

Abstract
Brown adipose tissue (BAT) plays a key role in energy expenditure and heat generation and is a promising target for diagnosing and treating obesity, diabetes and related metabolism disorders. While several nuclear and magnetic resonance imaging methods are established for detecting human BAT, there are no convenient protocols for high throughput imaging of BAT in small animal models. Here we disclose a simple but effective method for non-invasive optical imaging of interscapular BAT in mice using a micellar formulation of the commercially available deep-red fluorescent probe, SRFluor680. Whole-body fluorescence imaging of living mice shows extensive accumulation of the fluorescent probe in the interscapular BAT and ex vivo analysis shows 3.5-fold selectivity for interscapular BAT over interscapular WAT. Additional imaging studies indicate that SRFluor680 uptake is independent of mouse species and BAT metabolic state. The results are consistent with an unusual pharmacokinetic process that involves irreversible translocation of the lipophilic SRFluor680 from the micelle nanocarrier into the adipocytes within the BAT. Multimodal PET/CT and planar fluorescence/X-ray imaging of the same living animal shows co-localization of BAT mass signal reported by the fluorescent probe and BAT metabolism signal reported by the PET agent, 18F-FDG. The results indicate a path towards a new, dual probe molecular imaging paradigm that allows separate and independent non-invasive visualization of BAT mass and BAT metabolism in a living subject.
Keywords: brown adipose tissue, molecular imaging, in vivo fluorescence imaging, PET imaging, dual modality imaging
Introduction
Mammals utilize two types of adipose tissue for separate functions. White adipose tissue (WAT) stores excess triacylglycerols, performs endocrine signaling and represents 20-25% of human body mass.1 Brown adipose tissue (BAT) confers adaptive thermogenesis and is 5% of the infant body mass. It is also found in adults but in much smaller quantities.2, 3 BAT contains large amounts of mitochondria to dissipate chemical energy. Upon low temperature or pharmaceutical activation of the nervous system, BAT uses high levels of uncoupling protein-1 to generate heat instead of ATP production. The tissue is highly vascularized which promotes efficient heat transfer to the bloodstream and body temperature maintenance.4 Medical imaging studies have shown that BAT mass is inversely correlated with body mass index and other obesity parameters in human adults.3, 5 In recent years, multiple animal model studies and early stage clinical trials have explored strategies to activate and increase BAT mass.6-10 Stimulation of BAT metabolism has been achieved through exercise, low temperature, and pharmaceutical agents. WAT can be transformed to BAT in a process known as “browning” using the same strategies.11-16 BAT mass has also been increased through transplantation with promising results.17 Notably, BAT transplants have been reported to reverse type 1 diabetes by activating metabolism in the surrounding WAT.18 Most recently a clonogenic population of BAT stem cells has been identified in adult humans and shown to functionally differentiate into metabolically active brown adipocytes.19 Brown and beige adipocyte-specific cell surface markers have been discovered which may serve as tools for the selective delivery of drugs.16, 20 Taken together, these emerging results suggest that BAT is a very promising imaging and therapeutic target for clinical treatment of obesity, diabetes, and related metabolic disorders.21
Biomedical research on BAT and clinical translation is facilitated by non-invasive imaging methods that visualize BAT in living subjects.22 The most common in vivo imaging technique uses 2-deoxy-2-[18F]fluoro-D-glucose (18F-FDG) for positron emission tomography/computed tomography (PET/CT) of tissues with enhanced metabolism such as activated BAT.23 Several other nuclear probes have been investigated for BAT imaging and magnetic resonance imaging has also been utilized to monitor BAT morphology and chemical composition.24-30 While useful for the detection of human BAT, which requires deep tissue imaging, these methods employ radioactivity or expensive instruments and they are not convenient for preclinical research using small animal models. In contrast, optical imaging of small animals is highly attractive because it is safe to perform, relatively inexpensive, and amenable to high throughput.31 The shallow interscapular location of BAT in mice is perfectly suited for optical imaging protocols.32 To date, only two fluorescent molecular probes for BAT imaging have been reported; (1) IR786, a lipophilic cationic near-infrared dye with affinity for mitochondria,33 and (2) a fluorescently labeled nonapeptide that targets BAT vasculature.34 Both fluorescent probes are notable as pioneering lead molecular structures, but they exhibit modest in vivo imaging performance.
The objective of this study was to find an effective method for optical imaging interscapular BAT in living mice. We were attracted to the lipophilic and uncharged squaraine rotaxane probe, SRFluor680, which exhibits extremely bright, deep-red fluorescence that enables high-sensitivity in vivo optical imaging. 35-38 Cell microscopy studies have previously shown that SRFluor680 partitions rapidly and irreversibly into lipophilic intracellular sites such as lipid droplets and the endoplasmic reticulum.36 Here, we report that treatment of living mice with a micellar formulation of SRFluor680 leads to extensive and selective probe accumulation in the interscapular BAT. Furthermore, SRFluor680 uptake is independent of mouse species and BAT metabolic state. A series of experimental results are described and used to rationalize the selective molecular imaging.
Experimental Section
Ethics Statement
All animal experiments used protocols that were approved by the Notre Dame Institutional Animal Care and Use Committee (Protocol Number: 13-10-1304).
Materials
SRFluor680 is commercially available (Molecular Targeting Technologies Inc) or it can be synthesized as previously reported (Ε 297,780 cm-1 M-1, ex: 650 nm, em: 678 nm in DMSO; quantum yield 0.58 in 4:1 THF:water).36, 39 The following materials were purchased: DPPE-PEG2000, POPC (Avanti Polar Lipids); IR786, Nile Red, DAPI, hematoxylin and eosin, CL 316243 (Sigma); BODIPY 493/503, ProLong Gold Antifade Reagent, DiI (Invitrogen); 18F-FDG (Spectron MRC); Isoflurane (Butler Animal Health Supply).
Micellar Probe Formulations
Separate samples of SRFluor680, IR786, or Nile Red were dissolved in trichloromethane and combined with a trichloromethane solution of DPPE-PEG2000 or Cremophor El at a 1:40 probe:amphiphile molar ratio. The trichloromethane solvent was removed via rotary evaporation followed by drying for >1 hour on a high vacuum line. Deionized water was added to each film and the dispersions were sonicated for 15 minutes to produce the micellar formulations. Absorption and fluorescence spectra of the micellar formulations were obtained in Phosphate Buffered Saline (PBS) (10 mM sodium phosphate, 150 mM sodium chloride, pH 7.4).
Transmission Electron Microscopy (TEM)
TEM images of the micelles were obtained using a FEI Titan 80-300 microscope at an acceleration voltage of 80 kV. The samples were prepared by drop-coating micellar SRFluor680 onto an amorphous carbon coated 300 square mesh nickel grid (Electron Microscopy Sciences) followed by a single drop of 1% phosphotungstic acid (pH = 7) for negative staining. The grid was placed on filter paper to absorb excess solvent and dried overnight under vacuum.
Laser Transmission Spectroscopy (LTS)
The LTS instrumentation utilized for size measurements has been previously described.40, 41 LTS characterizes nanoparticles suspended in solution based on wavelength-dependent light extinction, which is capable of determining the size, shape, and density of particles with high precision. The measurement relies on the transmission of light through the nanoparticle suspension as a function of wavelength. LTS measurements were obtained by measuring the extinction of a sample and reference solution in standard 1 cm quartz cuvettes. Separate 1 mL samples of either, empty micelles (1 mM DPPE-PEG2000) or DPPE-PEG2000 micelles containing SRFluor680 (1 mM DPPE-PEG2000, 25 μM SRFluor680) were diluted 3-fold with deionized water in a quartz spectrometer cuvette. The diluted samples were measured with LTS to determine the particle size distribution and particle concentration. Deionized water was used as a reference for all samples.
