

Abstract
Objectives:
To determine the safety, pharmacokinetics (PK), and immunogenicity of the recombinant human monoclonal antibody MOR103 to granulocyte-macrophage colony-stimulating factor (GM-CSF) in patients with multiple sclerosis (MS) with clinical or MRI activity.
Methods:
In this 20-week, randomized, double-blind, placebo-controlled phase 1b dose-escalation trial (registration number NCT01517282), adults with relapsing-remitting MS (RRMS) or secondary progressive MS (SPMS) received an IV infusion of placebo (n = 6) or MOR103 0.5 (n = 8), 1.0 (n = 8), or 2.0 (n = 9) mg/kg every 2 weeks for 10 weeks. Patients had to have ≤10 gadolinium (Gd)-enhancing brain lesions on T1-weighted MRI at baseline. The primary objective was safety.
Results:
Most treatment-emergent adverse events (TEAEs) were mild to moderate in severity. The most frequent was nasopharyngitis. Between-group differences in TEAE numbers were small. There were no TEAE-related trial discontinuations, infusion-related reactions, or deaths. Nine patients experienced MS exacerbations: 3, 5, 1, and 0 patient(s) in the placebo, 0.5, 1.0, and 2.0 mg/kg groups, respectively. A few T1 Gd-enhancing lesions and/or new or enlarging T2 lesions indicative of inflammation were observed in all treatment groups. No clinically significant changes were observed in other clinical assessments or laboratory safety assessments. No anti-MOR103 antibodies were detected. PK evaluations indicated dose linearity with low/no drug accumulation over time.
Conclusions:
MOR103 was generally well-tolerated in patients with RRMS or SPMS. No evidence of immunogenicity was found.
Classification of evidence:
This phase 1b study provides Class I evidence that MOR103 has acceptable tolerability in patients with MS.
Therapy for multiple sclerosis (MS) is a rapidly advancing area in neurologic research. Although immunomodulatory agents are effective in reducing the frequency of exacerbations (relapses), a considerable proportion of patients achieves only a suboptimal response to medication1; thus, there remains an unmet need for effective and well-tolerated MS therapies, particularly for patients with progressive disease.
CNS infiltrating monocytes/macrophages and resident microglia contribute to neuroinflammation and neurodegeneration in MS through inflammatory mediator release and stimulation of leukocyte activity.2 Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a cytokine affecting functional activities of mature myeloid cells. Among these, GM-CSF promotes macrophage polarization and subsequent inflammatory mediator production.3,4 The frequency of GM-CSF–producing cells is higher in patients with MS than in patients with noninflammatory neurologic diseases.5
In experimental autoimmune encephalomyelitis (EAE), a widely used MS model,6 GM-CSF is required for disease induction and maintenance.7–9 GM-CSF −/− mice are resistant to EAE, and anti-GM-CSF antibody treatment in wild-type mice prevented the onset of clinical disease but also ameliorated disease score when administered therapeutically.7 It was observed that GM-CSF produced by autoreactive T cells serves a nonredundant function in the effector phase of EAE.10,11 In addition, GM-CSF is required for recruitment of peripheral myeloid cells into the CNS.9,10 Consequently, neutralizing antibodies against GM-CSF might provide a new therapeutic approach for MS.
MOR103 is a high-affinity recombinant human immunoglobulin (Ig) G1 antibody, blocking GM-CSF interaction with its receptor and thereby preventing subsequent signal transduction.12 MOR103 is a human IgG1 and, as with other IgGs, was therefore expected to cross the blood-brain barrier at low levels only. However, the blood-brain barrier becomes broken down in MS lesions,13 potentially leading to increased IgG penetration. Based on EAE data, there is evidence of GM-CSF having both a central role (e.g., GM-CSF production of CNS-infiltrating encephalitogenic T cells) and a peripheral role (mobilization in bone marrow and recruitment of peripheral myeloid cells into the CNS).9–11 However, in the EAE model, anti-GM-CSF monoclonal antibodies ameliorate the disease despite clear delineation of a central vs a peripheral action of GM-CSF in EAE. Positive results regarding tolerability and efficacy in patients with rheumatoid arthritis14 indicate the potential of MOR103 in the treatment of autoimmune diseases. We report results of a phase 1b trial of MOR103 in patients with MS.
