

Abstract
Neuromyelitis optica (NMO) and a related spectrum of inflammatory CNS disorders are unified by detection of a serum autoantibody specific for the aquaporin-4 (AQP4) water channel, which is abundant in astrocytic foot processes. The classic clinical manifestations of NMO are optic neuritis and longitudinally extensive transverse myelitis. Newly recognized manifestations of AQP4 autoimmunity include lesions of circumventricular organs and skeletal muscle. NMO is commonly relapsing, is frequently accompanied by other autoimmune disorders, and sometimes occurs in a paraneoplastic context. The goals of treatment are to minimize neurologic disability in the acute attack and thereafter to prevent relapses and cumulative disability. The disease specificity of AQP4 immunoglobulin (Ig) G approaches 100% using optimized molecular-based detection assays. Clinical, immunohistopathologic, and in vitro evidence support this antibody being central to NMO pathogenesis. Current animal models yield limited histopathologic characteristics of NMO, with no clinical deficits to date. Recent descriptions of a myelin oligodendrocyte glycoprotein autoantibody in a minority of patients with NMO spectrum phenotype who lack AQP4-IgG predict serologic delineation of additional distinctive disease entities.
Neuromyelitis optica (NMO) is an inflammatory autoimmune neurologic disease that typically presents as recurrent longitudinally extensive transverse myelitis and optic neuritis.1 The discovery of a serum immunoglobulin (Ig) G autoantibody specific for the aquaporin-4 (AQP4) water channel unified a spectrum of NMO-related disorders, NMO spectrum disorders (NMOSDs) and distinguished them from multiple sclerosis (MS).2–4 Appreciation of AQP4 distribution, both within and beyond the CNS (where it is largely concentrated on astrocytic foot processes), has enabled recognition of the broadening clinical NMOSD phenotype that we address in this review. An arising controversy concerns the relationship of idiopathic optic neuritis and transverse myelitis to NMO in AQP4-IgG–seronegative patients with alternative autoantibody markers for which CNS lesions are as yet undefined immunohistopathologically.5,6
CLINICAL CHARACTERISTICS
Epidemiology.
NMOSD prevalence has not been evaluated formally according to race/ethnicity in different populations, but in Western countries non-Caucasian patients are affected disproportionately.7–9 In Asia NMOSD diagnosis may be more frequent than MS diagnosis.10,11 Women are affected disproportionately, by 3.6:1 to 10.4:1.9,11–13 Initial symptoms begin around age 35–45 years, but 18% of cases occur in children or the elderly.11,13,14 Although most cases are sporadic, rare reports of familial AQP4-IgG–seropositive NMO with classic phenotype support a genetic component to NMO susceptibility.15
Neurologic manifestations.
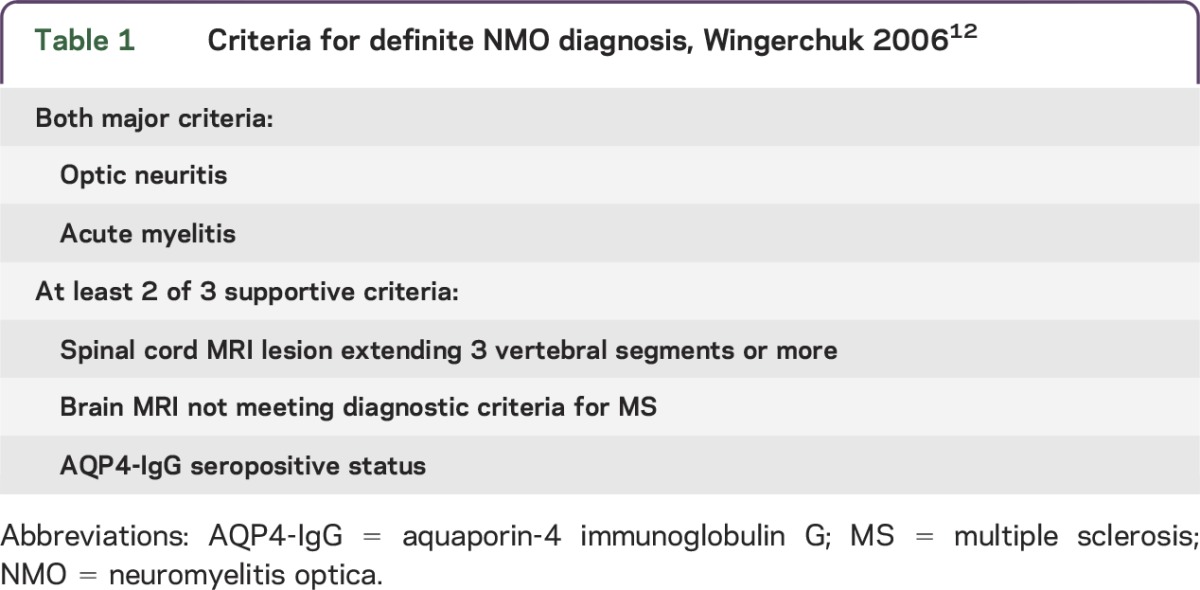
Table 1 summarizes 2006 criteria for definite NMO diagnosis.12 The patient described by Devic in 1894 was a woman with urinary retention, paraplegia, bilateral blindness, and papilledema. Autopsy revealed acute myelitis and bilateral optic neuritis.16 Simultaneous bilateral optic neuritis and transverse myelitis is a rare contemporary presentation for NMO. These cases generally follow a monophasic course, affect both sexes equally and are AQP4-IgG seronegative. They may not be synonymous with an autoimmune AQP4 channelopathy.14 A longer interval between initial clinical events, older age at disease onset, and female sex predict a relapsing NMO course.17 Unlike MS, a secondary progressive course is rare.14,18
Table 1.
Criteria for definite NMO diagnosis, Wingerchuk 200612
Unilateral or bilateral optic neuritis is generally more severe and recovery poorer in NMOSD than in MS, with complete remission in only 32%.14 Transverse myelitis in NMOSD is usually longitudinally extensive, with complete clinical remission in only 17% of attacks14 (figure 1). MRI lesions are short in 14% of first myelitis episodes (92% long at relapse).19 Conus medullaris lesions and lumbosacral myeloradiculitis are rare.20
Figure 1. Representative spinal cord MRIs from patients with neuromyelitis optica.

Longitudinally extensive transverse myelitis of the cervical (A) and cervicothoracic (B) region (T2-weighted images) with nonhomogeneous gadolinium enhancement in multiple levels of the spinal cord (C) (T1-weighted image with gadolinium injection) in aquaporin-4 immunoglobulin G–seropositive patients.
