

ABSTRACT
Potato virus A (PVA) is a single-stranded positive-sense RNA virus and a member of the family Potyviridae. The PVA coat protein (CP) has an intrinsic capacity to self-assemble into filamentous virus-like particles, but the mechanism responsible for the initiation of viral RNA encapsidation in vivo remains unclear. Apart from virion assembly, PVA CP is also involved in the inhibition of viral RNA translation. In this study, we show that CP inhibits PVA RNA translation in a dose-dependent manner, through a mechanism involving the CP-encoding region. Analysis of this region, however, failed to identify any RNA secondary structure(s) preferentially recognized by CP, suggesting that the inhibition depends on CP-CP rather than CP-RNA interactions. In agreement with this possibility, insertion of an in-frame stop codon upstream of the CP sequence led to a marked decrease in the inhibition of viral RNA translation. Based on these results, we propose a model in which the cotranslational interactions between excess CP accumulating in trans and CP translated from viral RNA in cis are required to initiate the translational repression. This model suggests a mechanism for how viral RNA can be sequestered from translation and specifically selected for encapsidation at the late stages of viral infection.
IMPORTANCE The main functions of the CP during potyvirus infection are to protect viral RNA from degradation and to transport it locally, systemically, and from host to host. Although virion assembly is a key step in the potyviral infectious cycle, little is known about how it is initiated and how viral RNA is selected for encapsidation. The results presented here suggest that CP-CP rather than CP-RNA interactions are predominantly involved in the sequestration of viral RNA away from translation. We propose that the cotranslational nature of these interactions may represent a mechanism for the selection of viral RNA for encapsidation. A better understanding of the mechanism of virion assembly may lead to development of crops resistant to potyviruses at the level of viral RNA encapsidation, thereby reducing the detrimental effects of potyvirus infections on food production.
INTRODUCTION
Plant viral coat proteins (CPs) are associated with surprisingly many functions during the infectious cycle. Besides their roles in encapsidation and movement, CPs are implicated in viral RNA translation and replication during the early stages of infection (see references 1, 2, and 3 for reviews). Some of these functions involve specific and nonspecific interactions with viral RNA (4–7). For several positive-sense RNA viruses, CP modulates translation and/or replication in a concentration-dependent manner (4, 5). In these viruses, higher CP concentrations repress RNA accumulation, whereas lower concentrations stimulate RNA accumulation and translation. Hence, depending on the amount of CP, the virus can regulate the progression of virus infection from genome replication and translation to virion assembly.
The potyviral CP is produced together with replication proteins as part of the polyprotein, but it is not required for replication (8). Despite this situation, part of the open reading frame encoding CP needs to be translated for genome amplification to be successful (8). This was proposed to ensure that only those viral genomes with an intact open reading frame are replicated. Potyviral CP inhibits viral RNA translation when present at high concentrations (9), which may be attributed to virion formation. CP phosphorylation is a mechanism which has been proposed to play a role in the prevention of premature particle assembly (10). This was based on the finding that phosphorylation of Potato virus A (PVA; genus Potyvirus) CP downregulates its RNA binding function (11). The phosphorylation site within PVA CP was found to localize to a predicted abCd structural motif (12) common to RNA binding proteins. In addition, a chaperone-mediated ubiquitin degradation pathway has been suggested to limit the amount of CP during the early stages of infection (9). The mechanism makes use of a J-domain HSP40 chaperone named CP interacting protein (CPIP) (13). A model was proposed in which CPIP delivers potyviral CP to the degradation pathway via HSP70 until the system is overpowered by a high level of CP accumulation, after which virion assembly may commence (9, 14).