METHODS
Patient selection.
Patients were aged 18–60 years with a neurologist-confirmed diagnosis of relapsing- remitting MS (RRMS) or secondary progressive MS (SPMS) according to the 2005 revised McDonald diagnostic criteria. They were also required to have a body mass index of 19.0–35.0 kg/m2 and to agree to use effective contraceptive methods and not to donate blood, sperm, or oocytes during and for ≥3 months after trial discontinuation. At screening, patients had to be ambulatory, have an Expanded Disability Status Scale (EDSS) score of ≥2.0 to ≤6.5, and have ≤10 gadolinium (Gd)-enhancing brain lesions on T1-weighted MRI. In addition, patients had to fulfill one of the following inclusion criteria: ≥1 documented exacerbation within 1 year, 2 documented exacerbations in the past 3 years, or a new T1 Gd-enhancing brain lesion or T2 brain lesion within the past year.
Patients were excluded if they had primary progressive MS or if they had not stabilized from an exacerbation occurring >30 days prior to trial enrollment. They were also excluded if they had received systemic glucocorticoids or adrenocorticotropic hormone within 1 month; any interferon (IFN) β within 2 months; glatiramer acetate, plasmapheresis, or IV gamma globulin (IVIg) within 3 months; mycophenolate mofetil within 6 months; or azathioprine, cyclosporine, or methotrexate within 12 months. Additional exclusion criteria were previous treatment with B cell– or T cell–depleting therapies, cytotoxic agents, any immunosuppressive/immunomodulating agents (other than those listed above), total lymphoid irradiation, stem cell therapy, or bone marrow transplantation.
Also excluded were patients with any medical condition or uncontrolled disease state other than MS requiring or likely to require systemic treatment with corticosteroids or other immunomodulators and those with a history of infection, tuberculosis, hepatitis B or C, myocardial infarction, Class III or IV heart disease, hepatic or renal dysfunction, severe pulmonary disease, psychiatric disorders, alcohol or drug abuse, or malignancy. Patients who had received a live vaccine in the previous 6 months, who had current or past clinically significant laboratory findings, or who were pregnant or lactating were also excluded from the trial.
Study design.
This was a multicenter, prospective, randomized, double-blind, placebo-controlled, ascending dose-escalation phase 1b trial evaluating 3 dose levels of MOR103 (MorphoSys AG, Martinsried/Planegg, Germany) for the treatment of RRMS and SPMS. The primary objective was to determine the safety and tolerability of MOR103 (level of evidence Class I); secondary objectives included the multiple-dose pharmacokinetics (PK) and potential immunogenicity of MOR103. Exploratory analyses assessed changes from baseline in cytokine levels and in T cell and monocyte subtypes.
An independent data monitoring committee (DMC) blinded to treatment group reviewed interim safety data from all patients in the 0.5 mg/kg and 1.0 mg/kg cohorts after the sixth dose of study drug. DMC approval was required before proceeding to the next higher dose level. A second safety review was performed when all patients had completed their week 10 visit.
Patient eligibility was determined at screening and confirmed at baseline before the first dose. Eligible individuals were randomized to receive IV MOR103 0.5, 1.0, or 2.0 mg/kg or placebo. Randomization was in a 4:1 ratio (active treatment:placebo) in each of the 3 active dose groups, according to an interactive Web response system randomization schedule. Each cohort was planned to include 10 patients. Investigators and patients were blinded to the randomization schedule and study drug. To avoid unblinding of study treatments (MOR103 doses and matching placebos), all relevant parts of the infusion set up had an opaque protective cover of foil, paper, or plastic. Study drug was administered over 1 hour every 2 weeks for 10 weeks (6 doses), and patients were followed up for a further 10 weeks. Study drug was discontinued due to adverse events (AEs) related to allergic-type reactions or at the discretion of the investigator.
Patients were assessed at screening (days −35 to −10), at baseline (day 1, first treatment day), on treatment days (days 15, 29, 43, 57, and 71), and during follow-up (days 85, 99, 113, and 141). In addition to inclusion/exclusion criteria evaluations, the following assessments were performed at screening: demographic characteristics, medical history, current/prior medications, physical examinations, vital signs, hematology, serum biochemistry, whole blood flow cytometry, urinalysis, and EKGs.
