

ABSTRACT
The nonenveloped simian polyomavirus (PyV) simian virus 40 (SV40) hijacks the endoplasmic reticulum (ER) quality control machinery to penetrate the ER membrane and reach the cytosol, a critical infection step. During entry, SV40 traffics to the ER, where host-induced conformational changes render the virus hydrophobic. The hydrophobic virus binds and integrates into the ER lipid bilayer to initiate membrane penetration. However, prior to membrane transport, the hydrophobic SV40 recruits the ER-resident Hsp70 BiP, which holds the virus in a transport-competent state until it is ready to cross the ER membrane. Here we probed how BiP disengages from SV40 to enable the virus to penetrate the ER membrane. We found that nucleotide exchange factor (NEF) Grp170 induces nucleotide exchange of BiP and releases SV40 from BiP. Importantly, this reaction promotes SV40 ER-to-cytosol transport and infection. The human BK PyV also relies on Grp170 for successful infection. Interestingly, SV40 mobilizes a pool of Grp170 into discrete puncta in the ER called foci. These foci, postulated to represent the ER membrane penetration site, harbor ER components, including BiP, known to facilitate viral ER-to-cytosol transport. Our results thus identify a nucleotide exchange activity essential for catalyzing the most proximal event before ER membrane penetration of PyVs.
IMPORTANCE PyVs are known to cause debilitating human diseases. During entry, this virus family, including monkey SV40 and human BK PyV, hijacks ER protein quality control machinery to breach the ER membrane and access the cytosol, a decisive infection step. In this study, we pinpointed an ER-resident factor that executes a crucial role in promoting ER-to-cytosol membrane penetration of PyVs. Identifying a host factor that facilitates entry of the PyV family thus provides additional therapeutic targets to combat PyV-induced diseases.
INTRODUCTION
Pathogens hijack protein quality control pathways of host cells to successfully cause infection. One pathway co-opted by pathogens during entry is endoplasmic reticulum (ER)-associated degradation (ERAD) (1–3). While ERAD is a surveillance system normally dedicated to the removal of misfolded ER proteins to the cytosol for proteasomal destruction, pathogens can co-opt elements of this pathway to gain entry into the host cytosol by disguising themselves as misfolded ER proteins. A clearer picture of the nature of this pathogen-host interaction is slowly emerging.
Entry of the nonenveloped polyomavirus (PyV) family, including the simian virus 40 (SV40) and the human BK PyVs, serves as a salient example of pathogens that co-opt the ERAD pathway during infection (4–6). Structurally, SV40 is composed of 360 copies of the VP1 major coat protein arranged as 72 pentamers, with each pentamer engaging either the VP2 or VP3 internal hydrophobic minor coat protein. The pentamers are assembled as a 45-nm-diameter icosahedral particle that in turn encapsulates its viral DNA genome (7, 8). To infect cells, SV40 undergoes receptor-mediated endocytosis and is sorted to the ER (9–13). There it co-opts components of the ERAD machinery to penetrate the ER membrane and reach the cytosol (4, 6, 14). From the cytosol, the virus enters the nucleus, where ensuing transcription and replication of the viral genome cause lytic infection or cell transformation.
In the ER, SV40 hijacks numerous ER chaperones that impart conformational changes to the viral particle to expose its hydrophobic VP2 and VP3 proteins (4–6, 15). This enables the resulting hydrophobic particle to integrate into and penetrate the ER membrane (4, 15, 16). Exposure of viral hydrophobic regions is a general principle observed during membrane penetration by many nonenveloped viruses (13, 17). However, prior to ER membrane transport, the ER-resident Hsp70 BiP forms a complex with the hydrophobic SV40 particle (4, 5), presumably to prevent it from aggregation by masking the exposed hydrophobic regions. When poised for membrane transport, BiP must be released from the hydrophobic SV40 so that the virus can bind to the ER membrane and initiate membrane penetration. How BiP disengages from SV40 is unclear.
BiP's ability to interact with substrates is tightly regulated by its ATP/ADP binding states (18): ATP-BiP displays a low affinity for the substrate, while ADP-BiP possesses high substrate binding affinity. These two opposing states are coordinately controlled by ER-resident J-proteins and nucleotide exchange factors (NEFs). Specifically, J-proteins stimulate BiP's ATPase activity, converting ATP-BiP to ADP-BiP. This reaction allows ADP-BiP to bind strongly to the substrate, which is often delivered to BiP by the J-proteins themselves (19, 20). NEFs recruited to the ADP-BiP-substrate complex induce an exchange of ADP for ATP to generate ATP-BiP. ATP-BiP in turn undergoes a conformational change that releases the substrate. While a J-protein called ERdj3 has been implicated in SV40 infection (5), NEFs have yet to be demonstrated to be involved in this process. In fact, more generally, very little is known regarding NEF functions during ERAD (21), in contrast to J-protein functions (22–25).
There are two reported ER-resident NEFs, the Grp170 ATPase and Sil1 (18, 26). In this report, we pinpoint the NEF activity of Grp170 but not Sil1 as specifically releasing SV40 from BiP. This step allows the hydrophobic virus to engage the ER membrane in order to initiate ER membrane penetration. Grp170 not only plays an important role during SV40 infection but also promotes BK PyV infection. These findings thus provide strong evidence that NEF activity performs a key role during ER-to-cytosol membrane transport of members of a nonenvelope virus family.
MATERIALS AND METHODS
Materials.
Polyclonal Hsp90 and SV40 large T-antigen antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), polyclonal BiP and monoclonal PDI antibodies from Abcam (Cambridge, MA), a monoclonal BiP antibody from BD (Franklin, NJ), monoclonal Grp170 and polyclonal Sil1 antibodies from GeneTex (Los Angeles, CA), monoclonal FLAG antibody and luciferase from Sigma-Aldrich (St. Louis, MO), monoclonal green fluorescent protein (GFP) antibody from Protein Tech Group (Chicago, IL), and monoclonal BAP31 antibody from Thermo Fisher Scientific (Waltham, MA). Monoclonal antibodies against SV40 VP1 were a generous gift from W. Scott (University of Miami). FLAG M2 antibody-conjugated agarose beads were purchased from Sigma-Aldrich (St. Louis, MO), S-protein-conjugated agarose beads and digitonin from EMD Millipore Chemicals (San Diego, CA), and protein G magnetic beads from Life Technologies (Carlsbad, CA). Purified phosphatidyl-choline, -ethanolamine, -serine, and -inositol were purchased from Avanti Polar Lipids (Alabaster, AL). Purified BK PyV and pAb416 antibodies against BK PyV large T-antigen were kindly provided by Michael Imperiale (University of Michigan).
DNA construction.
Sil1, Grp170, and BiP cDNAs were amplified from a 293T cDNA library. To generate FLAG-BiP, Grp170-FLAG, and FLAG-Sil1, the FLAG tag sequence was attached to the indicated positions by PCR, with the resulting PCR products inserted into pCDNA3.1(-). To generate G41L Grp170-FLAG, the corresponding mutation was inserted by overlapping PCR, with the resulting PCR product replacing the wild-type (WT) Grp170 coding sequence. For the small interfering RNA (siRNA)-resistant Grp170 constructs, the following silent mutations were introduced into each construct by overlapping PCR: 2716-CTGAACAAAGCTAAATTC-2733 (where the underlines denote the introduced silent mutations).
SV40 preparation, ER-to-cytosol transport, and infection assays.
SV40 preparation, ER-to-cytosol transport, and infection assays were performed as described previously in reference 14, except that COS-7 cells were semipermeabilized with 0.025% digitonin.