Neuromyelitis optica spectrum disorders.
AQP4-IgG seropositivity unifies partial and inaugural forms of NMO that do not fulfill all NMO diagnostic criteria. Clinical manifestations may include optic neuritis, transverse myelitis, circumventricular organ involvement, and autoimmune AQP4 myopathy (table 2).2,21 NMO and NMOSD can occur in a paraneoplastic context; however, detection of AQP4-IgG in a patient with cancer does not support NMOSD diagnosis unless neurologic findings are consistent.22
Table 2.
NMO spectrum disorders unified by AQP4-IgG positivity, 2015

Brain involvement.
Brain lesions are detectable by MRI at first relapse in 60% of patients with NMO. In 10% the brain lesions resemble those commonly seen in MS. Another 10% have “NMO-typical” lesions in regions of high AQP4 content (periventricular, hypothalamic)23,24 (figure 2). Brain lesions may be asymptomatic or a manifestation of circumventricular organ involvement: intractable nausea and vomiting (area postrema and medullary floor of the fourth ventricle), hiccups, inappropriate antidiuresis, narcolepsy or anorexia (hypothalamus), or posterior pituitary endocrinopathies.25–29 Brainstem signs occur in 31% of patients and are more common in non-Caucasians. These include vomiting (33%), hiccups (22%), oculomotor dysfunction (20%), and pruritus (12%); 3.3% have hearing loss, facial palsy, vestibulopathy, trigeminal neuralgia, or cranial neuropathies.29 Posterior reversible encephalopathy syndrome with MRI evidence of vasogenic edema was reported in 5 women.30 Clinical manifestations and lesions in children may resemble acute disseminated encephalomyelitis (ADEM).31
Figure 2. Representative brain MRIs from patients with neuromyelitis optica.

Lesions are localized at sites of high aquaporin-4 expression (white dots). Patient 1: around the 3rd ventricle and hypothalamus; patient 2: around the 4th ventricle; patient 3: around the 4th ventricle and aqueduct; patient 4: periependymal, around lateral ventricles, cervical lesion extends into the brainstem; patient 5: thalamus, hypothalamus, optic chiasm, around the 4th ventricle, subpial in cerebellar hemispheres; patient 6: around the 4th ventricle and cerebellar peduncles. Modified with permission from Pittock et al. Arch Neurol 2006. Copyright © 2006 American Medical Association. All rights reserved.
Coexisting autoimmune disorders.
Organ-specific and non-organ-specific autoimmune manifestations (i.e., coexisting disease or autoantibody markers) are frequent with NMOSD.32,33 However, AQP4-IgG is restricted to NMOSD. Sjögren syndrome and systemic lupus erythematosus are common NMOSD accompaniments.32 Transverse myelitis in either context is a manifestation of NMOSD and not a vasculitic complication of connective tissue disease.32 Autoimmune myasthenia gravis is 100 times more common in patients with NMOSD (2%) than in the general population (0.02%).34 Celiac disease, autoimmune gastritis, autoimmune thyroiditis, ulcerative colitis, and sclerosing cholangitis have also been reported.32,33,35,36
Skeletal muscle involvement.
HyperCKemia may precede or accompany NMOSD. An illustrative patient experienced corticosteroid-responsive episodes of proximal myalgia (serum creatine kinase exceeding 10,000 U/L) and attacks of optic neuritis and transverse myelitis. Biopsy of affected muscle revealed sarcolemmal AQP4 loss with linear deposits of IgG and complement products.21 As is characteristic of CNS NMO lesions, the lymphohistiocytic infiltrates in muscle contained scattered eosinophils. In contrast, healthy control muscle had abundant surface AQP4 immunoreactivity. Control biopsies of necrotizing autoimmune myopathy and dermatomyositis had patchy AQP4 loss without sarcolemmal immune complex deposition.
Paraneoplastic NMOSD.
The cancer frequency reported in large NMOSD cohorts is 4%–5%, and 15% in older patients (mean age 48.7 years).14,22,37 Reported neoplasms include thymoma, carcinomas (breast, lung, nasopharynx, cervix, bladder, gastrointestinal tract, thyroid, prostate, and skin), pituitary adenoma, carcinoids, and hematologic malignancies.14,22,37
Demonstration of AQP4 protein in thymoma tissue from patients with NMOSD but not in normal thymus suggests an etiologic association.38 AQP4 immunoreactivity was also demonstrated in breast carcinoma, intestinal carcinoid, and ovarian teratoma tissues from patients with and without NMOSD.39–41
Myelin oligodendrocyte glycoprotein autoimmunity.
Myelin oligodendrocyte glycoprotein (MOG)-IgG, once considered a candidate autoantibody for pediatric MS, is reproducibly found in pediatric cases of ADEM using a substrate of MOG-transfected cells.42 It is also found in some adult and pediatric cases of relapsing optic neuritis and transverse myelitis and in occasional AQP4-IgG–seronegative and AQP4-IgG–seropositive patients with NMO phenotype.5,42–44 MOG-IgG–positive cases tend to be monophasic, occur more frequently in men, have a younger age at onset, result in better outcome, and have more frequent MRI involvement of conus and brain deep gray matter.45,5 It is important to note that no MOG-IgG–positive case has been evaluated neuropathologically. A reported patient with clinically definite NMO, MOG-IgG positivity, and AQP4-IgG negativity had a high level of myelin basic protein (MBP) in CSF but undetectable glial fibrillary acidic protein (GFAP).46 This suggests demyelinating pathology rather than astrocytopathy.47 Until immunohistopathologic data are documented for MOG-IgG–positive cases, we advocate their classification as “autoimmune MOG optic neuritis, transverse myelitis, or encephalomyelitis” rather than “NMOSD.”48
Ancillary testing.
MRI findings.
Imaging of the acute myelitis of NMO typically reveals lesions longer than 3 vertebral segments,12 but lesions are short in 14% of initial attacks.19 Long lesions are edematous at onset, with nonhomogenous gadolinium enhancement and sometimes with central necrosis and cavitation.12,14 Late images show focal cord atrophy. Involvement of central gray matter aids in distinguishing NMO from other demyelinating diseases.49 Diffusion tensor imaging performed a year after the last NMO relapse revealed higher radial diffusivity within spinal white matter tracts than in MS, consistent with greater tissue destruction.50
Orbital MRI findings in the optic neuritis of NMO, unilateral or bilateral, are common to all optic neuritis. Optic nerve lesions more characteristic of NMO are bilateral and long and involve chiasm and posterior segments.51 Optical coherence tomography and visual evoked potentials complete the diagnostic evaluation.52 Brain lesions are typically periventricular and hypothalamic, but 10% are MS-like and sometimes fulfill Barkhof criteria.23,24
CSF findings.
