

Abstract
Objective:
To determine clinical and EEG features that might help identify patients with epilepsy harboring small, intrinsically epileptogenic, surgically treatable, bottom-of-sulcus dysplasias (BOSDs).
Methods:
Retrospective review of clinical records, EEG, MRI, and histopathology in 32 patients with drug-resistant epilepsy and MRI-positive (72% 3.0 tesla), pathologically proven (type 2B cortical dysplasia) BOSDs operated at our centers during 2005–2013.
Results:
Localization of BOSDs was frontal in 19, insula in 5, parietal in 5, and temporal in 3, on the convexity or interhemispheric surfaces. BOSDs were missed on initial MRI at our centers in 22% of patients. Patients presented with focal seizures during infancy in 9, preschool years in 15, and school years in 8 (median age 5 years). Seizures were stereotyped, predominantly nocturnal, and typically nonconvulsive, with semiology referable to the fronto-central or perisylvian regions. Seizures occurred at high frequency during active periods, but often went into prolonged remission with carbamazepine or phenytoin. Intellect was normal or borderline, except in patients with seizure onset during infancy. Scalp EEG frequently revealed localized interictal epileptiform discharges and ictal rhythms. Patients underwent lesionectomy (median age 14 years) guided by electrocorticography and MRI, with prior intracranial EEG monitoring in only one patient. Twenty-eight patients (88%) became seizure-free, and 20 discontinued antiepileptic medication (median follow-up 4.1 years).
Conclusions:
In patients with cryptogenic focal epilepsy, this clinical presentation and course should prompt review of or repeat MRI, looking for a BOSD in the frontal, parietal, or insula cortex. If a BOSD is identified, the patient might be considered for single-stage lesionectomy.
Bottom-of-sulcus dysplasia (BOSD)1 is a localized variety of type 2 focal cortical dysplasia (FCD) in which the dysplastic features are maximal at the sulcal depth, tapering to a relatively normal gyral crown.2,3 Pathologically, BOSDs are characterized by neuronal dyslamination and dysmorphology, with or without balloon cells, at the bottom of the sulcus, with hypomyelination in the underlying white matter.2,4,5
The characteristic MRI features of BOSD are cortical thickening, gray–white junction blurring, and subcortical T2 hyperintensity, often tapering to the ventricle as a “transmantle sign.”6–9 Straightening and elongation of the dysplastic sulcus, and depression or unusual sulcation of the overlying cerebral surface, may also be appreciated.8,10,11 BOSDs are typically found in the frontal and parietal lobes.12 Detection of BOSDs on MRI may be aided by coregistered fluorodeoxyglucose (FDG)-PET,13,14 morphometric analysis of cortical thickness or texture,7,15,16 and imaging at high field.17
BOSDs are clinically important lesions. They are intrinsically and focally epileptogenic, as evidenced by almost continuous, localized, rhythmic interictal epileptiform discharges (IEDs) and seizures often recorded on scalp EEG, electrocorticography, and stereo depth EEG.14,18–22 They seemingly contain no normal function, as suggested by their PET hypometabolism, perilesional fMRI activation, displaced motor functions on cortical stimulation, and absence of deficits following resection.13,23–26 In addition, seizure-free rates after surgery are as high as 90% if completely resected.9,13,27–34 Thus, patients with refractory epilepsy and BOSD are ideal candidates for epilepsy surgery.
The aim of this study was to identify electroclinical features that might aid detection of patients with BOSD among the drug-resistant, focal epilepsy population.
METHODS
Patient ascertainment.
Patients with drug-resistant epilepsy were identified from the epilepsy surgery databases of The Royal Children's Hospital and Austin Health in Melbourne, Australia, searching initially for patients with localized dysplasia on imaging or histopathology, operated during the period January 2005 to June 2013. The highest-quality preoperative MRI scans for all ascertained patients were reviewed by 5 authors together (A.S.H., S.A.M., W.J.M., R.J.L., G.D.J.), with consensus agreement reached on imaging diagnoses.3 Histopathology slides were reviewed by a single neuropathologist at each center (D.M., R.M.K.).
BOSD was diagnosed on MRI when cortical thickening and gray–white blurring were present and maximal at the depth of a sulcus, tapering in the banks of the sulcus, and not extending over the gyral surface beyond the adjacent sulcus, with or without cortical and subcortical signal change and a transmantle sign. For dysplasias that reached the gyral surface, distinction from “crown-of-gyrus” FCDs was made using the principle that BOSDs are longer in their depth than their width. Only patients with a single BOSD confined to one sulcus were included, although branching of a sulcus in its depth was allowed. The sample was strengthened further to include only patients with FCD type 2B histopathology,2 exhibiting both dysmorphic neurons and balloon cells, given the strong correlations among imaging findings, histopathology, and surgical outcome in this subgroup.9,13,32
This ascertainment strategy and case definition yielded 32 patients with MRI-positive, surgically treated, pathologically proven, single BOSDs with FCD type 2B histopathology. There were 19 children from the Royal Children's Hospital and 13 adults from Austin Health, 19 women and 13 men. Excluded from the initial search yield were 7 patients with crown-of-gyrus FCDs, 3 patients with multigyral/sulcal FCDs, and 1 patient with multiple BOSDs, all of whom had FCD type 2B histopathology. Five patients with MRI-negative FCDs were excluded, 2 with FCD type 2B and 3 with FCD type 2A histopathology.