Adipose Tissue Histology
Interscapular BAT and WAT from SKH1 hairless was excised and snap-frozen in OCT media. The tissue was sliced (5 μm thickness) at -17°C, and the slices adhered to Unifrost microscope slides (Azer Scientific), then fixed with chilled acetone for 5 minutes, and air-dried for an additional 20 minutes. In some experiments the tissue slices were washed three times in PBS, then submerged sequentially for 1 hour in micellar SRFluor680 (10 μM dye, 400 μM DPPE-PEG2000), BODIPY 493/503 (10 μg/mL) or DAPI (1 μg/mL) with three PBS washes between each staining. After staining, the slides were treated with ProLong Gold Antifade Reagent. Finally, a coverslip was adhered, and the slide was allowed to dry for at least 1 hour. Fluorescence microscopy was performed on a Nikon TE-2000U epifluorescence microscope equipped with the following filter sets; ex: 340/80, em: 435/85 (blue); ex: 450/90, em: 500/50 (green); ex: 535/50 nm, em: 610/75 (orange); and ex: 620/60, em: 700/75 (deep-red). Acquisition time for all images was 100 milliseconds.
Unstained BAT slices were imaged using the orange and green filter sets, and the autofluorescence intensities were subtracted as background from the images of stained BAT. Images were generated using NIS-Elements software (Nikon) and analyzed using ImageJ 1.40g.
Planar In Vivo Fluorescence Imaging
Athymic nude mice (strain NCr Foxn1nu, Taconic Farms) and immunocompetent SKH1 hairless mice (Charles River), Balb/C mice, and C57Bl/6 mice were housed at ambient room temperature and anesthetized with 2-3% isoflurane with an oxygen flow rate of 2 L/min. The mice were dosed with micellar SRFluor680, IR786, or Nile Red via the tail vein (100 μL). Following dosing, the living SKH1 and nude mice were immediately imaged at various time points, whereas, the BalbC and C57Bl/6 mice were anesthetized by isoflurane inhalation (2-3%), euthanized by cervical dislocation and the skin removed prior to imaging to reduce scattering and absorption by the hair. Planar fluorescence imaging employed an IVIS Lumina (Xenogen) equipped with a 150W Quartz Tungsten Halogen 21V bulb for excitation with the following fluorescence acquisition parameters: SRFluor680 fluorescence (ex: 615-665 nm, em: 695-770 nm, acquisition time: 5 seconds, binning: 2 × 2, F-stop: 2, field-of-view: 10 cm × 10 cm); IR786 fluorescence (ex: 710-760 nm, em: 810-875nm, acquisition time: 10 seconds, binning: 2 × 2, F-stop: 2, field-of-view: 10 cm × 10 cm); Nile Red fluorescence (ex: 500-550 nm, em: 575-650 nm, acquisition time: 10 seconds, binning: 2 × 2, F-stop: 1, field-of-view: 10 cm × 10 cm). After imaging studies, the mice were anesthetized by isoflurane inhalation (2-3%) and euthanized by cervical dislocation.The major organs were dissected, placed on low reflectance paper and imaged as described above.
Ex Vivo Imaging of BAT Digestion
The adipose tissue digestion procedure was adapted from literature.42 BAT was isolated from SKH1 mice (N=4) that had been treated with SRFluor680 (10 nmol) and sacrificed 6 hours after dosing. The adipose samples were separately washed with sterile PBS to remove debris and red blood cells, minced with scissors, and then placed into a 1.5 ml Eppendorf tube. The minced tissue was digested with 2 mg/ml Collagenase I (Worthington Biochemical Corporation) in Hanks' Balanced Salt Solution (HBSS) (1.3 mM CaCl2, 0.5 mM MgCl2•6H2O, 0.5 mM MgSO4•7H2O, 5.3 mM KCl, 0.40 mM KH2PO4, 4.2 mM NaHCO3, 140 mM, NaCl, 0.3 mM Na2HPO4, pH 7.5) at 37 °C for 2 hours. Following digestion, the samples were centrifuged at 400 g for 15 minutes and the tubes imaged with the Xenogen IVIS Lumina using the following parameters: SRFluor680 fluorescence (ex: 615-665 nm, em: 695-770 nm, acquisition time: 0.5 seconds, binning: 2 × 2, F-stop: 2, field-of-view: 5 cm × 5 cm)
Dye Leakage from FRET Pair Labeled Micelles
Micelles containing a FRET donor and acceptor pair (FRET pair labeled micelles) were prepared as follows: 0.28 mg of DiI (FRET donor) and 0.35 mg SRFluor680 (FRET acceptor) were dissolved in dichloromethane and combined with a dichloromethane solution containing 66 mg of PEG2000-DPPE. The trichloromethane solvent was removed via rotary evaporation followed by drying for >1 hour on a high vacuum line. Phosphate buffered saline (PBS: 137 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) was added to the film and the dispersion was sonicated for 15 minutes to produce the micellar formulation.
POPC liposomes were prepared using the thin film hydration method, followed by membrane extrusion using a Basic Liposo-Fast device (Avestin Inc., Ottawa, Canada). Ten milligrams of phosphatidylcholine (POPC) in trichloromethane were added to a clean and dry test tube and solvent was removed under a stream of nitrogen gas. The resulting lipid film was placed under high vacuum for at least 1 hour to remove residual organic solvent. Rehydration at room temperature with PBS formed a stock solution of multilamellar liposomes (10 mM total lipid) that were extruded twenty one times through a 19 mm polycarbonate Nucleopore filter with 200 nm diameter pores to form unilamellar liposomes.
FRET pair labeled micelles (4 μM SRFluor680, 4 μM DiI, 360 μM PEG2000-PE) and POPC liposomes (1.1 mM) were mixed under constant stirring in a quartz cuvette containing PBS at 37 °C. At regular time points over a 3 hour period, a fluorescence emission spectrum covering 540–750 nm was acquired with excitation of the DiI donor at 530 nm. The FRET efficiency ratio IA/(IA + ID) was calculated for each time point where IA and ID are the emission intensities for SRFLuor680 (em: 680 nm) and DiI (em: 560 nm) respectively.
Fluorescence Image Analysis
The 16-bit TIFF images of each living mouse at the different time points were sequentially opened using the ImageJ 1.40g software and then converted to an image stack using the “convert images to stack” software command. The stack of images was background subtracted using the rolling ball algorithm (radius 500 pixels). Next, the image stack was set to the “Fire” fluorescence intensity scale (under “Lookup Tables” menu) which color-codes the fluorescence counts contained in each pixel. The stack of images was converted into a montage using the “convert stack to montage” command. A calibration bar was added to the montage using the “calibration bar” command, and the resulting image saved as a TIFF file.
Probe biodistribution was measured by Region of Interest (ROI) analysis of the fluorescent images of the dissected tissues. Using ImageJ 1.40g software, a free-hand ROI was drawn around each tissue image and the mean pixel intensity was measured. The biodistribution analyses assume that probe fluorescence from a specific organ suffers the same amount of signal attenuation and thus, the mean pixel intensities reflect relative probe concentration in the organ. The tissue comparison in Figure 6 assumes that the BAT and WAT samples are equivalent tissue thicknesses, and that the mean pixel intensities reflect relative SRFluor680 concentration in the tissue.
Figure 6.