Live vaccines, plasmapheresis, oral and parenteral glucocorticosteroids, glatiramer acetate, IVIg, immunosuppressive/immunomodulating agents, and cytotoxic agents were prohibited during the trial. Oral (high-dose) or parenteral corticosteroid medication for exacerbations was permitted.
Standard protocol approvals, registrations, and patient consents.
This trial (registration number NCT01517282) was conducted from January 2012 to February 2014 at 5 centers in Germany, Poland, and the United Kingdom in accordance with the Declaration of Helsinki, Seoul 2008, the International Congress on Harmonization Good Clinical Practice Guidelines, Directive 2001/20/EC, and the appropriate regulatory policies and was approved by the independent ethics committee of each investigator site. Individuals gave written informed consent for participation and could voluntarily withdraw or be withdrawn for any reason at the investigator's discretion.
Safety and tolerability assessments.
The primary endpoint was the evaluation of the safety and tolerability of MOR103. On treatment days and during follow-up, AEs (Medical Dictionary for Regulatory Activities version 16.1) and concomitant medications (World Health Organization Drug Dictionary September 1, 2011) were reported and data were collected from physical examinations, vital signs, serum biochemistry, hematology, pregnancy tests, and EDSS. MS exacerbation was defined as acute worsening of one or more MS symptoms (e.g., numbness, loss of muscle function, tremor, and gait or balance problems) or the appearance of new symptoms lasting ≥24 hours and occurring ≥1 month from the conclusion of a previous exacerbation. Samples for laboratory analyses were collected before dosing, and analyses were performed either at the site (urinalysis) or at a central laboratory (INTERLAB GmbH, Munich, Germany). At intervals throughout the trial, coagulation parameters, whole blood flow cytometry (T cells [CD45RA, CD45RO, CD3, CD4] and monocyte subtypes [CD14, CD16]), urinalysis, EKGs, and MRI were evaluated. MRI was performed monthly to assess the number of T1 Gd-enhancing and T2 brain lesions and the volume of T1-hypointense and T2 lesions. MRI data were collected and analyzed at a centralized MRI institution (Medical Image Analysis Center, University Hospital Basel, Switzerland).
At baseline and at fixed intervals throughout the trial, serum surfactant protein D (SSP-D) levels were measured and pulmonary function tests (PFTs), including forced expiratory volume in the first second, vital capacity, forced vital capacity, and diffusing capacity of the lung for carbon monoxide (DLCO), were performed.
Serum levels of cytokines, including tumor necrosis factor α, interleukin (IL)-1β, IL-6, IL-8, IL-10, IL-12, IL-17, IL-23, IFN-γ, and GM-CSF, were measured and analyzed at a central laboratory (INTERLAB). Serum samples for anti-MOR103 antibodies were collected at baseline and at weeks 14, 16, and 20 and analyzed at a central bioanalytical laboratory (Eurofins Medinet BV, Breda, the Netherlands) for serum IgG, IgA, and IgM isotypes (INTERLAB).
Pharmacokinetic assessments.
Serum MOR103 levels were measured at baseline and week 10 before dosing and at 1 hour ± 5 minutes (i.e., end of infusion), 2 hours ± 5 minutes, and 4 hours ± 5 minutes after the start of infusion; at weeks 2 through 8 before dosing and at 1 hour ± 5 minutes after the start of infusion, and at weeks 12, 14, 16, and 20. Serum samples were analyzed at a central bioanalytical laboratory (Eurofins Medinet BV) using 2 different validated ligand binding assays detecting either free (bioactive) drug levels or levels of MOR103 bound to GM-CSF.
Statistical analysis.
Data were analyzed by descriptive statistics using SAS for Windows, release 9.3 by INC Research and summarized by dose level. Two analysis populations were defined: the safety population (all randomized patients who received ≥1 dose of study drug and had ≥1 safety assessment after dosing) and the PK population (all randomized patients who received study drug and had ≥1 PK sample after dosing). PK parameters were derived by noncompartmental analysis as data allowed. Dose proportionality was analyzed using the method proposed by Hummel et al.15
Missing data were not imputed but treated as missing with the following exceptions: if AE severity or relationship to study drug was missing, the event was considered to be severe and/or related to study drug (worst case principle); if the AE onset date was missing or incomplete, it was assumed to be a treatment-emergent AE (TEAE; unless otherwise indicated); and if the drug start and/or stop date was missing or incomplete, it was assumed that the drug was concomitant to study treatment (unless otherwise indicated).