Clinical, EEG, and imaging data review.
All 32 patients were imaged preoperatively with high-resolution, whole-brain MRI using volumetric T1-weighted sequences, axial and coronal T2-weighted sequences, volumetric or orthogonal fluid-attenuated inversion recovery (FLAIR) sequences, and diffusion-weighted sequences; recent patients also had volumetric double inversion recovery sequences. Volumetric images were reformatted in axial, coronal, and sagittal planes, and in some patients, oblique and curvilinear planes. Nine patients early in our series were imaged at 1.5 tesla (T) on either a GE Signa LX EchoSpeed scanner (GE Medical Systems, Milwaukee, WI) or a Magnetom Avanto scanner (Siemens, Erlangen, Germany). Twenty-three (72%) recent patients were imaged at 3.0T on either a Magnetom Trio or Magnetom Skyra (Siemens).
Outpatient clinic notes, inpatient records, clinician correspondence, surgery case conference notes, EEG reports, and neuropsychology reports present in the patients' hospital records were reviewed, to glean information about the presentation and evolution of each patient's seizure disorder. All routine EEGs and video-EEG monitoring (n = 29) recorded at our centers were reviewed by one of the epileptologist authors (A.S.H.). The location and extent of surgical resection was determined from operation notes, operative photographs, and postoperative MRI (n = 29). Although crucial to the detection and complete resection of BOSDs in these patients, PET scans (n = 21), ictal SPECT scans (n = 18), and intraoperative electrocorticographic recordings (n = 24) were not reviewed and analyzed for this clinical study.
Standard protocol approvals, registrations, and patient consents.
The study was approved by the human research and ethics committees at The Royal Children's Hospital and Austin Health.
RESULTS
MRI features.
MRI features of BOSDs included (1) cortical thickening and indistinct gray–white junction in 32, best appreciated on thin T1-weighted slices along the long axis of the dysplastic sulcus, (2) increased subcortical T2, FLAIR, and double inversion recovery signal in 30, and (3) a transmantle sign extending toward the ventricle in 20 (figures 1 and 2). Cortical thickening and subcortical T2/FLAIR hyperintensity were maximal at the depth of the sulcus in all patients. The dysplastic features extended up the sulcal banks and reached the gyral surface in 14 patients, but were confined to the sulcal depth and low banks in 18 patients. Dorsolateral frontal and parietal BOSDs were better appreciated on coronal slices. Medial frontal, insula, and perisylvian BOSDs were better appreciated on sagittal slices. Reformatted curvilinear slices and slices along and tangential to the plane of the dysplastic sulcus often highlighted the cortical thickening and gray–white blurring in the depth of subtle BOSDs. In 7 patients (22%), the BOSD was overlooked by the radiologists reporting the initial 1.5T MRI at our centers, and many more were not detected on MRI performed at referring centers. Subsequent imaging, at high field in 3 and supplemented with coregistered FDG-PET in 5, led to the detection of the BOSD in these 7 patients (figure 3).
