

ABSTRACT
Highly pathogenic H5N1 avian influenza viruses are associated with severe disease in humans and continue to be a pandemic threat. While vaccines are available, other approaches are required for patients that typically respond poorly to vaccination, such as the elderly and the immunocompromised. To produce a therapeutic agent that is highly efficacious at low doses and is broadly specific against antigenically drifted H5N1 influenza viruses, we developed two neutralizing monoclonal antibodies and combined them into a single bispecific Fc fusion protein (the Fc dual-affinity retargeting [FcDART] molecule). In mice, a single therapeutic or prophylactic dose of either monoclonal antibody at 2.5 mg/kg of body weight provided 100% protection against challenge with A/Vietnam/1203/04 (H5N1) or the antigenically drifted strain A/Whooper swan/Mongolia/244/05 (H5N1). In ferrets, a single 1-mg/kg prophylactic dose provided 100% protection against A/Vietnam/1203/04 challenge. FcDART was also effective, as a single 2.5-mg/kg therapeutic or prophylactic dose in mice provided 100% protection against A/Vietnam/1203/04 challenge. Antibodies bound to conformational epitopes in antigenic sites on the globular head of the hemagglutinin protein, on the basis of analysis of mutants with antibody escape mutations. While it was possible to generate escape mutants in vitro, they were neutralized by the antibodies in vivo, as mice infected with escape mutants were 100% protected after only a single therapeutic dose of the antibody used to generate the escape mutant in vitro. In summary, we have combined the antigen specificities of two highly efficacious anti-H5N1 influenza virus antibodies into a bispecific FcDART molecule, which represents a strategy to produce broadly neutralizing antibodies that are effective against antigenically diverse influenza viruses.
IMPORTANCE Highly pathogenic H5N1 avian influenza viruses are associated with severe disease in humans and are a pandemic threat. A vaccine is available, but other approaches are required for patients that typically respond poorly to vaccination, such as the elderly and the immunocompromised. The variability of the virus means that such an approach must be broad spectrum. To achieve this, we developed two antibodies that neutralize H5N1 influenza viruses. In mice, these antibodies provided complete protection against a spectrum of H5N1 influenza viruses at a single low dose. We then combined the two antibodies into a single molecule, FcDART, which combined the broad-spectrum activity and protective efficacy of both antibodies. This treatment provides a novel and effective therapeutic agent or prophylactic with activity against highly pathogenic H5N1 avian influenza viruses.
INTRODUCTION
Since 2003, a series of events that many believe to be the harbinger of an incipient influenza pandemic has occurred in Asia. The virus responsible for these events, an H5N1 influenza virus, has infected and killed millions of chickens and ducks and infected in excess of 650 people (www.who.int; reviewed in reference 1). Fortunately, this virus has yet to gain the ability to transmit from human to human; however, mammalian transmissibility can be conferred experimentally with relatively few mutations (2, 3). The virulence of this virus (4, 5), its wide geographic distribution (6), and the potential for human aerosol transmission (2, 3) urge the development of targeted therapeutic and prophylactic measures. Currently, two H5N1 vaccines, one of which is available worldwide, have been licensed, and these offer the best means of mass protection (reviewed in reference 7). However, it is likely that the first line of defense against an emerging pandemic will require additional options, including antivirals. Currently, the two options for treating influenza are the M2 ion channel blockers (e.g., amantadine) and the neuraminidase inhibitors (e.g., oseltamivir). Unfortunately, many of the current H5N1 viruses are resistant to amantadine (6), and for oseltamivir a dose higher than that used previously and a treatment schedule longer than that used previously are needed for protection in murine models (8), providing proof that new therapeutic options are very much needed.
Monoclonal antibodies (MAbs) against the influenza virus hemagglutinin (HA) protein have shown efficacy as therapeutic agents and prophylactics against H5N1 influenza virus infection in murine challenge models (9–13), and in mice, other antibodies have also shown efficacy against other strains of influenza viruses, including H1N1, H2N2, and H3N2 (14–19). The dosages at which these antibodies provided 100% protection against challenge in mice ranged from 5 to 50 mg/kg of body weight when delivered therapeutically and 1 to 30 mg/kg when delivered prophylactically. In ferrets, an anti-H5 antibody provided 100% protection against challenge with an H5N1 influenza virus when it was delivered at 10 mg/kg prophylactically and at 30 mg/kg therapeutically (20).
The variability of the HA protein, which contains the main antigenic sites of the virus, necessitates that any broadly utilized antibody be effective against antigenically diverse influenza viruses. The most conserved regions of HA are the stalk and the receptor-binding pocket, and antibodies directed at these sites are efficacious against antigenically diverse strains of influenza virus (14, 21, 22). However, the most exposed and antibody-accessible epitopes on HA are located on the globular head, although these regions are highly variable. Due to the variability of HA, it is likely that more than one neutralizing antibody will be required to provide broad protection against different lineages of H5N1 and to protect against antibody escape mutants. However, this presents certain problems for clinical testing and product development, as cocktails of antibodies are difficult and expensive to produce and test. Therefore, in this study we have developed a bispecific Fc fusion protein, the Fc dual-affinity retargeting (FcDART) molecule, that combines the neutralizing capacity and specificities of two different neutralizing monoclonal antibodies from human and murine sources that target the globular head of HA. Individually, the two monoclonal antibodies provided 100% protection against A/Vietnam/1203/04 (H5N1) (VN1203) challenge in mice and ferrets when they were used as therapeutic agents and prophylactics at low doses (2.5 mg/kg in mice and 1 mg/kg in ferrets). They also provided in mice 100% protection against challenge with A/Whooper swan/Mongolia/244/05 (H5N1) (Mon244), which is an antigenically drifted strain. Further, antibody escape mutants generated in vitro were not lethal in mice treated with a single therapeutic dose of antibody. The FcDART molecule that combines the antigen specificities of these two antibodies also provided 100% protection against challenge in mice when it was used as a therapeutic agent or prophylactic, and the strategy used to produce the FcDART molecule may be used to produce antibody-based therapeutic agents that are effective against antigenically diverse influenza viruses.
MATERIALS AND METHODS
Cell lines and culture conditions.
293T, Chinese hamster ovary (CHO), and Madin-Darby canine kidney (MDCK) cell lines were obtained from ATCC. 293T cells were grown in Opti-MEM medium (Invitrogen, CA) supplemented with 5% fetal calf serum (FCS; Gemini Bioproducts Inc., CA), CHO cells were grown in F12K medium (Invitrogen, CA) supplemented with 10% FCS, and MDCK cells were grown in Dulbecco's modified minimal essential medium (DMEM; Invitrogen, CA) supplemented with 10% FCS and 2 mM glutamine. For MDCK cell infections, viruses were diluted in infection medium (minimal essential medium supplemented with 5% (vol/vol) bovine serum albumin (BSA) and 2 mM glutamine (Sigma, MO).
Immunofluorescence assay (IFA).
MDCK cells were infected with VN1203 or Mon244 overnight. The cells were harvested by trypsinization and resuspended in phosphate-buffered saline (PBS) with 2% FCS. Aliquots containing 3 × 104 cells were spotted onto HTC Super Cured 24-spot slides (Erie Scientific Company, NH), dried, and fixed with 100% acetone for 10 min at room temperature. Fixed cells were incubated with hybridoma supernatants for 30 min at 37°C and washed for 5 min with PBS. The slides were then incubated for 30 min at 37°C with 50 ng/ml propidium iodide and fluorescein isothiocyanate (FITC)-conjugated goat anti-human IgG (Jackson ImmunoResearch Laboratories, West Grove, PA). Bound antibody was revealed by fluorescence microscopy.
HI assays.
Hemagglutination inhibition (HI) assays were conducted using standard methodologies. In brief, 25 μl of diluted antigen at four agglutination doses in PBS was added to wells of 96-well plates containing a 2-fold dilution series of the test antibody. After 30 min incubation at room temperature, 50 μl of 0.5% (vol/vol) chicken or horse red blood cells was added to each well and the plates were incubated at room temperature for another 30 min. Titers were recorded as the lowest dilution of antibody able to inhibit hemagglutination.
MN assays.