Each patient was randomly assigned to MOR103 or placebo in a ratio of 4:1 to minimize the number of patients with MS receiving placebo while providing sufficient data to assess safety and ascertain adequate MOR103 exposure. No statistical assumptions of power, error rate, or safety margins were used to determine sample size. Ten patients per cohort is a typical sample size for phase 1b studies.
Classification of evidence.
This study was designed to answer the following primary research question: is IV MOR103 0.5–2.0 mg/kg administered at 2-week intervals of acceptable safety and tolerance to patients with RRMS or SPMS? This phase 1b study provides Class I evidence that MOR103 has acceptable tolerability in patients with MS. The majority of TEAEs were of mild or moderate intensity, and TEAEs and those events considered to be related to study drug occurred at a comparable or lower rate in the MOR103 groups compared with the placebo group.
RESULTS
Patient disposition.
Of the 57 patients screened, 32 were randomized, 31 received ≥1 dose of study drug, and 27 (84.4%) completed the trial (figure 1). One randomized patient did not receive treatment and was excluded from the analyses. All 31 treated patients were included in the safety population and all 25 patients on active treatment were included in the PK population.
Figure 1. MOR103 sequential-dose trial flowchart of study population with multiple sclerosis.

aPatients received 2 doses of study drug before trial withdrawal. bOne patient became pregnant during the trial and therefore did not receive treatment at weeks 8 and 10 (received 4 doses of study drug).
All patients received all 6 doses of study drug except for 1 patient in the MOR103 1.0 mg/kg group and 2 in the 2.0 mg/kg group (due to exacerbation or withdrawal). The duration and volume of each infusion was generally within the predefined ranges (duration 58–62 minutes; volume 490–510 mL).
Baseline demographic and clinical characteristics of patients were generally comparable among the treatment groups (table 1). However, the MS types were not distributed equally over the treatment groups: although the majority of patients in all groups were diagnosed with RRMS, the placebo group consisted of only patients with RRMS and the MOR103 2.0 mg/kg group had 3 patients with SPMS. In addition, the mean time since diagnosis was longer in the higher MOR103 dose groups than in the MOR103 0.5 mg/kg and placebo groups. Also, the number of exacerbations, patients with Gd-enhancing lesions, median T1-hypointense lesion volume, and median T2 lesion volume (data not shown) were unequally distributed between the cohorts.
Table 1.
Baseline demographics and clinical characteristics

Safety and tolerability.
A total of 184 TEAEs were reported in 31 (96.8%) patients (table 2), with no overall indication of increased TEAE frequencies in the MOR103 groups compared with placebo. The most common TEAEs in all groups were nasopharyngitis, headache, and MS exacerbation. No cases of infusion-related reaction were reported.
Table 2.
Incidence of treatment-emergent adverse events (AEs) in patients in any treatment group

Eleven MS exacerbation events occurred in 9 patients. MS exacerbation occurred in 3 (50.0%), 5 (62.5%), 1 (12.5%), and 0 (0%) patients in the placebo, MOR103 0.5, 1.0, and 2.0 mg/kg groups, respectively. Events occurred in both the treatment and follow-up periods in the placebo and MOR103 0.5 mg/kg groups and during follow-up in the MOR 1.0 mg/kg group. All events either resolved/recovered or were resolving/recovering at the end of the follow-up period; recovery with sequelae was reported for 3 exacerbations. Four exacerbation events required medication, including 2 serious events requiring hospitalization (1 patient in the placebo group and 1 patient in the MOR103 1.0 mg/kg group). The latter were of moderate intensity, were considered not related to treatment, and resolved after medication. One exacerbation event was considered to be “probably” related to treatment (placebo) by the investigator; the remainder were considered to be possibly (4 events), unlikely (3 events), or not (3 events) treatment-related by investigators.
