

Abstract
Background and Purpose
Nuclear erythroid 2 related factor 2 (Nrf2) is an astrocyte-enriched transcription factor that has previously been shown to upregulate cellular antioxidant systems in response to ischemia. While resveratrol preconditioning (RPC) has emerged as a potential neuroprotective therapy, the involvement of Nrf2 in RPC-induced neuroprotection and mitochondrial reactive oxygen species (ROS) production following cerebral ischemia remains unclear. The goal of our study was to study the contribution of Nrf2 to RPC and its effects on mitochondrial function.
Methods
We used rodent astrocyte cultures and an in vivo stroke model with RPC. An Nrf2 DNA-binding ELISA and protein analysis via Western blotting of downstream Nrf2 targets were performed to determine RPC-induced activation of Nrf2 in rat and mouse astrocytes. Following RPC, mitochondrial function was determined by measuring ROS production and mitochondrial respiration in both wild-type (WT) and Nrf2−/− mice. Infarct volume was measured to determine neuroprotection, while protein levels were measured by immunoblotting.
Results
We report that Nrf2 is activated by RPC in rodent astrocyte cultures, and that loss of Nrf2 reduced RPC-mediated neuroprotection in a mouse model of focal cerebral ischemia. In addition, we observed that wild-type and Nrf2−/− cortical mitochondria exhibited increased uncoupling and ROS production following RPC treatments, Finally, Nrf2−/− astrocytes exhibited decreased mitochondrial antioxidant expression and were unable to upregulate cellular antioxidants following RPC treatment.
Conclusion
Nrf2 contributes to RPC-induced neuroprotection through maintaining mitochondrial coupling and antioxidant protein expression.
Keywords: preconditioning, resveratrol, mitochondria, reactive oxygen species
Introduction
In the United States, 1 in 20 deaths can be attributed to stroke, while more than 80% of the 800,000 people who suffer from stroke each year survive and require long-term rehabilitation1. Ischemic preconditioning (IPC) has emerged as a potential therapy that could mitigate the morbidity of cerebral ischemic injury; previous studies from our group have shown that IPC treatment induced neuroprotection in rodent models of global cerebral ischemia2, 3. This protection has been recapitulated using the polyphenolic compound resveratrol as a pharmacologic preconditioning agent4. The use of resveratrol as a preconditioning agent has been substantiated by numerous studies in a diverse range of in vitro and in vivo models5,6.
However, previous preconditioning studies have focused mainly on neuronal physiology and amelioration of neuronal cell death following cerebral ischemia. As a result, the role of astrocytes in mediating IPC is often neglected, despite the well-known functions of astrocytes in mediating several neuroprotective mechanisms7. Astrocytes have been suggested to have increased resistance to ischemic injury when compared to neurons8; however, astrocyte dysfunction has been shown to exacerbate various neurodegenerative conditions9–11 and increase susceptibility of neurons to ischemia12. Rodents have both a lower astrocyte:neuron ratio and fewer astrocytic processes compared to humans13. Indeed, the relative differences in cytoarchitecture between rodents and humans may have contributed to the relative plateau of clinical translatable neuroprotective.
One of the many neuroprotective functions of astrocytes includes supplying neurons with antioxidants, the production of which is primarily controlled by the transcription factor Nuclear erythroid 2 related factor 2 (Nrf2). Nrf2 has been previously suggested to be highly expressed in astrocytes as opposed to neurons, and has been shown to increase the antioxidant proteins thioredoxin and NAD(P)H-quinone oxidoreductase 1 (NQO-1)14. Since oxidative stress is a major consequence of cerebral ischemia, the function of Nrf2 to mitigate this stress makes Nrf2 and related downstream pathways attractive targets to combat cerebral ischemic injury.
In light of the aforementioned studies, the focus of our investigation was to determine if RPC treatment could induce neuroprotection through Nrf2 activation. We found that absence of functional Nrf2 protein reduced RPC-induced neuroprotection in a mouse model of focal cerebral ischemia. In addition, RPC treatment failed to increase mitochondrial and cellular antioxidants in cultured astrocytes when Nrf2 was absent. These studies highlight the contribution of astrocyte Nrf2 to RPC-induced protection and a novel role of Nrf2 in maintaining mitochondrial function.
Methods
Additional detailed methods are described in the supplemental section. All animal protocols were approved by the Animal Care and Use Committee of the University of Miami. Minimum Essential Medium (MEM), Hanks Balanced Salt Solution (HBSS) and Fetal Bovine Serum (FBS) were purchased from Life Technologies (Grand Island, NY). All other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
Preparation of primary cultures and in vitro preconditioning studies
Astrocyte cultures were prepared as previously described15. Cortical tissue was harvested from P2–P4 Sprague Dawley rats, WT mice, or Nrf2−/− mice Following treatment with 0.25% trypsin and 0.1% DNase, single cell suspensions were plated onto cell-culture dishes and maintained for 10–14 days prior to experimental use. After reaching approximately 80% confluency, cultures were trypsinized and passaged. Passages 1–3 were used for experiments. For RPC treatment, astrocyte cultures were exposed to 2 hr of resveratrol (25 μmol/L) or DMSO (Vehicle) 48 hrs prior to harvesting cell lysates or nuclear fractions for downstream analysis.