Purification of recombinant proteins.
All recombinant proteins used in this study were expressed in and isolated from HEK 293T cells transfected with the indicated DNA constructs. FLAG-tagged recombinant proteins were purified as described previously in reference 25 with minor modifications. For FLAG-SiL1 and WT and mutant Grp170-FLAG experiments, bound proteins on the beads were incubated in a buffer containing 20 mM HEPES (pH 7.5), 50 mM KCl, 0.1% Triton X-100, 2 mM ATP, and 2 mM MgCl2, washed extensively, and eluted with FLAG peptide.
In vitro release of SV40 from BiP.
ER-localized SV40 was isolated and captured with protein G magnetic beads (Life Technology) as described in reference 5, except that the Optiprep gradient centrifugation step was omitted. The virus-bound beads were resuspended in a buffer containing 20 mM HEPES (pH 7.5), 50 mM KCl, and 0.1% Triton X-100 and incubated with the indicated recombinant proteins in the presence or absence of ATP at 37°C. The beads were washed extensively, and the bound proteins were eluted with SDS sample buffer and separated by SDS-PAGE, followed by silver staining.
Liposome binding assay.
To form biotinylated liposomes, 32 μl of phosphatidyl-choline (10 mg/ml), 5 μl of phosphatidyl-ethanolamine (10 mg/ml), 5 μl of N-biotinyl cap-phosphatidyl-ethanolamine, 4.8 μl of phosphatidyl-inositol (10 mg/ml), and 1 μl of phosphatidyl-serine (10 mg/ml) (all dissolved in chloroform) were mixed. The mixture was dried and resuspended in 50 μl of a buffer containing 20 mM HEPES (pH 7.6) and 100 mM NaCl. The resuspended vesicles were sonicated in a water sonicator bath for 30 min, incubated at 4°C overnight, and conjugated with netravidin (Thermo Fisher Scientific) via the use of biotin. The virus-bound beads were prepared as described above, incubated with or without 2 mM ATP in the presence of 2 mM MgCl2 at 25°C for 20 min, washed extensively, and incubated with 20 μl of liposomes at 37°C for 30 min. The beads were washed, the bound liposomes solubilized by 1% Triton X-100, and the avidin-biotin complex recovered from the beads and analyzed by silver staining. Following recovery of the avidin-biotin complex, the virus were eluted from the beads by the use of SDS-sample buffer and analyzed by immunoblotting. When BiP-less ER-localized S40 prepared with Grp170-FLAG was used in the liposome binding assay, the virus-bound magnetic beads were incubated with 5 μl of liposomes at 25°C for 10 min.
In vitro binding assay.
Recombinant proteins were mixed, incubated at 37°C for 30 min, and subjected to immunoprecipitation with an anti-BiP antibody using protein G magnetic beads. The immune complexes were washed, eluted by SDS sample buffer, and subjected to SDS-PAGE followed by immunoblotting with the appropriate antibodies.
XBP1 splicing.
XBP1 splicing was performed as described in reference 27, except that CV-1 cells were used.
Knockdown of Grp170 and Sil1.
The target sequences of the siRNAs used in this study were as follows: for Grp170 no. 1 siRNA, 5′-GCUCAAUAAGGCCAAGUUU-3′ (Invitrogen); for Grp170 no. 2 siRNA, 5′-GCCUUUAAAGUGAAGCCAU-3′ (Invitrogen); for Sil1 no. 1 siRNA, 5′-GCUGAUCAACAAGUUCAAU-3′ (Invitrogen); and for Sil1 no. 2 siRNA, 5′-GCGCUCUUUGAUCUUGAAU-3′ (Invitrogen).
Duplex siRNA (10 nM) was reverse transfected into cells using Lipofectamine RNAiMAX (Life Technologies) according to the manufacturer's protocol. Allstar negative-control siRNA (Qiagen, Hilden, Germany) was used as a scrambled siRNA control.
SV40 infection under FLAG-tagged protein expression.
For a rescue experiment, approximately 2 × 105 CV-1 cells transfected with siRNA were further transfected with various DNA constructs using Fugene HD (Promega, Madison, WI), incubated for 24 h, and infected with SV40. Samples were processed using an immunofluorescence method as described previously in reference 14, except that cells were subjected to double staining with anti-T-antigen and FLAG antibodies; only T-antigen-positive nuclei in FLAG signal-positive cells were scored. For other experiments, cells were processed as described above except that siRNA transfection was omitted.
Luciferase aggregation assay.
Luciferase (1.4 μM) was incubated with bovine serum albumin (BSA), Grp170-FLAG, or G41L Grp170-FLAG (2.5 μM each) in a buffer containing 20 mM HEPES (pH 7.5), 50 mM KCl, 0.1% Triton X-100, and 1 mM dithiothreitol (DTT) at 42°C for 30 min and subjected to centrifugation at 16,000 × g for 10 min. The resulting supernatant and pellet fractions were subjected to SDS-PAGE and analyzed by Coomassie blue staining.
Nucleotide exchange assay.
A modified version of a nucleotide exchange assay was developed based on two published protocols (28, 29). Purified FLAG-BiP (5 μM) was incubated with 50 μCi of [α-32P]ATP (PerkinElmer) (3,000 Ci/mmol) in a final volume of 25 μl (50 μCi is equivalent to 0.66 μM ATP in this reaction) at 37°C for 30 min to form [α-32P]ADP FLAG-BiP, and the sample was subjected to a spin gel filtration column (GE Healthcare) to remove the free nucleotides. [α-32P]ADP FLAG-BiP (0.3 μM) was incubated with the indicated recombinant proteins (1 μM) in 23 μl of a buffer containing 20 mM HEPES (pH 7.5), 50 mM KCl, and 0.1% Triton X-100 at 23°C for 20 min. Following incubation, each reaction mixture was combined with unlabeled ATP (0.3 μM) and MgCl2 (2 mM) and further incubated at 23°C for 1 min. After removal of free nucleotides using another spin gel filtration column, 2 μl of the reaction was spotted onto a polyethyleneimine (PEI) cellulose plate (Sigma) and developed in 0.6 M KH2PO4 (pH 3.4) by the thin-layer chromatography (TLC) method.
Calculation of the number of BiP molecules bound to a SV40 particle.
ER-localized SV40 isolated from virus-infected cells is described above. Defined amounts of purified SV40 and FLAG-BiP used for standards were separated by SDS-PAGE. The bands were visualized by Coomassie staining for VP1 and silver staining for BiP. The VP1 and BiP bands in the ER-localized SV40 sample were quantified with ImageJ (NIH), and their levels were determined based on the standard VP1 and BiP bands, respectively. The number of BiP molecules per viral particle was calculated from the obtained protein amounts using 40 kDa and 78 kDa as the molecular masses for VP1 and BiP, respectively.
Visualization of SV40-induced focus formation.
Approximately 1 × 105 CV-1 cells transfected with the indicate DNA construct using Fugene HD were infected with SV40 (multiplicity of infection [MOI] = ∼50) for 16 h, fixed, and processed by the immunofluorescence method as described above, except that anti-FLAG and BAP31 antibodies were used.
RESULTS
Release of BiP from SV40 promotes viral membrane binding in vitro.