The CSF findings in NMO and MS differ. Only 16% of AQP4-IgG–seropositive NMO patients have oligoclonal Ig bands; 50% have pleocytosis (median leukocyte number, 19/μL), which is severe in 6.2% (>100 leukocytes/μL).53 Lymphocytes and monocytes are generally most frequent; neutrophils, eosinophils, and basophils may be admixed or predominate.1,53
Levels of GFAP, a marker of astrocytic damage, have been reported to be significantly higher in acute NMO than in MS and other neurologic diseases,47 with S100β cytoplasmic protein elevated more modestly; MBP and neurofilament H chain levels were similar for the 3 diagnoses. GFAP levels normalized rapidly following immunotherapy, but MBP remained high, presumably reflecting demyelination. Higher acute attack levels of GFAP, S100β, and MBP correlated with longer spinal lesion length and worse Expanded Disability Status Scale (EDSS) score.47 The EDSS outcome at 6 months correlated only with acute-phase GFAP level.
Autoantibody profile.
Multiple autoantibodies (organ-specific and non-organ-specific) are a serologic characteristic that aids in differentiating NMO from MS.54 Titers are generally relatively low. There may or may not be evidence of coexisting autoimmune disease1,32,33 (table 3).
Table 3.
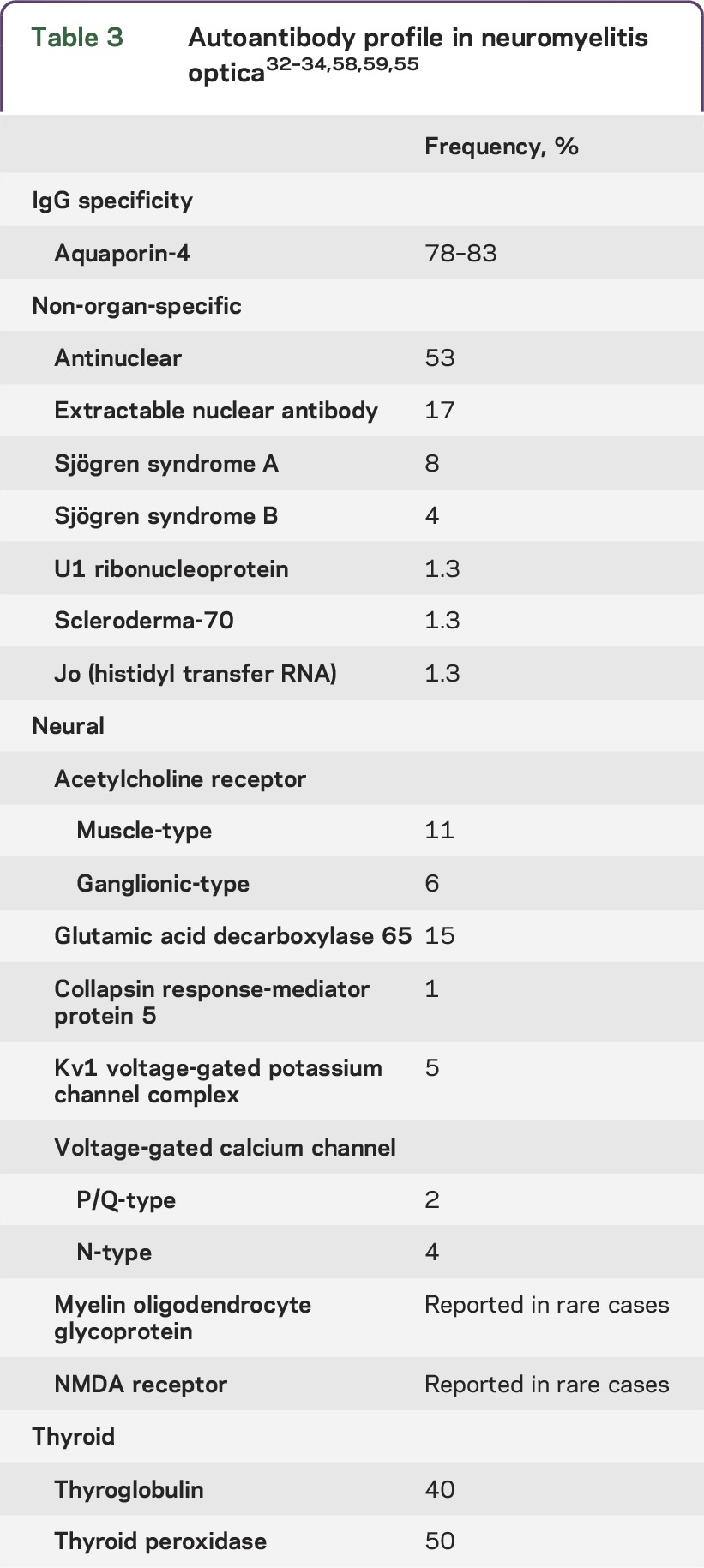
Muscle acetylcholine receptor (AChR) autoantibodies are detectable in 11% of patients with NMO.34 Other neural autoantibody specificities include ganglionic AChR, collapsin response-mediator protein 5, glutamic acid decarboxylase 65, NMDA receptor, and Kv1 voltage-gated potassium channel complex.34,55 Aquaporin-1-IgG has been reported in NMO, but clinical utility has not been established.6,56
The first-generation tissue-based immunofluorescence assay for detecting the AQP4-IgG marker of NMO is 91% specific but only 48% sensitive.57 Contemporary clinical practice uses molecular-based techniques, the most sensitive and specific being immunofluorescence applied to indicator cells expressing recombinant AQP4 (M1 or M23 isoform), either fixed or live.58,59 Fluorescence-activated sorting of live M1 isoform–transfected cells has proven to be the most sensitive and specific technique in Mayo Clinic experience.58
An ELISA based on bridging of soluble biotinylated AQP4 and plate-bound AQP4 by patient IgG is less sensitive and more prone to yield false-positive results.58 Immunoprecipitation of green fluorescence protein–tagged AQP4 is least sensitive (50%).57
Prognosis.
