

ABSTRACT
The major tegument protein of bovine herpesvirus 1 (BoHV-1), VP8, is essential for virus replication in cattle. VP8 is phosphorylated in vitro by casein kinase 2 (CK2) and BoHV-1 unique short protein 3 (US3). In this study, VP8 was found to be phosphorylated in both transfected and infected cells but was detected as a nonphosphorylated form in mature virions. This suggests that phosphorylation of VP8 is strictly controlled during different stages of the viral life cycle. The regulation and function of VP8 phosphorylation by US3 and CK2 were further analyzed. An in vitro kinase assay, site-directed mutagenesis, and liquid chromatography-mass spectrometry were used to identify the active sites for US3 and CK2. The two kinases phosphorylate VP8 at different sites, resulting in distinct phosphopeptide patterns. S16 is a primary phosphoreceptor for US3, and it subsequently triggers phosphorylation at S32. CK2 has multiple active sites, among which T107 appears to be the preferred residue. Additionally, CK2 consensus motifs in the N terminus of VP8 are essential for phosphorylation. Based on these results, a nonphosphorylated VP8 mutant was constructed and used for further studies. In transfected cells phosphorylation was not required for nuclear localization of VP8. Phosphorylated VP8 appeared to recruit promyelocytic leukemia (PML) protein and to remodel the distribution of PML in the nucleus; however, PML protein did not show an association with nonphosphorylated VP8. This suggests that VP8 plays a role in resisting PML-related host antiviral defenses by redistributing PML protein and that this function depends on the phosphorylation of VP8.
IMPORTANCE The progression of VP8 phosphorylation over time and its function in BoHV-1 replication have not been characterized. This study demonstrates that activation of S16 initiates further phosphorylation at S32 by US3. Additionally, VP8 is phosphorylated by CK2 at several residues, with T107 having the highest level of phosphorylation. Evidence for a difference in the phosphorylation status of VP8 in host cells and mature virus is presented for the first time. Phosphorylation was found to be a critical modification, which enables VP8 to attract and to redistribute PML protein in the nucleus. This might promote viral replication through interference with a PML-mediated antiviral defense. This study provides new insights into the regulation of VP8 phosphorylation and suggests a novel, phosphorylation-dependent function for VP8 in the life cycle of BoHV-1, which is important in view of the fact that VP8 is essential for virus replication in vivo.
INTRODUCTION
Bovine herpesvirus 1 (BoHV-1) is a herpesvirus belonging to the subfamily Alphaherpesvirinae and one of the most common pathogens in cattle. The major clinical symptoms caused by BoHV-1 are infectious bovine rhinotracheitis, conjunctivitis, vulvovaginitis, and balanoposthitis. The virus particle is composed of a capsid containing the double-stranded DNA genome, which is surrounded by a tegument layer and an envelope containing viral glycoproteins. The tegument layer is a stable macromolecular structure formed by tegument proteins, which provide critical functions, such as regulation of transcription (1), kinase functions (2), and virus assembly (3).
Viral protein 8 (VP8), a phosphoprotein encoded by the UL47 gene (4), is the most abundant tegument protein in BoHV-1. VP8 plays an indispensable role in BoHV-1 replication in host animals. A BoHV-1 mutant defective in expression of VP8 cannot establish a productive infection in cattle and poorly replicates in tissue culture (5). Herpesvirus tegument proteins can be posttranslationally modified in several ways (6, 7), and based on the studies with human herpesvirus 1 (HHV-1) (3, 6, 8), it is conceivable that phosphorylation regulates the function of tegument proteins in BoHV-1. The HHV-1 tegument protein VP22 is phosphorylated in infected cells, which promotes expression and packaging of ICP0 (3); the VP22 itself is packaged into mature virus particles only after dephosphorylation (9). In BoHV-1, however, phosphorylation of VP22 is required prior to its incorporation into virions (10). The phosphorylation of HHV-1 VP13/14, a VP8 homologue, initiates dissociation of the structural components of the tegument (6).
Phosphorylation, in addition to potentiating functions, may also play a role in altering the cellular localization of target proteins. For example, HHV-1 VP22 has the capacity to perform nuclear-cytoplasmic shuttling during infection, and the nonphosphorylated form localizes to the cytoplasm while the phosphorylated form localizes to the nucleus (9, 11). BoHV-1 VP8 has been found to shuttle between the nucleus and cytoplasm, and this is mediated by two nuclear localization signals (NLS) and one nuclear exportation signal (NES) (12, 13). However, the impact of phosphorylation on the cellular localization of VP8 is not known.
Promyelocytic leukemia (PML) protein is one of the components of PML nuclear bodies, also known as nuclear domain 10 (ND10). The PML protein contributes to the cellular defense by repressing viral lytic gene expression through modifying the viral genome (14) and thereby plays a key role in reducing herpesvirus replication (15). However, herpesviruses have developed a defense system against the PML-mediated antiviral response. This is supported by evidence that when ICP0 is mutated, HHV-1 replication is reduced by a failure to disrupt PML bodies (16). Further studies demonstrated that viral proteins disrupt PML bodies by interfering with the SUMOylation of PML protein (17). Some tegument proteins also remodel the PML protein. For example, human cytomegalovirus (HCMV) tegument protein UL35 forms nuclear bodies that subsequently recruit PML protein (18).
According to previous in vitro studies, VP8 is phosphorylated by at least two kinases, the unique short protein 3 (US3), a BoHV-1 kinase, and casein kinase 2 (CK2), a cellular kinase (19). The VP8 open reading frame (ORF) translates 741 amino acids, and 9.2% of them are serines and threonines, most of which are within consensus motifs for CK2 and US3. To better understand the role of VP8 phosphorylation during BoHV-1 infection, we investigated the phosphorylation events of VP8 at different stages of the virus life cycle and identified the active sites for US3 and CK2. We also showed that VP8 altered the distribution of PML protein in a phosphorylation-dependent manner.
MATERIALS AND METHODS
Cells and virus.
Madin-Darby bovine kidney (MDBK) cells, African green monkey fibroblast-like (COS-7) cells, and primary fetal bovine testis (FBT) cells were cultured in Eagle's minimum essential medium (MEM; Gibco, Life Technologies, Burlington, ON, Canada) supplemented with 10% fetal bovine serum (FBS; Gibco). Production of BoHV-1 strains 108 and Cooper was carried out in MDBK cells as previously described (20). Briefly, virus infections were accomplished by rocking 150-cm2 85 to 90% confluent cell monolayers with BoHV-1 in 10 ml of MEM at 37°C; the medium was replaced after 1 h with 10 ml of MEM supplemented with 2% FBS, followed by further incubation at 37°C. The virus titer was determined by plaque titration in 24-well plates overlaid with 8% low-melting-point agarose in MEM (20).
Antibodies and chemical reagents.
Monoclonal anti-VP8 antibody, polyclonal anti-VP8 antibody (20), and polyclonal anti-US3 antibody (21) have been generated previously. Polyclonal anti-CK2α (Abcam, Toronto, ON, Canada), monoclonal anti-FLAG (Sigma-Aldrich, St. Louis, MO, USA), polyclonal anti-nucleolin (Abcam), and polyclonal anti-PML (Santa Cruz Biotechnology, Dallas, TX, USA) antibodies are all commercial products. IRDye 680RD goat anti-rabbit IgG and IRDye 800CW goat anti-mouse IgG were purchased from Li-Cor Biosciences (Lincoln, NE, USA). Alexa 488-conjugated goat anti-mouse IgG and Alexa 633-conjugated goat anti-rabbit IgG were purchased from Life Technologies. The inhibitors SNS032 and AT7519 are products from Tocris Bioscience (Bristol, United Kingdom) and SelleckChem (Houston, TX, USA), respectively. Phos-tag acrylamide AAL-107 was purchased from Wako Pure Chemical Industries (Richmond, VA, USA).
Plasmid construction.