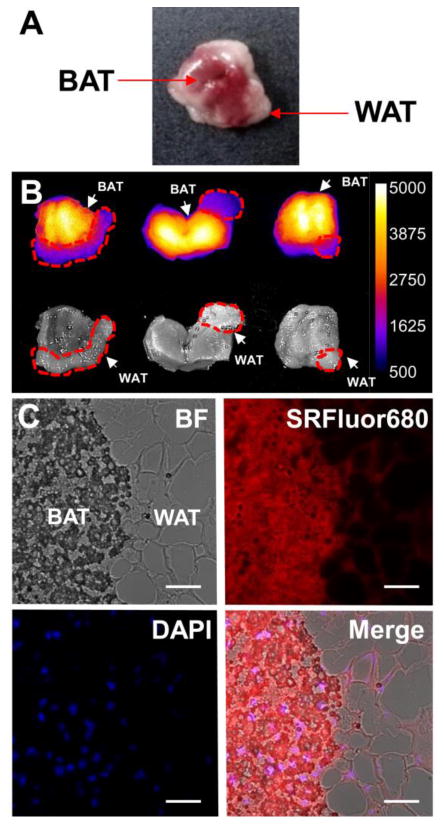
[A] Color photograph of a typical SKH1 mouse interscapular fat pad containing a mixture of brown (BAT) and white (WAT) adipose tissue. [B] Ex vivo imaging of interscapular fat pads excised from SKH1 mice treated with 10 nmol of micellar SRFluor680 and sacrificed after 6 hours. Dashed red lines indicate areas of visible WAT. [C] Representative micrographs of a histological section at the BAT/ WAT border removed from the interscapular fat pad (N = 3) that had been treated with an intravenous dose of micellar SRFluor680 (10 nmol) and sacrificed after 6 hours. Sections were stained with DAPI and imaged using fluorescence microscopy at 60x magnification, with DAPI observed in the blue channel and SRFluor680 observed in the red channel. Scale bar = 100 μm.
Effect of BAT Activation on Probe Biodistribution
A cohort of SKH1 mice (N=6) were anesthetized with 2-3% isoflurane with an oxygen flow rate of 2 L/min. Half the mice were given an intraperitoneal dose of CL 316243 (1 mg/kg, 100 μL) and five minutes later all of the mice were injected intravenously with 15 nmol (150 μL) of micellar SRFluor680 (in DPPE-PEG2000) via the tail vein. One hour later, the mice were sacrificed and fluorescent probe biodistribution determined by imaging the excised tissues using the Xenogen IVIS Lumina and the image acquisition parameters described above. As a positive control, the experiment was repeated with micellar IR786 (15 nmol) instead of SRFluor680.
Multimodal PET/CT and Fluorescence/X-ray Imaging
A cohort of SKH1 hairless mice (N=4) were dosed with 15 nmol of micellar SRFluor680 (in DPPE-PEG2000) via the tail vein (100 μL). Eighteen hours later, mice were dosed with 7 × 106 Bq of 18F-FDG and returned to their cages for one hour. The mice were subsequently anesthetized with 2-3% isoflurane with an oxygen flow rate of 2.5 L/min, and transferred to the Albira trimodal PET/SPECT/CT scanner (Bruker Molecular Imaging). PET scans were acquired with the Albira Acquisition Software Suite using the following parameters: 10 minute acquisition time with MLEM data reconstruction. CT scans were acquired with 400 projections, and reconstructed with a filtered back projection algorithm at a 250 μm isotropic voxel size. PET/CT overlay images were compiled on VolView v3.0, and automatically fused following reconstruction.
Following PET imaging, the anesthetized mice were rapidly transferred to the Xtreme 4MP In-Vivo Imaging System (Bruker Molecular Imaging) for whole-body planar fluorescence and X-ray imaging. Fluorescence imaging acquisition parameters: ex: 630 nm, em: 700, acquisition time: 20 seconds, binning: 1 × 1, F-stop: 1, field-of-view: 120 mm. X-ray acquisition parameters: 45 kVp, 0.8 mm Al, acquisition time 8 seconds, binning: 1 × 1, F-stop: 2, field-of-view: 120 mm. Fluorescence-X-ray overlay images were prepared with ImageJ v1.44p.
Statistical Analysis
Since the statistical analyses only compare two populations, the Student's t-test was applicable and the results are depicted as mean ± standard error of the mean.
Results
SRFluor680 is a lipophilic, uncharged, water-insoluble, fluorescent dye with a permanently interlocked molecular structure (Figure 1A). Shown in Figure 1B are the narrow and intense deep red absorption/emission bands for an aqueous formulation of SRFluor680 in micelles composed of DPPE-PEG2000. The micelle size distribution and morphological profile was determined using laser transmission spectroscopy (LTS)40,41 and transmission electron microscopy (TEM). LTS measurements of empty DPPE-PEG2000 micelles and DPPE-PEG2000 micelles containing SRFluor680 indicated a mean micelle size of approximately 27 nm and 30 nm respectively (Figure 2A). The slight increase in micelle size can be attributed to minor swelling caused by the lipophilic SRFluor680 dye. TEM imaging showed an almost spherical morphology with a near uniform size distribution of 30 nm (Figure 2B). The aqueous formulation was stable over a 6 hour period (Figure S1). Essentially the same spectral and stability results were obtained using Cremophor El as the micellar vehicle.
Figure 1.
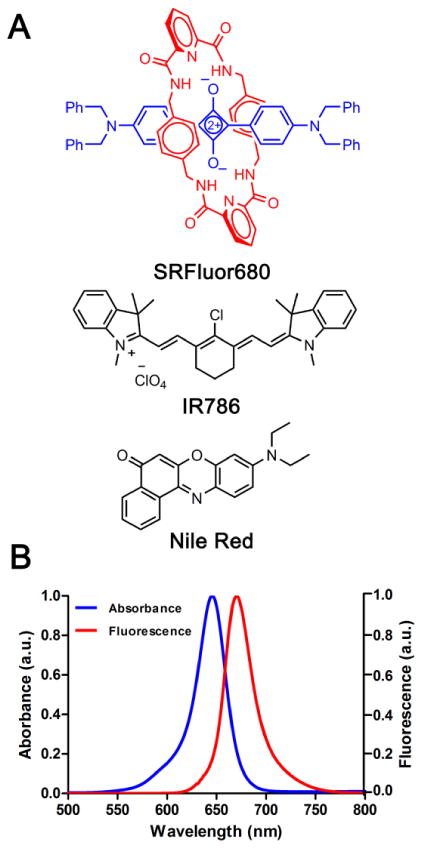
[A] Dye structures, and [B] spectral properties of micellar SRFluor680 in PBS (pH 7.4, excitation λ: 570 nm).
Figure 2.
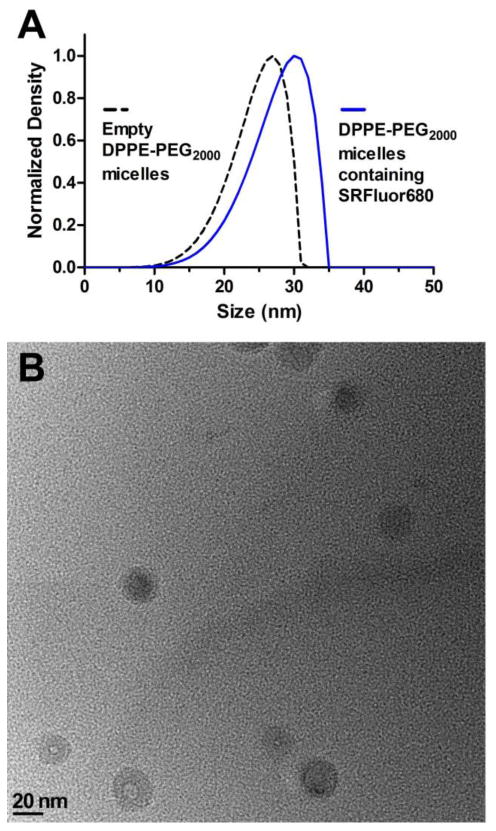
[A] Laser transmission spectroscopy (LTS) measurements of empty DPPE-PEG2000 micelles and DPPE-PEG2000 micelles containing SRFluor680, in deionized water, pH 7.4 at 25° C. [B] Representative transmission electron microscopy (TEM) image of DPPE-PEG2000 micelles containing SRFluor680.