NMO prognosis was very poor in reports predating the era of AQP4-IgG testing. Appropriate therapeutic options were uncertain. In a 1999 case series, Wingerchuk et al.1 reported that sequential index events of monophasic NMO occurred rapidly (median 5 days) with moderate recovery. With a relapsing course, the interval between initial myelitis and optic neuritis events was long (median 166 days), severe relapses clustered within 3 years, and severe disability developed in a stepwise manner; one-third of patients died of respiratory failure.1
In a 2012 study of AQP4-IgG–positive patients from the UK and Japan, permanent bilateral visual disability had developed in 18% of patients (median disease duration 75 months), permanent motor disability in 34% (inability to walk farther than 100 m unaided), and wheelchair dependency in 23%.60 At median disease duration of 99 months, 9% of patients had died after a mean of 4.5 attacks.60 In the UK cohort, Afro-Caribbean patients were younger at disease onset and had a higher likelihood of visual disability than Caucasian patients.60
In a 2013 report of AQP4-IgG–seropositive and AQP4-IgG–seronegative patients, 65% were blind a median of 8.3 years after disease onset (50% unilateral and 15% bilateral), and more than one-third could not walk without unilateral assistance.e1 These reports suggest a better outcome in the post–AQP4-IgG era, presumably because of earlier diagnosis and therapies that lower Ig levels or intercept complement activation or interleukin 6 action.
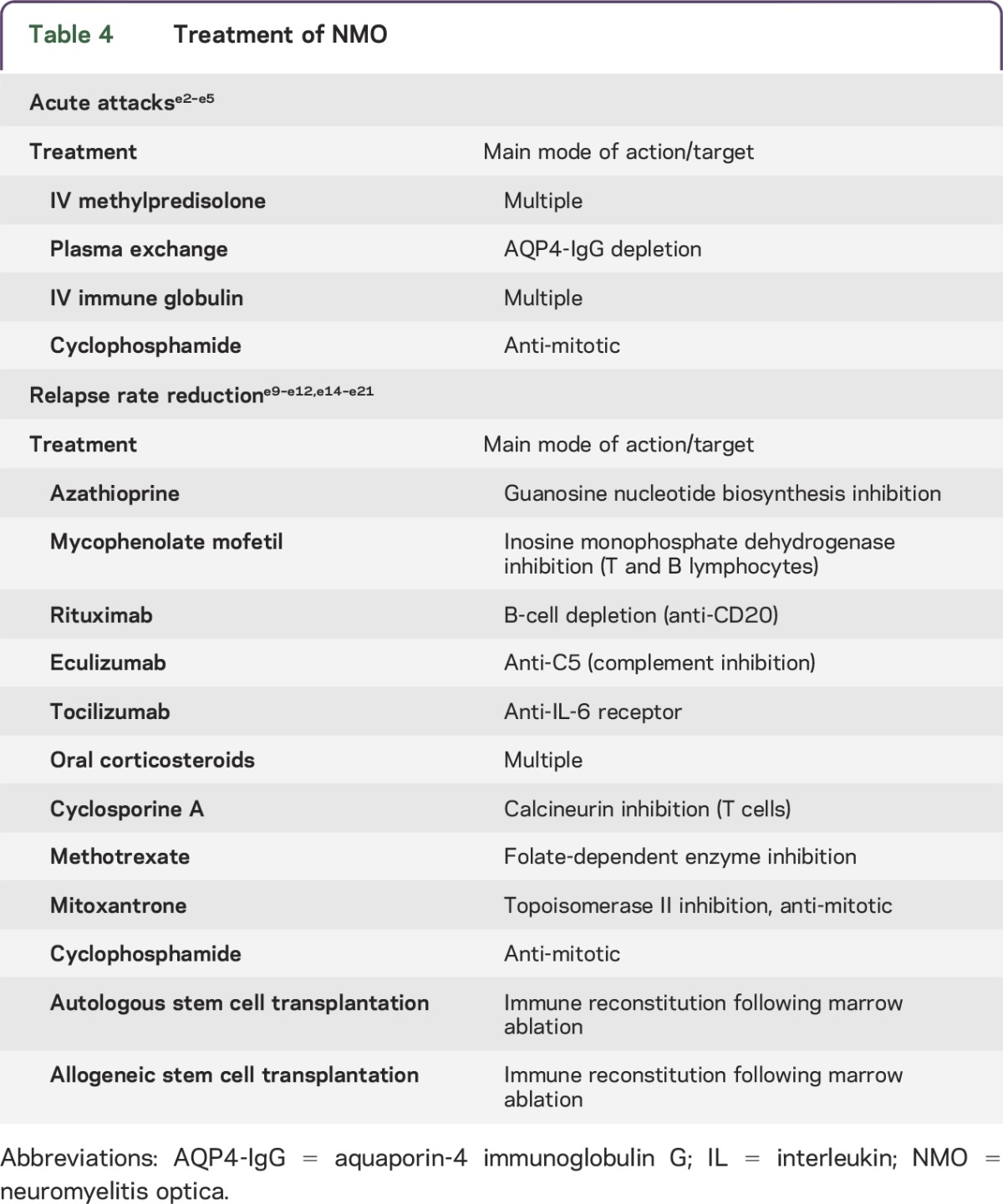
Treatment options.
NMO treatment has dual goals: (1) to stop the acute attack, thus minimizing neurologic disability and maximizing reversibility; and (2) to prevent relapses with cumulative disability. Therapeutic decisions should be individualized for each patient depending on the clinical situation and outcome and should not be based on AQP4-IgG levels given the current state of knowledge. No published prospective controlled trials have evaluated NMO treatments. Promising open-label pilot studies have spawned continuing randomized trials.e2 Table 4 summarizes current treatment options.
Table 4.
Treatment of NMO
For acute attacks.
As in other IgG-mediated autoimmune diseases, IV methylprednisolone (IVMP) and plasma exchange (PLEX) are generally considered optimal first-line therapy. Experts recommend 1 g IVMP for 3 to 5 days. A course of oral prednisone with a slow taper is usually undertaken to prevent early relapse and to serve as a bridge to immune suppression with a steroid-sparing drug.e2 For severe attacks, the IVMP response may not be satisfactory; PLEX (5–7 exchanges) is more efficacious in combination with IVMP.e3 In some severe cases cyclophosphamide has been reported to be efficacious as IV pulse therapy (750–1,000 mg/m2).e4
IVIg (with or without PLEX) was effective in a study of 10 patients (11 episodes) who did not respond to IVMP; 45% of attacks improved and no patient worsened.e5 Formal trials focusing on attacks are needed in order to determine optimal therapeutic options, taking into account the patient's age, comorbidities, and attack severity.
For relapse rate reduction.