The UL47 gene (GenBank accession no. AY530215.1) was cloned into the pFLAG-CMV-2 expression vector (where CMV is cytomegalovirus) (Sigma-Aldrich), as previously described (19). The yellow fluorescent protein (YFP) ORF was cloned into pFLAG-CMV-2 to give pFLAG-YFP. VP8 mutations and deletions were constructed by PCR amplification using primers designed to create either mutations or deletions after ligation of PCR fragments back into the constructs. All primers were synthesized by Life Technologies (Table 1). PCR amplifications were carried out with Q5 Hot Start High-Fidelity DNA Polymerase (New England BioLabs [NEB], Ipswich, MA, USA) according to the manufacturer's instructions. Briefly, PCR was carried out with a 50-μl mixture of 1 ng of template, 1 μM each primer pair, 200 μM concentrations of the deoxynucleoside triphosphates (dNTPs), and 2 U of DNA polymerase. The PCR amplification products were purified with a PCR purification kit (Qiagen, Germantown, MD, USA) and treated with DpnI (NEB). Escherichia coli DH5α competent cells were transformed with the DNA fragments ligated with T4 DNA ligase (NEB) and plated on selective LB agar plates. Plasmid purification was carried out according to the Qiagen miniprep protocol. The selected positive mutants were confirmed by DNA sequencing performed by the NRC-Plant Biotechnology Institute (Saskatoon, SK, Canada).
TABLE 1.
Primer list for plasmid construction using PCR
| Primer target in VP8 | Primer sequence (5′–3′) |
|
|---|---|---|
| Forward | Reverse | |
| Substitutions | ||
| S16A | GCCGGAACGTACCGCACGCAC | GCGGCGCGGGCGGCGCTCAG |
| S32A | GCCCTGCTGGACGCCCTGCG | CCGCCGGGCAGAGGGGCGCTGG |
| S65A | GCCAGTGAGGACGAGAACGTGTATGATTAC | GTCCTCGTCCGGGGGCCGCTG |
| S66A | ACCGCTGAGGACGAGAACGTGTATGATTAC | GTCCTCGTCCGGGGGCCGCTG |
| S79A | GCCAGCGACAGCGCCGACGACTATG | ATCGCCGTCGATGTAATCATACACGTTCTC |
| S80A | AGCGCCGACAGCGCCGACGACTATG | ATCGCCGTCGATGTAATCATACACGTTCTC |
| S82A | GCCGCCGACGACTATGATAGCGATTATTTTACTGC | GTCGCTGCTATCGCCGTCGATG |
| T92A | TTGCTGCTAACCGCGGCCCCAATCAC | AATAATCGCTATCATAGTCGTCGGCGCTG |
| T107A | ACCCGAGCGCGCCCCGGAAG | GGTGCGTCTGCGTCCATAGCATCGCC |
| Deletions | ||
| VP8(D65–110) | GCCGACGACTATGATAGCGATTATTT | ATCATAGTCGTCGGCGCTGTC |
| VP8(D65–125) | GCCGACGACTATGATAGCGATTATTT | CGTCAAGTAGTCTTGCGGGGCAC |
| VP8(D16–32) | CTGCTGGACGCCCTGCGCGCTGCGGAC | GCGGCGCGGGCGGCGCTCAGG |
| VP8(D79–92) | GCTAACCGCGGCCCCAATCAC | ATCGCCGTCGATGTAATCATACACGTTCTC |
| VP8(D65–82) | GCCGACGACTATGATAGCGATTATTT | GTCCTCGTCCGGGGGCCGCTGGAAG |
| VP8(D88–110) | CCCGAGCGCGCCCCGGAAGG | ATCATAGTCGTCGGCGCTGTC |
| VP8(D65–92) | GCCCACCTGCGCGCCATCGAG | ATCGCCGTCGATGTAATCATACACGTTCTC |
| Truncations | ||
| VP8(121–742) | GCGGTAGATCTGATTCAAGACTACTTGACGGCCCACCTG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8(219–742) | CGGTAGATCTGATTGAGCGGCTGTCGGAAGGG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8(343–742) | CGGTAGATCTGATTGGCGGCATGTACGTGGGCGCCCCTGAG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8(538–742) | GCGGTAGATCTGATTGCGGCGGCCTTCCGCGAAGTG | GGCAGTGAGCGCAACGCAATTAATG |
| VP8(1–120) | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCAGCCGTGATTGGGGCCGCGGTTAG |
| VP8(1–125) | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCACGTCAAGTAGTCTTGCGGGGCAC |
| VP8(1–258) | GAATCTAGAGCCACCATGGACTACAAAGACGATGAC | GAGCTCGAGTCACTCCCCCGCAGCCGCAGCG |
Immunoprecipitation.
BoHV-1-infected MDBK cells and plasmid-transfected COS-7 cells were pretreated with l-methionine-free or phosphate-free Dulbecco's modified Eagle's medium (DMEM; Life Technologies) for 3 h prior to incubation with [35S]methionine or [32P]orthophosphate (PerkinElmer, Woodbridge, ON, Canada). Cell lysates were precleared with protein G-Sepharose (GE Healthcare, Burnaby, BC, Canada) and then incubated with anti-VP8 monoclonal antibody and protein G-Sepharose overnight at 4°C. The protein G-Sepharose was washed three times with wash buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4) and boiled for 5 min with SDS-PAGE sample buffer. The samples were separated by SDS-PAGE in 10% gels. The gels were subsequently dried and exposed to Imaging Screen-K for visualization on a Molecular Imager FX (Bio-Rad, Mississauga, ON, Canada).
Protein purification and in vitro kinase assay.
COS-7 cells in six-well plates were transfected with plasmid (1.5 μg/well) using Lipofectamine with Plus reagent (Life Technologies). Cell lysates were collected at 48 h posttransfection (hpt). Twenty microliters of anti-FLAG M2 affinity gel (Sigma-Aldrich) or anti-hemagglutinin (HA)-agarose (Pierce, Rockford, IL, USA) was washed according to the manufacturer's instructions and incubated with 200 μl of the appropriate lysate overnight at 4°C. The beads were washed five times with 1 ml of wash buffer (50 mM Tris-HCl, 150 mM NaCl, pH 7.4). FLAG-VP8 (wild type [WT], mutations, and deletions) was eluted with 3×FLAG peptide (Sigma-Aldrich) according to the manufacturer's instructions. The proteins were stored at −80°C until used.
Kinase assays were performed following a procedure described previously (19) with a few optimized steps. Briefly, a 25-μl reaction mixture consisting of 0.3 mCi of [γ-32P]ATP (PerkinElmer), 1 μg of substrate protein, 0.5 ng of CK2 (EMD Millipore, Burlington, ON, Canada) or 5 μl of US3-HA on anti-HA-agarose, and 6.25 μl of 4× reaction buffer (80 mM HEPES, pH 7.6, 0.6 M NaCl, 0.4 mM EDTA, 20 mM dithiothreitol [DTT], 0.4% Triton X-100) was incubated at 30°C for 10 min. The reaction was stopped by boiling with SDS-PAGE sample buffer for 5 min, and then the proteins were separated by SDS-PAGE in a 10% gel. The gels were dried and exposed to Imaging Screen-K for visualization on a Molecular Imager FX.
Coimmunoprecipitation and Western blotting.
Whole-cell extracts were prepared by suspension of cell pellets in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% NP-40, 1% deoxycholate, 0.1% SDS) supplemented with protease inhibitor cocktail (Sigma-Aldrich) or phosphatase inhibitor cocktail (EMD Millipore). The cell lysates were clarified by centrifugation at 13,000 × g for 10 min at 4°C. Anti-FLAG M2 affinity gel or anti-HA-agarose was incubated with cell lysate (20 μl of resin per 400 μl) overnight at 4°C and washed three times with 1 ml of wash buffer (150 mM NaCl, 50 mM Tris-HCl, pH 7.4). Proteins were eluted by boiling in SDS-PAGE sample buffer for 5 min. In each experiment, 20 μl of each sample was subjected to SDS-PAGE in a 10% gel, and then the proteins were transferred to nitrocellulose membranes and incubated with the appropriate antibodies. After a washing step, the membranes were further incubated with IRDye 600RD/800CW-conjugated secondary antibodies at a 1:20,000 dilution and scanned with an Odyssey CLx infrared imaging system (Li-Cor Biosciences).
LC-MS.
