

Abstract
Joubert syndrome and related disorders comprise a subgroup of ciliopathies defined by the presence of the ‘molar tooth sign’, a midbrain-hindbrain malformation identifiable by neuroimaging. Characteristically, the corticospinal tract and superior cerebellar peduncles do not decussate. Epileptic seizures are uncommon. We present a case of a 28-year-old man with a background of Leber's congenital amaurosis with nephronophthisis, requiring kidney transplantation, and mental retardation, who developed epileptic seizures consisting of a short muffled cry and involuntary shaking movements of the extremities beginning in the left upper limb; these episodes lasted several seconds and occurred in clusters. Simultaneous video-EEG recording showed an ictal pattern in the left frontal lobe. Brain MRI revealed the pathognomonic ‘molar tooth sign’; diffusion tensor imaging (DTI)-tractography showed a lack of decussation of both corticospinal tracts. To the best of our knowledge, this is the first time that DTI-tractography has been used to uncover the anatomical substrate underlying the semiology of epileptic seizures.
Background
Originally, Joubert syndrome (JS) was described as a neurodevelopmental genetic disorder characterised by three primary findings: a midbrain-hindbrain malformation known as the ‘molar tooth sign’ (MTS); hypotonia with subsequent development of ataxia; and mental retardation. Additional clinical features in the past (hepatic fibrosis, ocular coloboma, retinal dystrophy, polydactyly, cystic kidney disease and nephronophthisis), contained in variants of classical JS, are now considered part of a clinical spectrum and classified under the wider term JS and related disorders (JSRD).1
The prevalence has been reported to be from 1/100 000 up to 1/80 000 newborns, though some authors believe it is underestimated. Autosomal recessive and X linked patterns of inheritance have been described. JSRD are caused by abnormalities in genes encoding for proteins of the primary cilium or its apparatus, resulting in loss of function. A total of 21 causative genes have been identified so far, accounting for about half of all cases.2 The genes involved in the remaining cases are yet to be identified. An algorithm for molecular genetic testing has been proposed, taking into account certain phenotypical characteristics.3
The diagnosis of JSRD is based on clinical and MRI findings. The mandatory neuroradiological hallmark of JSRD is the complex malformation at the midbrain–hindbrain junction called the MTS, characterised by aplasia or marked hypoplasia of the cerebellar vermis, elongation and thinning of the pontomesencephalic junction, thickened and elongated superior cerebellar peduncles and a deepened interpeduncular fossa. Neuropathological findings have also shown a complete or partial lack of decussation of the superior cerebellar peduncles as well as the corticospinal tracts at the medullary pyramids. About 13% of those patients show additional brain abnormalities such as hamartomas, periventricular nodular heterotopia, polymicrogyria, white matter cysts or cystic enlargement of the posterior fossa.
Clinically, JSRD is associated with neonatal breathing disorders, hypotonia evolving to ataxia, oculomotor apraxia, developmental delay, mental retardation and autistic features. Epileptic seizures are not a frequent symptom (3%) and are normally associated with cortical organisation defects.
Even though the clinical features of the syndrome are identifiable at birth, the diagnosis is often delayed several months or years due to the rareness of the disorder.
Case presentation
A 27-year-old man presented to the neurology department with a seven-day history of recurrent, stereotyped and paroxysmal episodes consisting of a short muffled cry and right oculocephalic deviation followed by flexion and involuntary shaking movements of the extremities, beginning in the left arm. The patient was able to respond immediately after. These episodes lasted an average of 8 s and occurred in clusters of 10–15 episodes over a 1 h period. The patient suffered from two or three clusters per day.
The patient displayed severe intellectual disability due to developmental delay. He had been diagnosed with Leber's congenital amaurosis, agenesia of the cerebellar vermis and nephronopthisis, the latter requiring kidney transplantation when the patient was 22 years of age. He was the only child of non-consanguineous parents, and none of his relatives up to the third degree had any relevant medical disorder. Usual medication included prednisone, sirolimus and mycophenolate mofetil as immunosuppressant treatment, as well as an ACE inhibitor and risperidone. The patient lived with his parents and required considerable help to carry out his daily activities.
Investigations
Blood and urine tests were unremarkable except for creatinine that was slightly above normal range but within the mean levels of the patient. Immunosuppressant drug blood levels were all within normal limits. A brain MRI was performed (figure 1), which showed an elongation and thinning of the pontomesencephalic junction, an anomalous configuration of the superior cerebellar peduncles and agenesia of the cerebellar vermis resulting in the MTS. No cortical malformation or migration defects were observed.
Figure 1.