Figure 1. Three MRI examples of BOSDs.
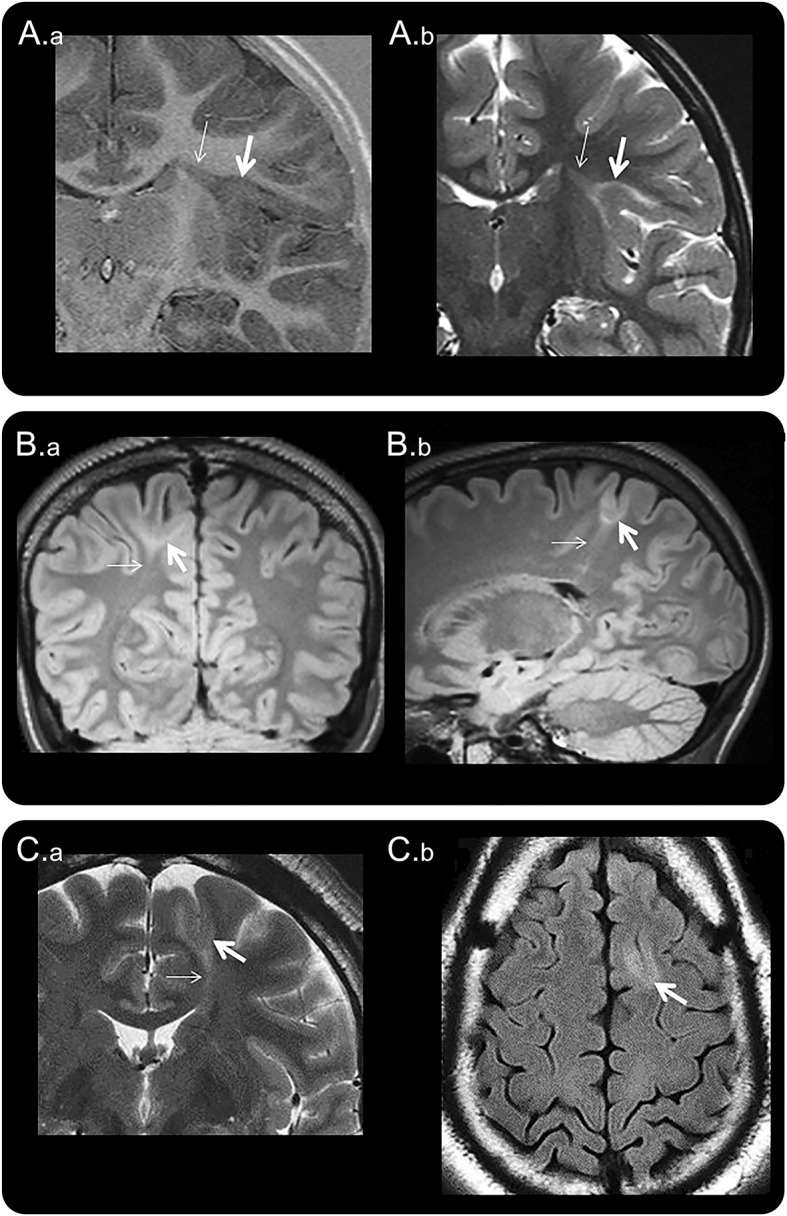
Coronal inversion recovery (A.a) and coronal T2-weighted (A.b) MRI scans at 3.0 tesla (T) showing a BOSD in the right circular sulcus in a 5-year-old girl with asymmetric tonic focal seizures. Coronal FLAIR (B.a) and sagittal FLAIR (B.b) MRI scans at 3.0T showing a BOSD in the right superior parietal lobule in a 17-year-old male with focal motor seizures. Coronal T2-weighted (C.a) and axial FLAIR (C.b) MRI scans at 1.5T showing a BOSD in the left superior frontal gyrus in a 35-year-old man with hypomotor focal seizures. Each example shows cortical thickening, blurred gray–white junction, and subcortical T2/FLAIR hyperintensity at the depth of the sulcus (thick arrow), with a “transmantle sign” extending through the deeper white matter and tapering to the ventricle (thin arrow). Resection of the BOSD identified focal cortical dysplasia type 2B pathology in each case. BOSD = bottom-of-sulcus dysplasia; FLAIR = fluid-attenuated inversion recovery.
Figure 2. Multisequence MRI of a right frontal pole BOSD.

Axial MRI scans in the same plane at 3.0 tesla with a 32-channel head coil and T1-weighted fast spoiled gradient recalled echo (A), T2-weighted fast spin echo (B), FLAIR (C), inversion recovery (D), DIR (E), and diffusion-weighted (F) sequences in a 16-year-old boy with hypermotor seizures and right anterior frontal epileptiform activity on scalp EEG. There is a subtle BOSD in the depth of the right superior frontal sulcus anteriorly with thickening of cortex, blurring of gray–white junction, and increased T2/FLAIR/DIR subcortical signal at the bottom of the sulcus (thick arrow) and a “transmantle sign” (thin arrow). Focal cortical dysplasia type 2B pathology was identified at the depth of the resected sulcus. BOSD = bottom-of-sulcus dysplasia; DIR = double inversion recovery; FLAIR = fluid-attenuated inversion recovery.
Figure 3. Coregistered PET and MRI of a right frontal BOSD.
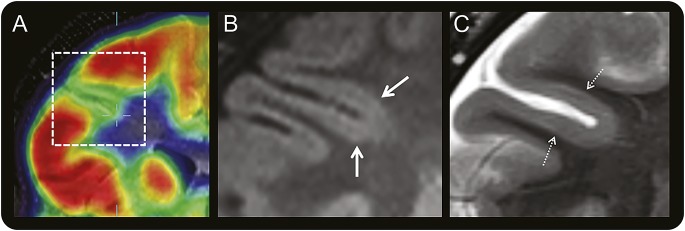
Coregistered coronal FDG-PET and T1-weighted MRI scans (A) through the right frontal lobe in a 10-year-old boy with versive seizures and right frontal epileptiform activity on scalp EEG showing localized cortical hypometabolism in the right inferior frontal sulcus. Magnified coronal FLAIR (B) and T2-weighted (C) MRI scans at 3.0 tesla with a 32-channel head coil showing subtle thickening of cortex with blurring of gray–white junction (thick arrow) and faint subcortical signal hyperintensity (hatched arrow) in the bottom of the hypometabolic sulcus, but no “transmantle sign.” The BOSD was not detected on this MRI scan until after coregistration with the PET scan and recognition that there was thickened gray matter deeper than the apparent depth of the hypometabolic sulcus on the PET scan, being more hypometabolic than the sulcal banks. Focal cortical dysplasia type 2B pathology was identified at the depth of the resected sulcus. BOSD = bottom-of-sulcus dysplasia; FDG = fluorodeoxyglucose; FLAIR = fluid-attenuated inversion recovery.
