

ABSTRACT
The minimum requirement for an active RNA-dependent RNA polymerase of respiratory syncytial virus (RSV) is a complex made of two viral proteins, the polymerase large protein (L) and the phosphoprotein (P). Here we have investigated the domain on P that is responsible for this critical P-L interaction. By use of recombinant proteins and serial deletions, an L binding site was mapped in the C-terminal region of P, just upstream of the N-RNA binding site. The role of this molecular recognition element of about 30 amino acid residues in the L-P interaction and RNA polymerase activity was evaluated in cellula using an RSV minigenome system and site-directed mutagenesis. The results highlighted the critical role of hydrophobic residues located in this region.
IMPORTANCE Respiratory syncytial virus (RSV) is the leading cause of lower respiratory tract illness in infants. Since no vaccine and no good antivirals against RSV are available, it is essential to better understand how the viral machinery functions in order to develop new antiviral strategies. Like all negative-strand RNA viruses, RSV codes for its own machinery to replicate and transcribe its genome. The core of this machinery is composed of two proteins, the phosphoprotein (P) and the large protein (L). Here, using recombinant proteins, we have mapped and characterized the P domain responsible for this L-P interaction and the formation of an active L-P complex. These findings extend our understanding of the mechanism of action of RSV RNA polymerase and allow us to define a new target for the development of drugs against RSV.
INTRODUCTION
Human respiratory syncytial virus (HRSV) is the leading cause of acute respiratory infections in infants worldwide and is the primary cause of infant hospitalization for respiratory infections (1). Moreover, RSV is increasingly recognized as a significant cause of disease in the elderly population and can often be fatal for patients with compromised immune systems (2). In parallel with its human counterpart, bovine RSV (BRSV) constitutes a major cause of respiratory disease in calves, resulting in substantial economic losses to the cattle industry worldwide (3). Despite the substantial health and economic burden caused by RSV illness, there is currently no human vaccine or antiviral drug available. The only significant preventive treatment available is prophylaxis with palivizumab (Synagis), a humanized monoclonal antibody that has provided about 50% protection to high-risk children. Therefore, there is an urgent need to discover compounds capable of blocking RSV infection. Protein-protein interactions are potential targets for antiviral chemotherapy (4). The viral RNA-dependent RNA polymerase (RdRp) complex represents an attractive target for drug discovery, because the different components have no cellular ortholog and are highly conserved between RSV strains. The mechanism of action of this complex involves highly specific and regulated protein-protein and RNA-protein interactions that we need to understand in order to facilitate drug design approaches.
RSV belongs to the Pneumovirus genus of the Paramyxoviridae family, order Mononegavirales. The RSV genome is a single-strand, negative-sense RNA of about 15 kb that contains 10 transcriptional units encoding 11 proteins. The RSV genomic RNA is packaged by the viral nucleoprotein (N) at all times, forming an N-RNA complex called the nucleocapsid (NC). The viral RdRp uses this ribonucleoprotein complex as a template for mRNA transcription and genomic or antigenomic RNA replication (5). The RdRp is composed of two multifunctional proteins, the large protein (L), harboring the capping, methyltransferase, and polyadenylation activities (5), and the tetrameric phosphoprotein (P). The RSV RdRp in transcription mode also associates with the viral protein M2-1, which acts as an antiterminator/elongation factor (6–8). All the components of the RdRp machinery can be found concentrated in cytoplasmic inclusion bodies (IBs), which are, by analogy with those of Rhabdoviridae (9, 10), considered to be viral factories where viral RNA synthesis takes place. Complete atomic structures of N and M2-1 are now available (11–13). No atomic structure has been resolved, even partially, for P or L. However, the crystal structure of the human metapneumovirus (HMPV) P oligomerization domain, formed by a tetrameric coiled-coil, has recently been resolved (14). Since the RSV and HMPV P proteins are highly similar, with 78% identical residues between them, the RSV P oligomerization domain should include at least residues 130 to 152, where a coiled-coil domain is also predicted (15).
The P protein is the main L cofactor and is essential for the formation of an active polymerase complex, allowing the L protein to gain access to the nucleocapsid, where the viral genome is sequestered; P interacts with both the L protein and the N protein simultaneously (16). The P protein has been shown to present multiple sites of phosphorylation, at threonine residues 46 and 108, serine residues 30, 39, 45, 54, 116, 117, 119, 156, 161, 232, and 237, and potentially also Ser86, Ser94, and Ser99 (17–26). However, the major phosphorylation sites of P are dispensable for RSV replication in vitro (24), and the exact role of phosphorylation in P activity is still debated. The RSV P protein forms highly stable tetramers and can be divided into three domains: an N-terminal domain (PNTD, comprising residues 1 to ∼120), a central oligomerization domain (POD, comprising residues ∼120 to 160), and a C-terminal domain (PCTD, comprising residues 161 to 241) (15, 22, 27, 28). PNTD and PCTD are predicted to be disordered regions (29), although some putative short α-helices have been predicted between residues 14 and 25 and between residues 220 and 228 (27). Although it is now well established that the last 9 C-terminal residues of PCTD are critical for binding to N-RNA complexes (30, 31), a second region encompassing residues 161 to 180 could also be involved in N binding (32). However, the L-binding domain of P is still debated. In this work, we have investigated the P-L interactions by using recombinant proteins. The RSV L protein was expressed using a baculovirus vector. This recombinant L protein was able to bind in vitro to recombinant P purified from bacteria. Serial deletions of P residues showed that the main L-binding domain extends from residue 212 to residue 239 in the C-terminal region of P. Using site-directed mutagenesis, we identified hydrophobic residues critical for the P-L interaction, which were also shown, by use of an RSV minigenome system, to be critical for the function of the polymerase. Taken together, our results reveal that the L-binding domain of P encompassing residues 212 to 239 constitutes a molecular recognition element (MoRE).
MATERIALS AND METHODS
Plasmid constructs for bacterial expression.
Plasmid pGEX-P has been described previously (15). The sequences of P with C-terminal deletions were produced by introducing stop codons at the appropriate sites in the coding sequence of pGEX-P using the QuikChange II site-directed mutagenesis kit (Agilent Technologies, Les Ulis, France). Constructs with N-terminal deletions of the P sequence were produced by PCR amplification using Phusion High-Fidelity DNA polymerase (Thermo Scientific) and were cloned into pGEX-4T-3 (GE Healthcare Life Sciences) at BamHI-XhoI sites. Constructs with internal deletions of the P sequence were produced by PCR amplification using a 5′-phosphorylated internal primer pair as described by Byrappa et al. in 1995 (33).
Expression and purification of recombinant proteins from Escherichia coli.
Escherichia coli BL21(DE3)pLysS chemically competent cells (Novagen, Madison, WI) transformed with pGEX-4T-3-derived constructs were grown at 37°C for 8 h in 100 ml of Luria-Bertani (LB) medium containing ampicillin (100 μg/ml). The same volume of fresh LB medium was then added, and protein expression was induced by the addition of 80 μg/ml isopropyl-β-d-thiogalactoside (IPTG) to the medium. The bacteria were incubated for 15 h at 28°C and were then harvested by centrifugation. Bacterial pellets were resuspended in lysis buffer (50 mM Tris-HCl [pH 7.8], 60 mM NaCl, 1 mM EDTA, 2 mM dithiothreitol [DTT], 0.2% Triton X-100, 1 mg/ml chicken egg lysozyme) supplemented with Complete protease inhibitor cocktail (Roche, Mannheim, Germany). Lysates were incubated for 1 h on ice, treated for 15 min with 1 U/ml Benzonase nuclease (Novagen, Madison, WI), and centrifuged at 4°C for 30 min at 10,000 × g. Glutathione-Sepharose 4B beads (GE Healthcare, Uppsala, Sweden) were added to clarified supernatants and were incubated at 4°C for 4 h. The beads were then washed twice in lysis buffer and once in phosphate-buffered saline (PBS) and were then stored at 4°C in an equal volume of PBS.