ELISA and Western blotting
Nuclear and cytoplasmic fractionations were prepared according to manufacturer’s protocol by using a nuclear extract kit (Active Motif Cat. #: 40010). Nuclear and cytoplasmic extracts were probed with both Lamin B and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to establish purity of the nuclear and cytoplasmic fractions, respectively. 10 μg of nuclear samples were used for the TransAM Nrf2 enzyme-linked immunosorbent assay (ELISA) kit (Active Motif) to measure DNA-binding of activated Nrf2 nuclear protein, as determined by absorbance measurements at 450nm. For immunoblotting, cells were lysed in Radioimmunoprecipitation assay (RIPA) buffer and immunoblotted for Nrf2, uncoupler protein 2 (UCP2), GAPDH, β-actin, manganese superoxide dismutase (MnSOD), or Lamin B. Proteins were detected using enhanced chemiluminescence system (Pierce, ThermoScientific) and densitometry was performed using ImageJ (National Institute of Health)16.
Animal model of Focal Cerebral Ischemia
For our focal cerebral ischemia model, the left middle cerebral artery (MCA) was occluded for 1 hr using an intraluminal filament model as previously described17. C57Bl/6J WT or Nrf2−/− male mice between 7–11 weeks were randomly assigned to 2 treatment groups: resveratrol (10 mg/kg i.p.) or DMSO (Vehicle). The investigator was blinded to the administration of these agents 48 hrs prior to MCA occlusion. Twenty-four hours following reperfusion, infarct volume was assessed with 2,3,5-triphenyltetrazolium chloride (TTC) and quantified using ImageJ software. Exclusion criteria for these studies included (1) greater than 30% of baseline MCA blood flow persisting through the occlusion phase of the MCA occlusion (MCAO) injury as measured by laser Doppler flowmetry; and (2) lack of detectable infarct following TTC staining.
Polarography
Mitochondrial respiration studies were conducted as previously described18. In brief, non-synaptic mitochondria were isolated from WT or Nrf2−/− mice 48 hrs following resveratrol or vehicle treatment. The ratio of State III/State IV respiration was measured using a Clark-type oxygen electrode. This ratio represented the respiratory control index (RCI), an established measure of mitochondrial coupling19.
Measurement of mitochondria ROS production
Mitochondrial ROS production was determined using a spectrophotometer following a previously established protocol20. Isolated non-synaptic mitochondria from Nrf2−/− or C57Bl/6J animals were added to a microplate containing horseradish peroxidase, Amplex Red Ultra, and superoxide dismutase. H2O2 emission was measured spectrophotometrically at 555nm excitation/590nm emission wavelengths. After establishing baseline measurements, respiratory substrates were added in a similar manner to the polarographic studies. Rates of H2O2 emission were recorded for each complex-specific substrate/inhibitor pair, and normalized to baseline H2O2 production for each sample.
Statistical Analysis
All data are expressed as mean±STDEV. Statistical analysis between two groups was performed using the unpaired Student’s t-test. Statistical analysis between more than two groups was performed using a one-way ANOVA with Bonferroni’s multiple comparison post-hoc test unless otherwise specified. p≤0.05 was considered statistically significant (GraphPad Prism v5.00 for Windows, GraphPad Software, San Diego, California USA).
Results
Loss of Nrf2 decreases RPC-induced neuroprotection following focal cerebral ischemia
Previous studies from our laboratory have shown that RPC can induce neuroprotection against cerebral ischemia in vivo4. Therefore, we wanted to determine if loss of functional Nrf2 decreased RPC-induced neuroprotection in a mouse model of focal cerebral ischemia. Nrf2−/− mice were verified by standard genotyping and immunoblotting for NQO-1 (supplemental figure I). WT and Nrf2−/− mice were subjected to MCAO injury 48 hrs following either RPC or vehicle treatment. Quantification of TTC-stained brain slices from each treatment group indicated that RPC treatment of WT mice significantly reduced infarct volume compared to vehicle treatment (44.43±13.90%, n=9 vs. 28.31±9.93%, n=6, respectively; p<0.05) (Figure 1A and 1B). However, no significant difference was observed in Nrf2−/− mice between RPC and vehicle-treated groups (40.86±17.10%, n=10 vs. 34.47±14.25%, n=9; p=0.39). In addition, there was no significant difference between WT and Nrf2−/− vehicle-treated groups. Therefore, the results from Figure 1 indicate that RPC-induced neuroprotection was decreased in the absence of functional Nrf2.
Figure 1. RPC-induced neuroprotection is decreased in Nrf2−/− mice following MCAO injury.