Our laboratory and those of others previously demonstrated that BiP is recruited to SV40 in the ER (4, 5), likely because the viral particle is rendered hydrophobic due to ER factor-induced conformational changes (4, 15, 30). We hypothesize that BiP recruitment shields the hydrophobic virus until it is ready to engage the ER membrane to initiate the penetration process. To test this idea, we asked if BiP-less SV40 preferentially binds to liposomes. We took advantage of our previously established semipermeabilized system and isolated ER-localized SV40 from virus-infected CV-1 cells (5, 14). As expected, BiP copurified with ER-localized SV40 (i.e., BiP-bound SV40) after immunopurification of the viral particles (Fig. 1A, top and bottom panels, lane 1). Quantification of the VP1 and BiP bands in the ER-localized virus based on comparisons to defined amounts of purified SV40 and BiP revealed that approximately 37 BiP molecules associated with each viral particle (see Materials and Methods). When a high (2 mM) ATP concentration was added to the isolated SV40-BiP complex followed by reimmunopurification of the virus and silver staining, BiP did not coprecipitate with the virus (i.e., BiP-less SV40) (Fig. 1B, top panel; compare lane 2 to lane 1). This finding demonstrates that a sufficiently high ATP concentration can cause release of SV40 from BiP, likely because ADP-BiP, which normally engages SV40, displayed a reduced affinity for the substrate once it was converted to the ATP-bound state.
FIG 1.
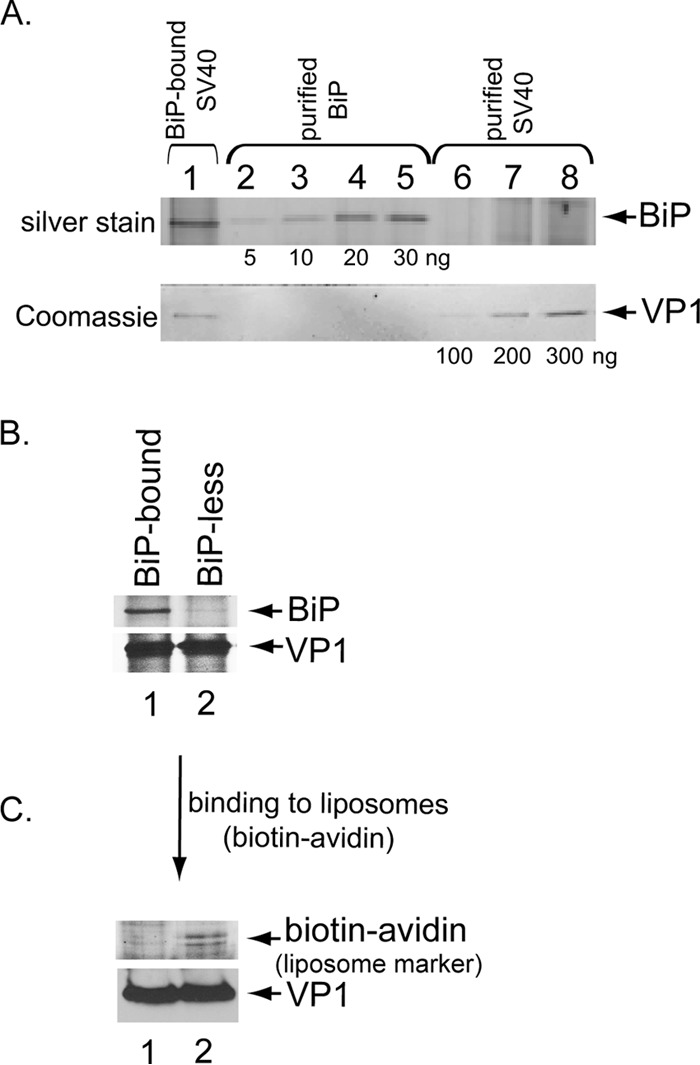
Release of BiP from SV40 promotes viral membrane binding in vitro. (A) ER-localized SV40 immunoprecipitated from infected CV-1 cells, along with the indicated amounts of purified SV40 (100, 200, and 300 ng of VP1) and purified recombinant BiP (5, 10, 20, and 30 ng), was separated by SDS-PAGE. The BiP band was visualized by silver staining and the VP1 band by Coomassie staining. The amounts of VP1 and BiP in ER-localized SV40 were quantified with ImageJ (NIH) using purified SV40 and BiP as standards in order to calculate the numbers of BiP molecules per viral particle. (B) ER-localized SV40 processed as described for panel A was incubated without ATP (i.e., BiP-bound SV40) or with ATP (2 mM) to generate BiP-less SV40. Following reprecipitation of the virus, the samples were subjected to SDS-PAGE and silver staining. (C) BiP-bound or BiP-less SV40 processed as described for panel B was incubated with liposomes labeled with avidin via a biotinylated phospholipid. Following reprecipitation of the virus, the samples were subjected to SDS-PAGE and analyzed by silver staining for the biotin-avidin complex and by immunoblotting with anti-VP1 antibodies.
If BiP masks hydrophobic regions of ER-localized SV40, release of SV40 from BiP should allow the viral particle to bind to membranes. Indeed, when BiP-bound or BiP-less ER-localized SV40 was incubated with liposomes (labeled with avidin via a biotinylated phospholipid) followed by reprecipitation, BiP-less SV40 but not BiP-bound SV40 pulled down the liposomes, as reflected by presence of the biotin-avidin complex (Fig. 1C, top panel; compare lane 2 to lane 1). This finding indicates that BiP release is essential for SV40 to interact with membranes because its hydrophobic regions are exposed. More importantly, it raises the issue of how BiP might be normally released from SV40 for efficient ER membrane penetration in cells.
Grp170 induces nucleotide-dependent release of ER-localized SV40 from BiP in vitro.
Grp170 and Sil1 are two ER-resident NEFs known to induce nucleotide exchange in the presence of BiP (28, 31–35). To analyze the mechanism by which SV40 is released from BiP, we first asked whether addition of recombinant Grp170 or Sil1 to the isolated SV40-BiP complex would promote release of the virus from BiP. Isolated SV40-BiP complex was incubated with a low (0.2 μM) ATP concentration and FLAG-tagged Grp170 (Grp170-FLAG), Sil1 (FLAG-Sil1), or the control GFP (GFP-FLAG) purified from mammalian cells (Fig. 2A), followed by reprecipitation of the virus and silver staining. We found that Grp170-FLAG but not GFP-FLAG or FLAG-Sil1 caused SV40 to disengage from BiP under this condition (Fig. 2B; compare lane 3 to lanes 2 and 4); in contrast, in the absence of ATP, Grp170 did not release SV40 from BiP (Fig. 2C; compare lane 3 to lane 2). These findings demonstrate that Grp170 but not Sil1 induces SV40 release from BiP and that the release reaction is ATP dependent, consistent with a previous report demonstrating that Grp170's NEF function requires ATP binding (36).
FIG 2.

Grp170 induces release of ER-localized SV40 from BiP in vitro. (A) Coomassie staining of C-terminally FLAG-tagged GFP (GFP-FLAG), C-terminally FLAG-tagged Grp170 (Grp170-FLAG), and N-terminally FLAG-tagged Sil1 (FLAG-Sil1) purified from 293T cells. (B) ER-localized SV40 immunoprecipitated from infected CV-1 cells was incubated without ATP or with ATP (0.2 μM) and the indicated recombinant protein (250 nM). Following reprecipitation of the virus, the samples were subjected to SDS-PAGE and silver staining. The black line indicates that intervening lanes have been spliced out. (C) The experiment was performed as described for panel B, except the immunoprecipitate was incubated with Grp170-FLAG in the absence of ATP where indicated. (D) Coomassie staining of purified C-terminally FLAG-tagged BiP (BiP-FLAG). (E) BiP-FLAG (300 nM) was incubated with the indicated recombinant protein (300 nM) and immunoprecipitated with an anti-BiP antibody. The immunoprecipitates were subjected to SDS-PAGE followed by immunoblotting with an anti-FLAG antibody. Input samples were also analyzed by immunoblotting with an anti-FLAG antibody. (F) Coomassie staining of purified N-terminally FLAG-tagged BiP (FLAG-BiP). (G) FLAG-BiP was incubated with [α-32P]ATP to form the radiolabeled ADP-BiP complex. The ADP-BiP complex was then incubated with the indicated proteins in the absence or presence of unlabeled ATP. ADP release from BiP was analyzed by TLC. (H) BiP-less ER-localized SV40 was prepared as described for panel B. The ability of BiP-bound or BiP-less ER-localized SV40 to bind to liposomes was assessed as described for Fig. 1C.