Plasmid constructs for mammalian expression.
Plasmids pN, pP, pM2-1, and pL, designed for the expression of RSV proteins N, P, M2-1, and L in BSRT7/5 cells, have been described previously (31, 34). They all contain a T7 transcription promoter, an encephalomyocarditis virus internal ribosome entry site (IRES) to enhance gene expression, and a T7 transcription terminator. The pL-HA and pL-EGFP plasmids have been described previously (34). The pM/Luc subgenomic replicon, which encodes the firefly luciferase (Luc) gene under the control of the M-SH gene start sequence, was derived from the pM/SH subgenomic replicon (8) and has been described previously (31). Point mutations were introduced into pP by site-directed mutagenesis as described above. For the expression of P fragments in BSRT7/5 cells, P fragments were PCR amplified and subcloned into pP in place of the P open reading frame (ORF) at KpnI-BamHI sites between the IRES and the T7 terminator. All sequences were from human RSV strain Long, ATCC VR-26 (GenBank accession no. AY911262.1). All constructs were verified by sequencing.
L protein constructs and expression in insect cells.
A codon-optimized sequence of the RSV L protein ORF (strain Long) was synthesized (ProteoGenix, Schiltigheim, France) and was cloned into the pFastBac Dual vector (Invitrogen) under the control of the polyhedrin promoter at BamHI-SacI sites. The synthetic L ORF was designed with a hemagglutinin (HA) epitope tag (YPYDVPDYASLGGP) inserted between residues 1738 and 1739 of the original sequence, and a hexahistidine tag was added at the 3′ end of the L ORF. For the coexpression of P and L, a codon-optimized sequence of the RSV P protein ORF (Long strain) was also synthesized (ProteoGenix, Schiltigheim, France) and cloned into the L ORF-containing pFastBac Dual vector under the control of the p10 promoter at KpnI-SmaI sites. Recombinant baculoviruses were recovered using the Bac-to-Bac baculovirus expression system (Invitrogen). A baculovirus expressing no foreign protein (Bac) was used as a control. Sf9 cells from Spodoptera frugiperda were infected at a multiplicity of infection (MOI) of 1 for 72 h and were then pelleted and stored at −80°C.
Pulldown assays.
Sf9 cells were infected with baculoviruses expressing L (Bac-L) or LNTD (Bac-LNTD) at an MOI of 1 for 72 h and were then harvested and resuspended in ice-cold lysis buffer (50 mM Tris-HCl [pH 7.4], 300 mM NaCl, 10% glycerol, 0.5% Triton X-100) supplemented with Complete protease inhibitor cocktail (Roche, Mannheim, Germany). The cells were lysed with a Dounce homogenizer, and the lysate was incubated on ice for 1 h. The lysates were clarified for 30 min at 10,000 × g and 4°C. Beads coated with purified recombinant glutathione S-transferase (GST)-tagged P protein (wild type [WT] or mutant) were incubated for 30 min at room temperature with Sf9 cell lysates. The beads were washed extensively with lysis buffer, boiled in an equal volume of Laemmli sample buffer, and analyzed by SDS-PAGE and silver staining or immunoblotting.
Immunoblotting.
Samples were boiled in Laemmli buffer, and proteins were resolved by SDS-PAGE and were transferred for 2 h at 100 V and 4°C to nitrocellulose membranes. The membranes were incubated in blocking solution (PBS–0.1% [vol/vol] Tween 20 supplemented with 5% [wt/vol] nonfat milk) for 1 h. The blots were rinsed with PBS–0.1% (vol/vol) Tween 20 and were incubated overnight at 4°C with primary antibodies in blocking solution. The membranes were rinsed as described above and were incubated for 30 min with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies diluted in blocking solution. The membranes were again rinsed as described above, and immunodetection was performed using the Clarity Western ECL substrate (Bio-Rad). Western blots were either exposed to X-ray film or imaged with a GeneGnome machine (Syngene).
Antibodies.
The following primary antibodies were used for immunofluorescence and/or immunoblotting: a mouse monoclonal anti-P protein antibody (clone 21/2P; kindly provided by José Melero, Madrid, Spain), rabbit anti-P and anti-N antisera described previously (15), a mouse monoclonal anti β-actin antibody (mAbcam 8224), and a rat monoclonal anti-HA antibody conjugated with horseradish peroxidase (Roche). Alexa Fluor 594-coupled secondary antibodies directed against rabbit IgG (Invitrogen) were used for immunofluorescence. HRP-coupled secondary antibodies directed against mouse and rabbit IgG (PARIS, Compiègne, France) were used for immunoblotting experiments.
Cell culture and transfections.
BHK-21 cells (clone BSRT7/5) constitutively expressing a cytoplasmic form of the T7 RNA polymerase (35) were grown in Dulbecco modified essential medium (DMEM) (Lonza, Cologne, Germany) supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, and antibiotics. Cells were transfected using Lipofectamine 2000 (Invitrogen, Cergy-Pontoise, France) as described by the manufacturer. Spodoptera frugiperda (Sf9) cells were cultured at 27°C in Grace's insect medium (Gibco) supplemented with 10% FCS. Sf9 transfection for baculovirus recovery was carried out using Lipofectin reagent (Invitrogen).
Minigenome transcription assay.
BSRT7/5 cells at 90% confluence in 24-well dishes were transfected with a plasmid mixture containing 0.25 μg of pM/Luc, 0.25 μg of pN, 0.25 μg of pP, 0.125 μg of pL, and 0.06 μg of pM2-1, as well as 0.06 μg of the p-β-Gal plasmid (Promega) to normalize transfection efficiencies. The dicistronic subgenomic replicon pM/Luc contains the authentic M-SH gene junction and the Luc reporter gene downstream of the gene start sequence present in this gene junction (34, 36, 37). The assay was performed three times, and each independent transfection was carried out in triplicate. Cells were harvested at 24 h posttransfection and were then lysed in luciferase lysis buffer (30 mM Tris [pH 7.9], 10 mM MgCl2, 1 mM DTT, 1% Triton X-100, and 15% glycerol). Luciferase activities were determined for each cell lysate with an Infinite 200 Pro microplate reader (Tecan, Männedorf, Switzerland) and were normalized to β-galactosidase (β-Gal) expression levels.
Fluorescence microscopy.
Immunofluorescence microscopy was performed with cells grown on coverslips and previously transfected with pN, pP, and pL-EGFP (34). At 24 h posttransfection, cells were fixed with 4% paraformaldehyde (PFA) for 25 min, made permeable, and blocked for 30 min with PBS containing 0.1% Triton X-100 and 3% bovine serum albumin (BSA). Cells were then successively incubated for 1 h at room temperature with primary and secondary antibody mixtures diluted in PBS containing 0.3% BSA. For nucleus labeling, cells were exposed to Hoechst 33342 stain (Invitrogen) during incubation with secondary antibodies. Coverslips were mounted with ProLong Gold antifade reagent (Invitrogen). Cells were observed with a Nikon TE200 microscope equipped with a CoolSNAP ES2 (Photometrics) camera, and images were processed with Meta-Vue software (Molecular Devices).
Coimmunoprecipitation assay.