FLAG-VP8 protein on anti-FLAG M2 affinity gel was treated with lambda protein phosphatase (NEB) at 30°C for 30 min and then eluted with 3×FLAG peptide. The dephosphorylated VP8 was rephosphorylated with the appropriate kinases in in vitro kinase assays and subsequently subjected to SDS-PAGE. The gels were stained with Coomassie brilliant blue (CBB) R250 in 40% methanol–10% acetic acid. Protein bands of interest were prepared and submitted to the University of Victoria Genome British Columbia Proteomics Centre (Victoria, BC, Canada) for analysis of the tryptic peptide molecular masses by liquid chromatography-mass spectrometry (LC-MS). Briefly, gel slices were manually cut into 1-mm pieces and transferred to a Genomics Solutions ProGest perforated digestion tray. The gel pieces were destained (50/45/5, vol/vol/vol; methanol-water-acetic acid) prior to reduction (10 mM dithiothreitol; Sigma-Aldrich) and alkylation (100 mM iodoacetamide; Sigma-Aldrich). Modified sequencing-grade porcine trypsin solution (20 ng/μl; Promega, Madison, WI, USA) was added to the gel slices at an enzyme/protein ratio of 1:50. Protein was then digested for 5 h at 37°C prior to collection of tryptic digests and acid extraction of the gel slices (50/40/10, vol/vol/vol; acetonitrile-water-formic acid). The peptide mixtures were separated by online reverse-phase chromatography using a Thermo Scientific EASY-nLC 1000 system. The chromatography system was coupled online with an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a Nanospray Flex NG source (Thermo Fisher Scientific).
Immunofluorescence staining.
COS-7 cells, cultured on Permanox two-well chamber slides (Thermo Fisher Scientific), were transfected with 1.5 μg of DNA per well and Lipofectamine with Plus reagent for 3 h. After incubation for 20 h, the cells were washed three times with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde for 20 min, and then washed three times with PBS. Subsequently, the cells were permeabilized with 0.1% Triton X-100 in PBS for 20 min, washed with PBS, and then incubated with 1% normal goat serum (Gibco) in PBS for 2 h at room temperature. The cells were incubated with primary antibodies at the appropriate dilutions for 2 h at room temperature, followed by washing with PBS and incubation with Alexa Fluor-conjugated antibodies (Life Technologies) at a 1:500 dilution for 1 h at room temperature. Finally, the cells were incubated with 0.5 μg/ml 4′,6′-diamidino-2-phenylindole (DAPI; Life Technologies) at 37°C for 10 min. Slides were washed with PBS, followed by deionized water, and then air dried and mounted using ProLong Gold Antifade Reagent (Life Technologies) prior to examination on a Zeiss LSM410 confocal microscope equipped with an external argon ion 488/633/461-nm laser.
Precision-cut lung slice (PCLS) preparation.
An ovine lung was perfused with 1.5% low-gelling-temperature agarose (Sigma-Aldrich) in RPMI medium (Gibco, Life Technologies) prior to sectioning. Sections (220 to 250 μm) were obtained by a Krumdieck tissue slicer (TSE Systems, Chesterfield, MO, USA). This procedure was approved by the University Council for Animal Care and Supply in accordance with the standards stipulated by the Canadian Council on Animal Care. They were washed in three changes of RPMI medium with antimycotic, enrofloxacin (Baytril), clotrimazole, and kanamycin and then incubated overnight at 37°C. The sections were infected with 106 PFU of the BoHV-1 Cooper strain for 24 h. The slides were analyzed by immunofluorescence staining using polyclonal anti-VP8 antibody and goat anti-rabbit Alexa Fluor-conjugated antibodies (Life Technologies) as described above.
RESULTS
VP8 is phosphorylated in BoHV-1-infected cells but not in virions.
VP8 has been identified as a 97-kDa phosphorylated tegument protein expressed during the later stage of BoHV-1 infection. By using [35S]methionine or [32P]orthophosphate to label the proteins, we confirmed VP8 to be extensively phosphorylated in BoHV-1-infected MDBK cells. The phosphate was completely removed by lambda protein phosphatase treatment (Fig. 1A). The in vitro dephosphorylation was confirmed by LC-MS as no phosphopeptide was detected in the lambda protein phosphatase-treated VP8 (data not shown).
FIG 1.
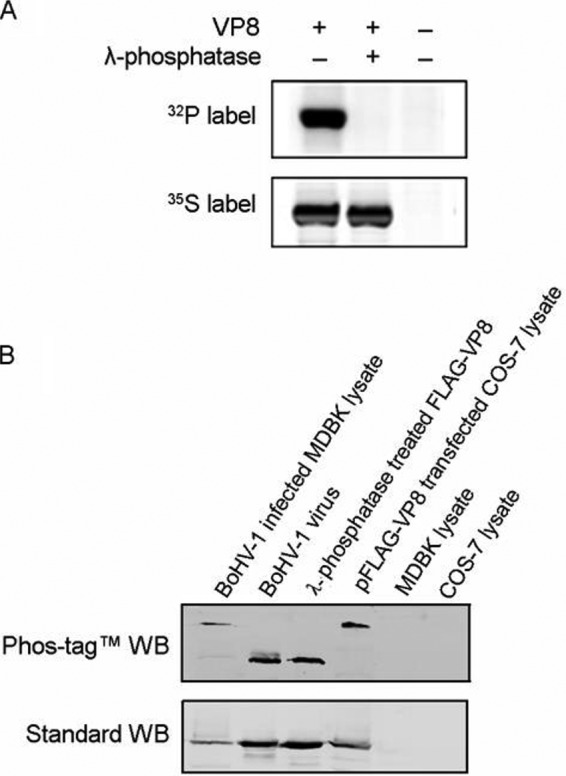
VP8 is phosphorylated in transfected and BoHV-1-infected cells but not phosphorylated in virions. (A) [32P]orthophosphate-labeled or [35S]methionine-labeled MDBK cells were infected with BoHV-1 at a multiplicity of infection of 10. Cell lysates were collected at 18 hpi and subsequently used for VP8 purification by incubation with anti-VP8 monoclonal antibody and protein G-Sepharose. A duplicate sample was treated with lambda protein phosphatase for 1 h at 30°C. The samples were separated by SDS-PAGE and exposed to Imaging Screen-K. (B) BoHV-1-infected MDBK cell lysate, purified virus lysate, lambda protein phosphatase-dephosphorylated FLAG-VP8, and pFLAG-VP8-transfected COS-7 cell lysate were analyzed in both Phos-tag and standard polyacrylamide gels, followed by Western blotting (WB). MDBK and COS-7 cell lysates were used as controls. A polyclonal anti-VP8 antibody and IRDye 800CW goat anti-mouse IgG were used to detect VP8. The upper panel shows migration of VP8 in the gel supplied with Phos-tag acrylamide, and the lower panel represents VP8 in a standard gel.
The VP8 phosphorylation status differed at different stages of infection. By using a Phos-tag acrylamide gel (22) followed by Western blotting, we showed that VP8 from transfected COS-7 cells and BoHV-1-infected MDBK cells migrates more slowly than VP8 from purified virus or a lambda protein phosphatase-treated protein sample (Fig. 1B, upper panel). Phosphorylated protein associates with the Phos-tag, which is a functional molecule and specifically binds phosphorylated ions, resulting in a larger molecular weight of phosphorylated protein than nonphosphorylated protein (22). As a result, the higher bands in the Phos-tag acrylamide gel represent phosphorylated VP8, and the lower bands are nonphosphorylated VP8. All samples had the same molecular weights in standard polyacrylamide gels (Fig. 1B, lower panel). This suggests that phosphorylation bestows different functions to VP8 during different stages of the viral life cycle.
Identification of US3 phosphorylation sites on VP8.
Previously we identified US3 as one of the kinases responsible for phosphorylation of VP8 in in vitro kinase assays (19). Interaction between VP8 and US3 was confirmed by coimmunoprecipitation. US3-HA was pulled down by FLAG-VP8 in cotransfected COS-7 cells, while FLAG-VP8 was pulled down by US3-HA (Fig. 2A, left panel). There was no interaction between US3-HA and the anti-FLAG beads when FLAG-VP8 was not present or between the FLAG-VP8 and the anti-HA beads when US3-HA was not present. pFLAG-YFP was used as a control plasmid. As shown in Fig. 2A (right panel), there was no interaction between FLAG-YFP and US3-HA in cotransfected COS-7 cells.
FIG 2.