We performed control experiments to assess the integrity of the recombinant NEFs used in our assay. First, as Grp170 and Sil1 must bind to BiP to execute their NEF activities against BiP, we asked whether recombinant Grp170 and Sil1 interact with BiP and found that Grp170-FLAG and FLAG-Sil1 bind to purified BiP-FLAG (Fig. 2D) with similar affinities (Fig. 2E; compare lane 5 to lane 6).
Second, we tested whether recombinant Grp170 and Sil1 retained their nucleotide exchange activities. Purified FLAG-BiP (Fig. 2F) was preloaded with [α-32P]-radiolabeled ATP and incubated for 30 min to allow conversion of ATP-BiP to ADP-BiP. The sample was subjected to an initial spin column gel filtration to remove the majority of the excess radiolabeled ATP. Radiolabeled ADP-BiP was then incubated with GFP-FLAG, Grp170-FLAG, or FLAG-Sil1 in the presence or absence of unlabeled ATP (0.3 μM) to induce nucleotide exchange. Samples were subjected to a second round of spin column gel filtration to remove released nucleotides. The level of radiolabeled ADP that remained bound to FLAG-BiP was monitored by thin-layer chromatography (TLC). Using this assay, we found that both Grp170-FLAG and FLAG-Sil1 but not GFP-FLAG promoted ADP release from BiP (Fig. 2G; compare lanes 3 and 4 to lane 2), demonstrating that Grp170 and Sil1 retain their nucleotide exchange activities. Hence, Sil1's inability to induce SV40 release from BiP is not a consequence of Sil1's being enzymatically inactive, and the results demonstrate that recombinant Sil1 was folded properly.
Interestingly, radiolabeled ATP was detected when Grp170-FLAG but not FLAG-Sil1 was used to induce ADP release from BiP (Fig. 2G; compare lanes 3 and 4). This signal is likely due to the presence of the residual free radiolabeled ATP (after the initial spin column filtration) that bound to Grp170, which is an ATPase, but not to Sil1, which, in contrast, is not an ATPase. While a low concentration of unlabeled ATP (0.3 μM) was used in the assay to prevent spontaneous ADP release from BiP, this ATP concentration was not sufficient to compete with the radiolabeled ATP and cause its removal from Grp170. Regardless, we conclude that Grp170 but not Sil1 directly triggers the release of ER-localized SV40 from BiP by converting ADP-BiP to ATP-BiP in vitro. Not surprisingly, Grp170-dependent release of BiP from SV40 also promoted association of the viral particle with liposomes, as the BiP-less ER-localized SV40 (generated by incubation with Grp170-FLAG) pulled down more avidin-labeled liposomes than the BiP-bound ER-localized SV40 (Fig. 2H, top panel; compare lane 2 to lane 1). Collectively, these data raise the possibilities that Grp170 might play a role in facilitating SV40 ER-to-cytosol membrane transport and that the observed substrate specificity of Grp170 and Sil1 might exist in cells.
Grp170 promotes SV40 ER-to-cytosol membrane transport.
To ascertain whether Grp170 and Sil1 perform any role in SV40 ER-to-cytosol membrane transport in cells, we silenced Grp170 and Sil1 in virus-infected CV-1 cells using two different siRNA oligonucleotides for each protein and monitored SV40 arrival to the cytosol from the ER using a cell-based ER-to-cytosol membrane transport assay established previously (14). A similar assay has also been reported (4). Briefly, SV40-infected cells transfected with a control siRNA (scrambled) and siRNAs directed against Grp170 (Grp170 siRNA no. 1 and no. 2) or Sil1 (Sil1 siRNA no. 1 and no. 2) were treated with a low concentration of digitonin to permeabilize the plasma membrane without damaging internal membranes. Cells were centrifuged to generate two fractions, a supernatant fraction that contains cytosolic proteins and virus that reaches the cytosol (i.e., cytosol) and a pellet fraction that harbors membranes that include the ER along with virus that are retained in membranes (i.e., membrane). ER-localized SV40 can be further isolated from the pellet by treating it with the detergent Triton X-100 and extracting the detergent-soluble material (i.e., Triton X-100). This is because SV40 normally recognizes its ganglioside GM1 receptor on the cell surface and remains bound to it until the virus-receptor complex reaches the ER, where the virus dissociates from the receptor. Because the receptor is highly concentrated in lipid rafts, SV40 remains associated with lipid rafts until it arrives to the ER. As lipid rafts are largely resistant to Triton X-100 extraction whereas ER lumenal components can be extracted by this detergent, SV40 extracted from a pellet by Triton X-100 thus represents virus that reaches the ER and is released into the lumen (37).
Immunoblot analyses revealed that Grp170 and Sil1 in the Triton X-100 fraction were effectively downregulated by their respective siRNAs compared to the results seen with scrambled siRNA (Fig. 3A, Triton X-100 fraction, first and second panels). VP1 in this fraction remained largely unperturbed when Grp170 or Sil1 was silenced (Fig. 3A, Triton X-100 fraction, fourth panel), indicating that Grp170 and Sil1 knockdown did not significantly affect SV40 ER arrival. Importantly, under this knockdown condition, cytosol-localized VP1 levels decreased markedly when Grp170 but not Sil1 was downregulated (Fig. 3A, cytosol fraction, top panel). Quantification of the cytosol-localized VP1 band intensity revealed that Grp170 siRNA no. 1 and no. 2 decreased VP1 by 75% and 50%, respectively, while Sil1 siRNA no. 1 and no. 2 produced a marginal effect, reducing VP1 levels by approximately 15% using either siRNA (Fig. 3B). Neither induction of BiP (Fig. 3A, Triton X-100 fraction, third panel) nor XBP-1 splicing (Fig. 3C) was observed when these NEFs were knocked down, suggesting that decreasing the levels of these factors did not induce massive ER stress. Thus, consistent with the in vitro results, the findings from our cell-based transport assay support the idea that Grp170 but not Sil1 promotes SV40 ER-to-cytosol membrane transport by releasing the virus from BiP. They also suggest that Grp170 and Sil1 have distinct substrate specificities in cells.
FIG 3.

Grp170 promotes SV40 ER-to-cytosol membrane transport. (A) Infected CV-1 cells transfected with the indicated siRNA were harvested and were subjected to the ER-to-cytosol membrane transport assay as described by Inoue and Tsai (14). Cytosol, membrane, and Triton X-100 fractions were subjected to SDS-PAGE followed by immunoblotting with the indicated antibodies. (B) The VP1 band intensity in the cytosolic fraction represented in panel A was quantified with ImageJ (NIH). Data represent means ± standard deviations (SD) of the results of at least 3 independent experiments. (C) Reverse transcription-PCR (RT-PCR) analysis of the unspliced (u) and spliced (s) forms of the XBP1 mRNA from cells transfected with the indicated siRNA or treated with DTT. (D) CV-1 cells initially transfected with scrambled or Grp170 siRNA no. 1 were subsequently transfected with BiP-S, infected with SV40 for 18 h, and harvested. Whole-cell extracts (WCEs) were incubated with S-protein-conjugated beads, and the isolated proteins subjected to SDS–PAGE followed by immunoblotting with anti-VP1 and anti-BiP antibodies. AP, affinity purification.