Knowledge of the mechanisms by which viruses package their genomes into structurally stable virions is an important prerequisite for understanding the infectious cycle. Furthermore, because viruses, including plant viruses, can be used as enzyme nanocarriers with potential applications in the medical and biotechnological fields (15, 16), understanding how these virus particles are assembled is of great importance. Research on the assembly mechanism of potyviruses, one of the largest class of plant viruses, is still in its infancy. Very little is known about how these viruses recruit their RNA for encapsidation. Similar to the regulation of viral RNA translation, virion assembly may rely on sequence-specific RNA-protein interactions. This is the case for plant virus genera such as Tobamovirus, Bromovirus, Cucumovirus, and Alfamovirus (17), in which virion formation requires that RNA functions as a nucleating agent, and hence assembly does not occur in the absence of viral RNA. In the genera Tymovirus and Comovirus, virion assembly is initiated via protein-protein interactions (18). The capsids of these viruses are predominantly stabilized by protein-protein interactions, so they also form capsid shells in the absence of RNA, but encapsidated viral RNA may further stabilize the capsids. Potyviruses are also well known for forming virus-like particles (VLPs) in the absence of full-length viral RNA (19). It is clear from previous literature that CP-CP interactions are essential for potyvirus VLP formation (20, 21). The model of potyvirus assembly based on an in vitro assembly study with Pepper vein banding virus (PVBV; genus Potyvirus) proposes that CP subunits interact electrostatically via their N- and C-terminal residues and, by doing so, form octameric ring-like structures (20). Subsequently, formation of helical aggregates starts and results in VLPs with the N and C termini of the CP subunits exposed on the surface of the particle.
In this study, we investigated the mechanisms of CP-mediated inhibition of viral RNA translation in the context of PVA infection. Our initial hypothesis was that viral RNA translation is regulated by the amount of CP and ceases in the presence of large amounts of CP, probably due to virion assembly. This hypothesis seemed plausible, as a PVA CP mutant similar to mutants of Tobacco etch virus (TEV; genus Potyvirus) (22) and Plum pox virus (PPV; genus Potyvirus) (22, 23), which were defective in assembly and movement, did not inhibit PVA RNA translation (9). We demonstrate here that the inhibition of potyviral RNA translation, and possibly initiation of assembly, occurs cotranslationally and may rely on CP-CP interactions to initiate the repressive effect on RNA translation, rather than specific binding of PVA CP to an RNA element. We present a model in which the role of CPIP during potyvirus infection is to prevent cotranslational CP-CP interaction until the correct timing for the shift from translation to assembly has been reached.
MATERIALS AND METHODS
Gene constructs.
All constructs used in this study were based on the full-length cDNA of PVA strain B11 (GenBank accession number AJ296311) carrying the Renilla luciferase (RLuc) reporter gene (24). The RLuc reporter gene (which enables quantification of viral gene expression) contains intron 1 of the ribulose-1,5-bisphosphate carboxylase/oxygenase (RubisCO) (RBC-1I) gene for inhibition of bacterial expression (25). Viral gene constructs used for Agrobacterium-mediated infection were all under the control of the Cauliflower mosaic virus (CaMV) 35S promoter, and all contained the transcription terminator from the nopalin synthase gene (nos) of Agrobacterium tumefaciens (26). Viral T-DNA clones were inserted into the pRD400 binary vector (27) via KpnI/SalI sites and nonviral clones into the pEAQ-HT vector via AgeI/XhoI sites (28). Viral constructs described previously and used in this study are shown in Table 1. Figure 1 presents additional viral and monocistronic gene constructs designed for use in this study.
TABLE 1.
Previously published PVA constructs used in this studya
| Viral/expression construct | Gene cassette | Phenotype | Reference |
|---|---|---|---|
| PVAwt | 35S-PVAwt::RLucint-nos | Wild-type PVA | 25 |
| PVAGDD | 35S-PVAGDD::RLucint-nos | Replication-deficient PVA | 25 |
| CPwt | 35S-CPwt-nos | Coat protein from PVA B11 | 9 |
| CPmut | 35S-CPmut-nos | Movement-deficient coat protein (R159D and Q160V) | 9 |
| FLuc | 35S-FLuc-nos | 25 | |
| 5′UTR-RLuc | 35S-5′UTR-RLuc-nos | 25 |
All constructs were created in the vector pRD400.
FIG 1.