There was a right hemisphere predominance of BOSDs (20 right, 12 left). Lobar localization was frontal in 19, insula in 5, parietal in 5, and temporal in 3; all were on the convexity or interhemispheric surfaces of the cerebrum (figure 4). No BOSDs were present in medial or basal temporal cortex or in the occipital lobe. The most common location of BOSDs was over the anterior or mid regions of the superior frontal gyrus or sulcus. Six BOSDs involved eloquent cortex anatomically, 2 in primary motor cortex and 4 in inferior frontal or lateral temporal language cortex.
Figure 4. Location of BOSDs.
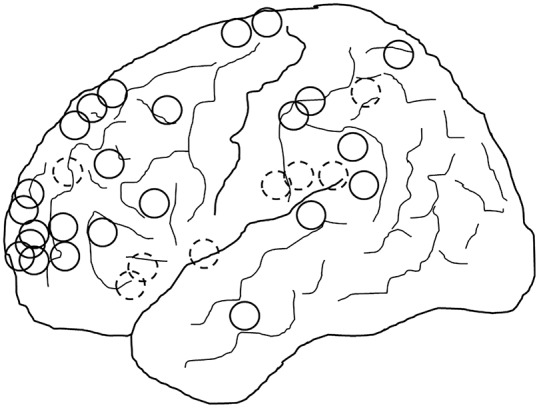
Schematic diagram showing the location of BOSDs in the 32 patients, all represented on the left hemisphere (20 right, 12 left). Full circles represent BOSDs on the cerebral convexity and hatched circles represent BOSDs on the interhemispheric surface and insula. The most common location of BOSDs was in the frontal, parietal, and insula cortex. BOSD = bottom-of-sulcus dysplasia.
Seizure presentation and evolution.
There were no significant antecedents to the patients' epilepsy. A single febrile seizure was reported in only one child, 2 months before the onset of habitual seizures. A family history of epilepsy in first- or second-degree relatives was reported in only 4 patients.
Median age at seizure onset was 5.0 years. Seizures commenced during infancy (1–18 months) in 9 patients, during the preschool years (3–6 years) in 15 patients, and during the school years (6–18 years) in 8 patients (figure 5).
Figure 5. Age at seizure onset in 32 patients with BOSD.
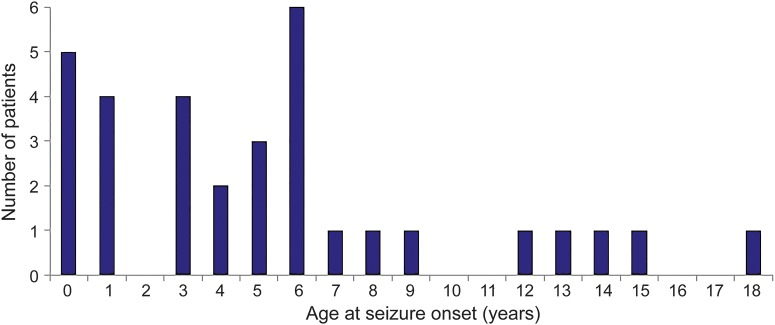
Age at seizure onset in 32 patients with bottom-of-sulcus dysplasia (BOSD).
All patients presented with focal seizures; none presented with epileptic spasms. Seizure semiology was referable to the temporal, frontal, and central cortical regions, with predominantly hypomotor/autonomic features in 9, versive features in 4, asymmetric tonic features in 6, focal sensorimotor features in 5, and hypermotor features in 6. Seizures occurred when awake, or both awake and asleep, in 30 patients; only 2 patients had exclusively nocturnal seizures at presentation. Bilateral convulsive seizures occurred in only 6 patients at presentation, typically leading to the diagnosis of epilepsy.
Epilepsy continued with highly stereotyped focal seizures, but with a change in semiology in 10 patients. Semiology when older and on treatment was more frequently referable to the frontal and central cortical regions, with predominantly hypermotor features in 7, focal sensorimotor features in 7, asymmetric tonic features in 11, and hypomotor/autonomic features in 6. Consistently lateralized motor manifestations were present in 21 patients. Seizure timing also changed, with seizures occurring exclusively or predominantly from sleep in 13 patients. Convulsive seizures were less frequent later in the course of epilepsy, on treatment, occurring in only 3 patients. One 6-year-old child developed a brief period of encephalopathy with epileptic spasms that ceased following a single course of oral prednisolone, and did not recur. One adult had bilateral tonic seizures but no other seizure types to make a definite diagnosis of Lennox-Gastaut syndrome.
Seizure onset was “explosive” in several patients, with rapidly escalating seizures resistant to initial antiepileptic medication (AEM) trials. Following commencement of AEM, many patients had control of seizures for long periods, only to have later seizure recurrence and a subsequent relapsing-remitting course. Fourteen patients (44%) had periods of complete seizure freedom lasting one or more years, 10 following commencement of AEM, and 9 later in the course of their epilepsy on AEM. Remissions lasted several years in some patients, such that 4 came off AEM. During active seizure periods, seizure frequency was multiple daily or nightly in 23 patients (72%), weekly in 5, and monthly in 4. Seizure clustering was reported in only 2 patients whose seizures occurred with monthly frequency. In addition, 7 patients had severe seizure exacerbations prompting hospitalization for multiple hourly seizures. In 13 patients, there seemed to be a striking but often temporary response to phenytoin or carbamazepine, with termination of seizure exacerbations or entering a seizure-free period for a year or more.