Forty-five minutes before transfection, BSRT7/5 cells were infected with modified vaccinia virus strain Ankara expressing T7 RNA polymerase (MVA-T7) (38, 39) at an MOI of 3. The medium was replaced with fresh DMEM, and the cells were cotransfected with pP (wild-type or mutant plasmid) and pL-HA by use of Lipofectamine 2000. After 16 h, the medium was replaced by PBS containing 2.5 mM EDTA for 5 min at room temperature. Cells were then resuspended and pelleted in microcentrifuge tubes for 5 min at 2,000 × g. Cells were lysed for 1 h at 4°C in ice-cold lysis buffer (50 mM Tris HCl [pH 7.4], 2 mM EDTA, 1 mM DTT, 150 mM NaCl, 0.5% Triton X-100, 10% glycerol) with Complete protease inhibitor cocktail (Roche), and coimmunoprecipitation experiments were performed on cytosolic extracts. Cell lysates were incubated for 1 h at 4°C with an anti-HA monoclonal antibody or an anti-P polyclonal antibody coupled to protein G-coated magnetic beads (Dynabeads; Invitrogen). The beads were then washed twice with lysis buffer and once with PBS, and proteins were first eluted in Laemmli buffer at 95°C for 5 min and then subjected to SDS-PAGE and immunoblotting as described above.
RESULTS
Expression of recombinant HA-tagged L protein.
On the basis of previous studies, it is known that RSV L protein can be expressed in insect cells by using a recombinant baculovirus and a codon-optimized L sequence (40). However, expression of L was conditioned by the presence of P. We determined previously that an L variant with the HA epitope tag inserted between residues 1738 and 1739 in variable region 2 (VR2) (amino acids [aa] 1718 to 1764) is functional and can be detected by immunofluorescence using anti-HA antibodies (34). Based on these data, a codon-optimized version of the L open reading frame containing the HA epitope tag inserted between residues 1738 and 1739 was used to engineer a recombinant baculovirus expression vector (Bac-L). A P/L dual baculovirus expression vector (Bac-L/P) was also engineered for coexpression of HA-tagged L (L-HA) and P. Lysates from cells infected with Bac-L, Bac-L/P, or control Bac were analyzed by Western blotting using anti-HA and anti-P antibodies. As shown in Fig. 1, P was observed only in Bac-L/P-infected cells, as expected. Two major bands were observed for L with lysates of Bac-L-infected cells, one migrating with an apparent mass of ∼250 kDa, as expected for full-length L, and the other corresponding to a truncated form (∼200 kDa) of L (designated L*). The same bands were observed with lysates of Bac-L/P-infected cells, but the expression levels of L were higher in the presence of P, as expected (Fig. 1). Analysis of these two bands from a representative gel by excision, trypsin digestion, and mass spectrometry revealed that the two bands contained L polypeptides and that the smaller L* fragment was truncated at the C-terminal end, after residue 1805 of HA-tagged L, corresponding to residue 1789 of the original L sequence. The relative abundance of this shorter form of L increased during the storage of purified complexes at 4°C. This observation confirmed the high instability of L and the fact that the presence of P was not sufficient to prevent L cleavage in this system.
FIG 1.
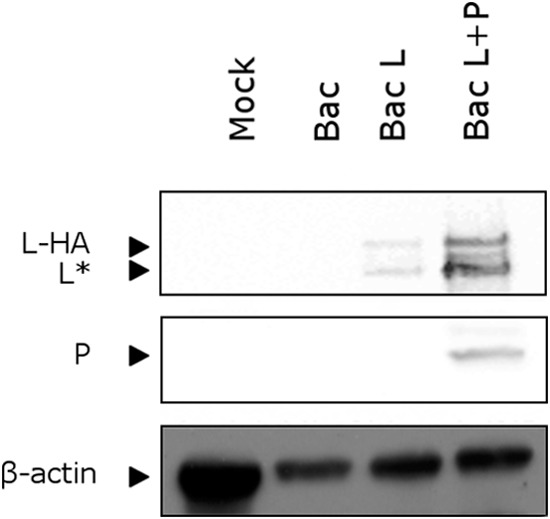
Expression of recombinant RSV HA-tagged L protein in insect cells. Recombinant baculoviruses expressing L-HA alone (Bac-L), coexpressing L-HA and P proteins (Bac-L/P), or expressing no foreign proteins (Bac) were used to infect Sf9 cells. At 72 h postinfection, cells were lysed, and the presence of L-HA and P in the soluble fraction was analyzed by Western blotting using anti-HA and anti-P antibodies. L* corresponds to the truncated form of L (residues 1 to 1789 of the original L sequence). A lysate from mock-infected Sf9 cells (Mock) was also included as a negative control. β-Actin was used as an internal standard. Western blots were imaged with a GeneGnome machine (Syngene).
In conclusion, although the expression level of L is lower when it is expressed alone in this system, coexpression of L and P would result in the formation of a P/L complex, which is obviously not suitable for the mapping of L-P binding sites. We therefore used Bac-L-infected cell lysates for further studies.
The C-terminal region of P contains an L binding site.
We first tried to determine whether the recombinant L produced in baculovirus-infected insect cells could bind to P in vitro by using GST pulldown assays in the presence of either full-length P or the truncated forms P[1-126], P[1-161], P[120-160], P[127-241], or P[161-241]. These P fragments were selected according to the molecular organization of RSV P (see the introduction) (Fig. 2A). A P construct with the tetramerization domain deleted (P[Δ122-160], or PΔOD) was also included in this first test. Full-length and truncated versions of the P protein fused to GST were expressed in bacteria, purified on glutathione-Sepharose beads, and incubated with lysates of Bac-L-infected cells. After extensive washing, complexes were resolved by SDS-PAGE. The presence of L was analyzed by silver staining and Western blotting, while GST-P fragments were revealed by Coomassie blue staining. As shown in Fig. 2B, L was pulled down by GST-P, GST-P[127-241], and GST-P[161-241]. No binding of L was observed with either PNTD (GST-P[1-126]), POD (GST-P[120-160]), or both domains together (GST-P[1-160]). Surprisingly, we observed that deletion of the P oligomerization domain (P[Δ122-160]) significantly and reproducibly increased the efficiency of L binding to P. In contrast to what was observed with the other constructs, the band corresponding to L was clearly visible after silver staining when GST-P[Δ122-160] was used for these binding assays. Bands corresponding to proteins with molecular weights higher than that of L were also present in all samples.
FIG 2.

A major L-binding domain is present in the C-terminal region of RSV P. (A) Schematic diagram of the GST-P fusion protein and of P deletion mutants used for P-L interaction studies. The names of the mutant GST-P proteins indicate the first and last remaining amino acids or deleted amino acids (GST-P[Δ122-160]), and the remaining sequences are shown as solid bars. The N-terminal (PNTD), oligomerization (POD), and C-terminal (PCTD) domains of P are indicated by arrows above the diagram. (B) P and truncated forms of P fused to GST and purified from bacteria on glutathione-Sepharose beads were incubated with lysates from Sf9 cells infected with the recombinant baculovirus expressing L-HA (Bac-L). After extensive washing, the presence or absence of L-HA was determined by SDS-PAGE and silver staining (top) or Western blotting (center) using anti-HA antibodies. The black star on the right of the silver-stained gel indicates contaminant proteins of higher molecular weight than L. GST-P proteins were visualized by SDS-PAGE and Coomassie blue staining (bottom).
Taken together, these results revealed that recombinant GST-P fusion proteins expressed in bacteria are capable of interacting with the recombinant L present in Bac-L-infected cell lysates and that an L-binding domain is located in PCTD.
Fine mapping of an L molecular recognition element (MoRE) in P.
Since we obtained the strongest L-P binding with P[Δ122-160], we used this oligomerization domain-deleted construct instead of full-length P for further GST pulldown assays and fine mapping of the L-binding domain(s). This construct was then named PΔOD. A total of 8 overlapping deletions of 20 or 30 amino acids each were generated throughout the 202 amino acids of the PΔOD protein. Complexes were resolved by SDS-PAGE; GST-P constructs were visualized by Coomassie blue staining; and the presence of L was determined by Western blotting. Figure 3 A shows that, of the 8 constructs tested, 2, GST-PΔOD[Δ197-226] and GST-PΔOD[Δ212-241], lost their capacity to bind L. These data confirmed that an L-binding domain is present at the C terminus of P.