T-DNA features of viral and nonviral constructs. The figure shows a schematic representation of the T-DNA features of viral and nonviral constructs used in this study, all under the control of the CaMV 35S promoter. PVAwt has been reported previously (25) and is included here as a reference to showcase the modifications made in the other constructs. Since all of the viral constructs contain the RLuc gene, it is not mentioned in their names.
Plants.
Nicotiana benthamiana plants were utilized for this study. They were grown under greenhouse conditions of 22°C for 18 h of light and 18°C for 6 h of darkness.
Agrobacterium transformation and infiltration.
Agrobacterium tumefaciens strain C58C1 (29) was transformed with viral and expression constructs by electroporation and grown on Luria-Bertani agar plates containing 50 μg/ml kanamycin and 50 μg/ml carbenicillin. Single colonies were inoculated into liquid medium containing the same antibiotics and grown overnight at 28°C. For Agrobacterium infiltration, cells were harvested by centrifugation at 3,000 × g for 5 min, washed once with double-distilled water, and resuspended in induction buffer (10 mM morpholineethanesulfonic acid [MES], pH 6.3, 10 mM MgCl2, and 150 μM acetosyringone). The optical density at 600 nm (OD600) was adjusted to 0.05 for full-length and truncated PVA constructs and to 0.3 for CPwt (wild-type coat protein), CPmut (movement-deficient CP mutant), and GUS (β-glucuronidase control). Agrobacterium carrying the firefly luciferase gene (Fluc), prepared as described above, was mixed with Agrobacterium carrying the reporter constructs at an OD600 of 0.005 and was used as an internal control. Samples were incubated for 2 h at room temperature before infiltration into Nicotiana benthamiana plant leaves. A 1-ml syringe was used to deliver the infiltrate to the abaxial side of the leaves. For transient expression, plants were always infiltrated by CPmut, CPwt, or GUS 1 day before the viral constructs were delivered.
Quantification of gene expression.
Viral gene expression was quantified via RLuc activity determination (25). Six leaf disks of about 5 mm were collected from infiltrated plants and immediately frozen in liquid nitrogen. A dual-luciferase kit (Promega) was used to prepare plant samples, and activity measurements were performed using a Luminoscan TL Plus instrument (Thermo Labsystems). FLuc was used as an internal control to normalize RLuc activity. This was done by first obtaining the average FLuc value from replicates, and the following formula was then used to calculate the normalized RLuc value: normalized RLuc activity = (FLuc activityaverage/FLuc activity per sample) × RLuc activity per sample. Normalized RLuc average and standard deviation values were calculated from values obtained from at least three parallel samples. Furthermore, the Student t test was employed to calculate the significance of the difference between the experimental and control samples, and significance is represented in the figures as follows: **, P < 0.01; and *, P < 0.05.
Recombinant protein expression and purification.
Expression and purification of recombinant PVA CP were performed as described earlier (10).
In vitro transcription and EMSA.
PCR products of similar sizes (∼800 bp), corresponding to the opposite ends of the PVA genome (the 5′-terminal P1-encoding region and the 3′-terminal CP-encoding region), were synthesized using the following primer pairs: 5′-GAAATTAATACGACTCACTATAGGGACTCAAAACGCAAGCATCAAT-3′ and 5′-ATTCTTTGCCCAGTCACCAG-3′ for the P1 region and 5′-GAAATTAATACGACTCACTATAGGGAAACTCTTGATGCAAGCGAAG-3′ and 5′-TTACACCCCCTTCACGCCTAA-3′ for the CP region. Each PCR product contained the 5′-terminal consensus T7 promoter sequence for in vitro transcription. Purified PCR products were used as templates for in vitro transcription with T7 RNA polymerase (Promega). RNA transcripts were column purified, adjusted to the same concentration, and refolded by heating to 85°C followed by slow cooling. Electrophoretic mobility shift assay (EMSA) mixtures were set up in a total volume of 20 μl containing 10 mM Tris-HCl (pH 8.0), 5 mM MgCl2, 0.1% (vol/vol) Tween 20, 450 ng RNA transcript, and purified recombinant PVA CP at the indicated molar ratios. After incubation for 30 min at room temperature, samples were loaded into a 0.8% Tris-borate-EDTA SeaKem GTG agarose gel (Lonza). Following electrophoresis, bands were visualized using a Gel Doc system (Bio-Rad Laboratories).
qRT-PCR.