Neurologic development.
Early developmental milestones were reached at a normal age in 25 patients (78%). Developmental delays, mainly in language, were reported in 7 children. However, at the time of surgery, cognitive impairments were present in 16 patients, including borderline intellect, executive dysfunction, and language deficits. Intellectual disability and global developmental delay were present in only 5 patients, all of whom had onset of seizures during infancy (5/9 patients with seizure onset younger than 18 months had intellectual disability compared with 0/23 with seizure onset 3 years and older; p = 0.0006, Fisher exact test).
Scalp EEG and seizure monitoring.
Interictal scalp EEG revealed focal IEDs in 26 patients (81%), with runs of or almost continuous, rhythmic, focal spike-wave in many; the remainder had either no IEDs or bilateral IEDs. Video-EEG monitoring revealed focal ictal rhythms in 20 of 29 (69%) monitored patients; the remainder had either no clear ictal rhythm or bifrontal slowing or low-voltage fast activity. One patient with bilateral tonic seizures had generalized spike-wave and low-voltage fast activity interictally, and generalized low-voltage fast activity ictally.
Surgery details.
Age at surgery was 2 to 42 (median 14) years. Epilepsy surgery was a single-stage procedure in 31 patients; only one adult underwent 2-stage surgery, with prior subdural EEG monitoring. Surgery was a lesionectomy in 30 patients and a lesionectomy plus surrounding corticectomy in 2 patients, one of whom underwent prior subdural EEG monitoring. In 2 patients, one with an insula BOSD and one with a medial parietal BOSD, a small resection of overlying cortex was necessary for access. Three patients were reoperated for persistent or recurrent seizures and residual dysplasia.
Extent and completeness of resection were determined from postoperative MRI in 30 patients. Lesionectomies were performed to neighboring (normal) sulcal boundaries in 19 patients, with overlying corticectomy for access in 2, and were confined to the dysplastic sulcus in 11. The cortical component of the BOSD was completely resected in 27 patients, including after reoperation in 3, and incompletely resected in 3. The “transmantle sign,” present on MRI in 20 patients, was resected to the level of the ventricle in 7, partially resected in 3, and not resected in 8.
Time from last surgery to most recent follow-up was 1.0 to 9.5 (median 4.1) years. Twenty-eight patients (88%) became seizure-free, including the 3 reoperated patients, 20 of whom were taken off AEM. Four patients have persistent seizures, at lower frequency and severity in 3. Three of the 4 have evidence of residual cortical dysplasia on postoperative MRI and have further surgery planned. Two patients had a monoparesis after surgery; one recovered almost completely.
DISCUSSION
Our patients with BOSD shared many features at presentation and during the course of their epilepsy, suggesting a recognizable epileptic phenotype. Those features included the following: (1) infant or early childhood onset of predominantly diurnal focal seizures; (2) chronic, unifocal epilepsy on AEM with highly stereotyped seizures, often nocturnal, having nonconvulsive semiology referable to the frontal, central, and insular regions; (3) high frequency of seizures during active periods, occasionally with hospitalization for severe exacerbations; (4) prolonged seizure remissions, often following treatment with carbamazepine or phenytoin; (5) normal or near-normal early development and intellectual functioning, unless onset of seizures during infancy, but with later executive dysfunction and/or language deficits in many; (6) prominent, localized IEDs and ictal rhythms on scalp EEG; and (7) absence of significant antecedents and family history of epilepsy.
These electroclinical findings are reported individually and collectively in numerous small series and several large studies of surgically treated patients with FCD type 2. These studies include mention of the childhood onset of epilepsy,13,14,21,28,31,32,35 the infrequency of infantile spasms,14,30,35 the rarity of significant antecedents such as febrile seizures,10,14,27,30 the frontoparietal location of small FCDs, the extratemporal semiology and nocturnal occurrence of seizures,36 the high frequency of seizures with tendency to status epilepticus,13,14,19,28,30–32,35,37 the occurrence of seizure remissions, often in response to sodium channel blocking AEM,19,35 the normal or near-normal intellect in many patients,14,30,32 and the prominence of localized and rhythmic IEDs on scalp EEG.14,18,19,31,32 These studies, however, included patients with a range of MRI-positive and MRI-negative, type 1 and type 2 FCDs of various sizes and locations, and were mostly focused on imaging or pathologic correlations, predictors of surgery outcome, or issues of classification. Patient heterogeneity and complex analyses in these studies made identification of clinicopathologic subsyndromes of FCD difficult.38
The report from the Centre Hospitalier Sainte-Anne group14 describes the same findings in a similar patient population as we report. Their study was largely concerned with the comparison of MRI-positive and MRI-negative patients with surgically treated FCD type 2, with MRI performed at 1.5T. Half of their MRI-negative patients were reported to have “unusual sulcus depth or gyral patterns,” many had localized epileptogenic regions on stereo-EEG and electrocorticography, and 88% became seizure-free after surgery, leading us to believe that they had MRI-occult BOSDs. In a follow-up study, these authors report improved detection of subtle FCD type 2 with 3T MRI,17 as performed in the majority of our patients.