FIG 3.

Mapping the region of P involved in L binding by serial deletions. (A and B) Internal overlapping deletions of 20 to 30 (A) or 10 (B) amino acids were made throughout GST-PΔOD. (C) Stop codons were introduced between residues 232 and 241 of the original P sequence. The diagrams of the P deletion mutants used in these assays are shown to the left of the corresponding gels. Numbers indicate the first and the last residues of the deleted region of P. The presence of L-HA after GST pulldown was analyzed by Western blotting using anti-HA antibodies and exposure to X-ray film. GST-PΔOD-derived complexes were visualized by SDS-PAGE and Coomassie blue staining.
Based on these results, and in order to map this L-binding domain with more accuracy, a second series of 12 constructs with internal overlapping deletions of 10 amino acids each in the C-terminal half of PΔOD between residues 177 and 241 was engineered. All the deletions in the C-terminal region extending from amino acid 216 to 241 abrogated the P-L interaction (Fig. 3B). These results revealed that the region of P encompassing amino acid residues 216 to 241 is necessary for L binding, at least in vitro.
It has been determined previously that the last 9 residues of P are necessary and sufficient for in vitro binding to ring-shaped N-RNA complexes and that the last Phe residue is particularly critical (31, 41). The results obtained with P deletions suggested that the L- and N-RNA-binding domains could overlap. We thus introduced a series of stop codons between residues 232 and 241 of the original P sequence in the GST-PΔOD construct and tested the interaction with L by GST pulldown. As shown in Fig. 3C, efficient L binding, similar to that observed with the full-length GST-PΔOD, was recovered with GST-PΔOD constructs lacking only the two last residues of P (Asp-Phe). These data showed that the domain extending from residue Leu216 to Leu239 of P, and thus partially overlapping the N-RNA-binding domain, is necessary for efficient binding to L.
Effects of targeted P gene mutations on RSV polymerase activity.
The formation of a P-L complex is necessary for RNA polymerase activity. We thus used a functional test to identify the residues critical for RdRp activity within the region encompassing amino acids 216 to 239. This functional assay uses an intracellular plasmid-based minireplicon system, which has been described previously, to study RSV RNA synthesis (12, 13, 31, 34, 41). In this system, BSRT7/5 cells expressing T7 RNA polymerase are cotransfected with the subgenomic replicon pM/Luc and plasmids pCMV-β-gal, pN, pL, pM2-1, and pP (WT or mutant), resulting in the replication and transcription of the minigenome and the expression of reporter luciferase. Hence, the production of Luc protein is dependent on these processes. Luciferase activities were determined and normalized based on β-galactosidase expression. In a first attempt to identify conserved residues among members of the genus Pneumovirus, the C-terminal regions of the P proteins of human RSV, BRSV, ovine RSV (ORSV) strains, pneumonia virus of mice (PVM), and canine pneumovirus (CPV) were aligned. Whereas the sequences of PCTD can be aligned until residue 230, two different alignments could be obtained for the 10 most C-terminal residues (Fig. 4A). Compared with all RSV strains, PVM and CPV have an insertion of 7 residues in this region corresponding to RSV P residues 230 to 241. Thus, from this sequence comparison, two stretches of residues can be distinguished: the first corresponds to RSV P residues 216 to 230 and the second to RSV P residues 231 to 241. These two domains are separated by a linker of variable length among pneumoviruses.
FIG 4.

Effects of P point mutations on RSV polymerase activity. (A) Sequence alignments of the C-terminal region of P of the pneumoviruses human RSV (HRSV), bovine RSV (BRSV), ovine RSV (ORSV), pneumonia virus of mice (PVM), and canine pneumovirus (CPV) (GenBank accession numbers AAX23990.1, NP_048051.1, Q83956.1, Q5MKM7.1, and AHF88957.1, respectively) were made by Clustal Omega and prepared with ESPript 3. Two possible alignments between the most C-terminal residues of the different P proteins are shown. (B) Analysis of RSV polymerase activity with WT P and P substitution mutants. BSRT7/5 cells were transfected with RSV pP, pN, pL, and pM2-1 plasmids and an RSV-specific minigenome containing the firefly luciferase reporter gene, together with p-β-Gal, constitutively expressing β-galactosidase. Luciferase activity, measured 24 h after transfection, was normalized to β-galactosidase activity, and the luciferase activity gained with WT P was set to 100%. The mean values and confidence intervals (error bars) result from 3 separate experiments performed in triplicate. A negative control without P was run. (C) The expression of the different mutants was controlled in cell lysates by Western blotting using a polyclonal anti-P rabbit serum after 24 h of transfection and was compared to the expression of α-tubulin, used as an internal standard.
Since residues 233 to 241 of RSV P constitute an N-RNA-binding domain (31), mutagenesis of this region could affect not only L-P but also P–N-RNA binding and therefore RNA synthesis. Based on these data, we restricted site-directed mutagenesis to amino acids located in the region encompassing amino acids 212 to 232. It is worth noting that sequence alignments revealed the presence of two residues highly conserved among pneumoviruses, L216 and L223, and the conservation of two other hydrophobic residues at positions 226 and 227 for RSV P (Fig. 4A). Residues were then replaced by Ala, and the effects on RNA polymerase activity were analyzed using the minigenome system. As shown in Fig. 4B, the single substitutions L216A, L223A, and L227A reduced RNA polymerase activity to background levels, while L226A had no significant effect. Double (L216A L223A, L216A L227A, and L223A L227A) or triple (L216A L223A L227A) mutants were also generated. In these cases, RdRp activity was no longer detectable. Among the 7 other Ala substitutions tested, 3, K222A, N230A, and D231A, induced a loss of 50% of the polymerase activity, whereas the P218A, E221A, N224A, and E228A mutations had no visible effect.
We replaced Ser and Thr residues representing potential targets for phosphorylation, which could modulate P function and viral RNA synthesis (T219 and S220), with either Ala (phosphoablatant) or Asp (phosphomimetic). S232, the main phosphorylation site of RSV P (20, 21), was also targeted for mutagenesis. Surprisingly, the T219A and S220A mutants exhibited slight increases in polymerase activity, whereas replacement of S220 and, to a lesser extent, T219 with Asp had an inhibitory effect on viral polymerase. Thus, although it is not known whether these two residues can be phosphorylated during the natural viral cycle, the presence of a negative charge in this region is deleterious for RSV polymerase activity. In contrast, with regard to the main P phosphorylation site S232, no effect on RNA polymerase was observed with S232A and S232D mutants, confirming previous reports (23, 24). Since the periodicity (i, i+3 and i, i+4) of critical residues identified suggests that this domain could fold into an α-helix (see Fig. 7), the N224 residue was replaced by Pro, a known α-helical breaker residue. This mutation was shown to reduce RNA polymerase activity to background levels (Fig. 4B), supporting the hypothesis of a structured domain of interaction. Finally, as assessed by Western blotting, all of the P mutants used in this study were expressed in similar amounts in BSRT7/5 cells (Fig. 4C).
FIG 7.