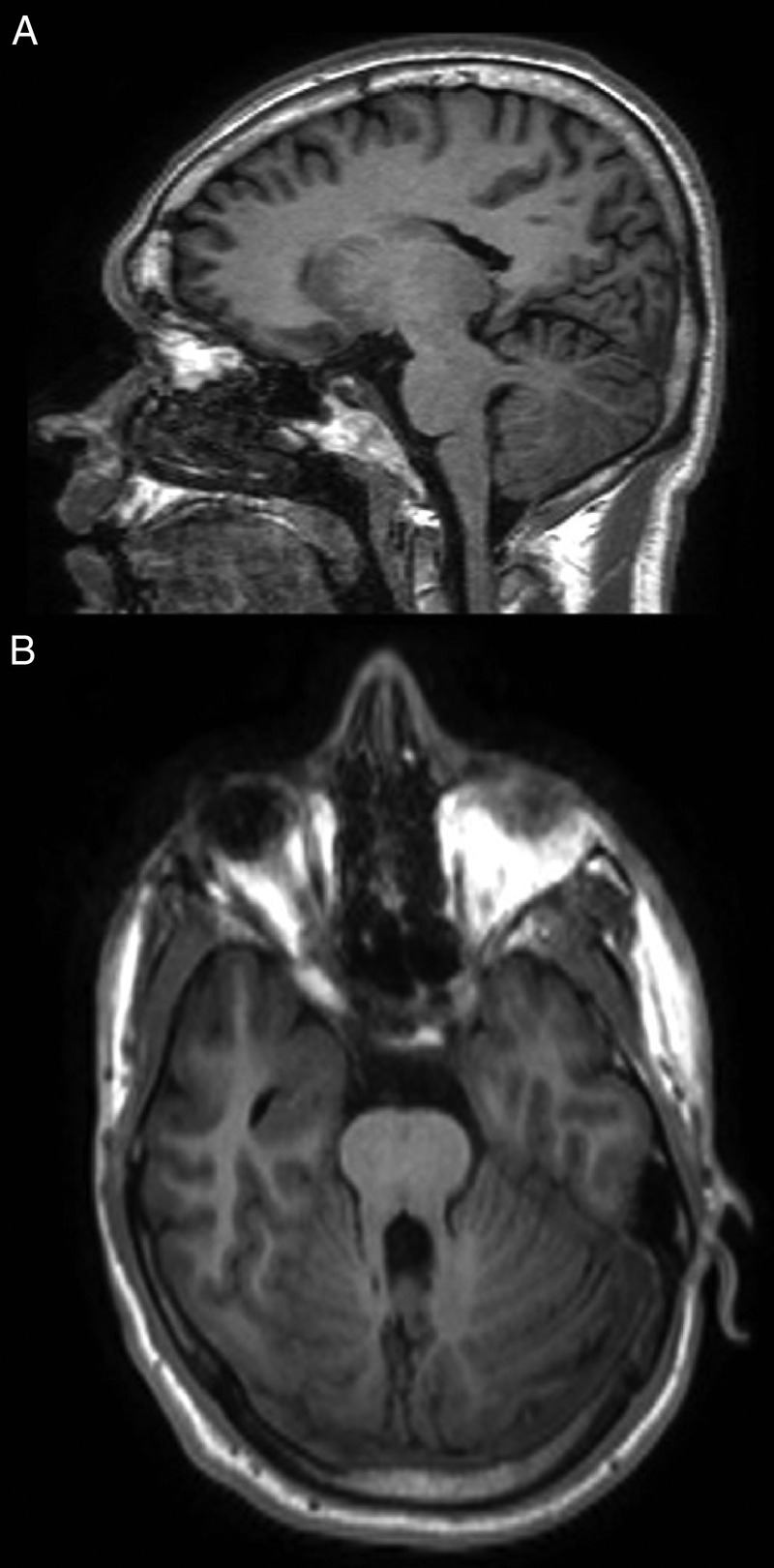
(A) Sagittal MRI T1-weighted images through the cerebellum and brain stem of the patient, depicting the horizontalisation of the superior cerebellar peduncles and other hindbrain anomalies. (B) Note the ‘molar tooth sign’ on the axial plane.
Twenty-four-hour video-EEG (VEEG) monitoring was performed, recording several of the patient's typical events. These episodes consisted of forced mouth opening, cry emission and left hand shaking. The shaking promptly spread to all four limbs and had a duration of less than 10 s (video 1 sequence). The simultaneous EEG showed a generalised flattening lasting 1–2 s followed by rhythmic δ activity in the left frontal area (figure 2). Interictal recordings showed spikes in the left central frontal region (maximum F3). Considering that JS was suspected and that the clinical symptomatology was ipsilateral to the ictal epileptiform activity seen in the VEEG (in the left frontal lobe), DTI-tractography was performed to reveal the disposition of the pyramidal tract. This technique highlighted a lack of decussation of the corticospinal tract at the medullary pyramids. Left hemispheric motor cortical efferences were aimed to the left side of the body, explaining the inconsistency between the ictal semiology and the VEEG findings.