VP8 is a substrate for US3, and S16 is a critical residue for phosphorylation. (A) FLAG-VP8 interacts with US3-HA. COS-7 cells were cotransfected with pFLAG-VP8 and pUS3-HA. Cell lysates were collected at 48 hpt. Protein was purified by incubating cell lysate with anti-FLAG beads or anti-HA beads for 12 h at 4°C and analyzed by Western blotting. pFLAG-YFP was used as a negative control for coimmunoprecipitation. FLAG-VP8, US3-HA, and FLAG-YFP were detected by monoclonal anti-VP8 antibody, polyclonal anti-US3 antibody, and monoclonal anti-FLAG antibody, respectively, followed by IRDye 680RD goat anti-rabbit IgG or IRDye 800CW goat anti-mouse IgG. (B) The presence of US3 increases phosphorylation of VP8 in vivo. COS-7 cells cotransfected with pFLAG-VP8 and pUS3-HA or transfected with pFLAG-VP8 were labeled with [32P]orthophosphate at 12 hpt. After a subsequent 3, 6, and 9 h of incubation, FLAG-VP8 was purified from the cell lysate by anti-FLAG beads. The samples were separated by SDS-PAGE, exposed to Imaging Screen-K, and stained with CBB. Band densities were scanned by the Quantity One program (CBB/32P-labeled band). (C) VP8 is phosphorylated by wild-type US3 but not by US3 mutants. VP8 and US3 (wild type or mutant) were analyzed by in vitro kinase assays with [λ-32P]ATP. TBCA was used to inhibit cellular kinases carried over by anti-FLAG beads and anti-HA beads during the protein purification process. Proteins were separated by SDS-PAGE and exposed to Imaging Screen-K. (D) VP8 mutants were generated by replacing the serine/threonines with alanines. The VP8 mutants were analyzed by an in vitro kinase assay. Protein expression was confirmed by Western blotting with monoclonal anti-VP8 antibody and IRDye 800CW goat anti-mouse IgG.
The phosphorylation of VP8 by US3 was further studied in vivo in pFLAG-VP8- and pUS3-HA-cotransfected, [32P]orthophosphate-treated COS-7 cells. Phosphorylated VP8 was detected as early as 3 h postlabeling, and phosphorylation increased in a time-dependent manner (Fig. 2B). At each time point, the intensity of phosphorylation was stronger in the presence of US3 than without US3 (Fig. 2B). The in vitro kinase assay confirmed that VP8 is a substrate of US3 (Fig. 2C). VP8 was intensely phosphorylated by wild-type US3 but not by the four US3 mutants, in which the two critical active sites for US3, K195 or K282, located in the catalytic loop and in the ATP-binding pocket, were mutated (21). These results indicate that US3 phosphorylates VP8 in vitro and in vivo.
To identify the active sites for US3 on VP8, single-site mutations were introduced into the potential target residues on VP8. Through aligning the VP8 sequence with a published minimal consensus sequence for US3 (21), four potential motifs (RRS16GTYR, RRS32LL, RRS173LR, and RRVT632VR) were identified. The serines and threonines within these motifs were replaced with alanines using site-directed mutagenesis. The six VP8 mutants were subsequently analyzed in the in vitro kinase assay in the presence of the CK2 inhibitor tetrabromocinnamic acid (TBCA), which showed that replacing S16 with an A residue (S16A) completely blocked phosphorylation by US3. Substitutions of S16 and T18 (S16A and T18A, respectively) had the same impact. However, other substitutions had no obvious effect on US3 phosphorylation (Fig. 2D). As observed previously, US3 performed auto-phosphorylation.
To further identify the US3 phosphorylation sites on VP8, purified VP8 phosphorylated by US3 was digested with trypsin and scanned by LC-MS. This method allowed the unambiguous assignment of a phosphorylation site on a phosphopeptide, RS32LLDALR (Table 2). This phosphopeptide was identified in two spectra, representing the frequency of detection of this peptide, with a confidence greater than 95%, indicating a positive match. A nonphosphorylated peptide with the same sequence was detected in the lambda protein phosphatase-treated sample. Six spectra were identified with the highest confidence of 92%, indicating that the phosphate group on the S32 site was removed by the phosphatase. However, due to the frequent presence of arginine in the VP8 sequence, especially within the US3 phosphorylation motifs, the tryptic LC-MS covered only 61% of VP8, and the S16 site could not be identified.
TABLE 2.
Peptide identified by LC-MS in the US3-phosphorylated VP8
| Sample treatment | Phosphorylation status of peptide RS32LLDALR |
|||
|---|---|---|---|---|
| Phosphorylated |
Nonphosphorylated |
|||
| Probability (%)a | No. of spectrab | Probability (%) | No. of spectra | |
| VP8 with US3 | 98 | 2 | 98 | 12 |
| VP8 without US3 | 0 | 0 | 92 | 6 |
The highest probability score is used.
Number of spectra in which the peptide was identified.
VP8 is phosphorylated by CK2.
Previously, we identified CK2 as a kinase phosphorylating VP8 in vitro (19). To confirm this and explore a potential role of other kinases, different kinase inhibitors were used to block VP8 phosphorylation. Phosphorylation was reduced by TBCA in a concentration-dependent manner but not by other two kinase inhibitors, SNS032 and AT7519 (Fig. 3A). TBCA, SNS032, and AT7519 specifically inhibit the activity of CK2, cyclin-dependent kinases (CDKs), and glycogen synthase kinase-3 beta (GSK-3β), respectively. To test the impact of TBCA on the phosphorylation of VP8 in vivo, pFLAG-VP8-transfected COS-7 cells were pretreated with TBCA at 80 μM for 3 h and then labeled with [32P]orthophosphate for 3, 6, or 9 h in the presence of TBCA. Phosphorylation was reduced in the TBCA-treated samples compared with the levels in samples not treated with TBCA (Fig. 3B). The phosphorylation of VP8 by CK2 was confirmed by subjecting CK2 and VP8 to an in vitro kinase assay (Fig. 3C). The result showed that VP8 was phosphorylated in the presence of CK2 and that CK2 performed auto-phosphorylation.
FIG 3.

VP8 is a substrate for CK2 and interacts with CK2. (A) Inhibition of VP8 phosphorylation in the in vitro kinase assay by TBCA. Different kinase inhibitors were applied to the in vitro kinase assays with VP8 and cellular kinases. (B) Reduction of VP8 phosphorylation by TBCA in pFLAG-VP8-transfected COS-7 cells. Cells were pretreated with TBCA at 80 μM for 3 h and then transfected with pFLAG-VP8. At 12 hpt, the cells were labeled with [32P]orthophosphate for 3, 6, and 9 h in the presence of TBCA. The whole-cell lysate was applied to the immunoprecipitation assay as described in the legend of Fig. 2B. (C) Phosphorylation of VP8 by CK2 and CK2α auto-phosphorylation. VP8 and CK2 were analyzed by an in vitro kinase assay. The 45-kDa protein, matching the molecular mass of CK2α, was phosphorylated. (D) Interaction of VP8 with CK2α in pFLAG-VP8-transfected COS-7 cells. Cell lysates were collected at 48 hpt and subjected to a coimmunoprecipitation (Co-IP) assay. The whole-cell lysates were analyzed to show protein expression in transfected cells. VP8, CK2α, and FLAG-YFP were detected by monoclonal anti-VP8 antibody, polyclonal anti-CK2α antibody, and monoclonal anti-FLAG antibody, respectively, followed by IRDye 680RD goat anti-rabbit IgG or IRDye 800CW goat anti-mouse IgG.
The interaction between CK2 and VP8 was confirmed by coimmunoprecipitation. The 45-kDa CK2α subunit was pulled down by FLAG-VP8 in pFLAG-VP8-transfected COS-7 cell lysate, while the CK2α subunit was not pulled down by anti-FLAG beads in nontransfected or pFLAG-YFP-transfected cell lysate (Fig. 3D).
CK2 phosphorylates VP8 at multiple residues.
To identify the phosphorylation sites for CK2, a series of truncated VP8 proteins was constructed and analyzed in the in vitro kinase assay. The VP8 constructs without residues 1 to 120 [VP8(121–741), VP8(219–741), VP8(343–741), and VP8(538–741)] were not phosphorylated by CK2, while the truncations containing residues 1 to 120 [VP8(1–120), VP8(1–125), and VP8(1–258)] were phosphorylated by CK2 (Fig. 4A). These results imply a critical role of residues 1 to 120 in phosphorylation. There are five consensus sequences matching a published CK2 motif within residues 1 to 120, which were named CK2 motif 1 (CM1) to CK2 motif 5 (CM5) (Fig. 4B). Single-site mutations of the S/T residues within these motifs reduced the phosphorylation to different degrees but did not achieve complete inhibition (Fig. 4C). Shorter deletions in VP8 were constructed to determine the involvement of these CMs (Fig. 4D). Deleting CM1 did not have an obvious impact; however, deleting CM2 to CM5 completely blocked phosphorylation, and deleting CM2 and CM3 or CM3 and CM4 almost eliminated phosphorylation. These results suggest that CM2, CM3, and CM4 play an important role. Indeed, deleting CM2, CM3, and CM4 (residues 65 to 92) eliminated phosphorylation, and substitutions of the S/T residues within these areas dramatically attenuated phosphorylation, demonstrating that they all contribute to VP8 phosphorylation. Substitutions of the S/T residues in the CM2, CM3, CM4 and CM5 all at once achieved complete inhibition (Fig. 4D). The above results reveal that CM2 to CM5 are critical for the phosphorylation of VP8 by CK2. To determine whether deleting CM2, CM3, and CM4 affects the interaction between the kinase and substrate, coimmunoprecipitation of VP8 with a deletion of residues 65 to 92 [VP8(D65–92)] and CK2 was performed. This showed that CK2α was pulled down with VP8(D65–92) (Fig. 4E), indicating that VP8 with deletions of CM2, CM3, and CM4 still associated with CK2 but was not phosphorylated. Thus, removal of any of these motifs had no effect in preventing formation of the kinase-substrate complex.