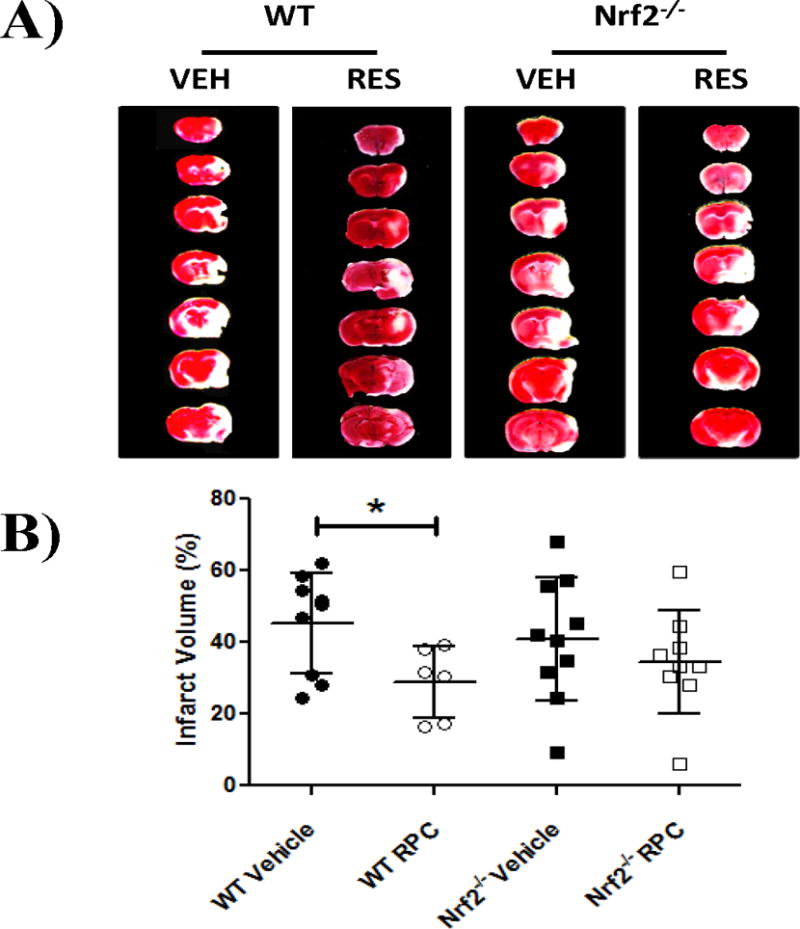
A) Representative image of TTC staining of brain slices to assess infarct volume of mice subjected to MCAO injury. Slices are distributed rostrocaudally. VEH: Vehicle. RES: resveratrol preconditioning. B) Quantification of infarct volume. Areas of cortical infarction at seven coronal levels were cumulated and normalized for edema. Number of subjects represented in inset of bar graph. Data are presented as mean ± STDEV; * p≤0.05, (multiple-comparison 1-way ANOVA followed by Bonferroni test).
Loss of RPC-induced neuroprotection in Nrf2−/− mice is not due to altered cerebral blood flow
A previous group has suggested that the cerebral blow flow is altered in Nrf2−/− mice, which led to their use of a modified in vivo model to achieve adequate middle cerebral artery occlusion21. To determine if the observed infarct volumes from Figure 1 were attributable to strain differences in cerebral blood flow, we analyzed blood flow throughout the MCAO injury via laser Doppler flowmetry for each treatment group of mice. The average blood flow for each group during the phases of the MCAO injury (baseline, occlusion, and reperfusion) were compared and expressed as a percentage of baseline laser Doppler Flowmetry measurements (Figure 2A); our results show that there was no statistically significant difference between blood flow of any of the treatment groups for each phase of the MCAO injury. In Figure 2B, the average age and weight for each treatment group were analyzed and once again there was no statistically significant differences observed. Therefore, the loss of RPC-induced neuroprotection in Nrf2−/− mice is not attributable to altered cerebral blood flow.
Figure 2. Reduced RPC-induced neuroprotection in Nrf2−/− mice is not due to altered cerebral blood flow during ischemia.
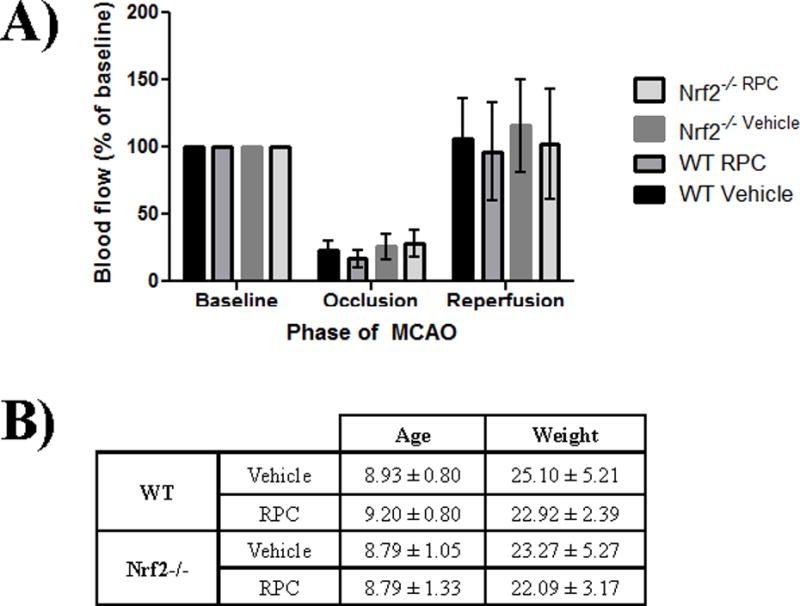
A) Blood flow measurements during the baseline, ischemic/occlusion, and reperfusion phases of the MCAO injury from the same treatment subjects as in Figure 1. Data represented as a percentage of baseline blood flow, as measured by laser Doppler flowmetry. B) Physiologic variables of WT and Nrf2−/− mice used for MCAO experiments.