The localized and prominent motor/premotor/prefrontal seizure semiology, the nocturnal occurrence of seizures, and the deficits in language and executive function are easily understood in the context of the predominant localization of BOSDs in frontal, insula, and pericentral cortex. Why these lesions have a predilection for these cortical regions, how such small lesions give rise to such a severe seizure disorder, and why they suddenly switch on and off is uncertain. Their intrinsic epileptogenicity, however, is unquestioned, given the prominent “pacemaker” activity recorded with intralesional EEG18–21 and the excellent response to complete resection.21,25,29,31–33,36
Regarding surgical treatment, our series contrasts with other series in which resections of small or MRI-occult type 2 FCDs were frequently performed with prior subdural grid or depth electrode monitoring, often with the extent of resection determined by intracranial EEG findings.36 Similar postoperative seizure outcomes were achieved in our patients (88% seizure-free) with imaging and electrocorticography-guided lesionectomies, confined to the dysplastic sulcus in 34%. Such localized, single-stage approaches to these small dysplastic lesions are sparsely reported.31,32,39 Single-stage resections in eloquent cortical regions, without prior cortical stimulation or awake functional monitoring, need to be undertaken with preservation of the cortex and vascular supply to the adjacent gyral crowns, avoidance of injury to the subcortical white matter tracts, and knowledge that FCD type 2A lesions (usually without a transmantle sign on MRI) may contain cortical function.
It is not possible to determine from our study whether the electroclinical phenotype we and others14,35 describe is unique, or overlaps with other causes of drug-resistant epilepsy. The phenotype is nevertheless different from several other refractory focal epilepsies in which MRI may appear normal, such as medial temporal lobe epilepsy, epileptic encephalopathies of childhood due to subtle malformations of cortical development, and “MRI-negative PET-positive” focal epilepsy due to FCD type 1.10,27,30,35,40
Recognition of this phenotype in patients with reportedly normal MRI should prompt (1) review of MRI with knowledge of seizure localization from the patient's semiology and scalp EEG, and awareness of the usual locations of BOSDs, and (2) repeat MRI at high field with postprocessing and coregistration with FDG-PET, looking for dysplastic, hypometabolic gray matter in the depth of a sulcus, and potentially a subtle transmantle sign. In patients in whom a BOSD is detected, referral for epilepsy surgery should be considered, given the excellent postoperative outcomes. Finally, epilepsy surgery for these exquisitely localized lesions might be possible with MRI-PET and electrocorticographic guidance, without need for extraoperative intracranial EEG monitoring.
Supplementary Material
GLOSSARY
- AEM
antiepileptic medication
- BOSD
bottom-of-sulcus dysplasia
- FCD
focal cortical dysplasia
- FDG
fluorodeoxyglucose
- FLAIR
fluid-attenuated inversion recovery
- IED
interictal epileptiform discharge
Footnotes
Editorial, page 2012
AUTHOR CONTRIBUTIONS
Dr. Harvey contributed to study concept and design, data acquisition, data analysis and interpretation, study coordination, and drafting and revising the manuscript. A/Prof. Mandelstam contributed to study concept and design, data acquisition, data analysis and interpretation, and revising the manuscript. Ms. Maixner contributed to study concept and design, data acquisition, data analysis and interpretation, and revising the manuscript. A/Prof. Leventer contributed to study concept and design, data acquisition, data analysis and interpretation, and revising the manuscript. Ms. Semmelroch contributed to data acquisition, study coordination, and revising the manuscript. A/Prof. MacGregor contributed to data acquisition and revising the manuscript. Dr. Kalnins contributed to data acquisition and revising the manuscript. Dr. Perchyonok contributed to data acquisition and revising the manuscript. Dr. Fitt contributed to data acquisition and revising the manuscript. Dr. Barton contributed to data acquisition and revising the manuscript. Mr. Kean contributed to data acquisition and revising the manuscript. Prof. Fabinyi contributed to data acquisition and revising the manuscript. Prof. Jackson contributed to study concept and design, data acquisition, data analysis and interpretation, and revising the manuscript.
STUDY FUNDING
No targeted funding reported.