Previous cell microscopy studies have demonstrated that SRFluor680 rapidly and irreversibly stains lipophilic intracellular sites without any evidence of cell toxicity.36 Treatment of cultured mammalian cells with SRFluor680 solubilized in either DMSO or an aqueous dispersion of micelles produces essentially the same intense intracellular staining patterns. Further evidence that SRFluor680 is able to translocate readily from the micellar vehicle into proximal lipophilic sites was gained by conducting experiments that monitored dye transfer into receiver liposomes using fluorescence resonance energy transfer (FRET). As illustrated in Figure 3A, FRET pair labeled micelles were created by encapsulating DiI and SRFluor680 into PEG2000-PE micelles. DiI is a cationic and highly lipophilic fluorophore that emits at 560 nm and is strongly retained by the anionic PEG2000-PE micelles. The DiI acts as an energy donor when paired with proximal SRFluor680 as the energy acceptor. SRFluor680 translocation from the micellar vehicle produces a change in the FRET efficiency which is detected by a change in the ratio IA/(IA + ID). In water the FRET pair labeled micelles are stable over time, but in the presence of receiver liposomes, which act as a lipophilic sink, there is a substantial change in FRET efficiency indicating dye translocation from the micelles into the liposome membranes (Figure 3B and C). The translocation half-life under these conditions is about 10 minutes.
Figure 3.
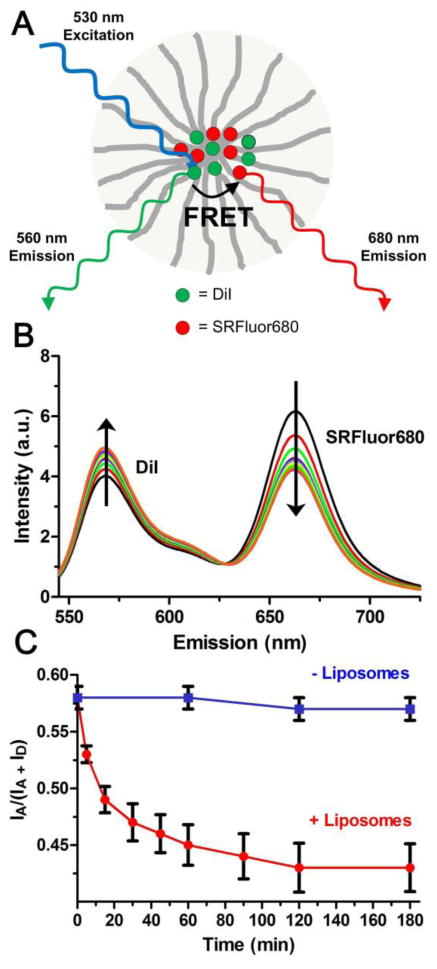
[A] Schematic picture of FRET pair labeled micelle with DiI as FRET donor and SRFluor680 as FRET acceptor. [B] Time dependent change in FRET efficiency for FRET pair labeled micelles after mixing with receiver liposomes, 37 °C. [C] Change in FRET efficiency ratio IA/(IA + ID) in the presence (red) and absence (blue) of receiver liposomes, 37 °C. (N = 3).
A comparative animal imaging study was performed using mice that were treated with the three lipophilic fluorescent dyes shown in Figure 1. Separate cohorts of immunocompetent SKH1 hairless mice (N=3) were anesthetized and given intravenous doses (100 μL) of SRFluor680, IR786 or Nile Red (10 nmol) solubilized in micelles composed of DPPE-PEG2000 (Figure S2). Each animal was imaged periodically using a whole-body, small animal imaging station that was configured for epifluorescence imaging with a deep-red filter set (ex: 615–665 nm, ex: 695–770 nm). A short while after dosage, there was clear evidence for selective SRFluor680 accumulation in the BAT deposits in the neck and interscapular area (Figure 4A).43 In comparison, analogous longitudinal images of mice treated with IR786 or Nile Red showed little or no evidence for BAT targeting (Figures 4B and 4C).
Figure 4.
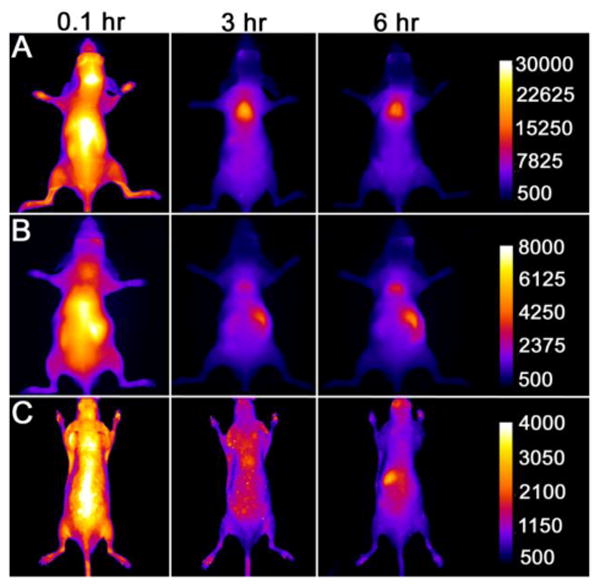
Fluorescence images of living SKH1 hairless mice (N = 3) given a single intravenous 10 nmol dose of SRFluor680 [A], IR780 [B] or Nile Red [C] and imaged periodically over a period of 6 hours. The fluorescence pixel intensity scale bar applies to all images in the same row (arbitrary units).
These in vivo imaging results were confirmed with an ex vivo imaging analysis of organs taken from the animals after they were sacrificed at 6 hours after probe injection. The biodistribution data in Figure 5 shows that micellar SRFluor680 produced high probe localization in the interscapular BAT, whereas, micellar IR786 led to extensive localization in the lungs and liver, with little BAT accumulation. Nile Red also accumulated strongly in the mouse clearance organs with very low accumulation at the interscapular BAT. The influence of the micellar vehicle on SRFluor680 biodistribution was assessed by repeating the mouse imaging experiments with a formulation of SRFluor680 in Cremophor El. The results (Figure S3) showed that SRFluor680 biodistribution is insensitive to the chemical structure of micellar vehicle. Athymic nude mice and immunocompetent Balb/C and C57Bl/6 mouse strains were also examined to determine if the selective accumulation of SRFluor680 in the interscapular BAT was universal among murine species. In vivo imaging of the nude mice and ex vivo imaging of the Balb/C and C57Bl/6 mice all showed extensive accumulation in the interscapular BAT and a similar biodistribution profile was observed (Figure 4, Figure S4, Figure S5). It is worth noting that in each case there was very little probe accumulation in the intraperitoneal (i.p.) WAT.
Figure 5.
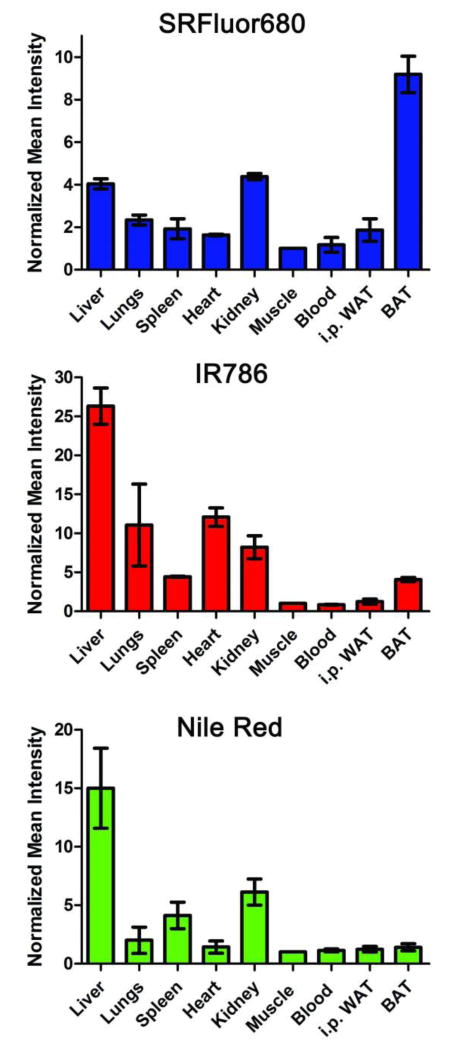
Biodistribution of fluorescent probe in organs taken from the mice (N = 3) in Figure 3 that were sacrificed 6 hours after a single intravenous dose of micellar SRFluor680, IR780 or Nile Red (10 nmol ). The mean pixel intensities are relative to the leg muscle and error bars are standard error of the mean.