TEAEs by system organ class generally occurred with similar frequencies in the 4 treatment groups. The most common were infections, occurring in 4 (66.7%), 5 (62.5%), 6 (75.0%), and 5 (55.6%) patients in the placebo, MOR103 0.5, 1.0, and 2.0 mg/kg groups, respectively. Nervous system disorders occurred in 5 (83.3%), 6 (75.0%), 4 (50.0%), and 3 (33.3%) patients in the placebo, MOR103 0.5, 1.0, and 2.0 mg/kg groups, respectively.
A total of 71 TEAEs considered to be possibly, probably, or definitely related to treatment were reported in 18 (58.1%) patients (table 3). MS exacerbation and headache occurred in the placebo and MOR103 0.5 mg/kg groups only, whereas a mild increase in C-reactive protein was reported for 1 patient in each of the higher MOR103 dose groups.
Table 3.
Incidence of treatment-emergent adverse events (AEs) considered to be possibly, probably, and definitely related to study drug in patients in any treatment group
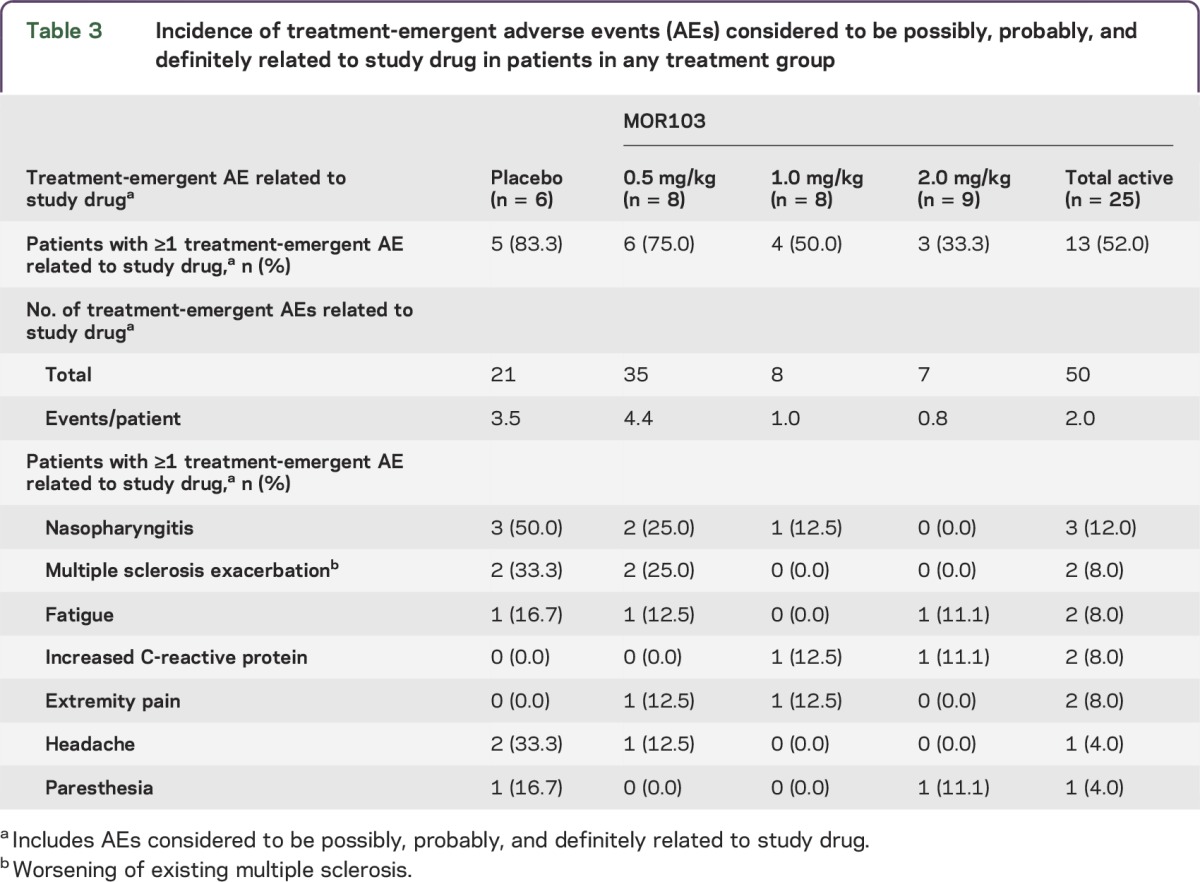
TEAEs were generally of mild or moderate intensity. There were 2 severe TEAEs (urinary tract infection, placebo group; decreased vibratory sense in right lower limb, MOR103 2.0 mg/kg group); both were considered unlikely to be related to treatment. No additional serious TEAEs (aside from the aforementioned MS exacerbations) were reported. No TEAEs led to death or trial discontinuation. One woman became pregnant and discontinued study drug after 4 doses but remained in the trial for observational follow-up (pregnancy terminated).