FIG 4.

Identification of the critical residues on VP8 for phosphorylation by CK2. (A) A series of VP8 truncations was constructed as listed. The bar indicates the VP8 portion, and the line indicates the deleted portion. The truncated proteins were purified from transfected COS-7 cell lysate and applied to in vitro kinase assays with CK2. Truncated proteins with deletions of residues 1 to 120 and beyond [VP8(121–741), VP8(219–741), and VP8(343–741)] were not phosphorylated, while those truncated at the carboxyl end after residue 120 [VP8(1–120), VP8(1–125), and VP8(1–258)] were phosphorylated by CK2 (upper panel). Protein expression is demonstrated by Western blotting using anti-FLAG antibody (lower panel). (B) Five CK2 consensus motifs (CM1 to CM5; underlined) were found within residues 1 to 127 by aligning the VP8 sequence with a published CK2 motif, T/S-Xn-E/D (n ≥ 0). The bar represents the VP8 sequence with 741 amino acids, and residues 1 to 127 are highlighted. (C) Single-site mutations of VP8 by replacing S/T residues with A residues within the identified CMs were analyzed by in vitro kinase assays with CK2. Replacing S66, S88, or T107 greatly reduced VP8 phosphorylation. Protein loading is shown by Western blotting (WB). The band density was scanned and analyzed by Quantity One software. (D) Analyses of the truncations and mutants of VP8 in in vitro kinase assays. VP8 deletions (D) and multiple site mutations (M) were constructed according to the description for panel B. Purified proteins were applied to in vitro kinase assays with CK2. Protein loading is shown by Western blotting using monoclonal anti-FLAG antibody and IRDye 800CW goat anti-mouse IgG. (E) Interaction of FLAG-VP8(D65–92), which has a deletion of CM2 to CM 4, with CK2 in the transfected COS-7 cells. pFLAG-VP8 and pFLAG-CMV-2 were used as positive and negative controls, respectively. The transfected COS-7 cell lysate was collected at 48 hpt and analyzed by coimmunoprecipitation (Co-IP). VP8 and CK2 were detected by polyclonal anti-VP8 antibody and polyclonal anti-CK2α antibody, followed by IRDye 680RD goat anti-rabbit IgG.
CK2-treated VP8 was analyzed by tryptic LC-MS, which detects any modification of a peptide by calculating the mass of the modification. A list of phosphopeptides was identified in the CK2 (3.3 ng/μl)-treated sample; specifically, peptide GPNGHAGDT107DAPPER was detected in 20 spectra with a confidence higher than 95% (Table 3). These results indicated that five residues (T107, S137, S221, S240, and S679) were phosphoreceptors, among which T107 had the highest level of phosphorylation. However, in the sample treated with a lower concentration of CK2 (≤3.3 pg/μl), two phosphopeptides containing residues T107 and S137 were detected, while the peptides containing S221, S240, and S679 were nonphosphorylated.
TABLE 3.
Peptides identified by LC-MS in the CK2-phosphorylated VP8
| CK2 concn | Site | Identified peptide | Peptide phosphorylation status |
|||
|---|---|---|---|---|---|---|
| Phosphorylated |
Nonphosphorylated |
|||||
| Probability (%)a | No. of spectrab | Probability (%) | No. of spectra | |||
| 3.3 ng/μl | T107 | GPNGHAGDT107DAPPER | 100 | 20 | 100 | 64 |
| GPNGHAGDT107DAPPERAPEGGAPQDYLTAHLR | 81 | 1 | 100 | 31 | ||
| S137 | AIEALPES137APHR | 100 | 6 | 100 | 37 | |
| S221 | LS221EGPPLLNMEAAAAAAGER | 98 | 1 | 100 | 136 | |
| DERLS221EGPPLLNMEAAAAAAGER | 100 | 3 | 0 | |||
| S240 | S240VVEELFTYAPAQPQVEVPLPR | 99 | 2 | 100 | 78 | |
| S679 | LRPVAS679PPLAGK | 100 | 2 | 100 | 14 | |
| ≤3.3 pg/μl | T107 | GPNGHAGDT107DAPPER | 100 | 2 | 100 | 35 |
| GPNGHAGDT107DAPPERAPEGGAPQDYLTAHLR | 0 | 100 | 18 | |||
| S137 | AIEALPES137APHR | 95 | 2 | 100 | 22 | |
| S221 | LS221EGPPLLNMEAAAAAAGER | 0 | 100 | 154 | ||
| DERLS221EGPPLLNMEAAAAAAGER | 0 | 100 | 5 | |||
| S240 | S240VVEELFTYAPAQPQVEVPLPR | 0 | 100 | 70 | ||
| S679 | LRPVAS679PPLAGK | 0 | 100 | 29 | ||
The highest probability score is used.
Number of spectra in which the peptide was identified.
Phosphorylation of VP8 contributes to BoHV-1 replication.
Based on the above results, a mutant VP8 (Mut-VP8), with all of the critical phosphorylation sites for US3 (S16) and CK2 (T65, S66, S79, S80, S82, S88, and T107) replaced by alanines, was constructed and analyzed in in vitro kinase assays. The Mut-VP8 was not phosphorylated by either CK2 or US3, while these two kinases phosphorylated the WT-VP8, as was expected (Fig. 5A).
FIG 5.
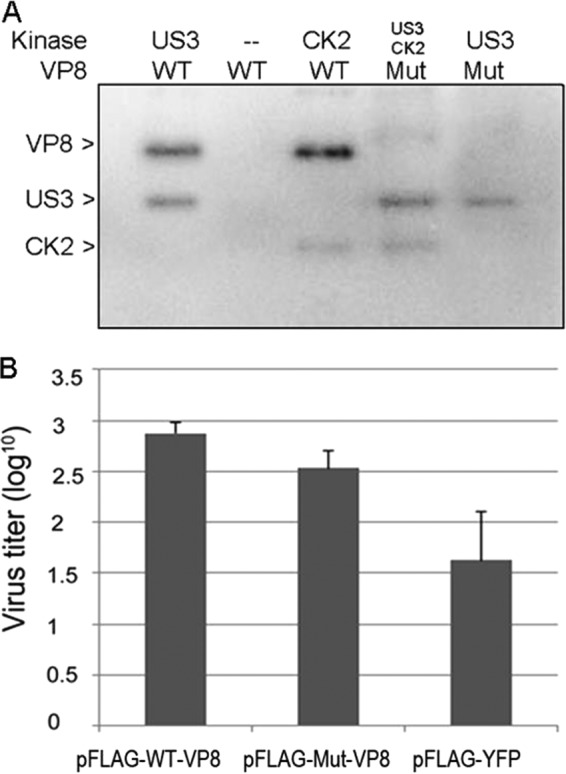
WT-VP8 benefits virus replication more than Mut-VP8, which is not phosphorylated by CK2 and US3. (A) Confirmation of nonphosphorylated Mut-VP8 in the in vitro kinase assay. Mut-VP8, which had all the critical phosphorylation sites for US3 (S16) and for CK2 (T65, S66, S79, S80, S82, S88, and T107) replaced by alanines, was constructed and analyzed in kinase assays with CK2 and US3. (B) BoHV-1 ΔUL47 replication in WT-VP8-expressing cells and in Mut-VP8-expressing cells. FBT cells were infected with BoHV-1 ΔUL47 at a multiplicity of infection of 0.3. At 4 hpi, cells were transfected with pFLAG-WT-VP8 or pFLAG-Mut-VP8. A control sample was transfected with pFLAG-YFP. Viruses were collected at 36 hpi and titrated on MDBK cells.