Microneutralization (MN) assays were conducted using MDCK cells according to standard methodologies. In brief, a 2-fold dilution series of each antibody was incubated with virus at 100 50% tissue culture infective doses (TCID50s)/50 μl for 1 h at 37°C. The antibody-virus solutions were then added to MDCK cells for an additional hour at 37°C and were then washed off, and the cells were incubated at 37°C for 72 h with 200 μl infection medium containing 1 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin. Neutralizing titers were read by incubating 50 μl of cell culture medium with 0.5% (vol/vol) chicken or horse red blood cells, followed by incubation at room temperature for another 30 min, and were expressed as the reciprocal of the serum dilution that inhibited 50% of the growth of 100 TCID50s of virus.
HMAb generation.
Plasma samples were obtained from individuals immunized with a recombinant, baculovirus-expressed HA protein from the A/Hong Kong/156/97 (H5N1) (HK156) virus (23), followed by the inactivated, subvirion vaccine against VN1203 (24). Approval for research involving human subjects was obtained from the Institutional Review Boards of Stanford University and St. Jude Children's Research Hospital. Samples were screened by HI or by IFA using H5N1 (VN1203) HA protein or H5N1-infected MDCK cells, respectively. In this study, a donor with a serum HI titer of 320 was used as the source of peripheral B cells for human hybridoma production, as previously described (25). Three hybridomas, designated BF1-1, BF1-10, and BF1-19, secreting neutralizing H5N1 human monoclonal antibodies (HMAbs) were identified by an HI assay. When they were tested against a panel of Southeast Asian H5N1 isolates, these three hybridomas showed reactivity to the antigenically distinct H5N1 virus HK156 but not to the clade 2.3.4 virus A/Common magpie/Hong Kong/645/06. BF1-32 was identified by IFA. Monoclonality was confirmed by sequencing the IgG genes isolated from 10 individual cell clones derived from each hybridoma cell line. Cloning and analysis of the variable light (VL) domain and the variable heavy (VH) domain of these clones were performed as previously described (26). HMAb production and purification were performed as described previously (25), and biotinylation of the antibodies was carried out according to the manufacturer's instructions (Pierce Biotechnology, Inc., Rockford, IL). The cell lines produced IgG1 antibody with the λ light chain for BF1-1 and κ light chains for BF1-10, BF1-19, and BF1-32 and secreted 10 to 40 μg human IgG per ml in the spent culture supernatant.
mMAb generation.
Generation of mouse monoclonal antibodies (mMAbs) was achieved via a vaccination regimen consisting of an initial 50% mouse lethal dose (MLD50) of 0.5 of a virus, generated by reverse genetics, bearing the HA and neuraminidase genes of VN1203 and the six internal genes of A/Puerto Rico/8/1934 (H1N1) (rgH5N1) delivered intranasally (i.n.), followed by two booster doses delivered intraperitoneally (i.p.) 4 weeks apart. Booster doses consisted of 10,000 hemagglutination units (HAU) of Mon244 delivered i.p., followed by 1,000 50% egg infectious doses (EID50s) of rgH5N1 delivered intravenously (i.v.). After vaccination, hybridomas were produced by Rockland Immunochemicals and were screened by the HI assay and IFA as described above.
Humanization of mMAbs.
mMAbs were humanized by first obtaining cDNA of the VH and VL segments from total RNA via 3′ rapid amplification of cDNA ends, followed by cloning into the pCR2.1-Topo vector (Life Technologies), which was then transformed into Escherichia coli, recovered, and sequenced. The VH and VL segments were also combined with human Cg1 and K constant region cDNA segments in the mammalian expression vectors pEE13.4 (light chain [LC]) and pEE6.4 (heavy chain [HC]) (Lonza) and then combined into a single expression vector, and the resulting chimeric HC and LC were then transfected into CHO-S cells (Life Technologies). Conditioned medium from transfected cells was assayed by enzyme-linked immunosorbent assay (ELISA) for human IgG production and anti-H5N1 activity. Human germ line genes with the highest homology to the VH and VL segments were identified using the Ig BLAST program, and humanized VH and VL segments were then designed using complementarity-determining region (CDR) grafting; VH1-18/VH1-69 was used for VH segment frameworks and VK-O12 was used for VL segment frameworks for the humanization of 10C3. Humanized VH and VL segments were then synthesized by gene synthesis (GenScript). The resulting fragments were then cloned into the mammalian expression vector pCIneo (Promega) with the appropriate C region, and clones were sequenced.
Generation of FcDART.
The antigen specificities of BF1-19 and humanized 10C3 (h10C3) were combined into a bispecific Fc fusion protein, termed FcDART. This was achieved by joining the two antibody chains such that each chain of FcDART comprised the VL segment from one antibody and the VH segment of the second antibody and vice versa, with a short linker (GGGSGGG) being placed between the VL and VH segments. One of the two chains was then fused to a human IgG1 hinge-Fc region (Fig. 1A). The two chains were transfected into CHO cells, and a stable pool of producing cells was selected. The material was purified over a protein A column, followed by size exclusion chromatography (Fig. 1B).
FIG 1.
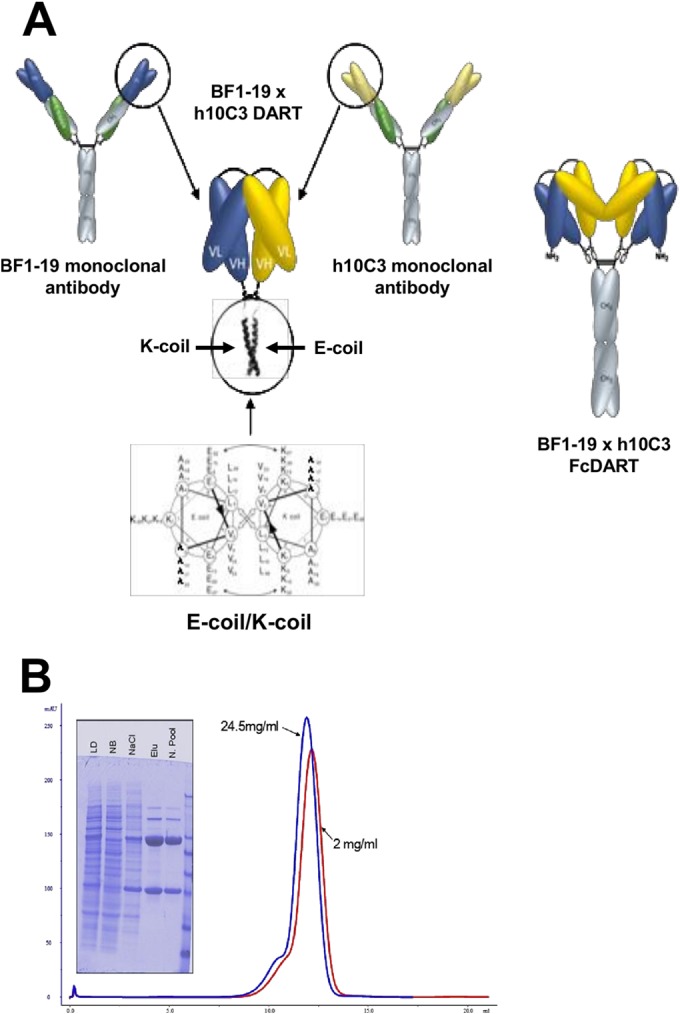
Structure and characterization of FcDART. (A) The VH and VL segments of monoclonal antibodies h10C3 and BF1-19 (yellow and blue, respectively) were combined into a single bispecific Fc fusion protein, FcDART. Shown in detail are the heterodimerization domains of FcDART, which were appended to the C terminus of each chain to promote heterodimeric assembly. These domains consisted of coiled coil-forming sequences where the hydrophobic core of the coiled coil interface is flanked by opposite charges (an E negative charge on one coil [E-coil] and a K positive charge on the other [K-coil]), which is believed to favor heterodimerization by repelling a homotypic association and preventing the initiation of zippering of the hydrophobic core. An Fc domain was also added to the C terminus of the coil with an E negative charge, causing two DART units to dimerize and form a tetravalent bispecific structure. (B) Characterization of FcDART by denaturing, reducing SDS-PAGE and size exclusion chromatography analysis. LD, load fraction on the protein A column; NB, nonbound fraction on the protein A column; NaCl, high-salt wash; Elu, material that eluted at low pH; N. Pool, material that eluted at low pH and that was neutralized. The size exclusion chromatography profiles of purified material at two concentrations (red, 2 mg/ml; blue, 24.5 mg/ml) show peak elution at about 12 ml. The slight difference in the peak elution volume is within the range of variability of manual loading and does not indicate a significant elution volume difference between the two samples.