Since Grp170 induces release of SV40 from BiP to promote SV40 ER-to-cytosol transport, we hypothesize that downregulating Grp170 should stabilize the SV40-BiP interaction. To test this, cells expressing S-tagged BiP (BiP-S) and transfected with scrambled or Grp170 no. 1 siRNA were infected with SV40 and subjected to S-tag affinity purification. As expected, Grp170 knockdown increased the level of VP1 that coprecipitated with BiP-S (Fig. 3D, top panel), demonstrating that Grp170 normally triggers release of SV40 from BiP to allow viral transport from the ER to the cytosol.
Grp170 supports polyomavirus infection.
Upon reaching the cytosol, SV40 transports to the nucleus to cause infection. Accordingly, we examined whether Grp170 or Sil1 regulates SV40 infection. CV-1 cells transfected with scrambled, Grp170 no. 1, Grp170 no. 2, Sil1 no. 1, or Sil1 no. 2 siRNA were infected with SV40, and the cells were stained for presence of the virally encoded large T-antigen in the host nucleus; large T-antigen expression is a hallmark of successful SV40 infection. In comparisons to scrambled siRNA, we found that Grp170 siRNA no. 1 and Grp170 siRNA no. 2 blocked SV40 infection by 75% and 50%, respectively, while neither Sil1 no. 1 siRNA nor Sil1 no. 2 siRNA exerted any statistically significant effect (Fig. 4A, black bars). Thus, in line with the in vitro virus release and cell-based ER-to-cytosol membrane transport assays, Grp170 but not Sil1 promoted SV40 infection. Grp170 but not Sil1 was required for infection of the human BK PyV, with a trend similar to that seen with SV40 (Fig. 4A, white bars), suggesting that Grp170 generally regulates PyV infection.
FIG 4.

Grp170 supports polyomavirus infection. (A) Large T-antigen-positive nuclei were scored in SV40- or human BK PyV-infected CV-1 cells transfected with the indicated siRNA. Data represent means ± SD of the results of at least 3 independent experiments. A Student two-tailed t test was used for the SV40 infection data. (B) The experiment was performed as described for Fig. 2G, except that G41L Grp170-FLAG was used. Purified G41L Grp170-FLAG results are also shown. (C) Grp170-FLAG or G41L Grp170-FLAG was immunoprecipitated (IP), and the samples were subjected to immunoblotting with the indicated antibodies. (D) Luciferase was incubated with BSA, Grp170-FLAG, or G41L Grp170-FLAG at 42°C for 30 min and centrifuged. The resulting supernatant and pellet fractions were subjected to SDS-PAGE and analyzed by Coomassie staining. The black line indicates that intervening lanes have been spliced out. (E) CV-1 cells initially transfected with scrambled or Grp170 siRNA no. 1 were subsequently transfected with GFP-FLAG, Grp170-FLAG, G41L Grp170-FLAG, or FLAG-Sil1. Cells were then infected with SV40 and subjected to immunofluorescence analyses using anti-FLAG and anti-T-antigen antibodies. FLAG and T-antigen doubly positive cells were counted and analyzed as described for panel A. Data represent means ± SD of the results of at least 3 independent experiments. A Student two-tailed t test was used. (F) WCEs derived from cells processed as described for panel E were analyzed by SDS-PAGE and immunoblotted with the indicated antibodies. (G) COS-7 cells transfected with the indicated DNA construct were infected with SV40 for 12 h and subjected to the ER-to-cytosol membrane transport assay as described for Fig. 3A. The cytosol fraction was subjected to SDS-PAGE, followed by immunoblotting with the indicated antibodies.
Grp170 functions not only as an ATPase-dependent NEF for BiP but also as an ATPase-independent holdase (29, 38). Interestingly, this holdase but not NEF activity was recently implicated in ERAD of the epithelial sodium channel α subunit, a BiP-independent ERAD substrate (39). Although recombinant Grp170-FLAG promoted release of the ER-localized SV40 from BiP in a nucleotide-dependent manner in vitro, whether Grp170 promotes SV40 infection by using its inherent nucleotide exchange or holdase activities in cells is unclear. To clarify these possibilities, we conducted a rescue experiment in which endogenous Grp170 silenced by Grp170 no. 1 siRNA was rescued by readdition of siRNA-resistant WT or mutant Grp170 or WT Sil1.
We performed a series of experiments to generate and characterize a NEF-defective Grp170 mutant. To this end, we took advantage of a previous study that mutated G28 in the Lhs1p yeast Grp170 homolog to leucine, generating the G28L mutant (36). This mutant cannot bind to ATP due to the presence of the bulky leucine side chain in the nucleotide binding pocket and fails to display any NEF activity. When the corresponding residue in human Grp170, G41, was mutated to leucine to generate G41L Grp170 (G41L Grp170-FLAG), we found that this purified protein (Fig. 4B, lane 1) did not induce ADP release from BiP (Fig. 4B; compare lanes 5 to 4). Additionally, G41L Grp170-FLAG did not trigger nucleotide-dependent release of SV40 from BiP (not shown). In cells expressing Grp170-FLAG or G41L Grp170-FLAG, endogenous BiP coprecipitated with Grp170-FLAG but not with G41L Grp170-FLAG (Fig. 4C, top panel; compare lanes 1 and 2). This finding indicates that Grp170 but not G41L Grp170 binds to BiP, consistent with a previous report demonstrating that nucleotide binding to Lhs1p is required for efficient Kar2p (yeast BiP homolog) interaction (36). We used a luciferase aggregation assay to further evaluate whether G41L Grp170's ATP-independent holdase activity remained intact. Luciferase aggregates at 42°C due to thermal instability and, upon centrifugation, partitions to the pellet but not the supernatant fraction. Whereas luciferase pelleted in the presence of the control protein BSA (Fig. 4D; compare lanes 4 to 1), it remained in the supernatant when incubated with either Grp170-FLAG or G41L Grp170-FLAG (Fig. 4D; compare lanes 2 and 3 to lanes 5 and 6). Thus, G41L Grp170 lacks a NEF activity but retains its holdase function.
To perform the rescue experiments, cells initially transfected with scrambled or Grp170 no. 1 siRNA were subsequently transfected with FLAG-GFP, the siRNA-resistant WT or G41L Grp170-FLAG, or FLAG-Sil1. Cells were then infected with SV40, fixed, and probed with antibodies directed against large T-antigen and FLAG. Only T-antigen-positive nuclei expressed in FLAG-positive cells (i.e., rescued cells) were scored. Compared to expression of control GFP-FLAG, only reexpression of Grp170-FLAG but not G41L Grp170-FLAG or FLAG-Sil1 under the Grp170 knockdown condition rescued SV40 infection (Fig. 4E). The reexpressed levels of Grp170-FLAG and G41L Grp170-FLAG were similar to that of endogenous Grp170 (Fig. 4F, top panel), while the reexpressed FLAG-Sil1 level was higher than the level seen with endogenous Sil1 (Fig. 4F, second panel). Consistent with this observation, overexpression of Grp170-FLAG but not G41L Grp170-FLAG in the CV-1-derived COS-7 cells in the context of the ER-to-cytosol transport assay increased the VP1 level in the cytosol fraction (Fig. 4G, top panel; compare lane 2 to lanes 3 and 1). (COS-7 cells were used in this experiment because they support the high transfection efficiency necessary for this study.) Thus, the NEF activity of Grp170 but not Sil1 is specifically required to promote the SV40 ER-to-cytosol transport that leads to successful infection, likely by inducing virus release from BiP via converting this chaperone to the low-affinity ATP-bound state.