To study the impact of VP8 phosphorylation on virus replication, we analyzed the replication of a UL47 gene deletion mutant (BoHV-1 ΔUL47), constructed previously (5), in WT-VP8- and Mut-VP8-expressing cells. FBT cells were infected with BoHV-1 ΔUL47 and then transfected with pFLAG-WT-VP8, pFLAG-Mut-VP8, or pFLAG-YFP. At 36 h postinfection (hpi), the virus titer from the pFLAG-WT-VP8-transfected cells was higher than that from the pFLAG-Mut-VP8-transfected cells, while the pFLAG-YFP-transfected cells showed the lowest titer (Fig. 5B). This indicated that BoHV-1 ΔUL47 replicated better in the WT-VP8-expressing cells than in the Mut-VP8-expressing cells.
Phosphorylation alters the intracellular localization of VP8 and PML protein.
To determine whether phosphorylation of VP8 influences its subcellular localization, as has been found for other proteins, the localization of WT-VP8 and Mut-VP8 was examined by immunofluorescence in transfected COS-7 cells. WT-VP8 displayed nuclear localization (Fig. 6A), which was consistent with previous results (13). Furthermore, the transfected cells developed circular bodies in the nucleus, with WT-VP8 being strongly labeled on these circular bodies. The size of these nuclear bodies extended in a time-dependent manner. At 12 h they were visible as small dots in the nucleus, and at later time points they became larger and circular (Fig. 6A, upper panel). In contrast, Mut-VP8 was evenly distributed throughout the nucleus and was not accumulated to the nuclear bodies (Fig. 6A, lower panel). Double staining using anti-nucleolin antibody and anti-VP8 antibody confirmed that these bodies were different from the nucleolus (data not shown). A similar pattern was observed in BoHV-1-infected lung tissue (Fig. 6B). Some cells in the infected tissue developed circular areas not stained with either DAPI or anti-VP8 antibody. A certain amount of VP8 accumulated to the edge of the circular areas.
FIG 6.

Cellular localization of WT-VP8 and Mut-VP8. (A) Localization and different patterns of WT-VP8 and Mut-VP8 in the nucleus. WT-VP8 and Mut-VP8 were expressed in COS-7 cells. VP8 was detected with polyclonal anti-VP8 antibody and Alexa-488-conjugated goat anti-rabbit IgG. DNA was labeled with DAPI. The cells were observed with a Zeiss LSM410 confocal microscope. (B) Localization of VP8 in BoHV-1-infected lung tissue slices. Lung tissue sections (220 to 250 μm) were infected with 106 PFU of BoHV-1 for 24 h. VP8 was detected with polyclonal anti-VP8 antibody and Alexa-488-conjugated goat anti-rabbit IgG. DNA was labeled with DAPI. The slides were observed with a Zeiss LSM410 confocal microscope.
To gain insight into the identity of these nuclear bodies, the transfected cells were stained with both anti-PML and anti-VP8 antibodies. While the PML bodies were evenly distributed as round dots or speckles in the nucleus of nontransfected cells, WT-VP8 altered the distribution of PML protein by recruiting it to the edge of the circles (Fig. 7). As is indicated by white arrowheads, PML aggregated to the nuclear bodies, where WT-VP8 was concentrated. Eventually, a large cluster of PML protein was formed around the VP8 bodies (Fig. 7). The formation of nuclear bodies and accumulation of VP8 might happen independently of PML; instead, the nuclear bodies might recruit PML protein through interactions between VP8 and PML protein or other PML body components. This is supported by the observation that not all VP8 nuclear bodies were associated with PML protein; especially at 6 hpt several PML bodies were distinct from the newly developed VP8 nuclear bodies. This observation suggests that the PML accumulation happened after the VP8 nuclear body development. Figure 8 shows the PML protein in the Mut-VP8-transfected COS-7 cells. Mut-VP8 did not accumulate to the nuclear bodies, and the PML protein had the same distribution in the transfected cells as in the control cells. No colocalization was detected between Mut-VP8 and the PML surrounding the nuclear bodies.
FIG 7.

PML protein accumulation to nuclear bodies and colocalization with WT-VP8. (A) pFLAG-VP8 was transfected into COS-7 cells. FLAG-WT-VP8 was detected with monoclonal anti-FLAG antibody and Alexa-488-conjugated goat anti-mouse IgG, and PML protein was detected with polyclonal anti-PML antibody and Alexa-633-conjugated goat anti-rabbit IgG. DNA was labeled with DAPI. The cells were observed with a Zeiss LSM410 confocal microscope. The white arrowheads indicate that at 6 hpt PML protein is recruited to the edge of nuclear bodies, where WT-VP8 accumulates, and that at 24 hpt nuclear PML accumulates to the bodies, resulting in protein clusters.
FIG 8.

The distribution of PML protein is not affected by Mut-VP8. pFLAG-Mut-VP8 was transfected into COS-7 cells. FLAG-Mut-VP8 was detected with monoclonal anti-FLAG antibody and Alexa-488-conjugated goat anti-mouse IgG, and PML protein was detected with polyclonal anti-PML antibody and Alexa-633-conjugated goat anti-rabbit IgG. DNA was labeled with DAPI. The cells were observed with a Zeiss LSM410 confocal microscope.
DISCUSSION
VP8 is known as a phosphoprotein and as a substrate for US3 and CK2 in an in vitro kinase assay (19). Based on this information, the objective of this study was to identify the active sites or areas for US3 and CK2 kinases, as well as a possible function of VP8 phosphorylation. This is the first evidence for a difference in the phosphorylation status of VP8 in host cells and mature virus and for phosphorylation as a critical modification for VP8 to recruit PML protein. We confirmed phosphorylation of VP8 by US3 and CK2 both in vitro and in vivo and specified the active phosphorylation sites though site-directed mutagenesis and LC-MS. Phosphorylated, but not nonphosphorylated, VP8 tended to accumulate PML protein, which plays a role as an antiviral protein (17).
Although VP8 contains four consensus sequences for US3, only mutation of S16 prevented VP8 phosphorylation by US3 (Fig. 2D), demonstrating that S16 is an essential residue for US3. It is not the only phosphoreceptor on VP8 because a phosphopeptide containing S32 was identified by LC-MS (Table 2). However, this site plays no decisive role in the phosphorylation at other sites since mutating S32 did not result in reduced protein phosphorylation (Fig. 2D). Both S16 and S32 are potential active sites for US3 and they are within the US3 consensus motif Rn-X-S/T-Y-Y (n ≥ 2; X is any residue; Y cannot not be aspartate, glutamate, or proline) (19). The phosphorylation of S16 might complete the kinase recognition motif for the neighboring residue S32 to be phosphorylated, resulting in a sequential phosphorylation cascade. We propose that US3 phosphorylates VP8 on S16 and that this triggers additional phosphorylation on S32. This contention is based on a previously described model, called primary phosphorylation, which proposes that the introduction of a phosphate group to a favorable site causes changes in the overall protein confirmation so that an otherwise unfavorable site is active for subsequent phosphorylation (23). Many other sequential phosphorylation examples have been described. For instance, a study of hepatitis C virus showed that there is a sequential and ordered cascade of phosphorylation events on the NS5A protein (24), where phosphorylation at S238 triggers phosphorylation at S235. Similarly, the regulatory phosphorylation of eukaryotic elongation factor 2 (eEF2) at S595 by CDK2 directly stimulates phosphorylation on T56 by eEF2 kinase (25). Conversely, phosphorylation of a primary site may downregulate further phosphorylation on other sites. For example, extracellular signal-regulated protein kinase (ERK) phosphorylates mitogen-activated protein kinase kinase 1 (MEK1) on T292, and this, in turn, blocks additional S298 phosphorylation (26).
Phosphorylation of VP8 by CK2 was confirmed by applying pure, active CK2 protein and VP8 to the in vitro kinase assay. TBCA, a relatively new CK2 inhibitor with better specificity and less toxicity than DMAT (2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole) (27), inhibited the phosphorylation of VP8 in vitro. This is in agreement with our previous observation that DMAT (19) blocks phosphorylation of VP8. CK2 is constitutively expressed and active in COS-7 cells, so TBCA was used to inhibit the CK2 activity. The reduction of VP8 phosphorylation in the TBCA-treated cells demonstrated that VP8 is phosphorylated by CK2 in vivo. Although no evidence has been reported that HHV-1 VP13/14 is phosphorylated by CK2, phosphorylation on this tegument protein might be mediated by cellular kinases, including CK2. This is supported by the observation that VP13/14 is phosphorylated in cells infected with an HHV-1 US3-dead mutant; furthermore, two potential consensus motifs for CK2 on VP13/14 have been reported (19). It has also been found that in certain circumstances HHV-1 US3 shares a similar amino acid content with protein kinase A (PKA) (28) and Akt (29), suggesting that these kinases may play a role in VP13/14 phosphorylation.