Affinity measurement.
The affinity of binding of each MAb to H5N1 HA was characterized by surface plasmon resonance (SPR) analysis. The experiments described below were carried out using a BIAcore 3000 system (Biacore). Affinity-purified anti-human IgG Fcγ (Jackson ImmunoResearch) was amine coupled (10,000 relative units [RU]) to all four flow cells of a CM5 sensor chip (Biacore). MAb samples at 0.5 to 1 μg/ml in running buffer were injected into flow cells 2 to 4, while flow cell 1 was kept as a reference. Injection of MAb samples led to the capture of roughly 150 RU of MAb in each flow cell. Association kinetics were observed by injecting purified H5N1 HA (Protein Sciences) in running buffer through all four flow cells at a flow rate of 30 μl/min for a duration of 90 s. Disassociation kinetics were then measured with running buffer flowing at a rate of 30 μl/min for a duration of 10 min. Flow cells were regenerated using a 15-μl injection of 100 mM H3PO4. Kinetics were tested at HA concentrations of 800, 400, 200, 100, 50, 25, 12.5, 6.25, 3.125, and 1.56 nM. After each binding cycle, the same protocol was run with running buffer substituted for the analyte to serve as a double reference. Data were analyzed using BIAevaluation software, version 4.1 (Biacore). The data were processed by subtracting the result for the double reference and trimming out the MAb loading and H3PO4 regeneration information. Kinetic values were obtained by applying the 1:1 (Langmuir) binding, simultaneous ka (association rate)/kd (dissociation rate) fit method.
Antibody binding profiles.
The direct ELISA format (the antigen down format) or the capture ELISA format (the antibody down format) was used to measure antibody binding to HA from different clades of H5N1 influenza viruses. In the antigen down format, HA protein was coated on an ELISA plate at a concentration of 2 μg/ml. HA protein was obtained from eEnzyme and Protein Sciences and was produced in 293T cells and an insect cell line, respectively. HA was uncleaved or cleaved at the furin cleavage site (Δfurin HA). The concentration of antibody indicated in Fig. 3 was then added to the plate, and detection was performed using a horseradish peroxidase (HRP)-conjugated antihuman antibody, followed by chemiluminescence detection (SuperSignal; Pierce). For the assays performed in an MAb down format, an antibody specific for CH1 (MAb 4A11; Abcam) was used to coat an ELISA plate at 2 μg/ml. The concentration of antibody indicated below was then captured. His-tagged antigen (100 ng/ml) was then applied to the plate, followed by application of an HRP-conjugated anti-His antibody and chemiluminescence detection (SuperSignal; Pierce). To detect binding of h10C3, this assay was performed in an antibody down format to maximize sensitivity using antibody captured with polyclonal anti-human IgG, and antigen binding was detected with streptavidin-HRP and developed colorimetrically with 3,3′,5,5′-tetramethylbenzidine (TMB; Sigma).
FIG 3.

Antibody binding to a hemagglutinin panel from diverse H5N1 influenza virus clades. Binding was assessed in an antigen down format, where HA was immobilized on a plate (A, C), or in an MAb down format, where the MAb was immobilized on a plate (B, D, E). Binding of the FcDART molecule (F) was measured in an antibody down format, with FcDART being captured with anti-human IgG Fc-specific polyclonal antibody. OD, optical density.
Prevention and therapeutic treatment in animals.
All animal studies were conducted under the applicable laws and guidelines of and after approval from the St. Jude Children's Research Hospital Animal Care and Use Committee. Female 6- to 8-week old BALB/c mice were housed in cages with 5 mice per cage. Mice received the amount of antibody indicated below in a total volume of 300 μl sterile PBS by i.p. injection. At various times before and after antibody administration, the mice were lightly anesthetized with isoflurane and inoculated intranasally with 100 MLD50s of VN1203 (1,000 50% EID50s) or 100 MLD50s (5 × 105 EID50s) of Mon244 in 30 μl of PBS.
Male 4- to 5-month-old ferrets were housed in cages with three animals per cage and were administered 1 mg/kg of the antibodies indicated below in 1 ml PBS intramuscularly, and on the next day the ferrets were lightly anesthetized with isoflurane and administered 3 × 104 EID50s of VN1203 in 1 ml of PBS i.n. For all animals, weight change and survival were monitored for 14 days following virus challenge.
Isolation and characterization of antibody escape mutants.
Antibody escape mutants were selected by inoculating 10-day-old specific-pathogen-free (SPF) embryonated chicken eggs with rgH5N1 incubated with neutralizing antibody essentially as described previously (27, 28). Eggs were screened for the presence of virus by hemagglutination assay of allantoic fluid, which was harvested from infected eggs, and viral RNA was extracted and used to generate cDNA by reverse transcription-PCR. The HA gene was then amplified and sequenced.
Data analysis and statistics.
The data collected were inputted and graphed using GraphPad Prism (version 5.03) software. Statistical analysis was performed using the log-rank (Mantel-Cox) test.
RESULTS
Characterization of H5N1 HMAbs.
To determine the genetic diversity of the antibodies identified, we sequenced CDRs 1, 2, and 3 of the antibody heavy chains. Sequence analysis of the 11 HI-positive hybridomas showed that they clustered into two different groups. Two HMAbs clustered into group 1, while the others clustered into the second group. One antibody representing group 1 (BF1-10) and two representing group 2 (BF1-1 and BF1-19) were chosen for further analysis. HI assays were performed with spent supernatants from human hybridomas to assess the cross-reactivity of the generated antibodies against a panel of H5N1 viruses isolated in Asia. All antibodies were reactive against VN1203, showing titers of 640 to 2,560 (Table 1). BF1-19 also showed reactivity against the antigenically drifted strain Mon244 (HI titer = 40), while BF1-1 and BF1-10 did not (Table 1). No antibodies showed reactivity against A/Japanese white eye/Hong Kong/1038/06 (HK1038), which is antigenically drifted from VN1203 to a greater degree than Mon244 (Table 1). BF1-19 showed strong reactivity against A/Hong Kong/156/97 (HK156) (HI titer = 1,280), while BF1-1 and BF1-10 showed only weak reactivity (HI titers = 10 and 20, respectively) (Table 1). All antibodies also showed strong neutralization activity in a microneutralization assay against VN1203, with titers of 640 to 10,240; however, no neutralization activity against Mon244 or HK1038 was observed (Table 2). BF1-19 showed strong microneutralization activity against HK156 (titer > 20,480), while BF1-1 and BF1-10 showed only weak activity (titers = 10 and 40, respectively) (Table 2).
TABLE 1.
HI reactivity profiles of HMAbs
| Antibody | HI antigen titer |
|||
|---|---|---|---|---|
| A/Hong Kong/156/97 | A/Vietnam/1203/04 | A/Whooper swan/Mongolia/244/05 | A/Japanese white eye/Hong Kong/1038/06 | |
| BF1-1 | 10 | 2,560 | 0 | 0 |
| BF1-10 | 20 | 640 | 0 | 0 |
| BF1-19 | 1,280 | 2,560 | 40 | 0 |
| h10C3 | 0 | 2,560 | 40 | 0 |
| FcDART | 640 | 640 | 40 | 0 |
TABLE 2.
MN reactivity profiles of HMAbs
| Antibody | MN titer |
|||
|---|---|---|---|---|
| A/Hong Kong/156/97 | A/Vietnam/1203/04 | A/Whooper swan/Mongolia/244/05 | A/Japanese white eye/Hong Kong/1038/06 | |
| BF1-1 | 10 | 10,240 | 0 | 0 |
| BF1-10 | 40 | 640 | 0 | 0 |
| BF1-19 | >20,480 | 2,560 | 0 | 0 |
| h10C3 | 0 | 2,560 | 0 | 0 |
| FcDART | 10,240 | 1,280 | 80 | 0 |
To determine the nature of the epitopes recognized by each HI-positive HMAb, Western blot analysis was performed using rgH5N1-infected MDCK cell lysates. None of the neutralizing HMAbs showed binding to denatured H5N1 antigens, suggesting that they recognized conformational epitopes. The one HI-negative HMAb that was included in this study, BF1-32, was Western blotting positive and was identified by IFA with VN1203-infected MDCK cells. This antibody appears to target HA1, as the most prominent bands corresponded to HA0 (82 kDa) and HA1 (58 kDa), with a less intense band being found at 78 kDa (Fig. 2). To assess the affinities of each of the HMAbs for the H5 HA protein, surface plasmon resonance studies were conducted using a BIAcore 3000 instrument. The KD values for the binding of HMAbs BF1-1, BF1-10, and BF1-19 to the VN1203 HA were 20.3 nM, 10.8 nM, and 48.2 nM, respectively, showing that each had similar affinities for their antigens. Following these studies, we selected BF1-19 for subsequent experiments.