Helical-wheel projection of the L-binding domain of P, encompassing residues 216 to 234. (A) Summary of the effects of amino acid substitutions in the region of P encompassing amino acids 216 to 239 on RdRp activity, L-P interaction (as assessed by coimmunoprecipitation [CoIP]), and the presence of L in IBs. The amino acid sequence of the region of P encompassing amino acids 210 to 241 is given at the top. The nucleocapsid binding domain (NCBD) and L-binding domain (LBD) are indicated, and the predicted α-helix is represented. For RdRp activity, a minus sign, a plus-or-minus sign, and a plus sign indicate activities of ≤5%, ≤50%, and ≥50%, respectively. nd, not determined. (B) Assuming that the MoRE of P adopts a helical conformation upon the binding of L, we made a helical-wheel representation of this region using the HeliQuest program. This projection shows three leucines (L216, L223, and L227), which most likely constitute the direct interface for interaction with L, clustered on one side of the P molecular recognition helix. Hydrophobic residues are shown in yellow, negatively charged residues in red, and positively charged residues in blue. Red letters N and C indicate the N and C termini, respectively. Amino acids for which RdRp activity was reduced to <50% or <5% are labeled with one or two stars, respectively. The residue N224, replacement of which by Pro abrogated polymerase activity, is labeled with a number sign (#).
Residues of P that are critical for interaction with L.
To confirm that the amino acid residues identified here as critical for RdRp activity were involved in direct P-L interaction, the P mutants generated as described above, and more specifically the L216A, L223A, and L227A single, double, or triple mutants, were tested for their abilities to bind L protein in mammalian cells. We also studied the interaction of L with PΔ[211-236] as a negative control of interaction, and with the N224A, N224P, and S220D mutants of P. BSRT7/5 cells were cotransfected with pP (WT or mutant) and pL-HA and were infected with MVA-T7 to enhance L expression. Before or after immunoprecipitation with a P-specific antiserum, proteins were separated and visualized by Western blotting (Fig. 5A). The L/P ratio was determined in all cases (Fig. 5B). Although L was still coimmunoprecipitated with the L216A, L223A, and L227A single mutants of P, L binding was strongly affected by double (L216A L223A, L223A L227A, and L216A L227A) or triple (L216A L223A L227A) mutations in P, by S220D and N224P substitutions, or by deletion of the region of P encompassing amino acids 211 to 236 (Fig. 5). In contrast, the N224A mutation had no visible effect. When L was coexpressed with P mutants for which L binding was reduced, the L expression levels in cell lysates before immunoprecipitation were also affected, reaching a minimum with the L216A L223A P mutant (∼40% of the level for WT P) (Fig. 5B). In conclusion, these results point to hydrophobic amino acid residues as critical for P-L interaction and indicate that L is protected from degradation when stable P-L complexes are formed. Moreover, the loss of interaction of L with the N224P mutant of P suggests that the L MoRE of P could fold into an α-helix when binding to L. These results also correlated with the minigenome assay results.
FIG 5.

Point mutations affecting the P-L interaction in mammalian cells. (A) BSRT7/5 cells infected with MVA-T7 were transfected with pP (WT or mutant) and pL-HA plasmids. P proteins were immunoprecipitated (IP) from cell lysates 16 h after transfection by using a rabbit anti-P antiserum, and the presence of L-HA in the samples was analyzed by Western blotting using anti-HA antibodies. (Top) Cell lysates; (bottom) IP products. (B) Western blots were imaged with a GeneGnome machine (Syngene), and the L/P ratio was calculated before (filled bars) or after (shaded bars) immunoprecipitation. The L/P ratio is expressed as a percentage of the value obtained for WT P.
The mechanisms of recruitment of RSV L polymerase to cytoplasmic inclusion bodies (IBs), where viral proteins involved in RNA synthesis are concentrated and RNA synthesis is believed to take place, are not well understood. Coexpression of the RSV P and N proteins is sufficient to induce the formation of IBs similar to those observed in RSV-infected cells (42). To investigate whether P mutations affecting P-L interaction could also affect the recruitment of L to IBs, BSRT7/5 cells were cotransfected with pL-EGFP, pN, and pP (WT or mutant). IBs were revealed by an anti-N antiserum, and the presence of enhanced green fluorescent protein (EGFP)-tagged L in these structures was analyzed by epifluorescence. As shown in Fig. 6, L-EGFP clearly colocalized with IBs when coexpressed with WT P and N proteins. In the presence of the L216A single mutant of P, the fluorescence of L-EGFP in IBs was attenuated. In contrast, L-EGFP was not detected in IBs when the L216A L223A double mutant of P was used. Similar results were obtained in the presence of the S220D and N224P mutants. It is noteworthy that IBs revealed by anti-N antibodies were observed with all P mutants considered (Fig. 6 and data not shown). Since the formation of IBs depends on N-P interactions (42), this revealed that the capacity of these P mutants to interact with N was not affected.
FIG 6.

Effects of point mutations targeting the L-binding domain of P on the recruitment of L to cytoplasmic inclusion bodies. BSRT7/5 cells were transfected with pP, pN, and pL-EGFP plasmids. Cells were fixed 24 h after transfection, and the presence of L-EGFP in cytoplasmic inclusion bodies was observed by epifluorescence. The RSV N protein was revealed by immunofluorescence using rabbit polyclonal anti-N (1:500) and Alexa Fluor 594-conjugated goat anti-rabbit (1:2,000) antibodies. Nuclei were stained with Hoechst 33342 stain. Bars, 10 μm.
In conclusion, these results revealed that P residues identified as critical for polymerase activity and located in the region encompassing amino acids 216 to 232 are involved in the interaction with the L protein as well as in the recruitment of L to IBs.
DISCUSSION
For two members of the order Mononegavirales, i.e., vesicular stomatitis virus (VSV), a rhabdovirus, and RSV, a paramyxovirus, it has been determined that the L polymerase is capable of RNA synthesis in vitro, using naked RNA as a substrate, with L initiating RNA synthesis de novo at the 3′ terminus of the leader (Le) RNA (40, 43). For VSV, it was also demonstrated that P serves as a processivity factor for L, even with naked RNA, and that full processivity of the P-L heterocomplex additionally requires the template-associated N protein (43). In living cells, the genomic RNA of RSV is not naked but wrapped by the N protein, forming an N-RNA complex in which RNA is sequestered and hidden. Reading of the RNA sequence by the polymerase requires (i) specific targeting to this template, which is mediated by the interaction between the nucleocapsid and the C terminus of P (31), and (ii) the local opening of the N-RNA complex, since RNA is sequestered in a basic groove located between the two α-helical lobes of N (11). It is now well established that in infected cells, an L-P complex is the minimum functional RdRp (44), with P mediating interactions between L and the N-RNA template. Although the mechanism by which N-RNA disassembles before RNA transcription and replication remains unknown, we can speculate that P, within the L-P complex, participates directly or indirectly in this mechanism. Thus, recently, the N-terminal domain of mumps virus P was shown to bind nucleocapsid and appeared to induce the uncoiling of the helical nucleocapsid (45). Understanding how P and L interact and function is critical not only for the elucidation of the mechanisms of viral replication but also for the design of compounds capable of interfering with these protein-protein interactions in order to block RSV replication. For RSV, these investigations have been hampered mainly by difficulties in expressing, visualizing, or purifying the L protein. Recently, a recombinant functional RSV L protein was obtained by using a codon-optimized L gene expressed with a baculovirus vector (40). However, attempts to express and purify L without P were unsuccessful, indicating that the RSV P protein might be necessary to stabilize L. Since our aim was to map the L-binding domain of P, we focused our efforts on the expression of L alone. A codon-optimized L gene was designed and expressed with a baculovirus expression vector. The insertion of an HA epitope tag into variable region 2 of L allowed us to analyze the expression of L in Bac-L-infected insect cells by Western blotting. Two major bands were observed, one corresponding to full-length L and the other to a shorter form, migrating with an apparent molecular mass of ∼200 kDa and including residues 1 to 1789 of L. This shorter band was also observed when L was coexpressed with P. These results are in agreement with those published by Noton et al. (40) and indicate that the C-terminal region of L located downstream of variable region 2 is more susceptible to degradation than the rest of the protein, even in the presence of P.