There were no clinically significant changes in vital signs, EKG, PFTs (including DLCO), or hematology, serum chemistry, or urine parameters during treatment. While whole blood flow cytometry (T cells [CD45RA, CD45RO, CD3, CD4] and monocyte subtypes [CD14, CD16]) was monitored, no differences or trends were observed in any of the flow cytometry parameters when comparing the active treatment groups with placebo. EDSS remained generally stable, with small changes indicating worsening (scores +0.5 to +1.0) usually associated with MS exacerbation. No clinically important differences between active treatment and placebo in these measures were observed.
A few T1 Gd-enhancing and/or new or enlarging T2 lesions indicative of inflammation were observed (table 4).
Table 4.
Number of new T1 gadolinium-enhancing (Gd+) lesions and number of new or enlarging T2 lesions on MRI

Given that GM-CSF and alveolar macrophages are regulators of lung surfactant homeostasis and that pulmonary alveolar proteinosis has been associated with neutralizing anti-GM-CSF antibodies,16 we measured SSP-D levels and performed PFTs (including DLCO) to evaluate potential lung toxicity. SSP-D levels showed some variability throughout the trial, although no consistent increase was observed for any cohort. There were no clinically relevant differences in PFTs in the MOR103 groups or reports of cough or dyspnea.
Peripheral blood cytokines, including GM-CSF, were usually near or below the respective lower limits of quantification and showed no notable or consistent changes related to MOR103 infusions throughout the trial. Serum samples did not test positive for anti-MOR103 antibodies, and there was no evidence of any treatment difference in serum IgG, IgA, and IgM isotype concentrations.
Pharmacokinetics.
Observed MOR103 serum concentrations over time are presented in figure 2. PK data analysis was based on free MOR103 serum levels. On day 1, median time of the apparent maximum concentration was 1.0–2.0 hours after the start of the infusion. The geometric mean apparent maximum concentration and apparent area under the serum concentration vs time curve (AUCtau) were dose proportional in the range of 0.5–2.0 mg/kg. Following noncompartmental data analysis, the mean apparent terminal half-life was approximately 17 days across all dose groups, and mean AUCtau accumulation as a result of multiple dosing was approximately 20%–32%. MOR103/GM-CSF complexes (bound drug levels) in serum were close to the limit of quantification at 20 pM or not detectable.
Figure 2. Mean MOR103 serum concentrations following 6 0.5, 1, or 2 mg/kg weekly IV infusions.

Mean MOR103 serum concentrations (conc.) per dosing cohort following 6 IV infusions once weekly of 0.5, 1, or 2 mg/kg (semilogarithmic scale, n = 7–9 individual patient values per sampling time point).
DISCUSSION
This randomized, double-blind, placebo-controlled, sequential ascending-dose trial was designed to evaluate the effects of neutralizing GM-CSF using MOR103 in patients with MS. The primary objective was to determine the safety and tolerability of MOR103 in patients with RRMS or SPMS. MOR103 administered at 3 different doses at 2-week intervals was well-tolerated in patients with MS.
The majority of TEAEs were of mild or moderate intensity, and only 2 events were considered to be serious. TEAEs and those events considered to be related to study drug occurred at a comparable or lower rate in the MOR103 groups compared with the placebo group. Of note, no increase in the incidences of nasopharyngitis, MS exacerbation, and headache (the most frequently reported AEs) were observed with higher MOR103 doses. The incidences of treatment-related nasopharyngitis, MS exacerbation, and headache were similar between the MOR103 0.5 mg/kg group and the placebo group. Nasopharyngitis was also reported to be the most common treatment-related AE in patients with rheumatoid arthritis treated with MOR103.14
Exacerbations occurred in both the treatment and follow-up periods in patients receiving MOR103 0.5 and 1.0 mg/kg. Whether this indicates a potential for a rebound effect at these doses remains to be determined. MRI examinations did not demonstrate any evidence of increased lesional activity or opportunistic CNS infections in patients treated with MOR103.