For quantitative purposes, six leaf disks of 4 mm each were collected from infiltrated plants 10 days after infiltration and immediately frozen in liquid nitrogen. Each sample set contained a pool of six biological replicates, from which eight technical replicates were made. Total RNAs were extracted from these leaves by use of an RNeasy plant minikit (Qiagen) according to the manufacturer's instructions. One microgram of RNA per sample was treated with DNase (Thermo Scientific) and then utilized for further downstream applications. First-strand cDNA synthesis from total RNA was performed according to the instructions of a RevertAid H Minus first-strand cDNA synthesis kit (Thermo Scientific), using random hexamers. Quantitative PCR (qPCR) was performed in 384-well plates, using a CFX384 Touch real-time system (Bio-Rad). Each 15-μl PCR mix contained 7.5 μl Maxima SYBR green qPCR master mix (Thermo Scientific), 0.5 μM (each) forward and reverse primers, 1 μl cDNA, and 4.5 μl nuclease-free water. RLuc was used as the target gene and was amplified with previously described primers (30), while the protein phosphatase 2A (PP2A) gene, a housekeeping gene, was used as a reference gene and was also amplified with previously described primers (31). The amplification parameters for qPCR were the same as those used earlier (30). The following three controls were included in each run: cDNA synthesized from mock plants and nuclease-free distilled water (dH2O) were used as nontemplate controls (NTC), while reverse transcription (RT) reaction mixtures lacking the reverse transcriptase enzyme were used as a non-RT (NRT) control. The specificity of each amplicon was assessed via a melting curve, and a subset of PCR products were confirmed through agarose gel electrophoresis.
The expression level of the target gene was calculated according to equation 2 of Q-Gene (32), which calculated the mean normalized expression (recommended for simplex PCRs). The reference gene was used to normalize the expression level of the target gene.
Immunocapture RT-PCR.
Plant samples were pulverized (1:5 [wt/vol]) in 100 mM sodium phosphate buffer, pH 7.4, containing 140 mM NaCl, 3 mM KCl, 0.5 μM polyvinylpyrrolidone (PVP-40), 0.03 μM bovine serum albumin (BSA), and 0.05% Tween 20. Debris was removed by centrifugation at 5,000 × g for 5 min. Four hundred microliters of the supernatant was added to a microcentrifuge tube which had been coated with polyclonal PVA CP antibody V042-C2 (AC Diagnostics) as described by Clark and Adams (33) and was then incubated overnight at 4°C. Microcentrifuge tubes were washed three times with washing buffer, once with phosphate-buffered saline (PBS) containing 0.05% Tween 20, and once with water. Reverse transcription (Superscript III reverse transcriptase; Invitrogen) was performed using the reverse primer TCTAGAGCGGCCGCTTTTTTTTTTTTTTTTTTTTCC. Five microliters of the resulting viral cDNA was utilized for PCR amplification with High Fidelity Phusion DNA polymerase (NEB). Part of the CP gene was amplified using primers GGATCCGCCGAAACTCTTGATGCAAGCG and AGGCCTCACCCCCTTCACGCCTAAAAGG. The 5′ region of the PVA genome was amplified using the 5′-untranslated region (5′UTR) forward primer CTCGAGGCGGCCGCAAAATAAACAAACTACAAAAC and the P1 reverse primer CCCTGTTGAATAGTGATGTGTTG. Underlining in the primer sequences indicates restriction sites. Resulting PCR products were analyzed in a 1% agarose gel.
Immunodetection analysis.
Leaf disks were homogenized in protein extraction buffer (100 mM Tris-HCl, pH 8.0, 1% SDS). Samples were spun down for 10 min at 13,000 × g. Proteins were analyzed via standard SDS-PAGE (34) followed by immunodetection. CP expression was detected using a 1/10,000 dilution of a sheep-derived polyclonal PVA CP antibody (Adgen Phytodiagnostics), while RLuc was detected using a 1/10,000 dilution of commercial mouse-derived monoclonal antibody clone 5B11.2 (Millipore). Immunodetection of the PVA cylindrical inclusion protein (CI) was done using a 1/10,000 dilution of a polyclonal antibody.