Practical considerations include the patient's age, access to care, cost, adverse events, previous treatments, and existing disability. Immunomodulatory treatments used for MS are generally not effective for NMO, and some are deleterious.e6–e8 Retrospective data are available for large numbers of patients treated with azathioprine, mycophenolate mofetil, or rituximab, which are the usual options considered for relapse prevention.
Azathioprine (2 mg/kg/day rather than lower dose) reduced the annual relapse rate in a study of 99 patients.e9 Hemogram and liver and renal function must be monitored during treatment, and it is desirable to ascertain thiopurine methyltransferase activity before initiating treatment.e10 Because azathioprine's efficacy is delayed, oral corticosteroids are usually continued for the first few months. Azathioprine reduced the relapse rate by 72% in a study of 32 patients but was ineffective for half the patients despite continuing prednisone.e11
Mycophenolate mofetil is another option for long-term immunosuppression. In a study of 28 patients, relapse rate was reduced up to 87% and failure rate was 36%.e11
Rituximab, an anti-CD20 monoclonal antibody that depletes B cells, has excellent efficacy for NMOSD. Open-label studies using different protocols for dosing and monitoring CD19-positive blood lymphocytes suggest that infusion should be repeated when B cells reappear. Annual relapse rates were reduced by 87%, and 60%–70% of patients became relapse-free for as long as 3 years following treatment. Studies reported improved or stabilized disability (80%–97% of patients).e11–e13
Eculizumab, a monoclonal antibody that neutralizes the C5 complement protein, rendered 12 of 14 patients relapse-free in a 12-month open-label pilot study.e14 During treatment, the median annualized pretreatment attack rate fell from 3 to 0 and the EDSS score improved from 4.3 to 3.5.e14
Tocilizumab, a monoclonal antibody targeting interleukin 6 receptor, showed efficacy in an open-label pilot study of 7 patients. After 1 year, the annualized relapse rate fell from 2.96 to 0.4. EDSS score, neuropathic pain, and general fatigue were significantly reduced.e15
Oral corticosteroids as monotherapy or adjunctive therapy have been reported to prevent relapses in a small retrospective study.e16 Addition of a steroid-sparing immunosuppressive agent minimizes the adverse events of extended corticosteroid therapy. Osteoporosis prophylaxis and monitoring of plasma glucose are recommended during long-term treatment.
Cyclosporine A has been used with oral corticosteroids. The annual relapse rate for 9 patients receiving this treatment fell from 2.7 to 0.38.e17
In a retrospective study of 14 patients, methotrexate therapy was associated with a significantly reduced median annualized relapse rate (0.18 during therapy vs 1.39 before); 43% of patients were relapse-free, and disability stabilized or improved in 79%.e18
In 20 patients with high relapse rate, mitoxantrone (IV monthly or every 3 months) was associated with a median annualized relapse rate reduction of 75%.e19 Disability improved or stabilized in all patients and 50% became relapse-free.e19 Laboratory and cardiac monitoring is necessary during treatment.
Cyclophosphamide was associated with a median EDSS improvement from 8.0 to 5.75 in 4 patients with longitudinally extensive transverse myelitis.e20
Autologous hemopoietic stem cell transplantation arrested progression in 3 of 16 patients who were refractory to conventional therapy. They remained treatment-free (median follow-up period: 47 months). The remaining 13 patients required continuing therapy due to disability progression or relapse.e21 Allogeneic hematopoietic stem cell transplantation was reported to be efficacious in 2 patients with aggressive NMO in whom previous treatments had failed, including autologous stem cell transplantation. Both patients remained relapse-free after 36 and 48 months.e22
IMMUNOPATHOGENESIS
Favorable response to PLEX and B-cell–depleting therapies provide clinical evidence for the pathogenicity of AQP4-IgG.e3,e11,e12
Immunopathology.
The distinctive immunopathology of NMO lesions supports a central role for AQP4-IgG in disease pathogenesis. Products of complement activation are prominent around thickened hyalinized blood vessels and coincide with Ig deposits. The predominance of polymorphonuclear inflammatory cells is consistent with involvement of IgG and complement in lesion genesis.e23 Loss of AQP4 in astrocytes surviving within sublytic CNS lesions distinguishes NMO from MS and may be indicative of potentially reversible histopathologic lesions.e24 Some of these lesions exhibit marked inflammation and astrocytic AQP4 loss but GFAP retention. Other lesions exhibit reduced astrocytic GFAP immunoreactivity but relative preservation of myelin. These findings support AQP4 loss as a precedent to astrocyte loss, with demyelination a later secondary event.e25,e26 In advanced NMO stages, spinal cord gray and white matter shows extensive demyelination, cavitation, necrosis, and axonal loss (spheroids).e23
Role of AQP4-IgG.
IgG responses to protein autoantigens reflect the interaction of helper and regulatory T cells with antigen-specific B cells. A role for AQP4-specific effector T cells has not been established, but they may contribute to initial blood-brain barrier disruption.e24 In established NMO lesions, CNS tissues contain IgG-producing plasma cells.e23 There is increasing evidence for intrathecal production of AQP4-IgG. High CSF levels correlate significantly with attack severity.e27–e30 Levels of interleukin 6, which enhances plasmablast survival, are higher than normal in the plasma of patients with NMO. Plasmablasts are detectable in NMO peripheral blood and are more numerous in attack phase. Interleukin 6 promotes in vitro production of IgG by AQP4-specific plasmablasts derived from blood and CSF of patients with NMO.e28,e31–e33
Target cell–specific pathogenicity requires the binding of IgG to the extracellular domain of AQP4.e34 AQP4-IgG is predominantly IgG1 class, a potent activator of complement. In the astrocytic plasma membrane, AQP4 exists as tetramers containing one or both of 2 major isoforms: full length (M1) and a shorter isoform (M23). M23-AQP4 spontaneously forms orthogonal arrays of particles; most homomeric M1 tetramers remain singlet.e35,e36 On exposure to AQP4-IgG, astrocytes rapidly internalize M1 tetramers. In contrast, AQP4-IgG cross-linking of M23-AQP4 arrays induces their coalescence into larger orthogonal arrays that exceed the astrocyte's endocytic capacity.e35 When complement becomes available locally, large AQP4 arrays displaying densely packed Fc tails of IgG1 would initiate complement activation explosively, promoting leukocyte chemotaxis, vascular permeability, and massive influx of plasma rich in IgG, complement, and rheumatoid factor–like IgM, which, on binding to the antigen-complexed IgG, would further enhance complement activation.e34 A reported positive in vitro correlate of NMO attack severity is the degree of complement-mediated injury to AQP4-expressing cells following exposure to IgG from patients with NMO (6 with severe attacks compared with 6 with mild attacks, p = 0.05).e37 Natural killer (NK) cell degranulation, activated as a complement-independent function of IgG binding to AQP4 (antibody-dependent cellular cytotoxicity), also causes astrocyte killing in vitro.e38
Autoantibody blockade of water flux would plausibly explain the prominent intramyelinic edema observed in early NMO lesions, which is a potential precursor of demyelination.e35,e39 Autoantibodies with potential to reduce water flux independent of AQP4 endocytosis (i.e., temperature independent) were demonstrated functionally in a Xenopus expression system using sera pooled from at least 50 NMO patients with high levels of AQP4-IgG.e35,e39 Data disputing this AQP4-IgG property were based on testing monoclonal AQP4-IgG and sera from relatively few individual patients by quantum dot technology.e40,e41
Disruption of glutamate homeostasis is another potentially pathogenic property of AQP4-IgG. The excitatory amino acid transporter 2 (EAAT2) is noncovalently linked to AQP4 on perisynaptic astrocytic membranes. EAAT2 accounts for 90% of CNS glutamate uptake and is co-internalized by AQP4-IgG. When applied to cultured astrocytes, AQP4-IgG reduces the functional uptake of 3H-glutamate. Furthermore, in sublytic lesions of NMO spinal cord, the central gray matter is profoundly deficient in EAAT2.e42 The particular vulnerability of oligodendrocytes to glutamate toxicity is another plausible explanation for demyelination in NMO.e43,e44
Animal models.