Generally, CK2 has more than one active site on substrates, and overall about 75% of the active sites match the CK2 motif S/T-X-X-D/E (30). Within the VP8 sequence, 10 threonines and 19 serines match the motif. To identify the active sites for VP8, we started by analyzing shorter forms of VP8. VP8 fragments without the C-terminal residues 1 to 120 were found not to be phosphorylated by CK2 (Fig. 4A), which has two possible explanations. The residues 1 to 120 maintain the protein structure for phosphorylation; alternatively, all the active sites are within this area. The fragments that encompass residues 1 to 120 were phosphorylated, but the fragment of residues 1 to 258 was more strongly labeled than shorter fragments, indicating the existence of active sites outside the region of residues 1 to 120 (Fig. 4A). There are five consensus sequences (Fig. 4B) for CK2 within this area. Although LC-MS did not detect any phosphopeptide that contains CM2, CM3, or CM4, these motifs appeared to be critical to maintain the catalytic domains without disrupting the formation of the kinase-substrate complex (Fig. 4D).
Two phosphopeptides containing CM5 were detected by LC-MS (Table 3), suggesting that T107 is a phosphoreceptor. However, it is not the only active site for CK2 since mutating this residue reduced the phosphorylation only to a certain level (Fig. 3C). Four additional phosphorylated sites (S137, S221, S240 and S679) were detected by tryptic LC-MS (Table 3). Among these sites, the T107 residue had the most abundant phosphorylation event as the level was 3 to 10 times higher than that of the other sites. This demonstrates that T107 is the preferred phosphorylation site for CK2. Moreover, the S221, S240, and S679 sites tended to be phosphorylated only at a high concentration (3.3 ng/μl) of CK2, while a low concentration (≤3.3 pg/μl) of kinase phosphorylated only T107 and S137.
The mutagenesis results allowed us to generate a mutant VP8 (Mut-VP8) that is not phosphorylated by US3 or CK2 (Fig. 5A). This allowed an investigation of the function of VP8 phosphorylation. Although phosphorylation by CK2 altered the subcellular localization of simian virus 40 (SV40) T antigen (31, 32), phosphorylation of VP8 had no obvious impact on its nuclear import. Both phosphorylated and nonphosphorylated VP8 were localized in the nucleus. In addition, CM5, which contains a CK2 phosphorylation site, did not alter the nuclear import function of the VP8-NLS (R11RPRR15) as fusion proteins NLS-GFP and NLS-CM5-GFP had similar distribution patterns in the transfected COS-7 cells (data not shown). These results suggest that phosphorylation does not change the nuclear localization of VP8. In contrast, phosphorylation by US3 on HHV-1 VP13/14 is needed for the NLS-mediated nuclear localization of VP13/14. VP13/14 contains a phosphorylation site for US3 at S77, which is close to the NLS. Phosphorylation at S77 by HHV-1 US3 is essential for the nuclear localization of VP13/14 as replacing S77 with an A residue results in accumulation of this protein to the nuclear rim (2).
The expression of VP8 generated DAPI-stain-negative nuclear bodies in COS-7 cells, and WT-VP8 accumulated around these bodies (Fig. 6A). A similar pattern was found in certain cells of infected lung tissue (Fig. 6B), indicating a potential role of these nonstained areas in the pathology and mechanism of virus-host interaction during BoHV-1 infection. Concomitantly, PML protein was gradually attracted to the edge of the nuclear bodies, resulting in colocalization with WT-VP8 (Fig. 7). Other investigators observed that PML bodies suppress virus replication in HHV-1 infection. To survive in spite of the suppression, HHV-1 uses a regulatory protein, ICP0, to degrade the PML bodies. This is supported by the finding that PML bodies perform stronger suppression against HHV-1 infection in the absence of the ICP0 protein (15). With respect to BoHV-1 infection, it is possible that VP8 plays a role in counteracting the PML-mediated cellular antiviral process. This is in agreement with our previous observation that BoHV-1 replication is dramatically reduced in the absence of VP8 (5).
The colocalization of WT-VP8 and PML protein in transfected cells suggests that VP8 might exert its effects on PML by remodeling PML bodies. This is in contrast to the results of another study on HCMV demonstrating that the formation of nuclear bodies and the recruitment of PML are enhanced by UL82 (33) through increasing the efficiency of UL35 nuclear body formation (18). However, other cellular proteins may be involved in the formation of VP8 nuclear bodies and remodeling of PML. We investigated the impact of phosphorylation of VP8 on its ability to recruit PML. A nonphosphorylated VP8 (Mut-VP8) did not attract PML protein although circular areas unstained by either DAPI or anti-VP8 antibody were observed (Fig. 8). The results show that phosphorylation is a critical modification for VP8 to accumulate to nuclear bodies, as well as for the subsequent remodeling of PML. It is possible that phosphorylation of VP8 may have a potential role in protecting BoHV-1 from the PML-mediated antiviral defense. This contention is also supported by evidence that BoHV-1 ΔUL47 replicated better in WT-VP8-transfected cells than in Mut-VP8-transfected cells. While deletion of VP8 reduces the virus titer by over 100-fold (5), this was partially amended by transiently expressing WT-VP8 and Mut-VP8, confirming the importance of VP8 in BoHV-1 replication. The fact that WT-VP8 increased the titer more than Mut-VP8 suggested that the phosphorylation of VP8 contributes to virus replication.
The changing phosphorylation status of VP8 during the different stages of viral infection suggests a regulatory role of this modification. Phosphorylation of the incoming VP8 might be an immediate event in BoHV-1 infection. It has been found that phosphorylation of VP13/14, a homologue of VP8 in HHV-1, occurs between virion penetration of the cell and the onset of viral protein synthesis (6). Our results showed that VP8 was extensively phosphorylated in BoHV-1-infected cells. CK2 likely phosphorylates VP8 after viral entry and during VP8 transport through the cell as this kinase is ubiquitous throughout the cell, including the plasma membrane, cytoplasm, and nucleus (34). The virion-associated VP8 might capture the cellular kinase, and the subsequent phosphorylation may help VP8 release, as was found for HHV-1 in that VP13/14 phosphorylation improves its dissociation from virions (6).
After release, the virion-associated VP8 has been shown to move to the nucleus as early as 2 h after infection (20), and this transport is mediated by the NLS independent of phosphorylation. The viral kinase US3 is synthesized later and is localized in the nucleus, where it can associate with VP8 (19). This indicates that the phosphorylation by US3 may affect a nuclear function of VP8 but not the tegument dissociation stage. This hypothesis is supported by a previous finding in HHV-1 that deletion of the viral kinase UL13 had no effect on VP13/14 release in an in vitro assay (6).
In summary, this study demonstrates that phosphorylation is a critical modification for VP8 accumulation to nuclear bodies and recruitment of PML, which is important for the cellular antiviral defense. This function of VP8 appears to depend on its phosphorylation status. At least two kinases target VP8. The phosphorylation by US3 is likely a cascade process, in which the activation of S16 triggers further phosphorylation at S32. CK2 phosphorylates VP8 on at least five active sites, among which T107 is the preferred residue.
ACKNOWLEDGMENTS
We thank Laura Latimer for kind assistance with immunoprecipitation experiments. We thank François Meurens for generating the PCLS sections.
This research was supported by grant 90887-2010 RGPIN from the Natural Sciences and Engineering Research Council of Canada. K.Z. was supported by a scholarship from the China Scholarship Council.
Footnotes
This article is VIDO manuscript number 727.