FIG 2.

Human monoclonal antibodies bind to conformational epitopes on hemagglutinin. Antibodies were used to probe a blot of lysed Madin-Darby canine kidney cells that were infected with A/Vietnam/03/2004 (H5N1). BF1-1, BF1-10, and BF1-19, which showed reactivity by the HI assay, showed no binding, indicating that they did not recognize linear epitopes of hemagglutinin. BF1-32, which did not show reactivity by the HI assay, recognized linear epitopes on what appeared to be HA1, as the most prominent bands corresponded to HA0 (82 kDa) and HA1 (58 kDa), with a less intense band being found at 78 kDa.
We next assessed the breadth of reactivity of BF1-19 to a panel of HAs from diverse H5N1 viruses via ELISA. We used antigens obtained from two commercial sources, eEnzyme and Protein Sciences, to investigate whether the antigen production system had an effect on the ability of the antibodies to bind, as the eEnzyme antigens were produced in 293T cells, while the Protein Sciences antigens were produced in insect cells. Further, we used both wild-type (wt) HA antigens and antigens from which the furin cleavage site was deleted (Δfurin antigens) to determine if cleavage had an impact on the exposure of the epitope recognized by these antibodies. All eEnzyme antigens with the exception of A/Indonesia/5/2005 (H5N1) were engineered to remove the furin cleavage site, resulting in a more homogeneous product (Fig. 3). The Protein Sciences antigens and the eEnzyme A/Indonesia/5/2005 (H5N1) antigen retained the furin cleavage site, resulting in oligomers that consisted of both cleaved and uncleaved antigens. Antigens obtained from Protein Sciences were tested in the antibody down format, where the antibody was bound to the solid phase via a capture antibody and the antigen was the soluble binding partner (Fig. 3). eEnzyme antigens contained a C-terminal His tag, which allowed them to be used as a soluble binding partner or to be immobilized on the solid phase using a His binding partner, and this format was termed the antigen down format (Fig. 3).
BF1-19 bound to clade 1 antigens from both suppliers in the antigen down format (Fig. 3A) and also bound to clade 2.2.1 and 2.3.4 antigens in the antibody down format (Fig. 3B). Some binding to clade 2.1.3.2 antigen was also detected at high concentrations (Fig. 3B). Given the oligomeric structure of the antigens, the ability to detect binding in the antibody down format may be the result of binding more avid than that in the antigen down format. BF1-19 also bound to clade 1 antigens with or without the furin cleavage site (Fig. 3A). While these data are not able to definitively demonstrate a quantitative difference between cleaved and uncleaved HA, they do clearly demonstrate that the recognized epitope is available on oligomers derived from both wt and Δfurin constructs.
Generation of mMAbs.
We used a three-dose vaccination regimen in mice, consisting of VN1203 delivered i.n. and then i.p., followed by the delivery of Mon244 i.v., to generate antibodies broadly specific to different clades of H5 influenza viruses. One of these antibodies, 10C3, bound to clade 1 and 2.2.1 antigens from both suppliers in the antigen down format and, like BF1-19, bound to both the wt and Δfurin constructs (Fig. 3C). Similar results were also observed in the antibody down format (Fig. 3D).
10C3 was then humanized to reduce potential immunogenicity by grafting the complementarity-determining regions of the antibody onto the framework regions from closely related human VH and VL segment gene sequences. Interestingly, there were indications of h10C3 binding to clade 2.1.3.2 and 2.3.4 antigens, which was not observed for 10C3 (Fig. 3E). While these data are not conclusive, they indicate that the humanization process broadened the coverage of this antibody (Fig. 3D and E). By HI, h10C3 was strongly reactive to VN1203 (HI titer = 2,560) and showed relatively weak reactivity against Mon244 (HI titer = 40) and no reactivity against HK1038. Unlike BF1-19, h10C3 did not show reactivity against HK156 (Table 1). In microneutralization assays, h10C3 showed neutralization activity only against VN1203 (titer = 2,560); no neutralization activity was observed against HK156, Mon244, or HK1038 (Table 2).
Design and development of FcDART.
The FcDART molecule consisted of two polypeptide chains that heterodimerized to form a bispecific molecule. Each chain contained the VL domain of one specificity and the VH domain of the other, such that heterodimerization of the two chains resulted in the assembly of matching VL and VH domains. In order to favor heterodimeric assembly, a strong heterodimerization domain was appended to the C terminus of each chain. These domains consisted of coiled-coil-forming sequences, where the hydrophobic core of the coiled coil interface was flanked by the opposite charges (an E negative charge on one coil and a K positive charge on the other). The E/K coiled-coil consisted of two peptides, each containing unique, repeating, heptad sequences. In the E coil, this sequence was E-V-S-A-L-E-K, while in the K coil, the sequence was K-V-S-A-L-E-K. These charges are believed to favor heterodimerization by repelling homotypic association and preventing the initiation of zippering of the hydrophobic core (Fig. 1A). These DART molecules were modified further by appending an Fc domain to the C terminus of the coil with the E negative charge, causing two DART units to dimerize and form a tetravalent bispecific structure (Fig. 1A). The addition of the Fc confers a long serum half-life on the molecules and restores the avidity associated with the bivalent parental MAbs. Further, the presence of the Fc confers protein A binding and makes these molecules amenable to platform purification processes routinely used for MAbs in large-scale cyclic GMP production.
Following expression in CHO cells and purification, analytical size exclusion chromatography showed that purified material predominantly consisted of one species that eluted at a volume consistent with the predicted molecular mass of FcDART. Only a small amount of higher-order aggregate was present, as indicated by the slight left shoulder on the peak. Increasing the concentration from 2 to 24.5 mg/ml did not cause a significant increase in the amount of aggregated material (Fig. 1B). These characteristics indicate that the molecule was properly assembled, homogeneously purified, and not prone to aggregation. We then tested the FcDART molecule against a panel of antigens in antibody down format and found that, with the exception of clade 2.1.3.2 antigens, the FcDART molecule was able to bind to the antigens in the panel and had a binding profile generally consistent with the binding profiles of BF1-19 and h10C3, indicating that FcDART indeed combined the binding profiles of BF1-19 and h10C3 (Fig. 3F). By HI, FcDART showed reactivity against VN1203 and HK156 (HI titer = 640), relatively weak reactivity against Mon244 (HI titer = 40), and no reactivity against HK1038, findings that are, again, consistent with a binding profile that is a combination of the binding profiles of BF1-19 and h10C3 (Table 1). The neutralization activity of FcDART was also consistent with an antibody with the combined specificities of h10C3 and BF1-19, as strong neutralization activity against both VN1203 (titer = 1,280) and HK156 (titer = 10,240) was observed (Table 2). Interestingly, FcDART also showed neutralization activity against Mon244 (titer = 80), which was not observed with any other antibody tested, suggesting an increased breadth of specificity (Table 2).
Escape mutant characterization.
To further characterize the epitopes recognized by these antibodies, we inoculated embryonated chicken eggs with rgH5N1 and antibody and sequenced the viruses present after incubation for 48 h. On the basis of the mutations in these viruses, MAbs primarily bound to antigenic sites on the globular head of HA (29). The L128S mutation in antigenic site C and the M265I mutation in antigenic site A were present in mutants that escaped binding to h10C3 (h10C3 escape mutants), the K139E mutation in antigenic site A was present in 10C3 escape mutants, the S140Y/F mutations in antigenic site A were present in BF1-19 escape mutants, the K188N/E mutations in antigenic site B were present in BF1-19 and FcDART escape mutants, and the T213A mutation in antigenic site D was present in FcDART escape mutants (Fig. 4). With the exception of the h10C3 escape mutants, in which the L128S and M265I mutations were present in one mutant, each escape mutant did not harbor more that one of the mutations indicated in Fig. 4. Further, escape mutants harboring the same mutation were seen in all eggs (n = 3) at the dilution of antibody that was permissive to the generation of escape mutants.
FIG 4.