Immunosorbent electron microscopy.
Leaf samples were collected from infected plants and immediately frozen in liquid nitrogen. Leaf samples (0.1 g per 100 ml buffer) were then homogenized in 0.1 M sodium phosphate buffer, pH 8.0. Leaf debris was removed by centrifugation for 10 min at +4°C. Carbon-coated grids were incubated for 1 h at room temperature in a 1/100 dilution of polyclonal PVA CP antibody (Adgen Phytodiagnostics) in preparation solution (0.1 M sodium phosphate buffer, pH 8.0, 0.1% Tween 20, 0.1% BSA). Grids were washed with 5 drops of preparation solution between each step. They were incubated in 50 μl of leaf extract overnight at +4°C and then in BSA for 1 h at room temperature, and after the last wash, they were stained for 30 s with 3% uranyl acetate solution and then dried. Virus particles were visualized using a JEOL JEM-1400 electron microscope (JEOL Ltd., Tokyo, Japan).
RESULTS
In our previous study, we observed inhibition of PVA RNA translation when the viral genome was coexpressed with wild-type PVA CP (9). Because inhibition of PVA gene expression by CP occurred for both replicating and nonreplicating viruses, with the amount of inhibition being 2 orders of magnitude higher for the latter (9), the inhibition most probably functioned through translational repression. As a continuation of this work, we aimed to identify putative high-affinity CP binding sites on viral RNA that are responsible for blocking viral RNA translation. A Renilla luciferase-based system was used to monitor the effects of CP on RNA translation from viral and nonviral constructs (25). The following two controls were used in these experiments: CPmut, an assembly and movement-deficient mutant which does not interact with viral RNA (9); and β-glucuronidase (GUS), an Escherichia coli enzyme which has no effect on PVA infection. Immunoblot analysis using anti-CP previously revealed that CPwt and CPmut are expressed at the same level in Nicotiana benthamiana (9).
CPwt inhibits translation in a dose-dependent manner.
We first tested if CP-mediated inhibition was concentration dependent. For this purpose, N. benthamiana leaves were coinfiltrated with a mixture of 35S-PVAwt and various amounts of Agrobacterium carrying 35S-CP-nos. We observed that the higher the OD600 of Agrobacterium containing CPwt in the infiltration mix, the higher was the inhibition of PVAwt gene expression (Fig. 2A). Furthermore, immunoblot analysis with anti-CP showed a correlation between the infiltrated Agrobacterium OD600 and the level of accumulated CP in the infiltrated leaves (Fig. 2B): the higher the OD600 of Agrobacterium, the higher was the expression level of CPwt. This led to the unequivocal conclusion that CP inhibits viral translation in a dose-dependent manner. However, in the case of PVAwt, the extent of the effect cannot be uncoupled from replication, so the amount of inhibition is likely increased due to a reduction in replication.
FIG 2.

CP inhibits translation in a dose-dependent manner. The dose of CP expressed in Nicotiana benthamiana plants is proportional to the level of inhibition of translation. (A) Plants were infiltrated with increasing amounts (OD600s of 0.05, 0.1, and 0.3) of Agrobacterium carrying a T-DNA to express CP. Twenty-four hours later, the same plants were infiltrated with PVAwt. GUS was used as a control, as it does not interfere with viral gene expression. GUS expression was initiated by Agrobacterium infiltration (OD600 = 0.3). Student's t test was carried out to study the significance of the difference between GUS and CPwt expression with PVAwt. **, P < 0.01. (B) CP expression was initiated similarly to that in panel A, with various amounts of Agrobacterium. Total protein from infiltrated leaves was extracted and analyzed via Western blotting. Anti-CP was used for immunodetection of CP. Mock-infected Nicotiana benthamiana plants were used as a negative control. A protein band corresponding to RubisCO on a Ponceau S-stained membrane was used as a loading control.