Further ex vivo analysis of the SKH1 mice treated with SRFluor680 examined the interscapular fat pad which consists of a depot of BAT surrounded by relatively small amounts of WAT (Figure 6A). Ex vivo imaging of the fat pads showed selective accumulation of SRFluor680 in the BAT compared to the peripheral WAT (Figure 6B). Moreover, the BAT fluorescence images indicate a symmetrical pattern of SRFluor680 distribution with highest accumulation on both sides of a midline. Histological sections were taken from the fat pad samples and subsequently stained with the blue nuclear stain DAPI. In Figure 6C are fluorescence micrographs of a section from the WAT and BAT border, showing that the SRFluor680 is broadly dispersed throughout the BAT with little accumulation in the WAT.
The in vivo probe selectivity for interscapular BAT over WAT was determined by imaging BAT and WAT samples of similar mass taken from SKH1 mice that had been treated with SRFluor680. ROI analysis of the fluorescence images (Figure 7A) indicated a 3.5-fold higher concentration of SRFluor680 in the BAT (Figure 7B). The tissue assignments as BAT and WAT were confirmed by taking histological slices and staining them with hematoxylin and eosin (H&E). The representative micrographs in Figure 7C show the characteristic features of small lipid droplets with extensive vasculature for BAT, and large lipid droplets with peripheral nuclei for WAT. A separate set of histological studies assessed the inherent affinity of SRFluor680 for BAT and WAT. Sections of BAT and WAT samples taken from untreated mice were stained by sequential bathing treatments with deep-red micellar SRFluor680, green lipid droplet stain BODIPY (493/503),44 and blue nuclear stain DAPI. The SRFluor680 fluorescence intensities for both tissue samples were very similar and they co-localized strongly with the green emission of the lipid droplet stain (Figure 8).
Figure 7.
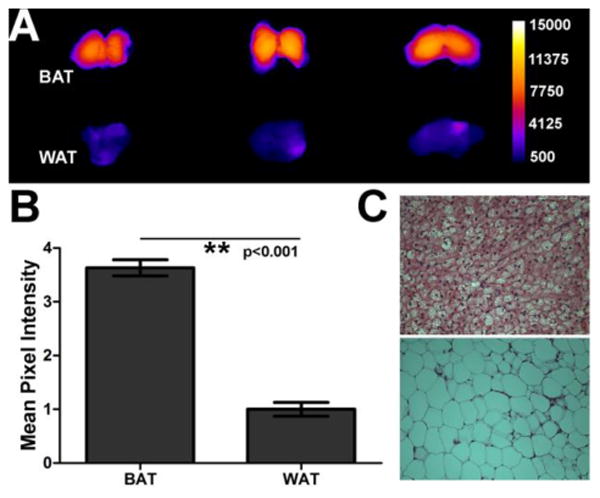
[A] Ex vivo imaging of BAT and WAT extracted from the interscapular fat pad of SKH1 mice (N=3) that were treated with a single 10 nmol intravenous dose of micellar SRFluor680 and sacrificed after 6 hours. The fluorescence intensity scale bar applies to all images (arbitrary units). [B] Normalized mean pixel intensities for SRFluor680 fluorescence in the two adipose tissues. Error bars are standard error of the mean. [C] Representative micrographs of histological sections of the two adipose tissues (BAT, top; WAT, bottom) after staining with hematoxylin and eosin (H&E).
Figure 8.
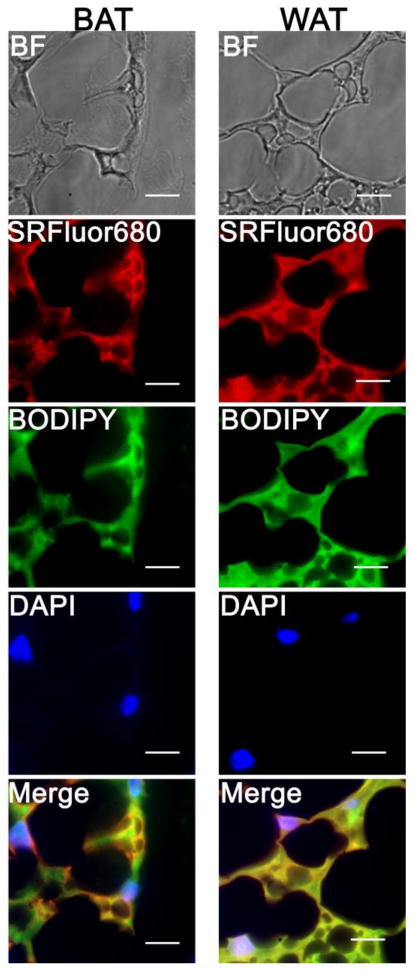
Micrographs of interscapular BAT (left) and interscapular WAT (right) sections from untreated SKH1 mice (N = 3). The sections were bathed with micellar SRFluor680 (red channel), lipid droplet stain BODIPY(493/503) (green channel), and nuclear stain DAPI (blue channel), then imaged using fluorescence microscopy at 100x magnification. Scale bar = 125 μm.
A standard tissue digestion process was used to separate the BAT samples into cellular fractions. Incubation with collagenase I and subsequent centrifugation created three fractions; a low density adipose fraction composed primarily of brown adipocytes, an infranatant fluid portion and a high density stromal vascular fraction containing preadipocytes, mesenchymal stem cells and endothelial cells (Figure 9A). Shown in Figure 9B are representative deep-red fluorescence images of digested and centrifuged samples originating from untreated control mice or mice that had been dosed with SRFluor680. The samples from the untreated mice produced a very low level of tissue autofluorescence. The intense deep-red fluorescence of the samples from the treated mice was primarily contained within the adipose fraction. ROI analysis of the images revealed ∼5-fold greater accumulation of SRFluor680 in the adipose fraction over the stroma fraction (Figure 9C).
Figure 9.
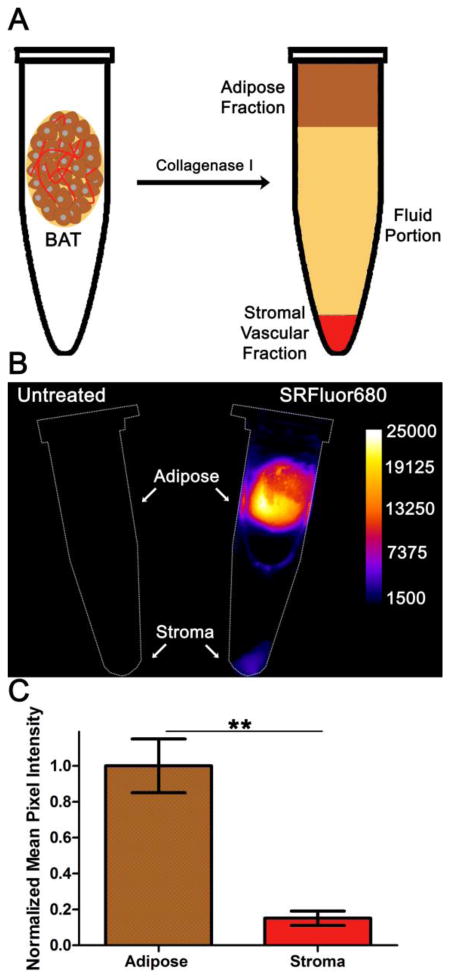
[A] Digestion of BAT with collagenase I produces an adipose fraction harboring brown adipocytes and a stromal fraction composed primarily of endothelium separated by a fluid portion. [B] Deep-red fluorescence intensity images of centrifugation tubes containing digested interscapular BAT samples from untreated control mice (left) or mice that had been dosed with 10 nmol of micellar SRFluor680 (right). [C] Region of Interest analysis of the fluorescence images in [B] (N = 4).