RPC treatments activate Nrf2 in rat astrocytes
Given that the reduction in RPC-induced neuroprotection in Nrf2−/− mice was not due to altered cerebral blood flow (Figure 2), the results from Figure 1 suggest that RPC-induced neuroprotection partially requires Nrf2. As there is greater Nrf2 protein abundance in astrocytes14, we next sought to determine if RPC can activate Nrf2 in vitro by utilizing astrocyte cultures. RPC treatment increased the amount of activated Nrf2 in nuclear astrocyte fractions at 48 hrs compared to vehicle-treated cultures as determined by absorbance measured at 450 nm (0.983±0.458 vs 0.625±0.352, p<0.05 n=3) (Figure 3A), which were not observed at earlier time points (1 or 24 hrs) following RPC treatment. To determine if downstream pathways of Nrf2 were increased following RPC, whole-cell astrocyte lysates were probed for NQO-1, an Nrf2-dependent gene target. 48 hrs following RPC treatments, NQO-1 protein levels were significantly increased in astrocyte cultures by approximately 1.6 fold compared to vehicle treatments (Figure 3B). The results indicate that RPC activates astrocyte Nrf2 by increasing Nrf2 DNA binding and Nrf2-dependent gene transcription.
Figure 3. RPC treatment activates Nrf2 in astrocyte cultures.
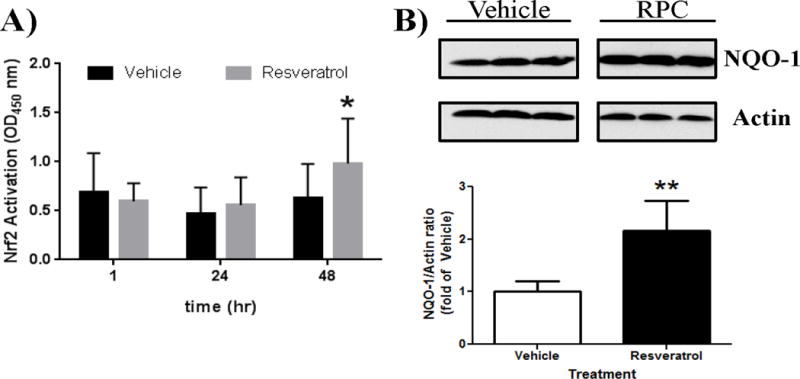
A) ELISA assay was used to measure Nrf2 DNA binding in rat astrocyte nuclear fractions harvested at 1, 24, or 48 hrs after RPC treatment. B) whole-cell astrocyte lysates were probed for NQO-1 expression following RPC treatment. n=4, *p≤0.05, **p≤0.01.
RPC induces uncoupling in WT and Nrf2−/− mitochondria
Resveratrol has previously been implicated in modifying cerebral mitochondrial function. Therefore, we investigated mitochondrial coupling (represented as respiratory coupling index or RCI) in isolated non-synaptic mitochondria from WT and Nrf2−/− mouse cortex following RPC treatment in vivo. We utilized non-synaptic mitochondria because this fraction contains more astrocyte-derived mitochondria than the synaptic fraction, and therefore would better represent functional changes due to the loss of Nrf2. RPC-induced treatment induced a mild uncoupling in both WT and Nrf2−/− mitochondria compared to vehicle treatments of each respective mouse strain (WT: Vehicle 5.84±0.55 vs RPC 3.93±0.42, p<0.05; Nrf2−/−: Vehicle 3.272±0.67 vs RPC 2.27±0.15, p<0.05) (Figure 4A). In addition, we observed a significant difference between Nrf2−/− and WT mice RCI values between vehicle groups, suggesting that Nrf2−/− mitochondria have an innate respiratory dysfunction. Therefore, RPC treatment had similar effects on the RCI in both mouse strains, with RPC treatment further reducing the already decreased RCI in Nrf2−/− mice.
Figure 4. RPC treatment increases mitochondrial uncoupling in Nrf2−/− and WT mice.