Animal models described to date reproduce some histopathologic characteristics of NMO but not clinical findings. NMO lesions do not ensue when serum IgG from patients with NMO is injected systemically into rats with an intact blood-brain barrier. However, when NMO-IgG is injected systemically into rats with incipient experimental autoimmune encephalomyelitis (EAE), Ig deposition, complement activation, and granulocyte influx occur.e28 Astrocytic damage similar to that of NMO was seen in spinal cord regions containing EAE-type lesions.e45 Neurologic impairment was worse in rats injected systemically with IgG from patients with NMO and did not occur in the absence of encephalitogenic T cells. Systemically injected NMO-IgG had no effect on juvenile rats with leaky CNS endothelial cells.e24 On the other hand, 2 groups have reported astrocytic damage and spinal cord AQP4 loss following systemic injection of IgG from patients with NMO in mice pretreated with complete Freund adjuvant, either alone or with pertussis toxin.e46,e47 The authors speculated that the mycobacterial adjuvant promoted blood-brain barrier leakiness.
Focal lesions characteristic of NMO developed in mice injected with IgG from patients with NMO directly into the cerebral hemisphere but only if fresh human serum was co-injected as a source of complement.e48 Similar findings in T-cell–deficient mice indicated that AQP4-IgG and locally available complement are sufficient for creating NMO-like pathologic lesions.e49 Astrocyte injury, defined as loss of AQP4 and GFAP, was produced in brain slice organ culture preparations in the absence of complement when IgG from patients with NMO was added to NK cells. Lesions consistent with antibody-dependent cellular cytotoxicity were also induced by injecting mice intracerebrally with IgG and NK cells from patients with NMO.e50 Optic nerve lesions resembling those of NMO were induced in mice by continuously infusing serum from patients with NMO for 3 days in the region of the optic chiasm.e51
The animal models described to date are based on the concept that AQP4-IgG produced in the periphery will penetrate the CNS when the blood-brain barrier is disrupted. It is evident, however, that AQP4-IgG can be produced intrathecally.e27–e30 The ideal animal model should reproduce typical clinical findings, including relapses and the immunohistopathologic phenotype of AQP4 autoimmunity.
CONCLUSIONS AND FUTURE CHALLENGES
In the decade since AQP4-IgG was described as a biomarker distinguishing NMO and related disorders from MS, our understanding of NMO immunopathogenesis has greatly advanced. Clinical dividends include sensitive molecular-based diagnostic assays and more effective therapies targeting pathogenic antibodies, B cells, complement, and an interleukin receptor. The initial trigger of the AQP4 autoimmune response remains an important question. Animal models have not yet reproduced clinical signs or full histopathologic manifestations of NMO lesions. The immunopathogenesis of optic neuritis and transverse myelitis occurring in the context of alternative autoantibodies and in the absence of detectable AQP4-IgG remains speculative and cannot be resolved without immunopathologic data. Symptoms and signs do not equate with molecular pathology.
GLOSSARY
- AChR
acetylcholine receptor
- ADEM
acute disseminated encephalomyelitis
- AQP4
aquaporin-4
- EAAT2
excitatory amino acid transporter 2
- EAE
experimental autoimmune encephalomyelitis
- EDSS
Expanded Disability Status Scale
- GFAP
glial fibrillary acidic protein
- Ig
immunoglobulin
- IVMP
IV methylprednisolone
- MBP
myelin basic protein
- MOG
myelin oligodendrocyte glycoprotein
- MS
multiple sclerosis
- NK
natural killer
- NMO
neuromyelitis optica
- NMOSD
NMO spectrum disorder
- PLEX
plasma exchange
Footnotes
Supplemental data at Neurology.org/nn
AUTHOR CONTRIBUTIONS
Anastasia Zekeridou: design, drafting/revising manuscript. Vanda A. Lennon: revising and editing manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
A. Zekeridou reports no disclosures. V. Lennon received research support from NIH. V. Lennon and Mayo Clinic have a financial interest in the following intellectual property: “Marker for Neuromyelitis Optica.” A patent has been issued for this technology, and it has been licensed to commercial entities. They have received cumulative royalties of greater than the federal threshold for significant financial interest from the licensing of these technologies. The author receives no royalties from the sale of these tests by Mayo Medical Laboratories; however, Mayo Collaborative Services, Inc. does receive revenue for conducting these tests. The Mayo Neuroimmunology Laboratory performs service testing for aquaporin-4 autoantibodies on behalf of Mayo Collaborative Services, Inc., an agency of Mayo Foundation. V. Lennon does not benefit financially from this testing. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Wingerchuk DM, Hogancamp WF, O'Brien PC, Weinshenker BG. The clinical course of neuromyelitis optica (Devic's syndrome). Neurology 1999;53:1107–1114. [DOI] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Lennon VA, Lucchinetti CF, Pittock SJ, Weinshenker BG. The spectrum of neuromyelitis optica. Lancet Neurol 2007;6:805–815. [DOI] [PubMed] [Google Scholar]
- 3.Lennon VA, Wingerchuk DM, Kryzer TJ, et al. A serum autoantibody marker of neuromyelitis optica: distinction from multiple sclerosis. Lancet 2004;364:2106–2112. [DOI] [PubMed] [Google Scholar]
