doi: 10.1215/15228517-2-4-229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson BA, Fram EK, Johnson PC, Jacobowitz R. The variable MR appearance of primary lymphoma of the central nervous system: comparison with histopathologic features. AJNR Am J Neuroradiol. 1997 Mar;18(3):563–572. [PMC free article] [PubMed] [Google Scholar]
- 22.Weller M, Martus P, Roth P, Thiel E, Korfel A. Surgery for primary CNS lymphoma? Challenging a paradigm. Neuro Oncol. 2012 Sep 14; doi: 10.1093/neuonc/nos159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sandor V, Stark-Vancs V, Pearson D, et al. Phase II trial of chemotherapy alone for primary CNS and intraocular lymphoma. J Clin Oncol. 1998 Sep;16(9):3000–3006. doi: 10.1200/JCO.1998.16.9.3000. [DOI] [PubMed] [Google Scholar]
- 24.Shah GD, Yahalom J, Correa DD, et al. Combined immunochemotherapy with reduced whole-brain radiotherapy for newly diagnosed primary CNS lymphoma. J Clin Oncol. 2007 Oct 20;25(30):4730–4735. doi: 10.1200/JCO.2007.12.5062. [DOI] [PubMed] [Google Scholar]
- 25.Thiel E, Korfel A, Martus P, et al. High-dose methotrexate with or without whole brain radiotherapy for primary CNS lymphoma (G-PCNSL-SG-1): a phase 3, randomised, non-inferiority trial. Lancet Oncol. 2010 Nov;11(11):1036–1047. doi: 10.1016/S1470-2045(10)70229-1. [DOI] [PubMed] [Google Scholar]
- 26.Choquet S, Leblond V, Herbrecht R, et al. Efficacy and safety of rituximab in B-cell post-transplantation lymphoproliferative disorders: results of a prospective multicenter phase 2 study. Blood. 2006 Apr 15;107(8):3053–3057. doi: 10.1182/blood-2005-01-0377. [DOI] [PubMed] [Google Scholar]
- 27.Liu CY, Teng HW, Lirng JF, Chiou TJ, Chen PM, Hsiao LT. Sustained remission and long-term survival of secondary central nervous system involvement by aggressive B-cell lymphoma after combination treatment of systemic high-dose chemotherapy and intrathecal rituximab. Leuk Lymphoma. 2008 Oct;49(10):2018–2021. doi: 10.1080/10428190802311375. [DOI] [PubMed] [Google Scholar]
- 28.Maximiano Alonso C, Sanchez Ruiz AC, Cantos Sanchez de Ibarguen B, Mendez Garcia M, Ronco IS, Provencio Pulla M. Ocular relapse of primary brain lymphoma in immunocompetent patient, treated with intrathecal rituximab. Clin Transl Oncol. 2010 Oct;12(10):701–703. doi: 10.1007/s12094-010-0580-y. [DOI] [PubMed] [Google Scholar]
- 29.Perissinotti AJ, Reeves DJ. Role of intrathecal rituximab and trastuzumab in the management of leptomeningeal carcinomatosis. Ann Pharmacother. 2010 Oct;44(10):1633–1640. doi: 10.1345/aph.1P197. [DOI] [PubMed] [Google Scholar]
- 30.Rubenstein JL, Fridlyand J, Abrey L, et al. Phase I study of intraventricular administration of rituximab in patients with recurrent CNS and intraocular lymphoma. J Clin Oncol. 2007 Apr 10;25(11):1350–1356. doi: 10.1200/JCO.2006.09.7311. [DOI] [PubMed] [Google Scholar]
- 31.van de Glind G, de Graaf S, Klein C, Cornelissen M, Maecker B, Loeffen J. Intrathecal rituximab treatment for pediatric post-transplant lymphoproliferative disorder of the central nervous system. Pediatr Blood Cancer. 2008 Apr;50(4):886–888. doi: 10.1002/pbc.21297. [DOI] [PubMed] [Google Scholar]
- 32.Haque T, Amlot PL, Helling N, et al. Reconstitution of EBV-specific T cell immunity in solid organ transplant recipients. J Immunol. 1998 Jun 15;160(12):6204–6209. [PubMed] [Google Scholar]
- 33.Khanna R, Bell S, Sherritt M, et al. Activation and adoptive transfer of Epstein-Barr virus-specific cytotoxic T cells in solid organ transplant patients with posttransplant lymphoproliferative disease. Proc Natl Acad Sci U S A. 1999 Aug 31;96(18):10391–10396. doi: 10.1073/pnas.96.18.10391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Savoldo B, Goss JA, Hammer MM, et al. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs) Blood. 2006 Nov 1;108(9):2942–2949. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haque T, Wilkie GM, Jones MM, et al. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood. 2007 Aug 15;110(4):1123–1131. doi: 10.1182/blood-2006-12-063008. [DOI] [PubMed] [Google Scholar]