The expression level of L in baculovirus-infected insect cells was sufficient for its detection by Western blotting, or by SDS-PAGE and silver staining after in vitro binding to GST-P. Since the L protein present in the cell lysates was soluble at low concentrations and was capable of binding specifically to GST-P, this result indicated that the L protein was correctly folded. We estimate that the concentration of L is approximately 1 to 10 ng per ml of cell lysate. However, our attempts to concentrate L were unsuccessful, resulting in protein aggregation (data not shown). Furthermore, when L was purified on beads charged with Ni2+, it was not possible to recover it as a soluble form after elution with imidazole. Our results are in agreement with the data obtained for human parainfluenza virus type 3, for which it was shown that in the absence of P, L tends to aggregate during the concentration step (46). Thus, it is likely that RSV P binds to or modifies the domains of L that are responsible for L aggregation at higher concentrations.
We thus used lysates from insect cells expressing this recombinant L protein to map the L-binding domain(s) of P by using truncated forms of P. We first observed that in our in vitro system using GST-P fusion proteins purified from E. coli, L-P binding was facilitated when the P oligomerization domain was removed (P[Δ122-160]). In addition, the isolated P oligomerization domain was not able to bind to L. Finally, an L-binding domain was mapped to the region encompassing residues 216 to 239 of P. However, because the P protein fragments were unstable when expressed in mammalian cells (data not shown), it was not possible to validate these results in mammalian cells.
Validation of the region of P encompassing amino acids 216 to 239 as an L MoRE, and identification of residues critical for the interaction with L, was further carried out by correlating functional assays and coimmunoprecipitation experiments from mammalian cells, using P point mutants. Based on a sequence alignment of the C termini of different paramyxoviruses, we focused on residues 216 to 232 of P. Among the 15 residues mutated and tested using a minigenome assay, 8 were shown to be critical for polymerase activity. More specifically, mutations of the three hydrophobic residues L216, L223, and L227 to Ala induced reductions of 80 to 90% in polymerase activity. It is also noteworthy that substitution of Asp for residues T219 and S220, which mimic phosphorylation, partially or totally impairs polymerase activity, respectively. These data suggest that although these residues have not been identified as P phosphorylation sites previously, phosphorylation of these residues could be implicated in the regulation of the P-L interaction. In order to study the role of these residues of P in the interaction with L, we thus performed immunoprecipitation assays of L in the presence of these mutants. Because L is expressed at low levels in BSRT7/5 cells transfected with a T7-driven L expression vector, we used the attenuated recombinant vaccinia virus MVA-T7, which overexpresses the T7 RNA polymerase with reduced cytopathic effects in mammalian cells (38, 39), to increase L expression levels. Using full-length P with mutations of Leu residues identified as critical for polymerase activity, we showed that coimmunoprecipitation of L was strongly reduced with these P mutants.
A short α-helix has been predicted in the region of P encompassing amino acids 220 to 228 (27). Our results are in agreement with this prediction, since substitution of Ala for L216, L223, and L227, which are clustered on the same side of the theoretical helix (Fig. 7), abolished polymerase activity. Furthermore, replacement of Asn224 by Pro, which is known to break α-helices, abrogated both RdRp activity and L-P interaction. Thus, it is likely that this region exists as, or folds into, an α-helix when binding to its L partner and that L-P interaction involves mainly hydrophobic interactions. When the L216A L223A, S220D, or N224P mutant of P was substituted for WT P for coexpression with L in mammalian cells, the L expression level was reduced to 40%, 75%, or 56%, respectively, and a strong reduction in P-L interactions was observed by immunoprecipitation, indicating that this putative α-helix is critical for L-P interaction and hence for L stabilization. These results were correlated with the absence of L-EGFP in cytoplasmic IBs when L-EGFP was coexpressed with the same P mutants. Although the stability of L-EGFP was probably also reduced by 25 to 60% in the presence of the three P mutants, these results suggest that the L-binding region of P encompassing amino acids 216 to 239 is also critical for the recruitment of L to cytoplasmic IBs.
Attempts to map the L-binding domain of RSV P have been made previously with human RSV (HRSV) and bovine RSV (BRSV). For BRSV, Khattar et al. mapped the L-binding site to a region of P encompassing amino acids 121 to 160 (32), which was further identified as the HRSV P oligomerization domain (POD) (15, 22, 27). On the other hand, Asenjo et al. observed that for HRSV, deletion of the POD reduced its capacity to interact with L by ∼50% but did not abolish that capacity, whereas deletion of amino acids 203 to 241 had a more pronounced negative effect on P-L interaction (47). In both studies, the authors used transfection in HEp-2 cells and infection with a T7-expressing vaccinia virus. Since no anti-L antibodies were available, the L protein was visualized by [35S]methionine radioactive labeling after coimmunoprecipitation with P. Despite this, the coimmunoprecipitation of L with P was poor and difficult to quantify. Since P or L sequence conservation between BRSV and HRSV is very high, it is unlikely that BRSV and HRSV use different P-L interaction domains. Our results are in agreement with those obtained by Asenjo et al., and it is more likely that destabilization of P oligomerization in vivo strongly lowers P-L avidity, which should be compensated for by the use of dimeric GST in vitro and high protein concentrations in the GST pulldown assays. Our results also indicate that the presence of GST at the N terminus of full-length P generates higher-order oligomers, which are less adapted for GST pulldown and protein-protein interaction studies.
Comparison with other members of the order Mononegavirales.
Due to technical difficulties, few reports describe the mapping and characterization of the L-binding domain of P. The L-binding domain within the P protein of Sendai virus has been mapped to aa 412 to 479 (48), in the C-terminal region of P and just downstream of the P tetramerization domain extending from amino acid 320 to 429 (49). For rabies virus (RV) and VSV, the L binding region has been mapped to the N-terminal region of P (50–52). Interestingly, for RV, this L binding site recovers the N binding domain that recognizes specifically the RNA-free form of N called N0. Our results show that for RSV, L- and nucleocapsid-binding domains overlap by 7 residues (aa 233 to 239). It is thus possible that L and N compete for binding to P. Given the respective extents of the L- and nucleocapsid-binding domains of P, which consist of 27 and 9 amino acid residues, respectively, L could have a higher affinity for P than N. Since L is produced in much smaller amounts than N in RSV-infected cells, this could also promote P-L interaction. However, since RSV P is a tetramer (15, 27, 53), it is also possible that P binds to L and to NC simultaneously, using only one or two protomers for the interaction with NC and the other protomer(s) for L binding. However, to address this issue, in vitro competition assays requiring higher concentrations of L protein should be performed.
ACKNOWLEDGMENTS
We thank David Hughes and Patrick England for critical readings of the manuscript, Damien Vitour (ANSES, Maisons-Alfort, France) for anti-N polyclonal rabbit serum, José Melero (Madrid, Spain) for providing monoclonal antibodies, Bruno Da Costa (INRA, Jouy-en-Josas, France) for providing Bac baculovirus, and Jean-François Vautherot (INRA, Tours-Nouzilly, France) for providing MVA-T7.