- 4.Lennon VA, Kryzer TJ, Pittock SJ, Verkman AS, Hinson SR. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J Exp Med 2005;202:473–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mao Z, Lu Z, Hu X, et al. Distinction between MOG antibody-positive and AQP4 antibody-positive NMO spectrum disorders. Neurology 2014;83:1122. [DOI] [PubMed] [Google Scholar]
- 6.Tzartos JS, Stergiou C, Kilidireas K, Zisimopoulou P, Thomaidis T, Tzartos SJ. Anti-aquaporin-1 autoantibodies in patients with neuromyelitis optica spectrum disorders. PLoS One 2013;8:e74773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mealy MA, Wingerchuk DM, Greenberg BM, Levy M. Epidemiology of neuromyelitis optica in the United States: a multicenter analysis. Arch Neurol 2012;69:1176–1180. [DOI] [PubMed] [Google Scholar]
- 8.Asgari N, Lillevang ST, Skejoe HP, Falah M, Stenager E, Kyvik KO. A population-based study of neuromyelitis optica in Caucasians. Neurology 2011;76:1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Collongues N, Marignier R, Zéphir H, et al. Neuromyelitis optica in France: a multicenter study of 125 patients. Neurology 2010;74:736–742. [DOI] [PubMed] [Google Scholar]
- 10.Apiwattanakul M, Asawavichienjinda T, Pulkes T. Diagnostic utility of NMO/AQP4-IgG in evaluating CNS inflammatory diseases in Thai patients. J Neurol Sci 2012;320:118–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagaishi A, Takagi M, Umemura A, et al. Clinical features of neuromyelitis optica in a large Japanese cohort: comparison between phenotypes. J Neurol Neurosurg Psychiatry 2011;82:1360–1364. [DOI] [PubMed] [Google Scholar]
- 12.Wingerchuk DM, Lennon VA, Pittock SJ, Lucchinetti CF, Weinshenker BG. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006;66:1485–1489. [DOI] [PubMed] [Google Scholar]
- 13.Quek AM, McKeon A, Lennon VA, et al. Effects of age and sex on aquaporin-4 autoimmunity. Arch Neurol 2012;69:1039–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarius S, Ruprecht K, Wildemann B, et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: a multicentre study of 175 patients. J Neuroinflammation 2012;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matiello M, Schaefer-Klein JL, Hebrink DD, Kingsbury DJ, Atkinson EJ, Weinshenker BG. Genetic analysis of aquaporin-4 in neuromyelitis optica. Neurology 2011;77:1149–1155. [DOI] [PubMed] [Google Scholar]
- 16.Devic E. Myélite subaiguë compliquée de névrite optique. Le Bulletin Médical 1894;8:1033–1034. [Google Scholar]
- 17.Wingerchuk DM, Weinshenker BG. Neuromyelitis optica: clinical predictors of a relapsing course and survival. Neurology 2003;60:848–853. [DOI] [PubMed] [Google Scholar]
- 18.Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007;68:603–605. [DOI] [PubMed] [Google Scholar]
- 19.Flanagan EP, Weinshenker BG, Krecke KN, et al. Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders. JAMA Neurol 2015;72:81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Takai Y, Misu T, Nakashima I, et al. Two cases of lumbosacral myeloradiculitis with anti-aquaporin-4 antibody. Neurology 2012;79:1826–1828. [DOI] [PubMed] [Google Scholar]
- 21.Guo Y, Lennon VA, Popescu BF, et al. Autoimmune aquaporin-4 myopathy in neuromyelitis optica spectrum. JAMA Neurol 2014;71:1025–1029. [DOI] [PubMed] [Google Scholar]
- 22.Pittock SJ, Lennon VA. Aquaporin-4 autoantibodies in a paraneoplastic context. Arch Neurol 2008;65:629–632. [DOI] [PubMed] [Google Scholar]
- 23.Pittock SJ, Lennon VA, Krecke K, Wingerchuk DM, Lucchinetti CF, Weinshenker BG. Brain abnormalities in neuromyelitis optica. Arch Neurol 2006;63:390–396. [DOI] [PubMed] [Google Scholar]
- 24.Pittock SJ, Weinshenker BG, Lucchinetti CF, Wingerchuk DM, Corboy JR, Lennon VA. Neuromyelitis optica brain lesions localized at sites of high aquaporin 4 expression. Arch Neurol 2006;63:964–968. [DOI] [PubMed] [Google Scholar]
- 25.Baba T, Nakashima I, Kanbayashi T, et al. Narcolepsy as an initial manifestation of neuromyelitis optica with anti-aquaporin-4 antibody. J Neurol 2009;256:287–288. [DOI] [PubMed] [Google Scholar]
- 26.Iorio R, Plantone D, Damato V, Paola Batocchi A. Anorexia heralding the onset of neuromyelitis optica. Intern Med 2013;52:489–491. [DOI] [PubMed] [Google Scholar]
- 27.Iorio R, Lucchinetti CF, Lennon VA, et al. Syndrome of inappropriate antidiuresis may herald or accompany neuromyelitis optica. Neurology 2011;77:1644–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vernant JC, Cabre P, Smadja D, et al. Recurrent optic neuromyelitis with endocrinopathies: a new syndrome. Neurology 1997;48:58–64. [DOI] [PubMed] [Google Scholar]
- 29.Kremer L, Mealy M, Jacob A, et al. Brainstem manifestations in neuromyelitis optica: a multicenter study of 258 patients. Mult Scler Epub 2013 Oct 7. [DOI] [PubMed]
- 30.Magaña SM, Matiello M, Pittock SJ, et al. Posterior reversible encephalopathy syndrome in neuromyelitis optica spectrum disorders. Neurology 2009;72:712–717. [DOI] [PubMed] [Google Scholar]
- 31.McKeon A, Lennon VA, Lotze T, et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008;71:93–100. [DOI] [PubMed] [Google Scholar]
- 32.Pittock SJ, Lennon VA, de Seze J, et al. Neuromyelitis optica and non organ-specific autoimmunity. Arch Neurol 2008;65:78–83. [DOI] [PubMed] [Google Scholar]
- 33.Long Y, Zheng Y, Chen M, et al. Serum thyroid-stimulating hormone and anti-thyroglobulin antibody are independently associated with lesions in spinal cord in central nervous system demyelinating diseases. PLoS One 2014;9:e100672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKeon A, Lennon VA, Jacob A, et al. Coexistence of myasthenia gravis and serological markers of neurological autoimmunity in neuromyelitis optica. Muscle Nerve 2009;39:87–90. [DOI] [PubMed] [Google Scholar]