Monoclonal antibody escape mutant viruses have amino acid changes located primarily in the globular head of hemagglutinin. (A) Amino acid changes shown on a cartoon of the hemagglutinin trimer of A/Vietnam/1203/04 (29). One subunit is highlighted for clarity. (B). Locations of amino acid changes. All mutations map to known antigenic sites (27). H5 numbering is used.
We next performed HI assays to determine if binding of the antibodies to these escape mutants was diminished or abrogated. BF1-19 showed little activity against the BF1-19 escape mutants with the K188N and S140F mutations (HI titers < 10; Table 3). 10C3 also showed little activity against the 10C3 escape mutant with the K139E mutation (HI titer < 10; Table 3). h10C3 showed little activity against the h10C3 escape mutant with the L128S/M265I mutations (HI titer < 10; Table 3). BF1-19, 10C3, and h10C3 were all inhibitory to wt virus VN1203 and to escape mutants with mutations that prevented binding to other antibodies (HI titers = 160 to 1,280; Table 3). Therefore, these antibodies bound to different sites on HA, and the mutant viruses could escape the antibodies that were used to select for them, at least in vitro. Two escape mutants with the K188E and T213A mutations, which prevented binding to FcDART, were generated. All antibodies, including FcDART, showed activity against the T213A mutant (HI titers = 160 to 320; Table 3), indicating that this escape mutation is unlikely to be effective in vivo. The K188E mutant, which could not bind to FcDART, contained a mutation at the same location as a BF1-19 escape mutant, the K188N mutant. Binding of FcDART to the K188E mutant was detected but was relatively weak (HI titer = 20; Table 3). BF1-19 showed little activity against the K188E mutant, which is consistent with the little activity that BF1-19 showed against the K188N escape mutant (HI titer < 10; Table 3). Both 10C3 and h10C3 showed activity against the K188E mutant (HI titers = 80; Table 3). Interestingly, FcDART showed greater activity against the BF1-19 escape mutant with the K188N mutations than against the mutant with the K188E mutation, despite the fact that the same residue was altered in both mutants (HI titers = 80 and 20, respectively). FcDART showed relatively strong activity against all other mutants and the wt virus (HI titers = 160 to 1,280; Table 3).
TABLE 3.
Profiles of HI reactivity of antibody escape mutants to MAbs
| Antibody used to generate escape | Virus HA mutationa | HI titer |
|||
|---|---|---|---|---|---|
| BF1-19 | 10C3 | h10C3 | FcDART | ||
| BF1-19 | K188N | <10 | 160 | 160 | 80 |
| BF1-19 | S140F | <10 | 1,280 | 1,280 | 1,280 |
| 10C3 | K139E | 320 | <10 | 160 | 160 |
| h10C3 | L128S and M265I | 320 | 320 | <10 | 160 |
| FcDART | K188E | <10 | 80 | 80 | 20 |
| FcDART | T213A | 320 | 160 | 160 | 320 |
| NAb | NA | 1,280 | 640 | 640 | 640 |
H5 numbering.
NA, not applicable.
In vivo protective efficacy of antibodies against homologous challenge.
To assess the potential for the isolated HMAbs to be used as prophylactics or therapeutic agents against H5N1 infection, we employed mouse infection models. To start, the prophylactic efficacy of neutralizing H5N1 MAbs was tested in conjunction with the homologous challenge virus, VN1203. Antibody was injected intraperitoneally at 2.5, 1.0, and 0.1 mg of antibody per kg of body weight 1 day prior to challenge with 103 EID50s of VN1203. A single 2.5-mg/kg dose of BF1-19 provided complete protection from the lethal effects of the virus, and the rate of survival was inversely proportional to the antibody dose. At 0.1 mg/kg, BF1-19 did not protect against challenge (Fig. 5A and B). The rates of survival of mice that received 2.5 and 1.0 mg/kg of BF1-19 were statistically significantly greater than those of mice that received PBS (100% and 80% survival, respectively, compared to 0% in the PBS group; P < 0.01; Fig. 5B). We next assessed the efficacy of h10C3 as a single 2.5-mg/kg prophylactic dose against VN1203 (Fig. 5C and D). h10C3 provided 80% protection against challenge, which was statistically significantly greater than that in mice that received PBS, which demonstrated 100% mortality (P < 0.01; Fig. 5D). FcDART also provided 100% protection against challenge as a prophylactic at a 2.5-mg/kg dose, and this protection was also statistically significantly greater than that in mice that received PBS, which demonstrated 100% mortality (P < 0.01; Fig. 5E and F).
FIG 5.

Antibodies provide 100% protection against challenge when administered prophylactically. Mice received a single intraperitoneal injection of antibody 24 h prior to challenge with 100 MLD50s of A/Vietnam/1203/04 (H5N1) administered intranasally, which caused 100% mortality in untreated mice within 10 days of infection (B, D, and F). BF1-19 provided 100%, 80%, and 0% protection against challenge when it was delivered at dosages of 2.5, 1, and 0.1 mg/kg, respectively (A and B). h10C3 provided 80% protection against challenge when it was delivered as a single 2.5-mg/kg dose (C and D), while FcDART provided 100% protection against challenge when it was delivered as a single 2.5-mg/kg dose 24 h before challenge (E and F). (A, C, and E) Data represent the mean percentage of the starting weight ± standard error of the mean for each mouse group (n = 5 mice per group). **, P < 0.01.
We next assessed the treatment efficacy of the antibodies against VN1203 infection at a dose of 2.5 mg/kg. When given at 24 h postchallenge, BF1-19, h10C3, and FcDART all provided 100% protection against challenge, whereas the rate of mortality was 100% in the PBS group; these differences were statistically significant (P < 0.01; Fig. 6). When administered at 2.5 mg/kg at 72 h postinfection, FcDART provided 100% protection against challenge, while BF1-19 and h10C3 provided 80% protection against challenge (Fig. 6). Compared to the 100% mortality observed in the groups treated with PBS, these differences were statistically significant (P < 0.01; Fig. 6). We next assessed the prophylactic efficacy of BF1-19 in ferrets, in which influenza virus infection is widely regarded to be more representative of influenza virus infection in humans than influenza virus infection in mice is. Three ferrets received 1 mg/kg of antibody 1 day prior to infection with 3 × 104 EID50s of VN1203, and 100% protection against challenge was observed. In contrast, 100% mortality was observed in the 2 ferrets that received PBS, which was a statistically significant difference (P < 0.05; Fig. 7).
FIG 6.

Antibodies provided 100% protection against challenge when administered at 2.5 mg/kg at 24 h postchallenge. Mice received a single intraperitoneal dose of antibody at 2.5 mg/kg 24 or 72 h after challenge with 100 MLD50s of A/Vietnam/1203/04 (H5N1) administered intranasally, which caused 100% mortality in untreated mice within 10 days of infection (B, D, and F). BF1-19 (A and B), h10C3 (C and D), and FcDART (E and F) provided 100% protection against challenge when administered at 24 h postchallenge. BF1-19 and h10C3 provided 80% protection against challenge when administered at 72 h postchallenge (B and D, respectively); however, FcDART provided 100% protection at 72 h postchallenge (F). (A, C, and E) Data represent the mean percentage of the starting weight ± standard error of the mean for each mouse group (n = 5 mice per group). hpi, hours postinfection; **, P < 0.01.
FIG 7.