Figure 2.

EEG recording showing a diffused flattening followed by rhythmic delta activity in the left frontal area. Sensitivity 7 uV/mm, high frequency filter 70 Hz, low frequency filter 1 Hz, Notch 50 Hz, time base 30 mm/s. (B and C) Left frontal spike (*) shown in bipolar (B) and referential (C) montage.
Video 1.
Audiovisual and EEG recordings of the typical seizures our patient presented with. Note that the limb shaking begins in the left arm and promptly propagates to the whole body, coincidentally with the left frontal spike after the diffuse flattening of the amplitude. A brief neurological exam follows, showing that the patient is not post-critical or intellectually impaired after the seizure. EEG has the visualisation settings described in Figure 2.
Treatment
The patient underwent several unsuccessful trials with clonazepam (10 mg/day), levetiracetam (2000 mg/day) and oxcarbazepine (600 mg/day) (dosage limited by kidney function and side effects). Finally, a combination of valproic acid (1500 mg/day), lacosamide (200 mg/day) and lamotrigine (200 mg/day) was effective in obtaining adequate control of the seizures.
Discussion
We present an unusual case of JSRD presenting with epileptic seizures, a rare manifestation of this syndrome. By means of DTI-tractography, we were able to establish the anatomical-clinical-electroencephalographic correlation of the seizures.
JSRD is a rare group of congenital disorders included in the expanding family of ciliopathies. The pathogenic basis of this disorder relates to the dysfunction of the primary cilium, a subcellular organelle involved in signalling pathways essential to cellular development, organogenesis and left-right axis formation. The ubiquity of this organelle sets the rationale for multiorgan involvement. Retinal dysfunction (from Leber congenital amaurosis to slowly progressive retinopathies) and renal defects (nephronophthisis) are some of the most common, both of which were present in our patient.3
Epilepsy occurs rarely, in 3% of all cases,1 and then too normally in association with central nervous system malformations such as hamartomas, periventricular nodular heterotopia or cortical organisation defects. These malformations are sometimes undetected in conventional neuroimaging; it is plausible that our patient’s epilepsy could arise from any of these structural anomalies, even if brain MRI revealed none.
The stereotypical and paroxysmal episodes beginning with a short supraglottic cry and involuntary shaking of the left arm were suggestive of seizures with a possible onset in the right frontal lobe. However, the simultaneous VEEG register showed that these episodes were preceded by a generalised attenuation followed by rhythmic delta activity in the left frontal region. The interictal EEG also highlighted persistent left frontal spikes with normal background activity. Taking this into account, we posited that the discrepancy between clinical symptoms and VEEG findings could be explained by a non-decussating corticospinal tract, a frequent finding in JSRD. The DTI-tractography, a powerful tool that maps in vivo axonal fibre tracts unidentifiable by conventional MRI, confirmed this hypothesis by showing a complete lack of decussation of the corticospinal tract at the medullary pyramids (figure 3) as well as an absence of crossing of the superior cerebellar peduncles (figure 4).
Figure 3.
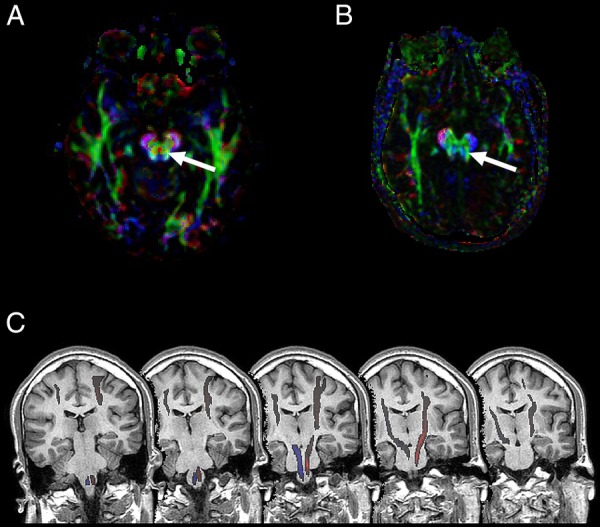
(A) Diffusion tensor imaging (DTI)-tractography showing normal decussation of the corticospinal tract as a red dot between the cerebral pyramids (arrow). (B) DTI-tractography of our patient, showing the absence of decussation. Colours represent tract orientation in space; red, green and blue represent the x, y and z-axis, respectively. (C) DTI-tractography over T1-weighted sequential coronal planes of the brainstem, illustrating lack of decussation at the medullar pyramids.