The 5′UTR and 3′UTR do not contain the CP RNA binding site.
Next, we examined the role of the 5′UTR in the CP-mediated inhibition of PVA RNA translation. We focused first on the 5′UTR, because virion assembly has been suggested to initiate from this region (35). We performed initial experiments using a replication-deficient mutant, PVAΔGDD (25), lacking the 5′UTR, i.e., PVAΔGDDΔ5UTR. We know from our previous study that the 5′UTR is indispensable for efficient viral RNA translation (36); however, PVAΔGDDΔ5UTR RNA translation remained quantifiable. When CPwt was coexpressed with PVAΔGDDΔ5UTR, we could not detect any RLuc activity above the background (Fig. 3A), which made it impossible to calculate a fold change in inhibition. However, because the inhibitory effect was not abolished, the 5′UTR could not be the main cause of the inhibition. Next, the 3′UTR was investigated. Expression of CPwt with a replication-deficient mutant virus with the 3′UTR deleted, PVAΔGDDΔ3UTR, resulted in a 100-fold decrease in reporter activity (Fig. 3B). This observation excluded the 3′UTR as being involved in this inhibition.
FIG 3.
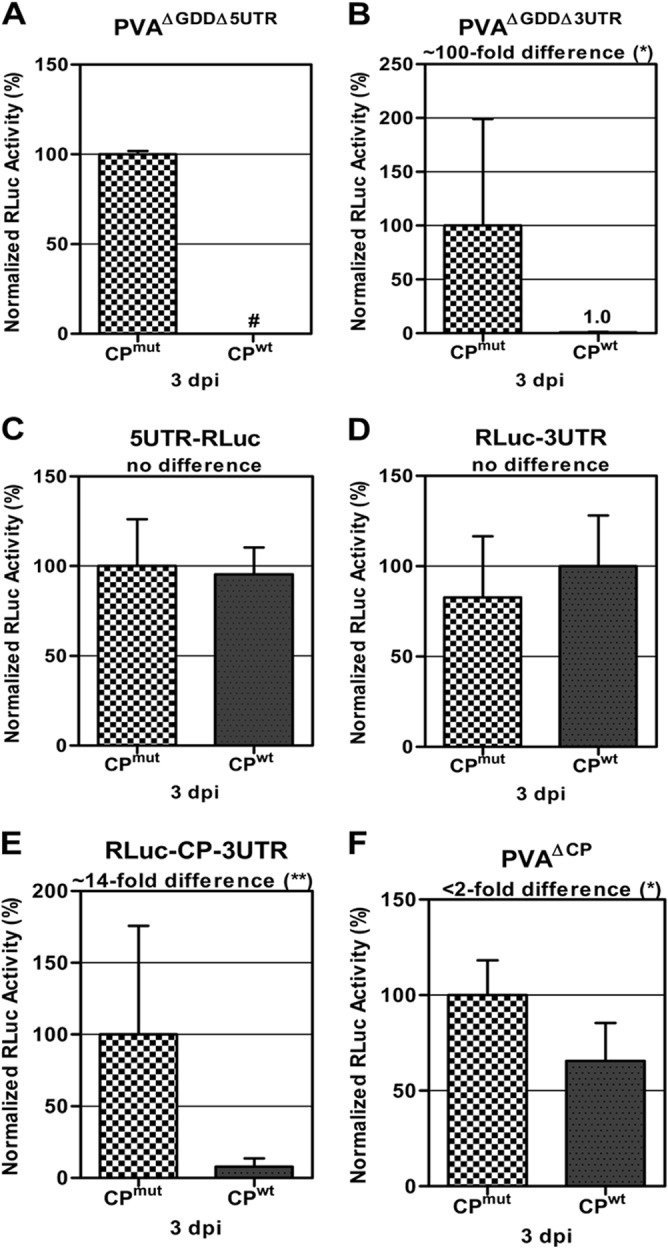
The PVA CP-encoding region is indispensable for inhibition. A PVA genome screen for a putative RNA element involved in RNA-CP interaction was performed. CPwt was expressed in trans with modified versions of PVA RNA carrying the RLuc reporter gene. RLuc activity was used to quantify viral RNA translation at 48 h postinfiltration and is given as the mean percentage for a minimum of three replicates per experiment. CPmut did not interfere with viral gene expression and hence was used as a control. The RLuc activity of the control experiments was taken as 100%. (A) No RLuc activity (#) above the background level could be detected when a nonreplicating PVAΔGDD mutant lacking the 5′UTR (PVAΔGDDΔ5UTR) was expressed with CPwt. (B) A 100-fold reduction in RLuc activity was detected when a nonreplicating PVAΔGDD mutant lacking the 3′UTR (PVAΔGDDΔ3UTR) was expressed with CPwt. (C and D) When monocistronic constructs 5′UTR-RLuc (C) and RLuc-3′UTR (D) were expressed with CPwt, no inhibition was detected. This confirmed that the untranslated regions of PVA did not carry the CP binding site. (E) When the bicistronic construct RLuc-CP-3′UTR was expressed with CPwt, a 14-fold reduction in RLuc activity was observed, suggesting a role in inhibition for the CP cistron. (F) Only a 2-fold decrease in RLuc activity was observed when a PVA mutant lacking the CP gene (PVAΔCP) was coexpressed with CPwt, which confirmed that the inhibition was due to the PVA CP-encoding region. Student's t test was carried out to study the significance of the differences between CPmut and CPwt expression with various constructs. **, P < 0.01; *, P < 0.05.