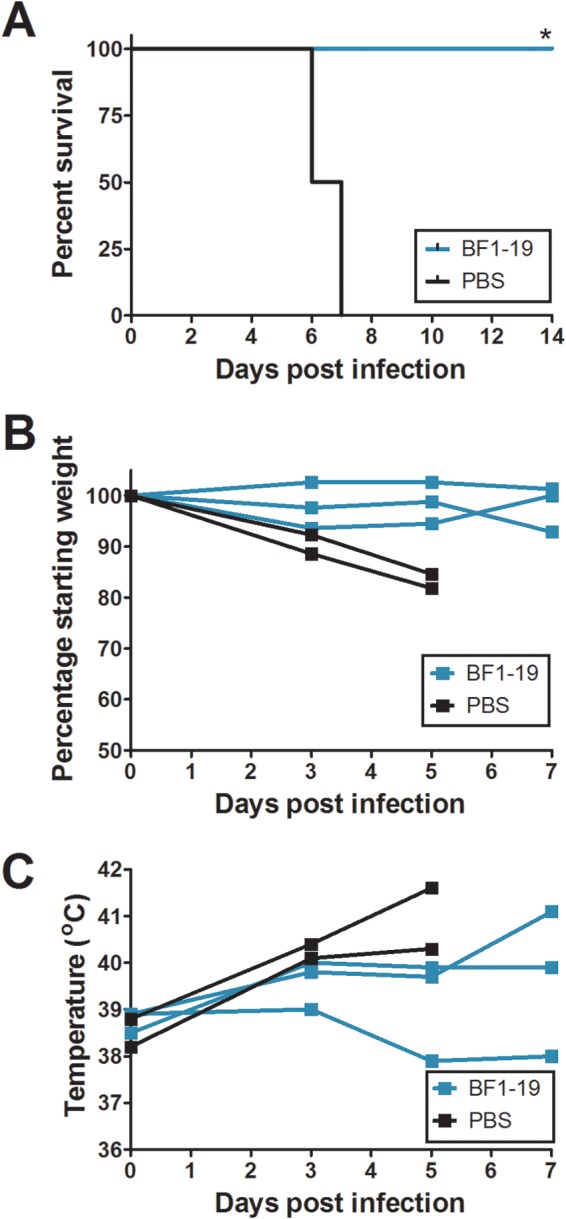
BF1-19 provides 100% protection against challenge by A/Vietnam/1203/04 (H5N1) in ferrets. BF1-19 was administered to ferrets as a single 1-mg/kg dose intraperitoneally at 24 h postchallenge with 3 × 104 EID50s of A/Vietnam/1203/04 (H5N1), which caused 100% mortality in ferrets injected with PBS but no mortality in ferrets that were injected with BF1-19 (A). Ferrets that were injected with BF1-19 also exhibited less weight loss and lower temperatures than ferrets injected with PBS (B and C, respectively); however, these differences were not significant. Three ferrets were administered BF1-19, and two ferrets were administered PBS. *, P < 0.05.
In vivo protective efficacy of antibodies against heterologous challenge.
HMAbs provided protection against challenge with VN1203. We next determined if BF1-19 could provide protection against Mon244, an antigenically drifted strain of H5N1. Surprisingly, BF1-19 was equally as effective at reducing Mon244-induced mortality in mice as it was at reducing VN1203-induced mortality when given 24 h before or after viral challenge, providing 100% protection against challenge, whereas 100% mortality was observed in the PBS group; these differences were statistically significant (P < 0.01; Fig. 8). When administered at 72 h postinfection, 80% survival was observed, which was also statistically significantly different from the rate of survival for the group that received PBS (P < 0.05; Fig. 8).
FIG 8.
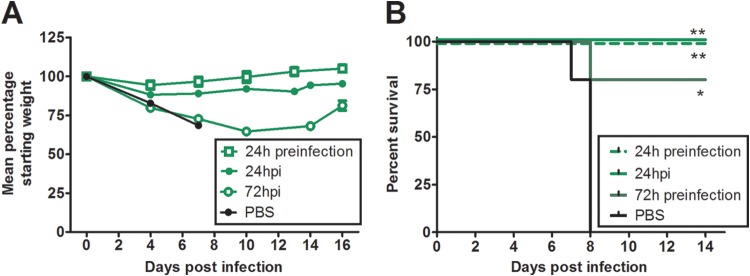
BF1-19 provides 100% protection against challenge from the antigenically drifted strain A/Whooper swan/Mongolia/244/05 (H5N1). A single 2.5-mg/ml intraperitoneal dose of BF1-19 administered at 24 h pre- or postchallenge provided 100% protection against challenge with 100 MLD50s of A/Whooper swan/Mongolia/244/05 (H5N1). When this dose of antibody was administered at 72 h postchallenge, 80% protection was observed (A and B). (A) Data represent the mean percentage of the starting weight ± standard error of the mean for each mouse group (n = 5 mice per group). *, P < 0.05; **, P < 0.01.
Antibody efficacy against escape mutants in vivo.
To determine if mutations that facilitate antibody escape in vitro are also effective in vivo, we used mutants that could escape binding to BF1-19 in a murine challenge model. We inoculated mice with 106 TCID50s of the S140F or K188N (H5 numbering) escape mutant in an rgH5N1 background and then administered 2.5 mg/kg of BF1-19 on the next day. All mice injected with BF1-19 were protected against challenge, while 80% of mice that did not receive antibody died following inoculation with the S140F mutant (P < 0.01; Fig. 9). Therefore, while these mutations allowed neutralization escape in vitro, they were not effective in vivo.
FIG 9.
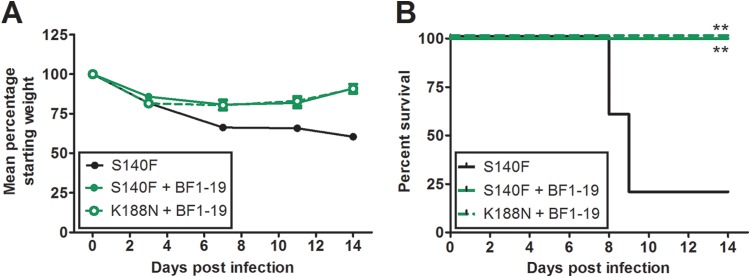
BF1-19 provides 100% protection against challenge from A/Vietnam/1203/04 (H5N1) in vitro escape mutants. BF1-19 escape mutants were generated using a virus based on A/Puerto Rico/8/34, generated by reverse genetics, containing the hemagglutinin and neuraminidase genes from A/Vietnam/1203/04 (H5N1). Mice were administered 106 TCID50s of the S145F or K144N escape mutant (in hemagglutinin) intranasally, followed by a single 2.5-mg/ml intraperitoneal dose of BF1-19 administered at 24 h postchallenge. All mice that received BF1-19 survived the challenge, while 80% of mice that did not receive antibody died (A and B). (A) Data represent the mean percentage of the starting weight ± standard error of the mean for each mouse group (n = 5 mice per group). **, P < 0.01.
DISCUSSION
Despite a number of promising clinical trial results with adjuvanted H5N1 vaccines, it is likely that a matched vaccine will not be available until at least 6 months after the emergence of a pandemic. During this period a number of other interventions are likely to be employed. These alternatives include stockpiled unmatched vaccine, nonpharmaceutical interventions, and antiviral drugs. Recent modeling studies have suggested that neuraminidase inhibitors, if administered soon after the initial spread of a pandemic, can have an impact on viral spread (30). Although their impact is promising, there are concerns over both classes of currently available antiviral drugs, including the development of resistance and uncertain clinical efficacy. Antibody-based therapeutic agents can be used prophylactically to protect at-risk populations, such as the immunocompromised and patients who do not develop immunity from vaccination. If targeted to conserved regions of HA, antibody-based therapeutic agents could potentially reduce the issue of resistance associated with chemotherapeutic agents; however, this remains to be seen. Antibodies have been developed and applied as therapeutic agents against several viral infections, including respiratory syncytial virus, hepatitis B virus, and vaccinia virus infections (reviewed in reference 31–33). Studies in animal models of influenza virus infection have demonstrated the potential for monoclonal antibodies as therapeutic agents and prophylactics (9–19). A phase I study (clinical trial number NCT01390025; http://clinicaltrials.gov/) assessing the efficacy of an anti-M2 antibody is also in progress, and a subvirion H5N1 vaccine is being used in a phase II study (clinical trial number NCT00383071; http://clinicaltrials.gov/) to generate antibody-based therapeutic agents. The results of these studies have not yet been published.
Perhaps the biggest theoretical issue facing a monoclonal antibody approach for treating influenza virus infection is antigenic drift. Unless well-conserved epitopes can be targeted, a cocktail approach is needed. Another strategy is the use of monoclonal antibodies specific for epitopes on the stem region of HA, which contains epitopes that are more conserved than those on the globular head (reviewed in reference 34). These antibodies can bind to the HAs of numerous influenza virus subtypes and neutralize them; however, they are not neutralizing in the classical sense, as they do not prevent cell binding of the viruses (14, 21, 22, 35). Rather, their action prevents the fusion of viral and cellular membranes, which is mediated by a conformational change in HA (17, 36). As such, the activity of these antibodies cannot be quantified by the standard hemagglutination inhibition assay. Therefore, assays that measure other inhibitory activities, such as cell-cell fusion assays and live-cell fluorescence microscopy, are required (36). As they do not prevent virus-cell interactions, antibodies directed against the HA stem are potentially not as potent as antibodies directed against the globular head. The strategy that we present here combines the specificities of two antibodies into a single molecule, which is advantageous, as a single therapeutic simplifies production and quality control and therefore reduces production costs. One caveat of this approach is that the protective dose of different FcDART molecules would not necessarily be equivalent. Therefore, individual testing of FcDART molecules is likely to be necessary to determine the optimal protective doses required. FcDART not only combined the specificities and efficacies of the monoclonal antibodies on which it was based, but also it showed a greater breadth of activity. Therefore, a therapy consisting of FcDART alone is likely to be equally as efficacious as a cocktail containing both monoclonal antibodies and may be more broadly efficacious against more antigenically diverse H5N1 viruses.