To determine if SRFluor680 targeting to BAT was influenced by changes in metabolic activity, a cohort of SKH1 mice (N=3) was pretreated with CL 316243 (1 mg/kg), a β3 adrenoceptor agonist that is known to activate BAT thermogenesis 45. Five minutes later the mice were administered an intravenous dose of micellar SRFluor680 or IR786. The latter optical probe is known to increase accumulation in metabolically activated BAT and thus served as a positive control.33 In each case, the animals were sacrificed at one hour after fluorescent probe injection and probe biodistribution in the excised tissues was determined. The data in Figure 10 show that BAT activation due to CL 316243 pretreatment produced the expected ∼2-fold increase in BAT uptake of IR786 but did not alter the BAT uptake of SRFluor680. There was no difference in SRFluor680 biodistribution between the BAT activated and unactivated cohorts.
Figure 10.
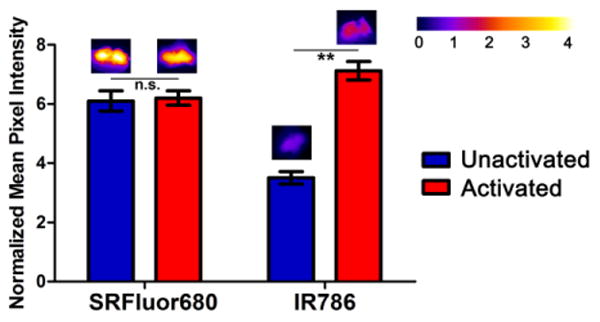
Effect of BAT metabolic activation on probe accumulation. The SKH1 mice were treated with adrenoceptor agonist CL 316243 (1 mg/kg) (activated BAT) or an equal volume of saline (unactivated BAT), followed 5 minutes later by a dose of micellar SRFluor680 or IR786 (15 nmol), and then sacrificed at 60 minutes. In each case, the deep-red fluorescence mean pixel intensities for the excised BAT are relative to a leg muscle value of 1 and error bars are standard error of the mean (N = 3 for each experiment). Representative fluorescence images of the excised BAT are shown above each of the bars.
A final experiment demonstrated multimodal PET/CT and fluorescence/X-ray imaging of BAT in a single living mouse. The PET agent was 18F-FDG and micellar SRFluor680 was the fluorescent probe. Each SKH1 mouse in a cohort (N=4) was injected intravenously with 15 nmol of micellar SRFluor680, and eighteen hours later dosed with 7 × 106 Bq of 18F-FDG. One hour after 18F-FDG dosing, whole-body PET/CT images were acquired, followed by planar fluorescence/X-ray images. In Figure 11A-C is a representative set of PET/CT and planar fluorescence/X-ray images of a single, living mouse showing strong co-localization of the 18F-FDG and SRFluor680 signals at the interscapular BAT.
Figure 11.
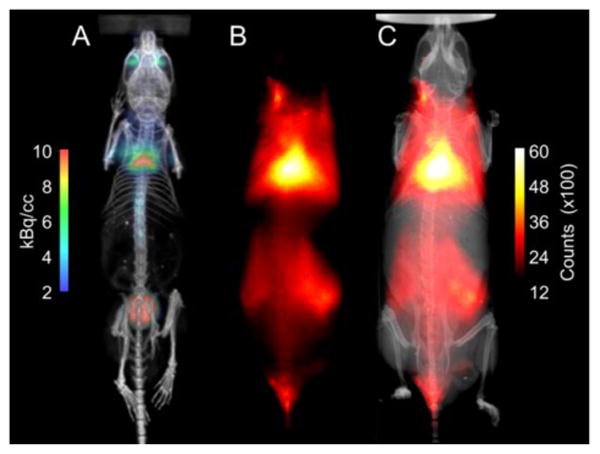
Representative set of multimodal PET/CT and fluorescence/X-ray images of a single living SKH1 mouse (N = 4). The mouse was injected with 15 nmol of micellar SRFluor680 followed by a 7 × 106 Bq dose of 18F-FDG 18 hours later. One hour after 18F-FDG dosing, whole-body PET/CT images were acquired followed by planar fluorescence/X-ray images. [A] PET/CT image showing 18F-FDG signal in the BAT and the bladder, [B] Fluorescence signal from SRFluor680 in the BAT, [C] Overlaid X-ray and SRFluor680 fluorescence images.
Discussion
Emerging research suggests that the amount BAT in a living subject and metabolic status of the BAT are important indicators and controllers of obesity and diabetes.1-20 Imaging methods that can quantify BAT in humans are likely to be important for personal health and treatment. Similarly, convenient high throughput methods that image BAT in small animals will facilitate preclinical research. Likely to be of great value are independent methods that non-invasively image BAT mass and BAT metabolic status. Quantification of BAT has been achieved using MRI techniques which exploit the greater water to fat ratio in BAT relative to WAT.46-48 Unfortunately, accurate BAT volume can be difficult to measure with MRI when the adipose tissue signal contrast from surrounding internal structures is poor.49 Although the metabolic probe 18F-FDG can locate BAT,50 it cannot easily measure BAT volume due to the fluctuating metabolic activity of BAT and the possibility of false-positive signals from other metabolically active tissues.51, 52 PET and Single-Positron Emission Tomography (SPECT) using 18F-fluorobenzyl triphenyl phosphonium,26 123I-metaiodobenzylguanidine27, 99mTc-tetrofosmin28, 99mTc-sestamibi,29 and 99mTc-methoxyisobutylisonitrile30 have all been used to detect the presence of BAT. However, nuclear and magnetic resonance imaging modalities are problematic for preclinical studies due to the expensive instrumentation and low throughput protocols. These imaging methods also require long scan times and large doses of anesthesia which are known to cause hypothermia and hypotension which may alter BAT metabolism.53, 54 A recent international workshop concluded that new imaging approaches are needed to quantify BAT physiology and its association to metabolic syndrome.22
The pharmaceutical challenge is to develop a molecular probe that will target the BAT and not the more abundant WAT. Two major differences between these tissue types are the extensive vasculature and high levels of mitochondria in BAT. Not surprisingly, one targeting strategy is to employ molecular probes that have selective affinity for mitochondria. An example of this approach is optical imaging using IR786, a cationic near-infrared fluorescent dye with affinity for mitochondria and BAT.33 An alternative strategy is to target the BAT vasculature, and a recent investigation uncovered a unique peptide sequence with affinity for the vasculature endothelium of BAT.34 The peptide was converted into a fluorescent probe that enabled optical imaging of BAT but relatively high probe dosages were required.