Figure 4.
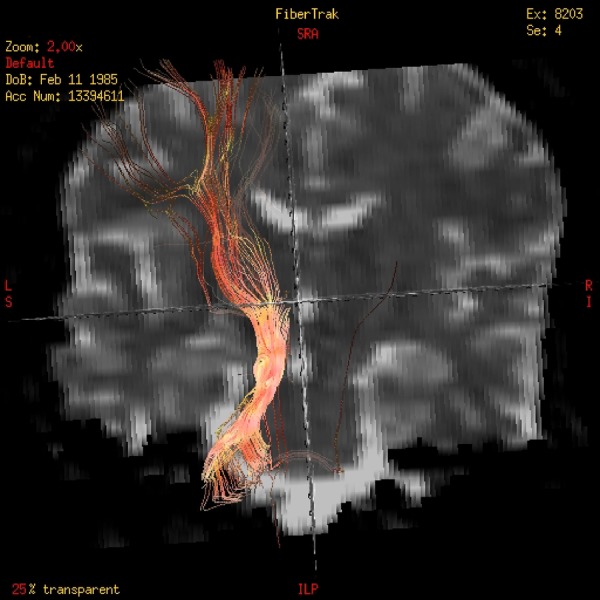
Diffusion tensor imaging (DTI)-tractography reconstruction of the tracts showing the lack of decussation of the superior cerebellar peduncle.
Complete and incomplete lack of decussation of both tracts has already been documented by different investigators using DTI-tractography.4–6 This defect in axon guidance is unique to JSRD and its presence, demonstrated by DTI-tractography, can be helpful in differentiating it from other ciliopathies or similar midbrain-hindbrain malformations.7
To the best of our knowledge, this is the first time this method has been used in a clinical setting as a means of explaining the anatomy underlying the semiology of epileptic seizures. No standard of practice exists for the management of seizures in these patients due to the insufficient number of cases reported, although antiepileptic drugs should be tailored with regard to kidney or liver function deficits.
Learning points.
Joubert syndrome is a rare disorder that can only be diagnosed through neuroimaging.
Under specific anatomical conditions, apparently false lateralising cloni can arise from the ipsilateral motor cortex.
Diffusion tensor imaging-tractography may provide insight into the anatomical substrate of seizure semiology.
Acknowledgments
The authors thank Miguel Yus Fuertes and Manuela Jorquera Moya (Radiology Department)—acquisition and interpretation of radiological studies.
Footnotes
Contributors: PLR taken care of the patient, planning, conducting and drafting of the manuscript, analysis of data. MEGG taken care of the patient, planning, conducting and drafting of the manuscript, analysis and acquisition of data. DGD was involved in planning and drafting of the manuscript, analysis and acquisition of data. AM-D taken care of the patient, final approval and revision of the manuscript.
Competing interests: None declared.
Patient consent: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Brancati F, Dallapiccola B, Valente EM. Joubert syndrome and related disorders. Orphanet J Rare Dis 2010;5:20 10.1186/1750-1172-5-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romani M, Micalizzi A, Valente EM. Joubert syndrome: congenital cerebellar ataxia with the molar tooth. Lancet Neurol 2013;12:894–905. 10.1016/S1474-4422(13)70136-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 2009;151C:326–40. 10.1002/ajmg.c.30229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poretti A, Boltshauser E, Loenneker T et al. Diffusion tensor imaging in Joubert syndrome. AJNR Am J Neuroradiol 2007;28:1929–33. 10.3174/ajnr.A0703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Poretti A, Meoded A, Rossi A et al. Diffusion tensor imaging and fiber tractography in brain malformations. Pediatr Radiol 2013;43:28–54. 10.1007/s00247-012-2428-9 [DOI] [PubMed] [Google Scholar]
- 6.Widjaja E, Blaser S, Raybaud C. Diffusion tensor imaging of midline posterior fossa malformations. Pediatr Radiol 2006;36:510–17. 10.1007/s00247-006-0146-x [DOI] [PubMed] [Google Scholar]
- 7.Poretti A, Singhi S, Huisman TAGM et al. Tecto-cerebellar dysraphism with occipital encephalocele: not a distinct disorder, but part of the Joubert syndrome spectrum? Neuropediatrics 2011;42:170–4. 10.1055/s-0031-1287763 [DOI] [PubMed] [Google Scholar]