DISCLOSURE
A. Harvey and S. Mandelstam report no disclosures relevant to the manuscript. W. Maixner receives royalties from Springer-Verlag for the books Pediatric Hydrocephalus and Spina Bifida. R. Leventer, M. Semmelroch, D. MacGregor, R. Kalnins, Y. Perchyonok, and G. Fitt report no disclosures relevant to the manuscript. S. Barton was supported by RCH1000, a unique arm of The Royal Children's Hospital Foundation devoted to raising funds for research at The Royal Children's Hospital. M. Kean reports no disclosures relevant to the manuscript. G. Fabinyi is section editor of ANZ Journal of Surgery. G. Jackson has received honoraria from UCB Pharma and receives royalties from Elsevier for the book Magnetic Resonance in Epilepsy. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Barkovich AJ, Kuzniecky RI, Bollen AW, Grant PE. Focal transmantle dysplasia: a specific malformation of cortical development. Neurology 1997;49:1148–1152. [DOI] [PubMed] [Google Scholar]
- 2.Blümcke I, Thom M, Aronica E, et al. The clinicopathologic spectrum of focal cortical dysplasias: a consensus classification proposed by an ad hoc Task Force of the ILAE Diagnostic Methods Commission. Epilepsia 2011;52:158–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barkovich AJ, Guerrini R, Kuzniecky RI, Jackson GD, Dobyns WB. A developmental and genetic classification for malformations of cortical development: update 2012. Brain 2012;135:1348–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mühlebner A, Coras R, Kobow K, et al. Neuropathologic measurements in focal cortical dysplasias: validation of the ILAE 2011 classification system and diagnostic implications for MRI. Acta Neuropathol 2011;123:259–272. [DOI] [PubMed] [Google Scholar]
- 5.Shepherd C, Liu J, Goc J, et al. A quantitative study of white matter hypomyelination and oligodendroglial maturation in focal cortical dysplasia type II. Epilepsia 2013;54:898–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colombo N, Tassi L, Galli C, et al. Focal cortical dysplasias: MR imaging, histopathologic, and clinical correlations in surgically treated patients with epilepsy. AJNR Am J Neuroradiol 2003;24:724–733. [PMC free article] [PubMed] [Google Scholar]
- 7.Besson P, Andermann F, Dubeau F, Bernasconi A. Small focal cortical dysplasia lesions are located at the bottom of a deep sulcus. Brain 2008;131:3246–3255. [DOI] [PubMed] [Google Scholar]
- 8.Hofman PA, Fitt GJ, Harvey AS, Kuzniecky RI, Jackson G. Bottom-of-sulcus dysplasia: imaging features. AJR Am J Roentgenol 2011;196:881–885. [DOI] [PubMed] [Google Scholar]
- 9.Colombo N, Tassi L, Deleo F, et al. Focal cortical dysplasia type IIa and IIb: MRI aspects in 118 cases proven by histopathology. Neuroradiology 2012;54:1065–1077. [DOI] [PubMed] [Google Scholar]
- 10.Krsek P, Maton B, Korman B, et al. Different features of histopathological subtypes of pediatric focal cortical dysplasia. Ann Neurol 2008;63:758–769. [DOI] [PubMed] [Google Scholar]
- 11.Bronen RA, Vives KP, Kim JH, Fulbright RK, Spencer SS, Spencer DD. Focal cortical dysplasia of Taylor, balloon cell subtype: MR differentiation from low-grade tumors. AJNR Am J Neuroradiol 1997;18:1141–1151. [PMC free article] [PubMed] [Google Scholar]
- 12.Kuzniecky R, Morawetz R, Faught E, Black L. Frontal and central lobe focal dysplasia: clinical, EEG and imaging features. Dev Med Child Neurol 1998;37:159–166. [DOI] [PubMed] [Google Scholar]
- 13.Chassoux F, Rodrigo S, Semah F, et al. FDG-PET improves surgical outcome in negative MRI Taylor-type focal cortical dysplasias. Neurology 2010;75:2168–2175. [DOI] [PubMed] [Google Scholar]
- 14.Chassoux F, Landré E, Mellerio C, et al. Type II focal cortical dysplasia: electroclinical phenotype and surgical outcome related to imaging. Epilepsia 2012;53:349–358. [DOI] [PubMed] [Google Scholar]
- 15.Bernasconi A, Bernasconi N, Bernhardt BC, Schrader D. Advances in MRI for “cryptogenic” epilepsies. Nat Rev Neurol 2011;7:99–108. [DOI] [PubMed] [Google Scholar]
- 16.Wagner J, Weber B, Urbach H, Elger CE, Huppertz HJ. Morphometric MRI analysis improves detection of focal cortical dysplasia type II. Brain 2011;134:2844–2854. [DOI] [PubMed] [Google Scholar]
- 17.Mellerio C, Labeyrie MA, Chassoux F, et al. 3T MRI improves the detection of transmantle sign in type 2 focal cortical dysplasia. Epilepsia 2014;55:117–122. [DOI] [PubMed] [Google Scholar]
- 18.Palmini A, Gambardella A, Andermann F, et al. Intrinsic epileptogenicity of human dysplastic cortex as suggested by corticography and surgical results. Ann Neurol 1995;37:476–487. [DOI] [PubMed] [Google Scholar]