The in vivo imaging and biodistribution data in Figures 4-7 show that SRFluor680 is able to strongly accumulate within interscapular BAT with 3.5-fold selectively over the adjacent interscapular WAT. Furthermore, the SRFluor680 targets the adipocytes within the BAT (Figure 9). Ex vivo tissue staining experiments show that the lipophilic SRFluor680 molecule has about the same inherent affinity for BAT and WAT (Figure 8); thus, the in vivo selectivity for BAT must be due to a selective pharmacokinetic effect. There is no evidence that the SRFluor680 is targeting a protein biomarker in the BAT. In contrast, cell fluorescence microscopy and liposome translocation studies (Figure 3) show that SRFluor680 transfers spontaneously from micelles into nearby lipophilic receiver sites, in agreement with literature expectations.55 Our results are consistent with a tissue partitioning process that involves translocation of the lipophilic SRFluor680 from the micellar vehicle in the bloodstream to the lipophilic interior of the adipocytes within the interscapular BAT. The selectivity for BAT over WAT is attributed to the extensive BAT vasculature and close proximity of the capillaries to the adipocytes. The process is reminiscent of the pharmacokinetic effect known as redistribution where lipid-soluble drugs rapidly enter highly perfused tissues after initial dosage and then slowly distribute back into the plasma. However, in this case the water-insoluble SRFluor680 does not redistribute from the BAT, in part because the micelle assembly in the bloodstream is destroyed by time-dependent dilution.
This hypothesis is supported by our observation that the selective interscapular BAT targeting exhibited by micellar SRFluor680 is insensitive to the molecular structure of micelle-forming amphiphiles (Figure S3). Furthermore analogous imaging studies of mice dosed with micellar Nile Red (a fluorescent probe used to stain lipophilic sites in cell culture) produced no significant probe accumulation in BAT. Nile Red is more water soluble than SRFluor680 and known to quickly escape from micelle nanocarriers and clear rapidly from the bloodstream.56 Strong partitioning of SRFluor680 into the lipophilic adipocytes within BAT explains why the in vivo accumulation is independent of BAT metabolic status (Figure 10); whereas, the observation of increased uptake of IR786 upon pharmacological activation of the BAT supports its known mitochondria targeting mechanism.33 Together these results suggest a new dual probe molecular imaging paradigm that allows separate and independent non-invasive visualization of BAT mass and BAT metabolism in a living subject. A prototype demonstration of the concept is the set of multimodal mouse images in Figure 10 showing interscapular BAT mass reported by the optical probe (SRFluor680) and enhanced BAT metabolism reported by the PET agent (18F-FDG). Additional studies are needed to determine the sensitivity of this specific dual probe imaging protocol to changes in BAT mass and metabolic state.
While fluorescence imaging of BAT is not likely to be directly useful for clinical studies of humans, it may be a valuable technique for preclinical imaging studies of living mice because it is safe to perform (no ionizing radiation), relatively inexpensive, involves short scan times (low anesthesia exposure), conducive to longitudinal studies, and has higher throughput compared to other imaging methods such MRI or PET. A drawback with the deep red absorption/emission wavelengths of SRFluor680 is modest tissue penetration and scattering of the light by mouse hair. Thus, in vivo imaging experiments are best conducted using nude mice, immunocompetent hairless strains, or other mice with appropriate sections of fur removed by standard methods. It should be possible to enhance the optical imaging performance of SRFluor680 by improving the fluorescence properties without losing BAT selectivity. Furthermore, it is worth noting that squaraine rotaxane dyes have unusually high two-photon cross sections (up to 10,000 GM) so it is possible that SRFluor680 can be used for BAT imaging using two-photon illumination methods.57
With SRFluor680 as a lead structure, it is likely that modified molecular probes can be designed to enable other types of in vivo imaging modalities, including radiolabeled versions for nuclear imaging and clinical translation. The previous community success at converting fluorescent molecules with unique targeting ability into clinically useful nuclear imaging probes is reason for optimism.58, 59
Supplementary Material
Acknowledgments
We are grateful for funding support from NIH grants R01GM059078 (B.D.S.) and T32GM075762 (D.R.R.), and the Notre Dame Integrated Imaging Facility (NDIIF). We thank S. Chapman, for technical assistance with histology and S. Ruggiero, C. Tanner, and S. Rouvimov for help with micelle characterization.
Abbreviations
- DiI
1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate
- DPPE-PEG2000
1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (ammonium salt)
- Isoflurane
2-chloro-2-(difluoromethoxy)-1,1,1-trifluoro-ethane
- LTS
Laser Transmission Spectroscopy
- PET/CT
positron emission tomography/computed tomography
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- SPECT
single photon emission tomography
- BAT
brown adipose tissue
- WAT
white adipose tissue
Footnotes
References
- 1.Haas B, Schlinkert P, Mayer P, Eckstein N. Diabetol Metab Syndr. 2012;4:43. doi: 10.1186/1758-5996-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carter BW, Schucany WG. Proceedings (BUMC) 2008;21:328–330. doi: 10.1080/08998280.2008.11928419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, Kolodny GM, Kahn CR. N Engl J Med. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enerback S. Cell Metab. 2010;11:248–252. doi: 10.1016/j.cmet.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 5.van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. N Engl J Med. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- 6.Yoneshiro T, Aita S, Matsushita M, Kayahara T, Kameya T, Kawai Y, Iwanaga T, Saito M. J Clin Invest. 2013;123:3404–3408. doi: 10.1172/JCI67803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orava J, Nuutila P, Lidell ME, Oikonen V, Noponen T, Viljanen T, Scheinin M, Taittonen M, Niemi T, Enerback S, Virtanen KA. Cell Metab. 2011;14:272–279. doi: 10.1016/j.cmet.2011.06.012. [DOI] [PubMed] [Google Scholar]
- 8.Cypess AM, Chen YC, Sze C, Wang K, English J, Chan O, Holman AR, Tal I, Palmer MR, Kolodny GM, Kahn CR. Proc Natl Acad Sci U S A. 2012;109:10001–10005. doi: 10.1073/pnas.1207911109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito M, Okamatsu-Ogura Y, Matsushita M, Watanabe K, Yoneshiro T, Nio-Kobayashi J, Iwanaga T, Miyagawa M, Kameya T, Nakada K, Kawai Y, Tsujisaki M. Diabetes. 2009;58:1526–1531. doi: 10.2337/db09-0530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Q, Zhang M, Ning G, Gu W, Su T, Xu M, Li B, Wang W. PLoS One. 2011;6:e21006. doi: 10.1371/journal.pone.0021006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL, Chin S, Tempst P, Lazar MA, Spiegelman BM. Genes Dev. 2008;22:1397–1409. doi: 10.1101/gad.1666108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson-Fritch L, Nicoloro S, Chouinard M, Lazar MA, Chui PC, Leszyk J, Straubhaar J, Czech MP, Corvera S. J Clin Invest. 2004;114:1281–1289. doi: 10.1172/JCI21752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Xu X, Ying Z, Cai M, Xu Z, Li Y, Jiang SY, Tzan K, Wang A, Parthasarathy S, He G, Rajagopalan S, Sun Q. Am J Physiol-Reg I. 2011;300:R1115–1125. doi: 10.1152/ajpregu.00806.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, Kajimura S, Zingaretti MC, Vind BF, Tu H, Cinti S, Hojlund K, Gygi SP, Spiegelman BM. Nature. 2012;481:463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosenwald M, Perdikari A, Rulicke T, Wolfrum C. Nat Cell Biol. 2013;15:659–667. doi: 10.1038/ncb2740. [DOI] [PubMed] [Google Scholar]