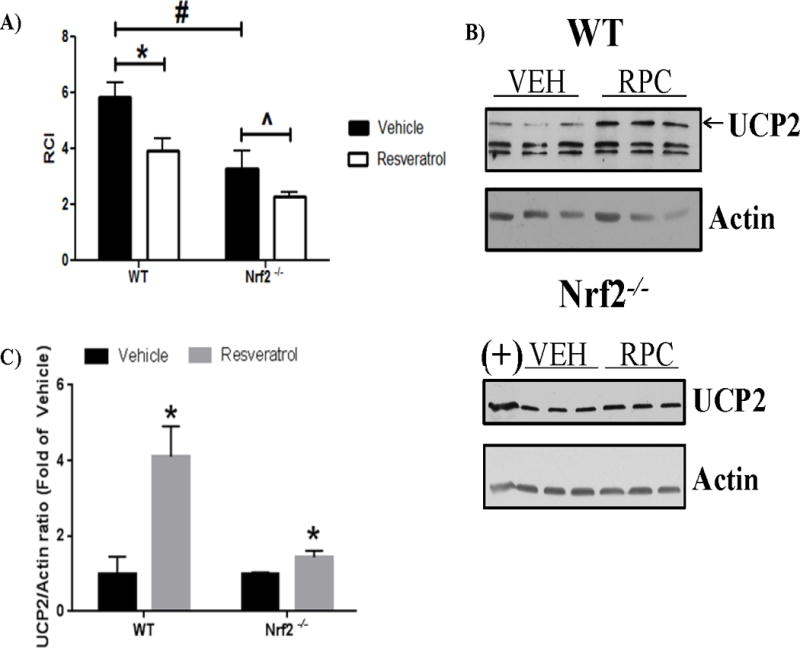
A) RCI of WT and Nrf2−/− non-synaptic mitochondria following RPC and vehicle treatment. n=4, *p≤0.05 (WT vehicle vs. WT RPC); #p≤0.05 (WT vehicle vs. Nrf2−/− vehicle); ˆp≤0.05 (Nrf2−/− vehicle vs. Nrf2−/− RPC. B) UCP2 and Actin protein levels from WT and Nrf2−/− astrocyte cultures treated with RPC and Vehicle. (+) positive control: mouse cortex tissue lysate. C) Quantification of UCP2 proteins levels normalized to Actin. n=3 *p≤0.05
RPC-induces increased UCP2 expression in Nrf2−/− astrocytes
Previous studies from our lab have suggested an interaction between resveratrol and UCP2, an uncoupler protein which if altered by RPC could explain RCI values and provide a molecular understanding for the results observed in Figure 4A. As isolation of astrocyte mitochondria from the brain was not feasible, we took advantage of highly-enriched astrocyte cultures to look at the effects of Nrf2 on UCP2 expression in vitro. Therefore, 48 hrs following RPC treatment, whole cell mouse WT and Nrf2−/− astrocyte lysates were prepared and probed for UCP2. Compared to vehicle treatments, UCP2 was significantly increased 48 hrs following RPC treatment in WT and Nrf2−/− astrocyte cultures (4.05±1.01 fold and 1.89±0.26 fold, respectively, p<0.05) (Figure 4B and 4C). Therefore, RPC treatment increased UCP2 protein expression in WT and Nrf2−/− mouse astrocyte cultures.
RPC treatment increases ROS production in WT and Nrf2−/− mitochondria
As Nrf2−/− mice are expected to have decreased antioxidant capacity, we also measured mitochondrial H2O2 production as a measure of ROS generation in non-synaptic mitochondria isolated from WT and Nrf2−/− mouse cortex following RPC treatment in vivo. RPC treatment significantly increased the fold of H2O2 production compared to baseline production following complex I (WT: Vehicle 3.66±1.19 vs RPC 8.29±1.56 fold of baseline p<0.05, Nrf2−/−: Vehicle 3.90±2.82 vs. RPC 8.35±2.65 p<0.05) and complex III (WT: Vehicle 7.54±2.43 vs RPC 31.06±10.17 p<0.005, Nrf2−/−: Vehicle 9.59±6.32 vs. RPC 23.25±10.05, p<0.05) for both strains of mice (Figure 5A). However, there were no significant differences between WT and Nrf2−/− mice for the same treatment group. These findings suggest a role of RPC in increasing hydrogen peroxide production at complex I and III in WT and Nrf2−/− cortical mitochondria.
Figure 5. RPC increases non-synaptic mitochondrial ROS production and only increases antioxidant expression in WT astrocytes.
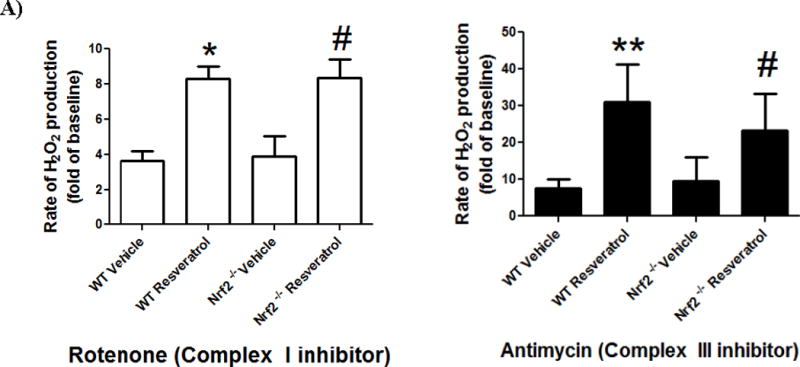
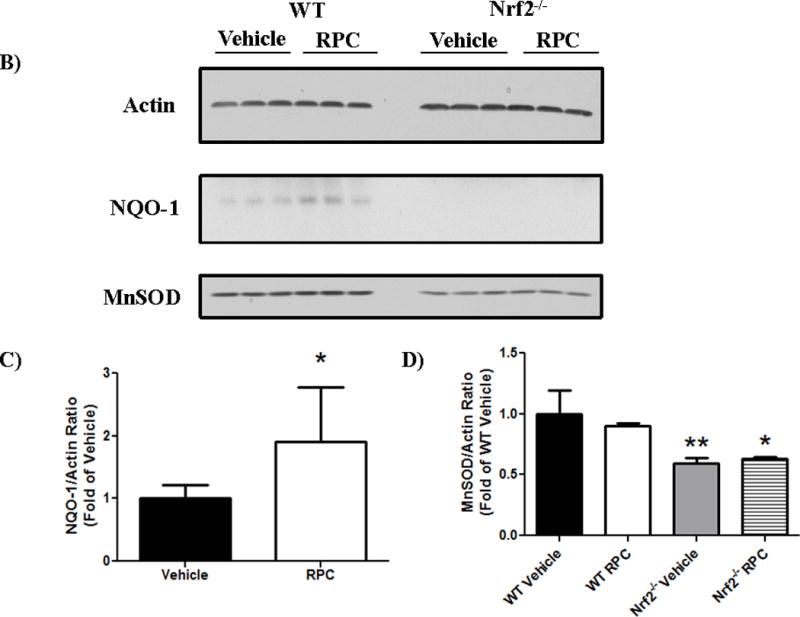
A) Complex I and Complex III H2O2 production rate in WT or Nrf2−/− non-synaptic mitochondria. n=4–6, *p≤0.05 (WT vehicle vs. WT RPC); #p≤0.05 (Nrf2 vehicle vs. Nrf2−/− RPC). B) Western blots of astrocyte cultures for MnSOD, NQO-1, and actin (loading control) 48 hrs after RPC or Vehicle treatment. C) Quantification of WT astrocyte NQO-1 levels and D) WT and Nrf2−/− astrocyte MnSOD protein levels, normalized to Actin protein levels. n=3–6. *p<0.05. **p<0.005.