SV40 triggers Grp170 to reorganize into foci in the ER.
Our laboratory and another group previously reported that SV40 induces ER lumenal proteins (e.g., BiP) and membrane proteins (e.g., BAP31) essential for viral infection to reorganize into discrete puncta (called foci) in the ER membrane (4, 40). To assess whether Grp170 moves into these foci upon SV40 infection, cells expressing FLAG-BiP or Grp170-FLAG were incubated with or without (mock treatment) SV40 for 16 h, fixed, and subjected to immunofluorescence staining. Positive focus formation can be visualized by staining for the endogenous ER membrane protein BAP31. Consistent with a previous report examining endogenous BiP (4), FLAG-BiP can be found to move into the BAP31-containing foci upon SV40 infection (Fig. 5A; compare second to first row). Importantly, a pool of Grp170-FLAG also mobilizes into BAP31-positive foci upon SV40 infection (Fig. 5B; compare second to first row). Interestingly, when Grp170 was knocked down, SV40 induced a larger amount of prominent BiP and BAP31 doubly positive foci than the scrambled control (Fig. 5C; compare second to first row). As Grp170 knockdown stabilized the BiP-SV40 interaction (Fig. 3D, top panel), this observation suggests that depletion of Grp170 causes the accumulation of BiP-bound SV40 to occur in the foci. Our finding that SV40 can trigger Grp170 to reorganize into the foci, similarly to the fate of other ER components involved in SV40 ER membrane transport, provides further independent evidence of this NEF's involvement during the virus ER-to-cytosol penetration process.
FIG 5.

SV40 triggers Grp170 to reorganize into foci in the ER. (A) Cells transfected with FLAG-BiP were mock infected or infected with SV40 for 16 h, fixed, and subjected to immunofluorescence staining with anti-FLAG and BAP31 antibodies. The bar represents 20 μm. (B) Cells were processed as described for panel A, except Grp170-FLAG was used. (C) Cells transfected with scrambled or Grp170 siRNA no. 1 were processed as described for panel A, except endogenous (endo) BiP antibodies were used.
DISCUSSION
Our studies identified the NEF activity of Grp170 but not Sil1 as crucial in facilitating host cell entry of PyVs, including SV40 and BK PyV. Specifically, our in vitro and coimmunoprecipitation binding experiments demonstrated that the NEF activity of Grp170 but not Sil1 induces SV40 release from BiP in the ER lumen. This step enables the hydrophobic virus to engage the ER membrane to initiate membrane penetration. Next, using cell-based assays, we found that Grp170 but not Sil1 promotes SV40 ER-to-cytosol transport and infection, consistent with results from the binding experiments. Grp170 but not Sil1 also supports BK PyV infection, suggesting that the PyV family generally uses this ER NEF activity to cross the ER membrane. Finally, imaging studies revealed that SV40 induces Grp170 to move to discrete foci in the ER, which is known to contain specific host components (including BiP) responsible for mediating viral ER-to-cytosol transport. This finding further strengthens the idea of the role of Grp170 in ER membrane transport of PyVs.
Grp170 and Sil1 are two established ER-resident NEFs. Although Grp170 is also a member of the Hsp70 protein family, it represents an evolutionarily distinct form of canonical Hsp70 proteins that harbor intrinsic ATPase activity. The ATPase activity of yeast Grp170 homolog Lhs1p controls its NEF activity against yeast BiP homolog Kar2p (36). Our data demonstrating that a Grp170 mutant which cannot bind to ATP is NEF defective, cannot stimulate virus ER-to-cytosol transport, and fails to restore SV40 infection in Grp170 knocked-down cells are in line with the notion that Grp170's ATPase activity is essential for its NEF function—this NEF activity is in turn required to release SV40 from BiP prior to viral ER membrane penetration leading to successful infection.
In addition to its recognized NEF activity, Grp170 can also act as an ATP-independent holdase to prevent protein aggregation (38, 41). In fact, Grp170's holdase but not NEF activity was implicated in ERAD in mammalian cells (39). In contrast, in part because it is not an ATPase, Sil1's NEF activity likely operates differently from that of Grp170. Biochemical and structural analyses of the yeast Sil1-Kar2p complex (42) indicate that Sil1 binding to ADP-Kar2p triggers a conformational change in Kar2p that releases ADP; whether this binding reaction is sufficient to induce substrate release from Kar2p, or requires subsequent ATP binding, is not clear. Note that deletion of SIL1 (21) but not LHS1 (39) resulted in a moderate ERAD phenotype, suggesting that Sil1 may exert a potential role in ERAD in yeast.
Interestingly, in yeast, Sil1 and Grp170/Lhs1 have been implicated in protein forward translocation, consistent with BiP's role in this process (31, 33, 34). In this context, the phenotypes caused by loss of either one of the NEFs can be rescued by overexpression of the other, suggesting that the activities of Sil1 and Grp170/Lhs1 are interchangeable, at least under certain circumstances (34, 43, 44). Nevertheless, this interchangeability was not observed in ER-to-cytosol transport of PyVs, even when Sil1 was exogenously overexpressed in Grp170-depleted cells. What then might account for this NEF specificity for PyV infection? Because Grp170 and Sil1 bind to substrate-free BiP with similar efficiencies in vitro, it is unlikely that their affinity for BiP per se dictates the specificity. This is consistent with previous observations that the two NEFs convert substrate-free ADP-BiP to ATP-BiP with similar efficiencies (29, 35). Instead, because BiP can reciprocally stimulate Grp170's ATPase activity (29), this reaction may increase Grp170's affinity for substrates. As indicated, Grp170 also harbors a unique holdase domain that interacts with hydrophobic proteins which is absent in Sil1. These properties observed in Grp170 might support the more stable Grp170-PyV-BiP complex necessary for the subsequent Grp170-triggered nucleotide exchange and substrate release reactions. We note that cytosolic NEF Hsp110 promotes dissociation of the clathrin basket from Hsc70 more efficiently than another cytosolic NEF, Bag1, in vitro (45). Thus, there is precedence for substrate discrimination during NEF-mediated cellular reactions.
An important issue is why BiP binding to and release from ER-localized SV40 are essential for viral ER-to-cytosol transport and infection. Because BiP recognizes hydrophobic moieties, it likely binds directly to the exposed hydrophobic regions of the VP2 and VP3 minor coat proteins. As there are 72 VP1 pentamers within each viral particle and as each pentamer harbors either a hydrophobic VP2 or VP3, we suspect that all of the minor coat proteins can be potentially exposed by conformational changes that SV40 experiences in the ER. Because our calculation estimates that 37 BiP molecules bind to one virion, it might be reasonable to assume that one BiP molecule recognizes and masks each hydrophobic segment in VP2/VP3. Alternatively, BiP may bind to the minor proteins via interaction with the VP1 pentamers. Regardless, the transient BiP interaction likely prevents the hydrophobic viral particle from nonspecifically engaging other hydrophobic proteins in the ER (or binding to each other), leading to massive aggregation. We envision that when the SV40-BiP complex reaches the proper membrane penetration site in the ER, BiP is fully released from SV40 to enable membrane penetration. In this case, it remains possible that BiP's presence on SV40 may simply serve to limit membrane association without fully preventing viral binding to membranes—complete release of BiP from the virus is necessary for translocation across the ER membrane. As stated above, BiP's ability to bind to and be released from SV40 is ultimately dictated by BiP's ATP/ADP states, which in turn are controlled by BiP's cochaperones, a J-protein and the Grp170 NEF. Hence, the efficiency with which a J-protein and Grp170 coordinately act on BiP to regulate its substrate binding determines the dynamic nature of this chaperone's association with SV40.