The assumption that MOR103 has a low immunogenic potential in patients with MS was confirmed by this trial, as no anti-MOR103–positive samples were detected throughout the complete trial.
The PK profile demonstrating dose linearity between MOR103 0.5 and 2 mg/kg and a terminal elimination half-life of approximately 17 days is comparable with previously reported results for other human IgG1 antibodies targeting proinflammatory cytokines.17,18
The limitations of this trial mostly apply to phase 1/2 clinical trials in general, including small sample size and limited duration. While not unexpected, the slight bias in the baseline characteristics needs to be acknowledged.
This trial demonstrates that MOR103 is well-tolerated with no unexpected safety concerns in the treatment of MS. These findings are consistent with prior safety data reported for patients with rheumatoid arthritis.14 Therefore, the use of GM-CSF antibodies may provide a novel approach to addressing the unmet need for MS therapies.
ACKNOWLEDGMENT
The authors thank all of the patients and investigators who participated in this trial. The authors thank the members of the Data Monitoring Committee: Professor C.H. Polman (VU University Medical Center, Amsterdam, the Netherlands), Professor Reinhard Hohlfeld (Ludwig Maximilians University, Munich, Germany), Professor Tomas Olsson (Karolinska University Hospital, Sweden), and Dr. Andres Floto (Addenbrooke's Hospital, Cambridge, UK). The authors also thank Dr. Steffen Stürzebecher and Dr. Stefan Härtle (MorphoSys AG, Martinsried/Planegg, Germany) for contributing to data analysis, interpretation, and manuscript review, and Susan Spruill, PhD (Applied Statistics and Consulting, Spruce Pine, NC) for critically reading the manuscript. The statistical analysis was conducted by Natalie Steigerwald, Lead Biostatistician, INC Research, Munich, Germany.
GLOSSARY
- AE
adverse event
- AUCtau
area under the serum concentration vs time curve
- DLCO
diffusing capacity of the lung for carbon monoxide
- DMC
data monitoring committee
- EAE
experimental autoimmune encephalomyelitis
- EDSS
Expanded Disability Status Scale
- Gd
gadolinium
- GM-CSF
granulocyte-macrophage colony-stimulating factor
- IFN
interferon
- IVIg
IV gamma globulin
- IL
interleukin
- MS
multiple sclerosis
- PFT
pulmonary function test
- PK
pharmacokinetics
- RRMS
relapsing-remitting MS
- SPMS
secondary progressive MS
- SSP-D
serum surfactant protein D
- TEAE
treatment-emergent AE
AUTHOR CONTRIBUTIONS
All authors were involved in drafting or revising the article critically for important intellectual content, and all authors approved the final version. C.S.C., R.P.K., and S.S. were involved in the trial conception and design. C.S.C., A.A., W.F., W.K., F.W., J.A., R.T., T.S., and E.W.R. were responsible for data acquisition. C.S.C., R.P.K., M.D.-H., S.S., T.S., and E.W.R. were involved in analysis and interpretation of data.
STUDY FUNDING
The trial was supported by MorphoSys AG, which provided funding for the trial, data analyses, and medical writing services.