The antibodies described here bind to conformational epitopes in antigenic sites located on the globular head of HA, as shown by escape mutant sequencing (Fig. 4). While the binding sites identified lie in antigenic sites of the HA and thus are subject to antigenic drift, we also show that escape mutants generated in vitro cannot produce a lethal infection in the presence of the same antibody in vivo. We demonstrated this by infecting mice with S140F and K188N (H5 numbering) escape mutants for binding to BF1-19 and then treating them with a protective dose of BF1-19. Our in vitro studies identified two amino acid changes, at positions 140 and 188 (H5 numbering), that facilitated escape from binding to BF1-19 (Fig. 4). Therefore, while a mutation at one of these sites may be sufficient for in vitro escape, multiple mutations may be required for escape in vivo. This may be a factor contributing to the activity of these antibodies against antigenically drifted H5N1 strains.
We observed two mutants that escaped binding to FcDART, the K188E and T213A mutants (Fig. 4). Residue 188 was also mutated in a BF1-19 escape mutant, but a mutated residue 213 was not observed in any other escape mutants, which was unexpected. However, FcDART bound strongly to the T213A mutant (HI titer = 320), as did the other antibodies (HI titers = 160 to 320) (Table 3), suggesting that this mutation is unlikely to allow effective escape in vivo. FcDART binding to the K188E mutant was not as strong (HI titer = 20), and BF1-19 also did not bind to the K188E mutant, as determined by the HI assay. This suggests that this mutant escaped binding by the BF1-19 arm of FcDART, and, possibly, the h10C3 arm was still able to bind to the escape mutant. However, further work is required to test this hypothesis.
To enhance the probability of generating cross-reactive HMAbs, we chose to use serum from a donor that had been vaccinated with vaccines derived from genetically and antigenically distinct H5N1 isolates, HK156 (23) and VN1203. Interestingly, vaccination with the VN1203 vaccine was able to boost the response to the HK156 vaccine (as determined by an enhanced response to the VN1203 vaccine in those that had received the HK156 vaccine compared with the response in those that had not), despite a period of 7 years between these vaccinations. Nevertheless, despite a good response to a number of H5N1 variants, serum from this individual was not able to react with the clade 2.3.4 virus A/Common magpie/Hong Kong/645/06 in the HI assay. The difficulty with fully extrapolating this result is that, similar to the work of Simmons et al. in 2007, we were able to show a substantial in vivo effect of an HMAb in the absence of supporting in vitro activity (13). This was particularly evident in our experiments with BF1-19 and the S140F escape mutant (H5 numbering), as the antibody showed no inhibitory activity by HI assay but could prevent mortality caused by the mutant in vivo. While we did not investigate the mechanism(s) behind this discrepancy in this study, it has been shown that components of the complement cascade, such as C1q, can enhance the in vivo activity of an antibody that is poorly reactive in vitro (37, 38).
Interestingly, we found that humanization of 10C3 increased the breadth of binding of this antibody, allowing it to bind to HA from clades 2.1.3.2 and 2.3.4. We did not investigate the possible mechanisms of this increase in specificity, but humanization has previously been shown to alter antibody binding specificity, possibly due to alterations in flexibility (39). This may have been a factor in the increased breadth of binding of h10C3 observed.
In this study, we have developed monoclonal antibodies from both human and murine sources that show activity against H5 influenza viruses of different clades and are efficacious as prophylactics and therapeutic agents at low doses in both mouse and ferret models of influenza virus infection. Further, we have developed FcDART, which combined the antigen binding domains of the two monoclonal antibodies. This represents a promising strategy by which to produce neutralizing antibodies that have the potential to be effective against a range of antigenically diverse influenza viruses.
ACKNOWLEDGMENTS
This work was supported by grant no. U01AI070373 from the U.S. Department of Health and Human Services, the National Institutes of Health, and the American Lebanese Syrian Associated Charities (ALSAC).
REFERENCES
- 1.Neumann G, Chen H, Gao GF, Shu Y, Kawaoka Y. 2010. H5N1 influenza viruses: outbreaks and biological properties. Cell Res 20:51–61. doi: 10.1038/cr.2009.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herfst S, Schrauwen EJ, Linster M, Chutinimitkul S, de Wit E, Munster VJ, Sorrell EM, Bestebroer TM, Burke DF, Smith DJ, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336:1534–1541. doi: 10.1126/science.1213362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imai M, Watanabe T, Hatta M, Das SC, Ozawa M, Shinya K, Zhong G, Hanson A, Katsura H, Watanabe S, Li C, Kawakami E, Yamada S, Kiso M, Suzuki Y, Maher EA, Neumann G, Kawaoka Y. 2012. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486:420–428. doi: 10.1038/nature10831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen H, Deng G, Li Z, Tian G, Li Y, Jiao P, Zhang L, Liu Z, Webster RG, Yu K. 2004. The evolution of H5N1 influenza viruses in ducks in southern China. Proc Natl Acad Sci U S A 101:10452–10457. doi: 10.1073/pnas.0403212101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Govorkova EA, Rehg JE, Krauss S, Yen HL, Guan Y, Peiris M, Nguyen TD, Hanh TH, Puthavathana P, Long HT, Buranathai C, Lim W, Webster RG, Hoffmann E. 2005. Lethality to ferrets of H5N1 influenza viruses isolated from humans and poultry in 2004. J Virol 79:2191–2198. doi: 10.1128/JVI.79.4.2191-2198.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.World Health Organization Global Influenza Program Surveillance Network. 2005. Evolution of H5N1 avian influenza viruses in Asia. Emerg Infect Dis 11:1515–1521. doi: 10.3201/eid1110.050644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keating GM, Plosker GL, Lyseng-Williamson KA. 2012. A/H5N1 prepandemic influenza vaccine (Vepacel®): a guide to its use. BioDrugs 26:425–430. doi: 10.2165/11209740-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 8.Yen HL, Monto AS, Webster RG, Govorkova EA. 2005. Virulence may determine the necessary duration and dosage of oseltamivir treatment for highly pathogenic A/Vietnam/1203/04 influenza virus in mice. J Infect Dis 192:665–672. doi: 10.1086/432008. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Luo W, Wu WL, Fang Z, Xia L, Gui X, Chen H, Shih JW, Xia N. 2010. Humanized antibodies with broad-spectrum neutralization to avian influenza virus H5N1. Antiviral Res 87:81–84. doi: 10.1016/j.antiviral.2010.04.012. [DOI] [PubMed] [Google Scholar]
- 10.Chen Y, Qin K, Wu WL, Li G, Zhang J, Du H, Ng MH, Shih JW, Peiris JS, Guan Y, Chen H, Xia N. 2009. Broad cross-protection against H5N1 avian influenza virus infection by means of monoclonal antibodies that map to conserved viral epitopes. J Infect Dis 199:49–58. doi: 10.1086/594374. [DOI] [PubMed] [Google Scholar]