- 16.Villarroya F, Vidal-Puig A. Cell Metab. 2013;17:638–643. doi: 10.1016/j.cmet.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 17.Gunawardana SC. Adipocyte. 2012;1:250–255. doi: 10.4161/adip.21042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunawardana SC, Piston DW. Diabetes. 2012;61:674–682. doi: 10.2337/db11-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva FJ, Holt DJ, Vargas V, Yockman J, Boudina S, Atkinson D, Grainger DW, Revelo MP, Sherman W, Bull DA, Patel AN. Stem Cells. 2014;32:572–581. doi: 10.1002/stem.1595. [DOI] [PubMed] [Google Scholar]
- 20.Ussar S, Lee KY, Dankel SN, Boucher J, Haering MF, Kleinridders A, Thomou T, Xue R, Macotela Y, Cypess AM, Tseng YH, Mellgren G, Kahn CR. Sci Transl Med. 2014;6:247ra103. doi: 10.1126/scitranslmed.3008490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boss O, Farmer SR. Front Endocrinol. 2012;3:14. doi: 10.3389/fendo.2012.00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cypess AM, Haft CR, Laughlin MR, Hu HH. Cell Metab. 2014;20:408–415. doi: 10.1016/j.cmet.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirbolooki MR, Constantinescu CC, Pan ML, Mukherjee J. EJNMMI Res. 2011;1:30. doi: 10.1186/2191-219X-1-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lau AZ, Chen AP, Gu Y, Ladouceur-Wodzak M, Nayak KS, Cunningham CH. Int J Obes. 2014;38:126–131. doi: 10.1038/ijo.2013.58. [DOI] [PubMed] [Google Scholar]
- 25.Chen YI, Cypess AM, Sass CA, Brownell AL, Jokivarsi KT, Kahn CR, Kwong KK. Obesity. 2012;20:1519–1526. doi: 10.1038/oby.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Madar I, Isoda T, Finley P, Angle J, Wahl R. J Nucl Med. 2011;52:808–814. doi: 10.2967/jnumed.110.084657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okuyama C, Sakane N, Yoshida T, Shima K, Kurosawa H, Kumamoto K, Ushijima Y, Nishimura T. J Nucl Med. 2002;43:1234–1240. [PubMed] [Google Scholar]
- 28.Fukuchi K, Ono Y, Nakahata Y, Okada Y, Hayashida K, Ishida Y. J Nucl Med. 2003;44:1582–1585. [PubMed] [Google Scholar]
- 29.Kyparos D, Arsos G, Georga S, Petridou A, Kyparos A, Papageorgiou E, Mougios V, Matziari C, Karakatsanis C. Physiol Res. 2006;55:653–659. doi: 10.33549/physiolres.930890. [DOI] [PubMed] [Google Scholar]
- 30.Goetze S, Lavely WC, Ziessman HA, Wahl RL. J Nucl Med. 2008;49:752–756. doi: 10.2967/jnumed.107.048074. [DOI] [PubMed] [Google Scholar]
- 31.Balas C. Meas Sci Technol. 2009;20:104020. [Google Scholar]
- 32.Zhang X, Kuo C, Moore A, Ran C. PLoS One. 2013;8:e62007. doi: 10.1371/journal.pone.0062007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakayama A, Bianco AC, Zhang CY, Lowell BB, Frangioni JV. Mol Imaging. 2003;2:37–49. doi: 10.1162/15353500200303103. [DOI] [PubMed] [Google Scholar]
- 34.Azhdarinia A, Daquinag AC, Tseng C, Ghosh SC, Ghosh P, Amaya-Manzanares F, Sevick-Muraca E, Kolonin MG. Nat Commun. 2013;4:2472–2482. doi: 10.1038/ncomms3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.White AG, Fu N, Leevy WM, Lee JJ, Blasco MA, Smith BD. Bioconjug Chem. 2010;21:1297–1304. doi: 10.1021/bc1000998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson JR, Fu N, Arunkumar E, Leevy WM, Gammon ST, Piwnica-Worms D, Smith BD. Angew Chem Int Ed Engl. 2007;46:5528–5531. doi: 10.1002/anie.200701491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith BA, Harmatys KM, Xiao S, Cole EL, Plaunt AJ, Wolter W, Suckow MA, Smith BD. Mol Pharm. 2013;10:3296–3303. doi: 10.1021/mp300720k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xiao S, Turkyilmaz S, Smith BD. Tetrahedron Lett. 2013;54:861–864. doi: 10.1016/j.tetlet.2012.11.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fu N, Gassensmith JJ, Smith BD. Supramol Chem. 2009;21:118–124. doi: 10.1080/10610270802468454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li F, Schafer R, Hwang CT, Tanner CE, Ruggiero ST. Appl Opt. 2010;49:6602–6611. doi: 10.1364/AO.49.006602. [DOI] [PubMed] [Google Scholar]
- 41.Li F, Mahon AR, Barnes MA, Feder J, Lodge DM, Hwang CT, Schafer R, Ruggiero ST, Tanner CE. PLoS One. 2011:6. doi: 10.1371/journal.pone.0029224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Craig JC, Williams GJ, Jones M, Codarini M, Macaskill P, Hayen A, Irwig L, Fitzgerald DA, Isaacs D, McCaskill M. BMJ. 2010;340:c1594. doi: 10.1136/bmj.c1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rothwell NJ, Stock MJ. Comp Biochem Phys A. 1985;82:745–751. doi: 10.1016/0300-9629(85)90477-3. [DOI] [PubMed] [Google Scholar]
- 44.Spangenburg EE, Pratt SJ, Wohlers LM, Lovering RM. J Biomed Biotechnol. 2011;2011:598358. doi: 10.1155/2011/598358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshida T, Sakane N, Wakabayashi Y, Umekawa T, Kondo M. Life Sci. 1994;54:491–498. doi: 10.1016/0024-3205(94)00408-0. [DOI] [PubMed] [Google Scholar]
- 46.Rasmussen JM, Entringer S, Nguyen A, van Erp TG, Burns J, Guijarro A, Oveisi F, Swanson JM, Piomelli D, Wadhwa PD, Buss C, Potkin SG. PLoS One. 2013;8:e77907. doi: 10.1371/journal.pone.0077907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holstila M, Virtanen KA, Gronroos TJ, Laine J, Lepomaki V, Saunavaara J, Lisinen I, Komu M, Hannukainen JC, Nuutila P, Parkkola R, Borra RJ. Metabolism. 2013;62:1189–1198. doi: 10.1016/j.metabol.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 48.Hu HH, Perkins TG, Chia JM, Gilsanz V. Am J Roentgenol. 2013;200:177–183. doi: 10.2214/AJR.12.8996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu HH, Nayak KS, Goran MI. Obes Rev. 2011;12:e504–e515. doi: 10.1111/j.1467-789X.2010.00824.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Minze LJ, Shi ZZ. J Vis Exp. 2012;69:4060. doi: 10.3791/4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fueger BJ, Czernin J, Hildebrandt I, Tran C, Halpern BS, Stout D, Phelps ME, Weber WA. J Nucl Med. 2006;47:999–1006. [PubMed] [Google Scholar]
- 52.Tatsumi M, Engles JM, Ishimori T, Nicely OB, Cohade C, Wahl RL. J Nucl Med. 2004;45:1189–1193. [PubMed] [Google Scholar]
- 53.Sancho AR, Dowell JA, Wolf W. Cancer Chemother Pharmacol. 1997;40:521–525. doi: 10.1007/s002800050696. [DOI] [PubMed] [Google Scholar]
- 54.Hildebrandt IJ, Su H, Weber WA. ILAR J. 2008;49:17–26. doi: 10.1093/ilar.49.1.17. [DOI] [PubMed] [Google Scholar]
- 55.Chen H, Kim S, Li L, Wang S, Park K, Cheng JX. Proc Natl Acad Sci U S A. 2008;105:6596–6601. doi: 10.1073/pnas.0707046105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schadlich A, Rose C, Kuntsche J, Caysa H, Mueller T, Gopferich A, Mader K. Pharm Res. 2011;28:1995–2007. doi: 10.1007/s11095-011-0426-5. [DOI] [PubMed] [Google Scholar]
- 57.Podgorski K, Terpetschnig E, Klochko OP, Obukhova OM, Haas K. PLoS One. 2012;7:e51980. doi: 10.1371/journal.pone.0051980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Murthy N. Sci Transl Med. 2014;6:259fs243. doi: 10.1126/scitranslmed.3010746. [DOI] [PubMed] [Google Scholar]
- 59.Ni R, Gillberg PG, Bergfors A, Marutle A, Nordberg A. Brain. 2013;136:2217–2227. doi: 10.1093/brain/awt142. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.