DISCLOSURE
C.S. Constantinescu is on the scientific advisory boards for Biogen Idec, Teva, and MESEMS trial; received travel funding and/or speaker honoraria from Biogen Idec, Novartis, and Teva; is on the editorial board for Multiple Sclerosis International, European Neurological Review, Biomedical Journal, and CML MS; has consulted for Teva and MorphoSys; and received research support from Biogen Idec, Merck Serono, Bayer Schering, MorphoSys, Roche, Sanofi Pasteur MSD, and Multiple Sclerosis Society of Great Britain and Northern Ireland. A. Asher reports no disclosures. W. Fryze received travel funding and speaker honoraria from Teva, Biogen Idec, Bayer, and Merck Serono and received research support from Biogen Idec, Sanofi-Aventis, Teva, Novartis, and Roche. W. Kozubski has consulted for Assessment of Medical Technologies. F. Wagner, J. Aram, and R. Tanasescu report no disclosure. R.P. Korolkiewicz is employed by MorphoSys. M. Dirnberger-Hertweck reports no disclosures. S. Steidl holds patents and patent applications for MOR103 and is employed by and holds stock options in MorphoSys AG. Susan E. Libretto reports no disclosures. T. Sprenger is on the scientific advisory boards for Genzyme, Novartis, Actelion, Electrocore, and Mitsubishi Pharma; received speaker honoraria and/or travel funding from Biogen, Genzyme, Teva, and Novartis; and received research support from Novartis Pharma, Swiss National Science Foundation, and Swiss MS Society. E.W. Radue received travel funding and speaker honoraria from Bayer Schering, Biogen, Genzyme, Novartis, Merck Serono, MorphoSys, and Synthon; has consulted for Bayer Schering, Biogen, Genzyme, Novartis, Merck Serono, Synthon, and Morphosys; and received research support from Novartis, Biogen Idec, Actelion, Basilea, SAKK, and Synarc. Go to Neurology.org/nn for full disclosure forms.
REFERENCES
- 1.Waubant E. Overview of treatment options in multiple sclerosis. J Clin Psychiatry 2012;73:e22. [DOI] [PubMed] [Google Scholar]
- 2.Bogie JF, Stinissen P, Hendriks JJ. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol 2014;128:191–213. [DOI] [PubMed] [Google Scholar]
- 3.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008;8:533–544. [DOI] [PubMed] [Google Scholar]
- 4.van Nieuwenhuijze A, Koenders M, Roeleveld D, Sleeman MA, van den Berg W, Wicks IP. GM-CSF as a therapeutic target in inflammatory diseases. Mol Immunol 2013;56:675–682. [DOI] [PubMed] [Google Scholar]
- 5.Hartmann FJ, Khademi M, Aram J, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun 2014;5:5056. [DOI] [PubMed] [Google Scholar]
- 6.Constantinescu CS, Farooqi N, O'Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol 2011;164:1079–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McQualter JL, Darwiche R, Ewing C, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med 2001;194:873–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol 2007;178:39–48. [DOI] [PubMed] [Google Scholar]
- 9.King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009;113:3190–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Codarri L, Gyülvészi G, Tosevski V, et al. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol 2011;12:560–567. [DOI] [PubMed] [Google Scholar]
- 11.El-Behi M, Ciric B, Dai H, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol 2011;12:568–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steidl S, Ratsch O, Brocks B, Dürr M, Thomassen-Wolf E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol Immunol 2008;46:135–144. [DOI] [PubMed] [Google Scholar]
- 13.Minagar M, Alexander S. Blood-brain barrier disruption in multiple sclerosis. Mult Scler 2003;9:540–549. [DOI] [PubMed] [Google Scholar]
- 14.Behrens F, Tak PP, Ostergaard M, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomized, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis Epub 2014 Feb 17. [DOI] [PMC free article] [PubMed]
- 15.Hummel J, McKendrick S, Brindley C, French R. Exploratory assessment of dose proportionality: review of current approaches and proposal for a practical criterion. Pharm Stat 2009;8:38–49. [DOI] [PubMed] [Google Scholar]
- 16.Huizar I, Kavuru MS. Alveolar proteinosis syndrome: pathogenesis, diagnosis, and management. Curr Opin Pulm Med 2009;15:491–498. [DOI] [PubMed] [Google Scholar]
- 17.Weisman MH, Moreland LW, Furst DE, et al. Efficacy, pharmacokinetic, and safety assessment of adalimumab, a fully human anti-tumor necrosis factor-alpha monoclonal antibody, in adults with rheumatoid arthritis receiving concomitant methotrexate: a pilot study. Clin Ther 2003;25:1700–1721. [DOI] [PubMed] [Google Scholar]
- 18.Zhou H, Jang H, Fleischmann RM, et al. Pharmacokinetics and safety of golimumab, a fully human anti-TNF-alpha monoclonal antibody, in subjects with rheumatoid arthritis. J Clin Pharmacol 2007;47:383–396. [DOI] [PubMed] [Google Scholar]