- 35.Bergamaschi R, Jarius S, Robotti M, Pichiecchio A, Wildemann B, Meola G. Two cases of benign neuromyelitis optica in patients with celiac disease. J Neurol 2009;256:2097–2099. [DOI] [PubMed] [Google Scholar]
- 36.Jarius S, Paul F, Ruprecht K, Wildemann B. Low vitamin B12 levels and gastric parietal cell antibodies in patients with aquaporin-4 antibody-positive neuromyelitis optica spectrum disorders. J Neurol 2012;259:2743–2745. [DOI] [PubMed] [Google Scholar]
- 37.Ontaneda D, Fox RJ. Is neuromyelitis optica with advanced age of onset a paraneoplastic disorder? Int J Neurosci 2014;124:509–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan KH, Kwan JS, Ho PW, et al. Aquaporin-4 water channel expression by thymoma of patients with and without myasthenia gravis. J Neuroimmunol 2010;227:178–184. [DOI] [PubMed] [Google Scholar]
- 39.Armağan H, Tüzün E, Içöz S, et al. Long extensive transverse myelitis associated with aquaporin-4 antibody and breast cancer: favorable response to cancer treatment. J Spinal Cord Med 2012;35:267–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Figueroa M, Guo Y, Tselis A, et al. Paraneoplastic neuromyelitis optica spectrum disorder associated with metastatic carcinoid expressing aquaporin-4. JAMA Neurol 2014;71:495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frasquet M, Bataller L, Torres-Vega E, et al. Longitudinally extensive transverse myelitis with AQP4 antibodies revealing ovarian teratoma. J Neuroimmunol 2013;263:145–147. [DOI] [PubMed] [Google Scholar]
- 42.Reindl M, Di Pauli F, Rostásy K, Berger T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat Rev Neurol 2013;9:455–461. [DOI] [PubMed] [Google Scholar]
- 43.Kitley J, Waters P, Woodhall M, et al. Neuromyelitis optica spectrum disorders with aquaporin-4 and myelin-oligodendrocyte glycoprotein antibodies: a comparative study. JAMA Neurol 2014;71:276–283. [DOI] [PubMed] [Google Scholar]
- 44.Höftberger R, Sepulveda M, Armangue T, et al. Antibodies to MOG and AQP4 in adults with neuromyelitis optica and suspected limited forms of the disease. Mult Scler Epub 2014 Oct 24; pii: 1352458514555785. [DOI] [PMC free article] [PubMed]
- 45.Kitley J, Woodhall M, Waters P, et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype. Neurology 2012;79:1273–1277. [DOI] [PubMed] [Google Scholar]
- 46.Ikeda K, Kiyota N, Kuroda H, et al. Severe demyelination but no astrocytopathy in clinically definite neuromyelitis optica with anti-myelin-oligodendrocyte glycoprotein antibody. Mult Scler 2015;21:656–659. [DOI] [PubMed] [Google Scholar]
- 47.Takano R, Misu T, Takahashi T, Sato S, Fujihara K, Itoyama Y. Astrocytic damage is far more severe than demyelination in NMO: a clinical CSF biomarker study. Neurology 2010;75:208–216. [DOI] [PubMed] [Google Scholar]
- 48.Zamvil SS, Slavin AJ. Does MOG Ig-positive AQP4-seronegative opticospinal inflammatory disease justify a diagnosis of NMO spectrum disorder? Neurol Neuroimmunol Neuroinflamm 2015;2:e62. 10.1212/NXI.0000000000000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nakamura M, Miyazawa I, Fujihara K, et al. Preferential spinal central gray matter involvement in neuromyelitis optica. An MRI study. J Neurol 2008;255:163–170. [DOI] [PubMed] [Google Scholar]
- 50.Klawiter EC, Xu J, Naismith RT, et al. Increased radial diffusivity in spinal cord lesions in neuromyelitis optica compared with multiple sclerosis. Mult Scler 2012;18:1259–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Storoni M, Davagnanam I, Radon M, Siddiqui A, Plant GT. Distinguishing optic neuritis in neuromyelitis optica spectrum disease from multiple sclerosis: a novel magnetic resonance imaging scoring system. J Neuroophthalmol 2013;33:123–127. [DOI] [PubMed] [Google Scholar]
- 52.Petzold A, Wattjes MP, Costello F, et al. The investigation of acute optic neuritis: a review and proposed protocol. Nat Rev Neurol 2014;10:447–458. [DOI] [PubMed] [Google Scholar]
- 53.Jarius S, Paul F, Franciotta D, et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: results from 211 lumbar punctures. J Neurol Sci 2011;306:82–90. [DOI] [PubMed] [Google Scholar]
- 54.Brickshawana A, Hinson SR, Romero MF, et al. Investigation of the KIR4.1 potassium channel as a putative antigen in patients with multiple sclerosis: a comparative study. Lancet Neurol 2014;13:795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Titulaer MJ, Höftberger R, Iizuka T, et al. Overlapping demyelinating syndromes and anti–N-methyl-D-aspartate receptor encephalitis. Ann Neurol 2014;75:411–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Long Y, Zheng Y, Shan F, et al. Development of a cell-based assay for the detection of anti-aquaporin 1 antibodies in neuromyelitis optica spectrum disorders. J Neuroimmunol 2014;273:103–110. [DOI] [PubMed] [Google Scholar]
- 57.Waters PJ, McKeon A, Leite MI, et al. Serologic diagnosis of NMO: a multicenter comparison of aquaporin-4-IgG assays. Neurology 2012;78:665–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fryer JP, Lennon VA, Pittock SJ, et al. AQP4 autoantibody assay performance in clinical laboratory service. Neurol Neuroimmunol Neuroinflamm 2014;1:e11. 10.1212/NXI.0000000000000011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jarius S, Wildemann B. Aquaporin-4 antibodies (NMO-IgG) as a serological marker of neuromyelitis optica: a critical review of the literature. Brain Pathol 2013;23:661–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitley J, Leite MI, Nakashima I, et al. Prognostic factors and disease course in aquaporin-4 antibody-positive patients with neuromyelitis optica spectrum disorder from the United Kingdom and Japan. Brain 2012;135:1834–1849. [DOI] [PubMed] [Google Scholar]