RPC-induced antioxidant enzyme expression in WT and Nrf2−/− astrocytes
While RPC-induced increase in mitochondrial ROS production occurred in both WT and Nrf2−/− mice, we hypothesized that this phenomenon was detrimental in Nrf2−/− mice and could explain loss of RPC-induced neuroprotection in this population. Therefore, we immunoblotted WT and Nrf2−/− astrocyte-culture lysates for the antioxidants MnSOD and NQO-1 48 hrs following RPC treatment. (Figure 5B). RPC treatment significantly increased NQO-1 protein expression (normalized to actin) in WT astrocytes compared to vehicle treatment (1.91±0.35 fold increase, n=6, p<0.05). As expected, we were unable to observe or measure the Nrf2-dependent protein NQO-1 in Nrf2−/− astrocyte lysates (Figure 5B and 5C). Next, we measured the mitochondrial antioxidant MnSOD proteins levels in WT and Nrf2−/− astrocyte cultures via Western blotting. We found no statistically significant difference in MnSOD protein levels between vehicle and RPC-treated groups of either strain of mice (Figure 5D). However, Vehicle and RPC-treated Nrf2−/− astrocytes had significantly less MnSOD protein compared to WT Vehicle MnSOD levels (40.3% and 37.2% less MnSOD protein, respectively; p≤0.005). These results suggest that Nrf2−/− astrocytes have depressed mitochondrial and cellular antioxidants, and are unable to upregulate these proteins in response to RPC-induced ROS production observed in Figure (5A).
Discussion
In the present study, we investigated the contribution of Nrf2 to RPC-induced neuroprotection and the effect of Nrf2 on cortical mitochondrial function. We investigated the role of Nrf2 in RPC-induced neuroprotection in a rodent model of focal cerebral ischemia. Our MCAO studies suggest that in the absence of Nrf2 protein, RPC-induce neuroprotection is not effective in significantly ameliorating cerebral infarction in mice (Figure 1). Given that RPC treatment increased ROS production in both WT and Nrf2−/− cortical mitochondria (Figure 4B), and that RPC was unable to induce antioxidant expression in Nrf2−/− mice (Figure 5), we believe this evidence suggests that RPC induced neuroprotection can be attributed to ROS-mediated signaling pathways, which ultimately activates astrocytic Nrf2 and confers cerebral ischemic tolerance.
The role of astrocyte pathways have not been fully elucidated in preconditioning research, but their importance to neuronal disease has been extensively studied. Previous studies by Belle et. al22 highlighted that astrocytic Nrf2 was necessary for ischemic tolerance in murine neuronal cultures. In direct contrast, Layon-Haskew et. al determined that physiologic levels of H2O2 could induce ischemic tolerance in neurons independent of Nrf223. Although our study is a different preconditioning paradigm (i.e., RPC), our current studies indicate that RPC is indeed able to increase Nrf2 DNA-binding and downstream expression of NQO-1, an Nrf2-dependent gene.
Similar to a previous study21, we did not observe any difference in infarct volume between WT and Nrf2−/− mice at 24 hrs without a preconditioning treatment. In contrast, we observed significant neuroprotection with RPC treatment in WT mice following MCAO, but there was no significant reduction of infarct volume in RPC-treated Nrf2−/− mice. While resveratrol has been shown to activate a multitude of pathways, resveratrol was still unable to significantly ameliorate infarct injury in Nrf2−/− mice following MCAO. This suggests that Nrf2 activation is a key pathway for RPC-neuroprotection.
Our current investigation has also shown that resveratrol induced an increase in ROS production in WT and Nrf2−/− mitochondria, which can be detected 48 hr post-treatment. This increase in ROS following resveratrol treatment has been seen previously in yeast cells24, human adipocytes24, and cancer cell-lines25. In addition, the increase in ROS following preconditioning treatments is a well observed phenomenon, and inhibition of ROS production has been shown to ameliorate preconditioning-induced neuroprotective effects26.