- 19.Chassoux F, Devaux B, Landre E, et al. Stereoelectroencephalography in focal cortical dysplasia: a 3D approach to delineating the dysplastic cortex. Brain 2000;123:1733–1751. [DOI] [PubMed] [Google Scholar]
- 20.Tassi L, Colombo N, Garbelli R, et al. Focal cortical dysplasia: neuropathological subtypes, EEG, neuroimaging and surgical outcome. Brain 2002;125:1719–1732. [DOI] [PubMed] [Google Scholar]
- 21.Aubert S, Wendling F, Régis J, et al. Local and remote epileptogenicity in focal cortical dysplasias and neurodevelopmental tumours. Brain 2009;132:3072–3086. [DOI] [PubMed] [Google Scholar]
- 22.Varotto G, Tassi L, Franceschetti S, Spreafico R, Panzica F. Epileptogenic networks of type II focal cortical dysplasia: a stereo-EEG study. Neuroimage 2012;61:591–598. [DOI] [PubMed] [Google Scholar]
- 23.Marusic P, Najm IM, Ying Z, et al. Focal cortical dysplasias in eloquent cortex: functional characteristics and correlation with MRI and histopathologic changes. Epilepsia 2002;43:27–32. [DOI] [PubMed] [Google Scholar]
- 24.Burneo JG, Kuzniecky RI, Bebin M, Knowlton RC. Cortical reorganization in malformations of cortical development: a magnetoencephalographic study. Neurology 2004;63:1818–1824. [DOI] [PubMed] [Google Scholar]
- 25.Sarkis RA, Jehi LE, Bingaman WE, Najm IM. Surgical outcome following resection of rolandic focal cortical dysplasia. Epilepsy Res 2010;90:240–247. [DOI] [PubMed] [Google Scholar]
- 26.Barba C, Montanaro D, Frijia F, et al. Focal cortical dysplasia type IIb in the rolandic cortex: functional reorganization after early surgery documented by passive task functional MRI. Epilepsia 2012;53:e141–e145. [DOI] [PubMed] [Google Scholar]
- 27.Widdess-Walsh P, Kellinghaus C, Jeha L, et al. Electro-clinical and imaging characteristics of focal cortical dysplasia: correlation with pathological subtypes. Epilepsy Res 2005;67:25–33. [DOI] [PubMed] [Google Scholar]
- 28.Lawson JA, Birchansky S, Pacheco E, et al. Distinct clinicopathologic subtypes of cortical dysplasia of Taylor. Neurology 2005;64:55–61. [DOI] [PubMed] [Google Scholar]
- 29.Krsek P, Maton B, Jayakar P, et al. Incomplete resection of focal cortical dysplasia is the main predictor of poor postsurgical outcome. Neurology 2009;72:217–223. [DOI] [PubMed] [Google Scholar]
- 30.Krsek P, Pieper T, Karlmeier A, et al. Different presurgical characteristics and seizure outcomes in children with focal cortical dysplasia type I or II. Epilepsia 2009;50:125–137. [DOI] [PubMed] [Google Scholar]
- 31.Lerner JT, Salamon N, Hauptman JS, et al. Assessment and surgical outcomes for mild type I and severe type II cortical dysplasia: a critical review and the UCLA experience. Epilepsia 2009;50:1310–1335. [DOI] [PubMed] [Google Scholar]
- 32.Wang DD, Deans AE, Barkovich AJ, et al. Transmantle sign in focal cortical dysplasia: a unique radiological entity with excellent prognosis for seizure control. J Neurosurg 2013;118:337–344. [DOI] [PubMed] [Google Scholar]
- 33.Rowland NC, Englot DJ, Cage TA, Sughrue ME, Barbaro NM, Chang EF. A meta-analysis of predictors of seizure freedom in the surgical management of focal cortical dysplasia. J Neurosurg 2012;116:1035–1041. [DOI] [PubMed] [Google Scholar]
- 34.Salamon N, Kung J, Shaw SJ, et al. FDG-PET/MRI coregistration improves detection of cortical dysplasia in patients with epilepsy. Neurology 2008;71:1594–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fauser S, Huppertz HJ, Bast T, et al. Clinical characteristics in focal cortical dysplasia: a retrospective evaluation in a series of 120 patients. Brain 2006;129:1907–1916. [DOI] [PubMed] [Google Scholar]
- 36.Kral T, von Lehe M, Podlogar M, et al. Focal cortical dysplasia: long term seizure outcome after surgical treatment. J Neurol Neurosurg Psychiatry 2007;78:853–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Palmini A, Andermann F, Olivier A, et al. Focal neuronal migration disorders and intractable partial epilepsy: a study of 30 patients. Ann Neurol 1991;30:741–749. [DOI] [PubMed] [Google Scholar]
- 38.Sisodiya SM, Fauser S, Cross JH, Thom M. Focal cortical dysplasia type II: biological features and clinical perspectives. Lancet Neurol 2009;8:830–843. [DOI] [PubMed] [Google Scholar]
- 39.Wellmer J, Kopitzki K, Voges J. Lesion focused stereotactic thermo-coagulation of focal cortical dysplasia IIB: a new approach to epilepsy surgery? Seizure 2014;23:475–478. [DOI] [PubMed] [Google Scholar]
- 40.Carne RP, O'Brien TJ, Kilpatrick CJ, et al. MRI-negative PET-positive temporal lobe epilepsy: a distinct surgically remediable syndrome. Brain 2004;127:2276–2285. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.