Finally, our analyses also revealed that Grp170 mobilizes into foci in the ER upon SV40 infection. Because the foci selectively harbor ER components known to promote ER-to-cytosol transport of SV40 (4, 40), reorganization of this NEF into the foci further supports the idea of its role in facilitating viral ER membrane penetration. This reorganization of Grp170 into the foci might clarify how SV40, upon release from BiP, is able to fully interact with the ER lipid bilayer to initiate membrane penetration and how it avoids repeated cycles of reengaging BiP. By concentrating this NEF into the foci, SV40 released from BiP is likely to bind to ER membrane components (which are also concentrated in the foci) that assist the virus in engaging the ER lipid bilayer. As these foci have been postulated to represent the actual site from where PyVs exit the ER to enter the cytosol (4, 40), additional experiments are required to clarify how PyVs released from BiP are mechanistically coupled to its ER membrane penetration in a step presumably aided by a membrane component(s) that initiates retrotranslocation within the foci.
Viral pathogens are known to hijack fundamental cellular processes in order to facilitate their entry, replication, and assembly. Emerging findings demonstrate that viruses use the host ER to promote each of these steps (17). The mechanism by which SV40 interacts with elements of the ER protein quality control pathway called ERAD elegantly illustrates how a virus uses the ER during entry. In addition to viruses, bacterial toxins such as cholera and Shiga toxin, as well as the plant toxin ricin, also co-opt ERAD during entry (37). Interestingly, while the BiP chaperone cycle promotes ER-to-cytosol transport of cholera and Shiga toxin (25, 46, 47), it appears to antagonize ricin's ER membrane transport (48). Whether the mechanism by which Grp170 exerts its role during PyV ER membrane penetration also operates on toxin membrane transport awaits further investigations.
ACKNOWLEDGMENTS
We thank Tom Rapoport (Harvard Medical School) and Chris Walczak (University of Michigan) for critically reading the manuscript.
This work was supported by NIH grant RO1 AI064296.
REFERENCES
- 1.Brodsky JL. 2012. Cleaning up: ER-associated degradation to the rescue. Cell 151:1163–1167. doi: 10.1016/j.cell.2012.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Olzmann JA, Kopito RR, Christianson JC. 10 December 2012, posting date The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb Perspect Biol doi: 10.1101/cshperspect.a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith MH, Ploegh HL, Weissman JS. 2011. Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334:1086–1090. doi: 10.1126/science.1209235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geiger R, Andritschke D, Friebe S, Herzog F, Luisoni S, Heger T, Helenius A. 2011. BAP31 and BiP are essential for dislocation of SV40 from the endoplasmic reticulum to the cytosol. Nat Cell Biol 13:1305–1314. doi: 10.1038/ncb2339. [DOI] [PubMed] [Google Scholar]
- 5.Goodwin EC, Lipovsky A, Inoue T, Magaldi TG, Edwards AP, Van Goor KE, Paton AW, Paton JC, Atwood WJ, Tsai B, DiMaio D. 2011. BiP and multiple DNAJ molecular chaperones in the endoplasmic reticulum are required for efficient simian virus 40 infection. mBio 2:e00101-11. doi: 10.1128/mBio.00101-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schelhaas M, Malmstrom J, Pelkmans L, Haugstetter J, Ellgaard L, Grunewald K, Helenius A. 2007. Simian virus 40 depends on ER protein folding and quality control factors for entry into host cells. Cell 131:516–529. doi: 10.1016/j.cell.2007.09.038. [DOI] [PubMed] [Google Scholar]
- 7.Liddington RC, Yan Y, Moulai J, Sahli R, Benjamin TL, Harrison SC. 1991. Structure of simian virus 40 at 3.8-A resolution. Nature 354:278–284. doi: 10.1038/354278a0. [DOI] [PubMed] [Google Scholar]
- 8.Stehle T, Gamblin SJ, Yan Y, Harrison SC. 1996. The structure of simian virus 40 refined at 3.1 A resolution. Structure 4:165–182. doi: 10.1016/S0969-2126(96)00020-2. [DOI] [PubMed] [Google Scholar]
- 9.Damm EM, Pelkmans L, Kartenbeck J, Mezzacasa A, Kurzchalia T, Helenius A. 2005. Clathrin- and caveolin-1-independent endocytosis: entry of simian virus 40 into cells devoid of caveolae. J Cell Biol 168:477–488. doi: 10.1083/jcb.200407113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engel S, Heger T, Mancini R, Herzog F, Kartenbeck J, Hayer A, Helenius A. 2011. Role of endosomes in simian virus 40 entry and infection. J Virol 85:4198–4211. doi: 10.1128/JVI.02179-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kartenbeck J, Stukenbrok H, Helenius A. 1989. Endocytosis of simian virus 40 into the endoplasmic reticulum. J Cell Biol 109:2721–2729. doi: 10.1083/jcb.109.6.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pelkmans L, Kartenbeck J, Helenius A. 2001. Caveolar endocytosis of simian virus 40 reveals a new two-step vesicular-transport pathway to the ER. Nat Cell Biol 3:473–483. doi: 10.1038/35074539. [DOI] [PubMed] [Google Scholar]
- 13.Tsai B. 2007. Penetration of nonenveloped viruses into the cytoplasm. Annu Rev Cell Dev Biol 23:23–43. doi: 10.1146/annurev.cellbio.23.090506.123454. [DOI] [PubMed] [Google Scholar]
- 14.Inoue T, Tsai B. 2011. A large and intact viral particle penetrates the endoplasmic reticulum membrane to reach the cytosol. PLoS Pathog 7:e1002037. doi: 10.1371/journal.ppat.1002037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rainey-Barger EK, Magnuson B, Tsai B. 2007. A chaperone-activated nonenveloped virus perforates the physiologically relevant endoplasmic reticulum membrane. J Virol 81:12996–13004. doi: 10.1128/JVI.01037-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Daniels R, Rusan NM, Wadsworth P, Hebert DN. 2006. SV40 VP2 and VP3 insertion into ER membranes is controlled by the capsid protein VP1: implications for DNA translocation out of the ER. Mol Cell 24:955–966. doi: 10.1016/j.molcel.2006.11.001. [DOI] [PubMed] [Google Scholar]
- 17.Inoue T, Tsai B. 2013. How viruses use the endoplasmic reticulum for entry, replication, and assembly. Cold Spring Harb Perspect Biol 5:a013250. doi: 10.1101/cshperspect.a013250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kampinga HH, Craig EA. 2010. The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11:579–592. doi: 10.1038/nrm2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin Y, Awad W, Petrova K, Hendershot LM. 2008. Regulated release of ERdj3 from unfolded proteins by BiP. EMBO J 27:2873–2882. doi: 10.1038/emboj.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Petrova K, Oyadomari S, Hendershot LM, Ron D. 2008. Regulated association of misfolded endoplasmic reticulum lumenal proteins with P58/DNAJc3. EMBO J 27:2862–2872. doi: 10.1038/emboj.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. 2000. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101:249–258. doi: 10.1016/S0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 22.Dong M, Bridges JP, Apsley K, Xu Y, Weaver TE. 2008. ERdj4 and ERdj5 are required for endoplasmic reticulum-associated protein degradation of misfolded surfactant protein C. Mol Biol Cell 19:2620–2630. doi: 10.1091/mbc.E07-07-0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hagiwara M, Maegawa K, Suzuki M, Ushioda R, Araki K, Matsumoto Y, Hoseki J, Nagata K, Inaba K. 2011. Structural basis of an ERAD pathway mediated by the ER-resident protein disulfide reductase ERdj5. Mol Cell 41:432–444. doi: 10.1016/j.molcel.2011.01.021. [DOI] [PubMed] [Google Scholar]
- 24.Massey S, Burress H, Taylor M, Nemec KN, Ray S, Haslam DB, Teter K. 2011. Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect Immun 79:4739–4747. doi: 10.1128/IAI.05503-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams JM, Inoue T, Banks L, Tsai B. 2013. The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell 24:785–795. doi: 10.1091/mbc.E12-07-0522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodsky JL, Bracher A. 2000. Nucleotide exchange factors for Hsp70 molecular chaperones. Madame Curie Bioscience Database. http://www.ncbi.nlm.nih.gov/books/NBK5987/.