REFERENCES
- 1.Yu X, Li W, Liu L, Che Y, Cun W, Wu W, He C, Shao C, Li Q. 2008. Functional analysis of transcriptional regulation of herpes simplex virus type 1 tegument protein VP22. Sci China C Life Sci 51:966–972. doi: 10.1007/s11427-008-0127-4. [DOI] [PubMed] [Google Scholar]
- 2.Kato A, Liu Z, Minowa A, Imai T, Tanaka M, Sugimoto K, Nishiyama Y, Arii J, Kawaguchi Y. 2011. Herpes simplex virus 1 protein kinase Us3 and major tegument protein UL47 reciprocally regulate their subcellular localization in infected cells. J Virol 85:9599–9613. doi: 10.1128/JVI.00845-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Potel C, Elliott G. 2005. Phosphorylation of the herpes simplex virus tegument protein VP22 has no effect on incorporation of VP22 into the virus but is involved in optimal expression and virion packaging of ICP0. J Virol 79:14057–14068. doi: 10.1128/JVI.79.22.14057-14068.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carpenter DE, Misra V. 1991. The most abundant protein in bovine herpes 1 virions is a homologue of herpes simplex virus type 1 UL47. J Gen Virol 72:3077–3084. doi: 10.1099/0022-1317-72-12-3077. [DOI] [PubMed] [Google Scholar]
- 5.Lobanov VA, Maher-Sturgess SL, Snider MG, Lawman Z, Babiuk LA. 2010. A UL47 gene deletion mutant of bovine herpesvirus type 1 exhibits impaired growth in cell culture and lack of virulence in cattle. J Virol 84:445–458. doi: 10.1128/JVI.01544-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morrison EE, Wang Y-F, Meredith DM. 1998. Phosphorylation of structural components promotes dissociation of the herpes simplex virus type 1 tegument. J Virol 72:7108–7114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meredith DM, Lindsay JA, Halliburton IW, Whittaker GR. 1991. Post-translational modification of the tegument proteins (VP13 and VP14) of herpes simplex virus type 1 by glycosylation and phosphorylation. J Gen Virol 72:2771–2775. doi: 10.1099/0022-1317-72-11-2771. [DOI] [PubMed] [Google Scholar]
- 8.Ottosen S, Herrera FJ, Doroghazi JR, Hull A, Mittal S, Lane WS, Triezenberg SJ. 2006. Phosphorylation of the VP16 transcriptional activator protein during herpes simplex virus infection and mutational analysis of putative phosphorylation sites. Virology 345:468–481. doi: 10.1016/j.virol.2005.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Geiss BJ, Tavis JE, Metzger LM, Leib DA, Morrison LA. 2001. Temporal regulation of herpes simplex virus type 2 VP22 expression and phosphorylation. J Virol 75:10721–10729. doi: 10.1128/JVI.75.22.10721-10729.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren X, Harms JS, Splitter GA. 2001. Tyrosine phosphorylation of bovine herpesvirus 1 tegument protein VP22 correlates with the incorporation of VP22 into virions. J Virol 75:9010–9017. doi: 10.1128/JVI.75.19.9010-9017.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pomeranz LE, Blaho JA. 1999. Modified VP22 localizes to the cell nucleus during synchronized herpes simplex virus type 1 infection. J Virol 73:6769–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verhagen J, Donnelly M, Elliott G. 2006. Characterization of a novel transferable CRM-1-independent nuclear export signal in a herpesvirus tegument protein that shuttles between the nucleus and cytoplasm. J Virol 80:10021–10035. doi: 10.1128/JVI.01322-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zheng C, Brownlie R, Babiuk LA, van Drunen Littel-van den Hurk S. 2004. Characterization of nuclear localization and export signals of the major tegument protein VP8 of bovine herpesvirus-1. Virology 324:327–339. doi: 10.1016/j.virol.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 14.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J Virol 84:5594–5604. doi: 10.1128/JVI.00348-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Everett RD, Rechter S, Papior P, Tavalai N, Stamminger T, Orr A. 2006. PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J Virol 80:7995–8005. doi: 10.1128/JVI.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Everett R, O'Hare P, O'Rourke D, Barlow P, Orr A. 1995. Point mutations in the herpes simplex virus type 1 Vmw110 RING finger helix affect activation of gene expression, viral growth, and interaction with PML-containing nuclear structures. J Virol 69:7339–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J Virol 86:11209–11222. doi: 10.1128/JVI.01145-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salsman J, Wang X, Frappier L. 2011. Nuclear body formation and PML body remodeling by the human cytomegalovirus protein UL35. Virology 414:119–129. doi: 10.1016/j.virol.2011.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Labiuk SL, Babiuk LA, van Drunen Littel-van den Hurk S. 2009. Major tegument protein VP8 of bovine herpesvirus 1 is phosphorylated by viral US3 and cellular CK2 protein kinases. J Gen Virol 90:2829–2839. doi: 10.1099/vir.0.013532-0. [DOI] [PubMed] [Google Scholar]
- 20.van Drunen Littel-van den Hurk S, Garzon S, van den Hurk JV, Babiuk LA, Tijssen P. 1995. The role of the major tegument protein VP8 of bovine herpesvirus-1 in infection and immunity. Virology 206:413–425. doi: 10.1016/S0042-6822(95)80057-3. [DOI] [PubMed] [Google Scholar]
- 21.Labiuk SL, Lobanov V, Lawman Z, Snider M, Babiuk LA. 2010. Bovine herpesvirus-1 US3 protein kinase: critical residues and involvement in the phosphorylation of VP22. J Gen Virol 91:1117–1126. doi: 10.1099/vir.0.016600-0. [DOI] [PubMed] [Google Scholar]
- 22.Komis G, Takac T, Bekesova S, Vadovic P, Samaj J. 2014. Affinity-based SDS-PAGE identification of phosphorylated Arabidopsis MAPKs and substrates by acrylamide pendant Phos-Tag. Methods Mol Biol 1171:47–63. doi: 10.1007/978-1-4939-0922-3_5. [DOI] [PubMed] [Google Scholar]
- 23.Fior CJ, Roach PJ. 1996. Hierarchal phosphorylation of proteins, p 285–296. In Marks F. (ed), Protein phosphorylation. VCH, Weinheim, Germany. [Google Scholar]
- 24.Ross-Thriepland D, Harris M. 2014. Insights into the complexity and functionality of hepatitis C virus NS5A phosphorylation. J Virol 88:1421–1432. doi: 10.1128/JVI.03017-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hizli AA, Chi Y, Swanger J, Carter JH, Liao Y, Welcker M, Ryazanov AG, Clurman BE. 2013. Phosphorylation of eukaryotic elongation factor 2 (eEF2) by cyclin A–cyclin-dependent kinase 2 regulates its inhibition by eEF2 kinase. Mol Cell Biol 33:596–604. doi: 10.1128/MCB.01270-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eblen ST, Slack-Davis JK, Tarcsafalvi A, Parsons JT, Weber MJ, Catling AD. 2004. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol Cell Biol 24:2308–2317. doi: 10.1128/MCB.24.6.2308-2317.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pagano MA, Poletto G, Di Maira G, Cozza G, Ruzzene M, Sarno S, Bain J, Elliott M, Moro S, Zagotto G. 2007. Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. Chembiochem 8:129–139. doi: 10.1002/cbic.200600293. [DOI] [PubMed] [Google Scholar]
- 28.Benetti L, Roizman B. 2004. Herpes simplex virus protein kinase US3 activates and functionally overlaps protein kinase A to block apoptosis. Proc Natl Acad Sci U S A 101:9411–9416. doi: 10.1073/pnas.0403160101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chuluunbaatar U, Roller R, Feldman ME, Brown S, Shokat KM, Mohr I. 2010. Constitutive mTORC1 activation by a herpesvirus Akt surrogate stimulates mRNA translation and viral replication. Genes Dev 24:2627–2639. doi: 10.1101/gad.1978310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Joughin BA, Liu C, Lauffenburger DA, Hogue CW, Yaffe MB. 2012. Protein kinases display minimal interpositional dependence on substrate sequence: potential implications for the evolution of signalling networks. Philos Trans R Soc Lond B Biol Sci 367:2574–2583. doi: 10.1098/rstb.2012.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rihs H-P, Jans D, Fan H, Peters R. 1991. The rate of nuclear cytoplasmic protein transport is determined by the casein kinase II site flanking the nuclear localization sequence of the SV40 T-antigen. EMBO J 10:633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hübner S, Xiao C-Y, Jans DA. 1997. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J Biol Chem 272:17191–17195. doi: 10.1074/jbc.272.27.17191. [DOI] [PubMed] [Google Scholar]
- 33.Schierling K, Stamminger T, Mertens T, Winkler M. 2004. Human cytomegalovirus tegument proteins ppUL82 (pp71) and ppUL35 interact and cooperatively activate the major immediate-early enhancer. J Virol 78:9512–9523. doi: 10.1128/JVI.78.17.9512-9523.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Faust M, Montenarh M. 2000. Subcellular localization of protein kinase CK2. A key to its function? Cell Tissue Res 301:329–340. doi: 10.1007/s004410000256. [DOI] [PubMed] [Google Scholar]