We also present findings that RPC treatment increased UCP2 expression in astrocyte cultures, and that this increase was several fold more in WT vs. Nrf2−/− astrocytes (Figure 4). While our previous findings described an RPC-induced decrease in UCP2 protein expression in adult rat hippocampal mitochondria, our current studies investigated UCP2 in cortical post-natal mouse astrocyte cultures. This discrepancy could be due to the use of different models and cell types, and future studies may serve to elucidate the dependence of cell-type and brain region on the regulation of UCP2 by RPC treatment. Taken together, we propose that ROS production from RPC treatment induces UPC2 expression, leading to mild uncoupling and subsequent protection against oxidative stress. Furthermore, RPC-induced increase in UCP2 (Figure 4), basal MnSOD, and NQO-1 (Figure 5) protein levels were all decreased several fold in Nrf2−/− mice compared to WT mice; we believe this implicates Nrf2 as a critical pathway in which RPC-mediated mitochondrial ROS production activates Nrf2, thus promoting the induction of antioxidant pathways and subsequent neuroprotection against focal cerebral ischemia.
Following RPC treatment, WT and Nrf2−/− exhibited reductions in their RCI, suggestive of a resveratrol-induced uncoupling effect. By further decreasing the coupling of Nrf2−/− mitochondria, resveratrol may exacerbate already dysfunctional cortical mitochondria in Nrf2−/− mice. Thus, decreased mitochondrial coupling, increased ROS production, and decreased antioxidant defenses could be plausible explanations as to explain why RPC was less effective in Nrf2−/− mice than WT mice. Our findings that Nrf2−/− non-synaptic mitochondria exhibited decreased coupling were in line with previous studies which suggested that Nrf2−/− brain, heart and liver mitochondria exhibit decreased RCI27. Interestingly, studies by Fiskum et al. did not describe any changes to brain non-synaptic28 mitochondrial respiration following activation of Nrf2 with sulforaphane. Future studies may serve to understand the relationship of Nrf2 to cortical non-synaptic and synaptic mitochondria.
In conclusion, our investigation provides new insight into the mechanism of RPC-induced protection, and implicates Nrf2 as an important pathway to induce RPC’s neuroprotective effects in the context of stroke.
Supplementary Material
Acknowledgments
None
Disclosures: Dr. Perez-Pinzon was supported in part by the University of Miami Miller School of Medicine. All authors were supported by grants from the National Institutes of Health, National Institute of Neurological Disease and Stroke (NINDS) NS45676, NS054147, NS34773, F31NS080344.
Sources of Funding: This work was supported by grants from the National Institutes of Health, National Institute of Neurological Disease and Stroke (NINDS) NS45676, NS054147, NS34773, F31NS080344, and the Lois Pope Life Science Research Award.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics-2015 update: A report from the american heart association. Circulation. 2014;131:e29–e322. doi: 10.1161/CIR.0000000000000152. [DOI] [PubMed] [Google Scholar]
- 2.Della-Morte D, Dave KR, DeFazio RA, Bao YC, Raval AP, Perez-Pinzon MA. Resveratrol pretreatment protects rat brain from cerebral ischemic damage via a sirtuin 1-uncoupling protein 2 pathway. Neuroscience. 2009;159:993–1002. doi: 10.1016/j.neuroscience.2009.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dave KR, Lange-Asschenfeldt C, Raval AP, Prado R, Busto R, Saul I, et al. Ischemic preconditioning ameliorates excitotoxicity by shifting glutamate/gamma-aminobutyric acid release and biosynthesis. Journal of neuroscience research. 2005;82:665–673. doi: 10.1002/jnr.20674. [DOI] [PubMed] [Google Scholar]
- 4.Raval AP, Dave KR, Perez-Pinzon MA. Resveratrol mimics ischemic preconditioning in the brain. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2006;26:1141–1147. doi: 10.1038/sj.jcbfm.9600262. [DOI] [PubMed] [Google Scholar]
- 5.Yan H, Jihong Y, Feng Z, Xiaomei X, Xiaohan Z, Guangzhi W, et al. Sirtuin 1-mediated inhibition of p66shc expression alleviates liver ischemia/reperfusion injury. Critical care medicine. 2014;42:e373–381. doi: 10.1097/CCM.0000000000000246. [DOI] [PubMed] [Google Scholar]
- 6.Cavdar Z, Egrilmez MY, Altun ZS, Arslan N, Yener N, Sayin O, et al. Resveratrol reduces matrix metalloproteinase-2 activity induced by oxygen-glucose deprivation and reoxygenation in human cerebral microvascular endothelial cells. International journal for vitamin and nutrition research. Internationale Zeitschrift fur Vitamin- und Ernahrungsforschung. Journal international de vitaminologie et de nutrition. 2012;82:267–274. doi: 10.1024/0300-9831/a000119. [DOI] [PubMed] [Google Scholar]
- 7.Perez-Alvarez A, Araque A. Astrocyte-neuron interaction at tripartite synapses. Current drug targets. 2013;14:1220–1224. doi: 10.2174/13894501113149990203. [DOI] [PubMed] [Google Scholar]