REFERENCES
- 1.Tregoning JS, Schwarze J. 2010. Respiratory viral infections in infants: causes, clinical symptoms, virology, and immunology. Clin Microbiol Rev 23:74–98. doi: 10.1128/CMR.00032-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins PL, Melero JA. 2011. Progress in understanding and controlling respiratory syncytial virus: still crazy after all these years. Virus Res 162:80–99. doi: 10.1016/j.virusres.2011.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Valarcher JF, Taylor G. 2007. Bovine respiratory syncytial virus infection. Vet Res 38:153–180. doi: 10.1051/vetres:2006053. [DOI] [PubMed] [Google Scholar]
- 4.Loregian A, Marsden HS, Palu G. 2002. Protein-protein interactions as targets for antiviral chemotherapy. Rev Med Virol 12:239–262. doi: 10.1002/rmv.356. [DOI] [PubMed] [Google Scholar]
- 5.Morin B, Kranzusch PJ, Rahmeh AA, Whelan SP. 2013. The polymerase of negative-stranded RNA viruses. Curr Opin Virol 3:103–110. doi: 10.1016/j.coviro.2013.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collins PL, Hill MG, Camargo E, Grosfeld H, Chanock RM, Murphy BR. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc Natl Acad Sci U S A 92:11563–11567. doi: 10.1073/pnas.92.25.11563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fearns R, Collins PL. 1999. Role of the M2-1 transcription antitermination protein of respiratory syncytial virus in sequential transcription. J Virol 73:5852–5864. http://jvi.asm.org/content/73/7/5852.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hardy RW, Wertz GW. 1998. The product of the respiratory syncytial virus M2 gene ORF1 enhances readthrough of intergenic junctions during viral transcription. J Virol 72:520–526. http://jvi.asm.org/content/72/1/520.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lahaye X, Vidy A, Pomier C, Obiang L, Harper F, Gaudin Y, Blondel D. 2009. Functional characterization of Negri bodies (NBs) in rabies virus-infected cells: evidence that NBs are sites of viral transcription and replication. J Virol 83:7948–7958. doi: 10.1128/JVI.00554-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinrich BS, Cureton DK, Rahmeh AA, Whelan SP. 2010. Protein expression redirects vesicular stomatitis virus RNA synthesis to cytoplasmic inclusions. PLoS Pathog 6:e1000958. doi: 10.1371/journal.ppat.1000958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tawar RG, Duquerroy S, Vonrhein C, Varela PF, Damier-Piolle L, Castagne N, MacLellan K, Bedouelle H, Bricogne G, Bhella D, Eléouët JF, Rey FA. 2009. Crystal structure of a nucleocapsid-like nucleoprotein-RNA complex of respiratory syncytial virus. Science 326:1279–1283. doi: 10.1126/science.1177634. [DOI] [PubMed] [Google Scholar]
- 12.Blondot ML, Dubosclard V, Fix J, Lassoued S, Aumont-Nicaise M, Bontems F, Eléouët JF, Sizun C. 2012. Structure and functional analysis of the RNA- and viral phosphoprotein-binding domain of respiratory syncytial virus M2-1 protein. PLoS Pathog 8:e1002734. doi: 10.1371/journal.ppat.1002734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanner SJ, Ariza A, Richard CA, Kyle HF, Dods RL, Blondot ML, Wu W, Trincao J, Trinh CH, Hiscox JA, Carroll MW, Silman NJ, Eléouët JF, Edwards TA, Barr JN. 2014. Crystal structure of the essential transcription antiterminator M2-1 protein of human respiratory syncytial virus and implications of its phosphorylation. Proc Natl Acad Sci U S A 111:1580–1585. doi: 10.1073/pnas.1317262111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leyrat C, Renner M, Harlos K, Grimes JM. 2013. Solution and crystallographic structures of the central region of the phosphoprotein from human metapneumovirus. PLoS One 8:e80371. doi: 10.1371/journal.pone.0080371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castagné N, Barbier A, Bernard J, Rezaei H, Huet JC, Henry C, Da Costa B, Eléouët JF. 2004. Biochemical characterization of the respiratory syncytial virus P-P and P-N protein complexes and localization of the P protein oligomerization domain. J Gen Virol 85:1643–1653. doi: 10.1099/vir.0.79830-0. [DOI] [PubMed] [Google Scholar]
- 16.Ruigrok RW, Crepin T. 2010. Nucleoproteins of negative strand RNA viruses; RNA binding, oligomerisation and binding to polymerase co-factor. Viruses 2:27–32. doi: 10.3390/v2010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Navarro J, Lopez-Otin C, Villanueva N. 1991. Location of phosphorylated residues in human respiratory syncytial virus phosphoprotein. J Gen Virol 72(Part 6):1455–1459. doi: 10.1099/0022-1317-72-6-1455. [DOI] [PubMed] [Google Scholar]
- 18.Mazumder B, Barik S. 1994. Requirement of casein kinase II-mediated phosphorylation for the transcriptional activity of human respiratory syncytial viral phosphoprotein P: transdominant negative phenotype of phosphorylation-defective P mutants. Virology 205:104–111. doi: 10.1006/viro.1994.1624. [DOI] [PubMed] [Google Scholar]
- 19.Mazumder B, Adhikary G, Barik S. 1994. Bacterial expression of human respiratory syncytial viral phosphoprotein P and identification of Ser237 as the site of phosphorylation by cellular casein kinase II. Virology 205:93–103. doi: 10.1006/viro.1994.1623. [DOI] [PubMed] [Google Scholar]
- 20.Barik S, McLean T, Dupuy LC. 1995. Phosphorylation of Ser232 directly regulates the transcriptional activity of the P protein of human respiratory syncytial virus: phosphorylation of Ser237 may play an accessory role. Virology 213:405–412. doi: 10.1006/viro.1995.0013. [DOI] [PubMed] [Google Scholar]
- 21.Dupuy LC, Dobson S, Bitko V, Barik S. 1999. Casein kinase 2-mediated phosphorylation of respiratory syncytial virus phosphoprotein P is essential for the transcription elongation activity of the viral polymerase; phosphorylation by casein kinase 1 occurs mainly at Ser215 and is without effect. J Virol 73:8384–8392. http://jvi.asm.org/content/73/10/8384.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Asenjo A, Villanueva N. 2000. Regulated but not constitutive human respiratory syncytial virus (HRSV) P protein phosphorylation is essential for oligomerization. FEBS Lett 467:279–284. doi: 10.1016/S0014-5793(00)01171-6. [DOI] [PubMed] [Google Scholar]
- 23.Villanueva N, Hardy R, Asenjo A, Yu Q, Wertz G. 2000. The bulk of the phosphorylation of human respiratory syncytial virus phosphoprotein is not essential but modulates viral RNA transcription and replication. J Gen Virol 81:129–133. http://vir.sgmjournals.org/content/81/1/129.long. [DOI] [PubMed] [Google Scholar]
- 24.Lu B, Ma CH, Brazas R, Jin H. 2002. The major phosphorylation sites of the respiratory syncytial virus phosphoprotein are dispensable for virus replication in vitro. J Virol 76:10776–10784. doi: 10.1128/JVI.76.21.10776-10784.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asenjo A, Rodriguez L, Villanueva N. 2005. Determination of phosphorylated residues from human respiratory syncytial virus P protein that are dynamically dephosphorylated by cellular phosphatases: a possible role for serine 54. J Gen Virol 86:1109–1120. doi: 10.1099/vir.0.80692-0. [DOI] [PubMed] [Google Scholar]
- 26.Asenjo A, Calvo E, Villanueva N. 2006. Phosphorylation of human respiratory syncytial virus P protein at threonine 108 controls its interaction with the M2-1 protein in the viral RNA polymerase complex. J Gen Virol 87:3637–3642. doi: 10.1099/vir.0.82165-0. [DOI] [PubMed] [Google Scholar]