- 11.Hanson BJ, Boon AC, Lim AP, Webb A, Ooi EE, Webby RJ. 2006. Passive immunoprophylaxis and therapy with humanized monoclonal antibody specific for influenza A H5 hemagglutinin in mice. Respir Res 7:126. doi: 10.1186/1465-9921-7-126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu H, Voss J, Zhang G, Buchy P, Zuo T, Wang L, Wang F, Zhou F, Wang G, Tsai C, Calder L, Gamblin SJ, Zhang L, Deubel V, Zhou B, Skehel JJ, Zhou P. 2012. A human antibody recognizing a conserved epitope of H5 hemagglutinin broadly neutralizes highly pathogenic avian influenza H5N1 viruses. J Virol 86:2978–2989. doi: 10.1128/JVI.06665-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simmons CP, Bernasconi NL, Suguitan AL, Mills K, Ward JM, Chau NV, Hien TT, Sallusto F, Ha DQ, Farrar J, de Jong MD, Lanzavecchia A, Subbarao K. 2007. Prophylactic and therapeutic efficacy of human monoclonal antibodies against H5N1 influenza. PLoS Med 4:e178. doi: 10.1371/journal.pmed.0040178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ekiert DC, Kashyap AK, Steel J, Rubrum A, Bhabha G, Khayat R, Lee JH, Dillon MA, O'Neil RE, Faynboym AM, Horowitz M, Horowitz L, Ward AB, Palese P, Webby R, Lerner RA, Bhatt RR, Wilson IA. 2012. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 489:526–532. doi: 10.1038/nature11414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kashyap AK, Steel J, Rubrum A, Estelles A, Briante R, Ilyushina NA, Xu L, Swale RE, Faynboym AM, Foreman PK, Horowitz M, Horowitz L, Webby R, Palese P, Lerner RA, Bhatt RR. 2010. Protection from the 2009 H1N1 pandemic influenza by an antibody from combinatorial survivor-based libraries. PLoS Pathog 6:e1000990. doi: 10.1371/journal.ppat.1000990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sakabe S, Iwatsuki-Horimoto K, Horimoto T, Nidom CA, Le M, Takano R, Kubota-Koketsu R, Okuno Y, Ozawa M, Kawaoka Y. 2010. A cross-reactive neutralizing monoclonal antibody protects mice from H5N1 and pandemic (H1N1) 2009 virus infection. Antiviral Res 88:249–255. doi: 10.1016/j.antiviral.2010.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, Wan H, Murakami A, Yammanuru A, Han T, Cox NJ, Bankston LA, Donis RO, Liddington RC, Marasco WA. 2009. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat Struct Mol Biol 16:265–273. doi: 10.1038/nsmb.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Throsby M, van den Brink E, Jongeneelen M, Poon LL, Alard P, Cornelissen L, Bakker A, Cox F, van Deventer E, Guan Y, Cinatl J, ter Meulen J, Lasters I, Carsetti R, Peiris M, de Kruif J, Goudsmit J. 2008. Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM+ memory B cells. PLoS One 3:e3942. doi: 10.1371/journal.pone.0003942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshida R, Igarashi M, Ozaki H, Kishida N, Tomabechi D, Kida H, Ito K, Takada A. 2009. Cross-protective potential of a novel monoclonal antibody directed against antigenic site B of the hemagglutinin of influenza A viruses. PLoS Pathog 5:e1000350. doi: 10.1371/journal.ppat.1000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesen RH, Koudstaal W, Koldijk MH, Weverling GJ, Brakenhoff JP, Lenting PJ, Stittelaar KJ, Osterhaus AD, Kompier R, Goudsmit J. 2010. New class of monoclonal antibodies against severe influenza: prophylactic and therapeutic efficacy in ferrets. PLoS One 5:e9106. doi: 10.1371/journal.pone.0009106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. 2011. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333:850–856. doi: 10.1126/science.1205669. [DOI] [PubMed] [Google Scholar]
- 22.Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251. doi: 10.1126/science.1171491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treanor JJ, Wilkinson BE, Masseoud F, Hu-Primmer J, Battaglia R, O'Brien D, Wolff M, Rabinovich G, Blackwelder W, Katz JM. 2001. Safety and immunogenicity of a recombinant hemagglutinin vaccine for H5 influenza in humans. Vaccine 19:1732–1737. doi: 10.1016/S0264-410X(00)00395-9. [DOI] [PubMed] [Google Scholar]
- 24.Treanor JJ, Campbell JD, Zangwill KM, Rowe T, Wolff M. 2006. Safety and immunogenicity of an inactivated subvirion influenza A (H5N1) vaccine. N Engl J Med 354:1343–1351. doi: 10.1056/NEJMoa055778. [DOI] [PubMed] [Google Scholar]
- 25.Hadlock KG, Lanford RE, Perkins S, Rowe J, Yang Q, Levy S, Pileri P, Abrignani S, Foung SK. 2000. Human monoclonal antibodies that inhibit binding of hepatitis C virus E2 protein to CD81 and recognize conserved conformational epitopes. J Virol 74:10407–10416. doi: 10.1128/JVI.74.22.10407-10416.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keck ZY, Sung VM, Perkins S, Rowe J, Paul S, Liang TJ, Lai MM, Foung SK. 2004. Human monoclonal antibody to hepatitis C virus E1 glycoprotein that blocks virus attachment and viral infectivity. J Virol 78:7257–7263. doi: 10.1128/JVI.78.13.7257-7263.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Caton AJ, Brownlee GG, Yewdell JW, Gerhard W. 1982. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 31:417–427. doi: 10.1016/0092-8674(82)90135-0. [DOI] [PubMed] [Google Scholar]
- 28.Yewdell JW, Webster RG, Gerhard WU. 1979. Antigenic variation in three distinct determinants of an influenza type A haemagglutinin molecule. Nature 279:246–248. doi: 10.1038/279246a0. [DOI] [PubMed] [Google Scholar]
- 29.Yamada S, Suzuki Y, Suzuki T, Le MQ, Nidom CA, Sakai-Tagawa Y, Muramoto Y, Ito M, Kiso M, Horimoto T, Shinya K, Sawada T, Usui T, Murata T, Lin Y, Hay A, Haire LF, Stevens DJ, Russell RJ, Gamblin SJ, Skehel JJ, Kawaoka Y. 2006. Haemagglutinin mutations responsible for the binding of H5N1 influenza A viruses to human-type receptors. Nature 444:378–382. doi: 10.1038/nature05264. [DOI] [PubMed] [Google Scholar]
- 30.Longini IM Jr, Nizam A, Xu S, Ungchusak K, Hanshaoworakul W, Cummings DA, Halloran ME. 2005. Containing pandemic influenza at the source. Science 309:1083–1087. doi: 10.1126/science.1115717. [DOI] [PubMed] [Google Scholar]
- 31.Berry JD, Gaudet RG. 2011. Antibodies in infectious diseases: polyclonals, monoclonals and niche biotechnology. N Biotechnol 28:489–501. doi: 10.1016/j.nbt.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burton DR, Poignard P, Stanfield RL, Wilson IA. 2012. Broadly neutralizing antibodies present new prospects to counter highly antigenically diverse viruses. Science 337:183–186. doi: 10.1126/science.1225416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yamada T. 2011. Therapeutic monoclonal antibodies. Keio J Med 60:37–46. doi: 10.2302/kjm.60.37. [DOI] [PubMed] [Google Scholar]
- 34.Han T, Marasco WA. 2011. Structural basis of influenza virus neutralization. Ann N Y Acad Sci 1217:178–190. doi: 10.1111/j.1749-6632.2010.05829.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Margine I, Krammer F, Hai R, Heaton NS, Tan GS, Andrews SA, Runstadler JA, Wilson PC, Albrecht RA, Garcia-Sastre A, Palese P. 2013. Hemagglutinin stalk-based universal vaccine constructs protect against group 2 influenza A viruses. J Virol 87:10435–10446. doi: 10.1128/JVI.01715-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brandenburg B, Koudstaal W, Goudsmit J, Klaren V, Tang C, Bujny MV, Korse HJ, Kwaks T, Otterstrom JJ, Juraszek J, van Oijen AM, Vogels R, Friesen RH. 2013. Mechanisms of hemagglutinin targeted influenza virus neutralization. PLoS One 8:e80034. doi: 10.1371/journal.pone.0080034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng JQ, Mozdzanowska K, Gerhard W. 2002. Complement component C1q enhances the biological activity of influenza virus hemagglutinin-specific antibodies depending on their fine antigen specificity and heavy-chain isotype. J Virol 76:1369–1378. doi: 10.1128/JVI.76.3.1369-1378.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mozdzanowska K, Feng J, Eid M, Zharikova D, Gerhard W. 2006. Enhancement of neutralizing activity of influenza virus-specific antibodies by serum components. Virology 352:418–426. doi: 10.1016/j.virol.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Bumbaca D, Wong A, Drake E, Reyes AE II, Lin BC, Stephan JP, Desnoyers L, Shen BQ, Dennis MS. 2011. Highly specific off-target binding identified and eliminated during the humanization of an antibody against FGF receptor 4. MAbs 3:376–386. doi: 10.4161/mabs.3.4.15786. [DOI] [PMC free article] [PubMed] [Google Scholar]