- 8.Gurer G, Gursoy-Ozdemir Y, Erdemli E, Can A, Dalkara T. Astrocytes are more resistant to focal cerebral ischemia than neurons and die by a delayed necrosis. Brain pathology. 2009;19:630–641. doi: 10.1111/j.1750-3639.2008.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pehar M, Beeson G, Beeson CC, Johnson JA, Vargas MR. Mitochondria-targeted catalase reverts the neurotoxicity of hsod1g(9)(3)a astrocytes without extending the survival of als-linked mutant hsod1 mice. PloS one. 2014;9:e103438. doi: 10.1371/journal.pone.0103438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alvarez MI, Rivas L, Lacruz C, Toledano A. Astroglial cell subtypes in the cerebella of normal adults, elderly adults, and patients with alzheimer’s disease: A histological and immunohistochemical comparison. Glia. 2014;63:287–312. doi: 10.1002/glia.22751. [DOI] [PubMed] [Google Scholar]
- 11.Miao SH, Sun HB, Ye Y, Yang JJ, Shi YW, Lu M, et al. Astrocytic jwa expression is essential to dopaminergic neuron survival in the pathogenesis of parkinson’s disease. CNS neuroscience & therapeutics. 2014;20:754–762. doi: 10.1111/cns.12249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ouyang YB, Voloboueva LA, Xu LJ, Giffard RG. Selective dysfunction of hippocampal ca1 astrocytes contributes to delayed neuronal damage after transient forebrain ischemia. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:4253–4260. doi: 10.1523/JNEUROSCI.0211-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: Redefining the functional architecture of the brain. Trends in neurosciences. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 14.Bell KF, Fowler JH, Al-Mubarak B, Horsburgh K, Hardingham GE. Activation of nrf2-regulated glutathione pathway genes by ischemic preconditioning. Oxidative medicine and cellular longevity. 2011;2011:689524. doi: 10.1155/2011/689524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaech S, Banker G. Culturing hippocampal neurons. Nature protocols. 2006;1:2406–2415. doi: 10.1038/nprot.2006.356. [DOI] [PubMed] [Google Scholar]
- 16.Degasperi A, Birtwistle MR, Volinsky N, Rauch J, Kolch W, Kholodenko BN. Evaluating strategies to normalise biological replicates of western blot data. PloS one. 2014;9:e87293. doi: 10.1371/journal.pone.0087293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke; a journal of cerebral circulation. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 18.Dave KR, DeFazio RA, Raval AP, Torraco A, Saul I, Barrientos A, et al. Ischemic preconditioning targets the respiration of synaptic mitochondria via protein kinase c epsilon. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:4172–4182. doi: 10.1523/JNEUROSCI.5471-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. The Biochemical journal. 2011;435:297–312. doi: 10.1042/BJ20110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Starkov AA. Measurement of mitochondrial ros production. Methods in molecular biology. 2010;648:245–255. doi: 10.1007/978-1-60761-756-3_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shih AY, Li P, Murphy TH. A small-molecule-inducible nrf2-mediated antioxidant response provides effective prophylaxis against cerebral ischemia in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:10321–10335. doi: 10.1523/JNEUROSCI.4014-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bell KF, Al-Mubarak B, Fowler JH, Baxter PS, Gupta K, Tsujita T, et al. Mild oxidative stress activates nrf2 in astrocytes, which contributes to neuroprotective ischemic preconditioning. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:E1–2. doi: 10.1073/pnas.1015229108. author reply E3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haskew-Layton RE, Payappilly JB, Smirnova NA, Ma TC, Chan KK, Murphy TH, et al. Controlled enzymatic production of astrocytic hydrogen peroxide protects neurons from oxidative stress via an nrf2-independent pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:17385–17390. doi: 10.1073/pnas.1003996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Escote X, Miranda M, Menoyo S, Rodriguez-Porrata B, Carmona-Gutierrez D, Jungwirth H, et al. Resveratrol induces antioxidant defence via transcription factor yap1p. Yeast. 2012;29:251–263. doi: 10.1002/yea.2903. [DOI] [PubMed] [Google Scholar]
- 25.Sun W, Wang W, Kim J, Keng P, Yang S, Zhang H, et al. Anti-cancer effect of resveratrol is associated with induction of apoptosis via a mitochondrial pathway alignment. Advances in experimental medicine and biology. 2008;614:179–186. doi: 10.1007/978-0-387-74911-2_21. [DOI] [PubMed] [Google Scholar]
- 26.Puisieux F, Deplanque D, Bulckaen H, Maboudou P, Gele P, Lhermitte M, et al. Brain ischemic preconditioning is abolished by antioxidant drugs but does not up-regulate superoxide dismutase and glutathion peroxidase. Brain research. 2004;1027:30–37. doi: 10.1016/j.brainres.2004.08.067. [DOI] [PubMed] [Google Scholar]
- 27.Holmstrom KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, et al. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biology open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Greco T, Fiskum G. Brain mitochondria from rats treated with sulforaphane are resistant to redox-regulated permeability transition. Journal of bioenergetics and biomembranes. 2010;42:491–497. doi: 10.1007/s10863-010-9312-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.