- 27.Llorente MT, Garcia-Barreno B, Calero M, Camafeita E, Lopez JA, Longhi S, Ferron F, Varela PF, Melero JA. 2006. Structural analysis of the human respiratory syncytial virus phosphoprotein: characterization of an alpha-helical domain involved in oligomerization. J Gen Virol 87:159–169. doi: 10.1099/vir.0.81430-0. [DOI] [PubMed] [Google Scholar]
- 28.Esperante SA, Paris G, de Prat-Gay G. 2012. Modular unfolding and dissociation of the human respiratory syncytial virus phosphoprotein P and its interaction with the M(2-1) antiterminator: a singular tetramer-tetramer interface arrangement. Biochemistry 51:8100–8110. doi: 10.1021/bi300765c. [DOI] [PubMed] [Google Scholar]
- 29.Simabuco FM, Asara JM, Guerrero MC, Libermann TA, Zerbini LF, Ventura AM. 2011. Structural analysis of human respiratory syncytial virus P protein: identification of intrinsically disordered domains. Braz J Microbiol 42:340–345. doi: 10.1590/S1517-83822011000100043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Slack MS, Easton AJ. 1998. Characterization of the interaction of the human respiratory syncytial virus phosphoprotein and nucleocapsid protein using the two-hybrid system. Virus Res 55:167–176. doi: 10.1016/S0168-1702(98)00042-2. [DOI] [PubMed] [Google Scholar]
- 31.Tran TL, Castagne N, Bhella D, Varela PF, Bernard J, Chilmonczyk S, Berkenkamp S, Benhamo V, Grznarova K, Grosclaude J, Nespoulos C, Rey FA, Eléouët JF. 2007. The nine C-terminal amino acids of the respiratory syncytial virus protein P are necessary and sufficient for binding to ribonucleoprotein complexes in which six ribonucleotides are contacted per N protein protomer. J Gen Virol 88:196–206. doi: 10.1099/vir.0.82282-0. [DOI] [PubMed] [Google Scholar]
- 32.Khattar SK, Yunus AS, Samal SK. 2001. Mapping the domains on the phosphoprotein of bovine respiratory syncytial virus required for N-P and P-L interactions using a minigenome system. J Gen Virol 82:775–779. http://vir.sgmjournals.org/content/82/4/775.long. [DOI] [PubMed] [Google Scholar]
- 33.Byrappa S, Gavin DK, Gupta KC. 1995. A highly efficient procedure for site-specific mutagenesis of full-length plasmids using Vent DNA polymerase. Genome Res 5:404–407. doi: 10.1101/gr.5.4.404. [DOI] [PubMed] [Google Scholar]
- 34.Fix J, Galloux M, Blondot ML, Eléouët JF. 2011. The insertion of fluorescent proteins in a variable region of respiratory syncytial virus L polymerase results in fluorescent and functional enzymes but with reduced activities. Open Virol J 5:103–108. doi: 10.2174/1874357901105010103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J Virol 73:251–259. http://jvi.asm.org/content/73/1/251.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu Q, Hardy RW, Wertz GW. 1995. Functional cDNA clones of the human respiratory syncytial (RS) virus N, P, and L proteins support replication of RS virus genomic RNA analogs and define minimal trans-acting requirements for RNA replication. J Virol 69:2412–2419. http://jvi.asm.org/content/69/4/2412.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tran TL, Castagné N, Dubosclard V, Noinville S, Koch E, Moudjou M, Henry C, Bernard J, Yeo RP, Eléouët JF. 2009. The respiratory syncytial virus M2-1 protein forms tetramers and interacts with RNA and P in a competitive manner. J Virol 83:6363–6374. doi: 10.1128/JVI.00335-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sutter G, Ohlmann M, Erfle V. 1995. Non-replicating vaccinia vector efficiently expresses bacteriophage T7 RNA polymerase. FEBS Lett 371:9–12. doi: 10.1016/0014-5793(95)00843-X. [DOI] [PubMed] [Google Scholar]
- 39.Wyatt LS, Moss B, Rozenblatt S. 1995. Replication-deficient vaccinia virus encoding bacteriophage T7 RNA polymerase for transient gene expression in mammalian cells. Virology 210:202–205. doi: 10.1006/viro.1995.1332. [DOI] [PubMed] [Google Scholar]
- 40.Noton SL, Deflube LR, Tremaglio CZ, Fearns R. 2012. The respiratory syncytial virus polymerase has multiple RNA synthesis activities at the promoter. PLoS Pathog 8:e1002980. doi: 10.1371/journal.ppat.1002980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Galloux M, Tarus B, Blazevic I, Fix J, Duquerroy S, Eléouët JF. 2012. Characterization of a viral phosphoprotein binding site on the surface of the respiratory syncytial nucleoprotein. J Virol 86:8375–8387. doi: 10.1128/JVI.00058-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.García J, Garcia-Barreno B, Vivo A, Melero JA. 1993. Cytoplasmic inclusions of respiratory syncytial virus-infected cells: formation of inclusion bodies in transfected cells that coexpress the nucleoprotein, the phosphoprotein, and the 22K protein. Virology 195:243–247. doi: 10.1006/viro.1993.1366. [DOI] [PubMed] [Google Scholar]
- 43.Morin B, Rahmeh AA, Whelan SP. 2012. Mechanism of RNA synthesis initiation by the vesicular stomatitis virus polymerase. EMBO J 31:1320–1329. doi: 10.1038/emboj.2011.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grosfeld H, Hill MG, Collins PL. 1995. RNA replication by respiratory syncytial virus (RSV) is directed by the N, P, and L proteins; transcription also occurs under these conditions but requires RSV superinfection for efficient synthesis of full-length mRNA. J Virol 69:5677–5686. http://jvi.asm.org/content/69/9/5677.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cox R, Pickar A, Qiu S, Tsao J, Rodenburg C, Dokland T, Elson A, He B, Luo M. 2014. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc Natl Acad Sci U S A 111:15208–15213. doi: 10.1073/pnas.1413268111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chattopadhyay S, Banerjee AK. 2009. Phosphoprotein, P of human parainfluenza virus type 3 prevents self-association of RNA-dependent RNA polymerase, L. Virology 383:226–236. doi: 10.1016/j.virol.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Asenjo A, Mendieta J, Gomez-Puertas P, Villanueva N. 2008. Residues in human respiratory syncytial virus P protein that are essential for its activity on RNA viral synthesis. Virus Res 132:160–173. doi: 10.1016/j.virusres.2007.11.013. [DOI] [PubMed] [Google Scholar]
- 48.Smallwood S, Ryan KW, Moyer SA. 1994. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202:154–163. doi: 10.1006/viro.1994.1331. [DOI] [PubMed] [Google Scholar]
- 49.Tarbouriech N, Curran J, Ruigrok RW, Burmeister WP. 2000. Tetrameric coiled coil domain of Sendai virus phosphoprotein. Nat Struct Biol 7:777–781. doi: 10.1038/79013. [DOI] [PubMed] [Google Scholar]
- 50.Chenik M, Schnell M, Conzelmann KK, Blondel D. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J Virol 72:1925–1930. http://jvi.asm.org/content/72/3/1925.long. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emerson SU, Schubert M. 1987. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc Natl Acad Sci U S A 84:5655–5659. doi: 10.1073/pnas.84.16.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rahmeh AA, Morin B, Schenk AD, Liang B, Heinrich BS, Brusic V, Walz T, Whelan SP. 2012. Critical phosphoprotein elements that regulate polymerase architecture and function in vesicular stomatitis virus. Proc Natl Acad Sci U S A 109:14628–14633. doi: 10.1073/pnas.1209147109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Llorente MT, Taylor IA, Lopez-Vinas E, Gomez-Puertas P, Calder LJ, Garcia-Barreno B, Melero JA. 2008. Structural properties of the human respiratory syncytial virus P protein: evidence for an elongated homotetrameric molecule that is the smallest orthologue within the family of paramyxovirus polymerase cofactors. Proteins 72:946–958. doi: 10.1002/prot.21988. [DOI] [PubMed] [Google Scholar]