- 27.Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B. 2010. The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell 21:140–151. doi: 10.1091/mbc.E09-07-0586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung KT, Shen Y, Hendershot LM. 2002. BAP, a mammalian BiP-associated protein, is a nucleotide exchange factor that regulates the ATPase activity of BiP. J Biol Chem 277:47557–47563. doi: 10.1074/jbc.M208377200. [DOI] [PubMed] [Google Scholar]
- 29.Steel GJ, Fullerton DM, Tyson JR, Stirling CJ. 2004. Coordinated activation of Hsp70 chaperones. Science 303:98–101. doi: 10.1126/science.1092287. [DOI] [PubMed] [Google Scholar]
- 30.Magnuson B, Rainey EK, Benjamin T, Baryshev M, Mkrtchian S, Tsai B. 2005. ERp29 triggers a conformational change in polyomavirus to stimulate membrane binding. Mol Cell 20:289–300. doi: 10.1016/j.molcel.2005.08.034. [DOI] [PubMed] [Google Scholar]
- 31.Boisramé A, Beckerich JM, Gaillardin C. 1996. Sls1p, an endoplasmic reticulum component, is involved in the protein translocation process in the yeast Yarrowia lipolytica. J Biol Chem 271:11668–11675. doi: 10.1074/jbc.271.20.11668. [DOI] [PubMed] [Google Scholar]
- 32.Craven RA, Egerton M, Stirling CJ. 1996. A novel Hsp70 of the yeast ER lumen is required for the efficient translocation of a number of protein precursors. EMBO J 15:2640–2650. [PMC free article] [PubMed] [Google Scholar]
- 33.Kabani M, Beckerich JM, Gaillardin C. 2000. Sls1p stimulates Sec63p-mediated activation of Kar2p in a conformation-dependent manner in the yeast endoplasmic reticulum. Mol Cell Biol 20:6923–6934. doi: 10.1128/MCB.20.18.6923-6934.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tyson JR, Stirling CJ. 2000. LHS1 and SIL1 provide a lumenal function that is essential for protein translocation into the endoplasmic reticulum. EMBO J 19:6440–6452. doi: 10.1093/emboj/19.23.6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weitzmann A, Baldes C, Dudek J, Zimmermann R. 2007. The heat shock protein 70 molecular chaperone network in the pancreatic endoplasmic reticulum—a quantitative approach. FEBS J 274:5175–5187. doi: 10.1111/j.1742-4658.2007.06039.x. [DOI] [PubMed] [Google Scholar]
- 36.de Keyzer J, Steel GJ, Hale SJ, Humphries D, Stirling CJ. 2009. Nucleotide binding by Lhs1p is essential for its nucleotide exchange activity and for function in vivo. J Biol Chem 284:31564–31571. doi: 10.1074/jbc.M109.055160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inoue T, Moore P, Tsai B. 2011. How viruses and toxins disassemble to enter host cells. Annu Rev Microbiol 65:287–305. doi: 10.1146/annurev-micro-090110-102855. [DOI] [PubMed] [Google Scholar]
- 38.Park J, Easton DP, Chen X, MacDonald IJ, Wang XY, Subjeck JR. 2003. The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry 42:14893–14902. doi: 10.1021/bi030122e. [DOI] [PubMed] [Google Scholar]
- 39.Buck TM, Plavchak L, Roy A, Donnelly BF, Kashlan OB, Kleyman TR, Subramanya AR, Brodsky JL. 3 May 2013. The Lhs1/GRP170 chaperones facilitate the endoplasmic reticulum associated degradation of the epithelial sodium channel. J Biol Chem doi: 10.1074/jbc.M113.469882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walczak CP, Ravindran MS, Inoue T, Tsai B. 2014. A cytosolic chaperone complexes with dynamic membrane J-proteins and mobilizes a nonenveloped virus out of the endoplasmic reticulum. PLoS Pathog 10:e1004007. doi: 10.1371/journal.ppat.1004007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Easton DP, Kaneko Y, Subjeck JR. 2000. The hsp110 and Grp1 70 stress proteins: newly recognized relatives of the Hsp70s. Cell Stress Chaperones 5:276–290. doi:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yan M, Li J, Sha B. 2011. Structural analysis of the Sil1-Bip complex reveals the mechanism for Sil1 to function as a nucleotide-exchange factor. Biochem J 438:447–455. doi: 10.1042/BJ20110500. [DOI] [PubMed] [Google Scholar]
- 43.Yi M, Chi MH, Khang CH, Park SY, Kang S, Valent B, Lee YH. 2009. The ER chaperone LHS1 is involved in asexual development and rice infection by the blast fungus Magnaporthe oryzae. Plant Cell 21:681–695. doi: 10.1105/tpc.107.055988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao L, Rosales C, Seburn K, Ron D, Ackerman SL. 2010. Alteration of the unfolded protein response modifies neurodegeneration in a mouse model of Marinesco-Sjogren syndrome. Hum Mol Genet 19:25–35. doi: 10.1093/hmg/ddp464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schuermann JP, Jiang J, Cuellar J, Llorca O, Wang L, Gimenez LE, Jin S, Taylor AB, Demeler B, Morano KA, Hart PJ, Valpuesta JM, Lafer EM, Sousa R. 2008. Structure of the Hsp110:Hsc70 nucleotide exchange machine. Mol Cell 31:232–243. doi: 10.1016/j.molcel.2008.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Winkeler A, Godderz D, Herzog V, Schmitz A. 2003. BiP-dependent export of cholera toxin from endoplasmic reticulum-derived microsomes. FEBS Lett 554:439–442. doi: 10.1016/S0014-5793(03)01217-1. [DOI] [PubMed] [Google Scholar]
- 47.Yu M, Haslam DB. 2005. Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun 73:2524–2532. doi: 10.1128/IAI.73.4.2524-2532.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gregers TF, Skånland SS, Wälchli S, Bakke O, Sandvig K. 2013. BiP negatively affects ricin transport. Toxins (Basel) 5:969–982. doi: 10.3390/toxins5050969. [DOI] [PMC free article] [PubMed] [Google Scholar]