To confirm these results, the 5′UTR was fused upstream of the RLuc gene for examination of its effect. When this construct, 5′UTR-RLuc, was coexpressed with CPwt, there was no negative effect on RNA translation (Fig. 3C). Similarly, a construct containing only the 3′UTR and the RLuc reporter gene upstream, RLuc-3′UTR, was also analyzed. No effect was detected when this construct was coexpressed with CPwt (Fig. 3D). Taken together, our results indicate that the 5′UTR and 3′UTR are not the causative factors of CP-mediated inhibition.
CP-mediated inhibition comes from the CP mRNA.
We next dissected the rest of the PVA genome to find the site responsible for CP-mediated inhibition of translation. For this purpose, viral RNA fragments were coexpressed with CPwt, and RLuc reporter activity was monitored. When CPwt was coexpressed with a viral construct containing the CP gene and 3′UTR downstream of the RLuc reporter gene, i.e., RLuc-CP-3′UTR, a 14-fold decrease in reporter activity was observed compared to that with CPmut and the same construct under the same conditions (Fig. 3E). Since the 3′UTR had no effect, as shown in Fig. 3D, this result indicated that the CP cistron is probably responsible for the CP-mediated inhibition. To further investigate if CP negatively modulates gene expression via its own RNA coding sequence, it was coexpressed with a PVA mutant devoid of the CP gene, i.e., PVAΔCP. CPwt inhibited RNA translation <2-fold with this construct (Fig. 3F), indicating that the negative effect was alleviated in the absence of the CP-encoding region. The 2-fold inhibition observed might have come from unspecific interactions between CPwt and the viral genome. Based on these results, we envisaged a scenario wherein CP interferes with viral RNA translation by interacting with its coding sequence on the viral RNA genome.
CP's interaction with its RNA coding sequence is not specific.
In order to determine whether CP has a binding preference for a secondary structure(s) within its RNA coding sequence, we performed comparative EMSAs with synthetic RNA transcripts corresponding to the CP-encoding region and the P1-encoding region. The results of EMSA showed that the two RNA transcripts bound CP with approximately equal affinities (Fig. 4), indicating that CP binds RNA in a cooperative but sequence-nonspecific manner. Similar results were obtained when EMSA incubation mixtures were supplemented with N. benthamiana extract (data not shown), suggesting that the presence of host factors does not change the sequence-nonspecific nature of CP binding to RNA. Taken together, these results support a model in which the initiation of cooperative CP binding to viral RNA does not depend on the presence of a specific RNA secondary structure within the CP-encoding region of the viral genome.
FIG 4.
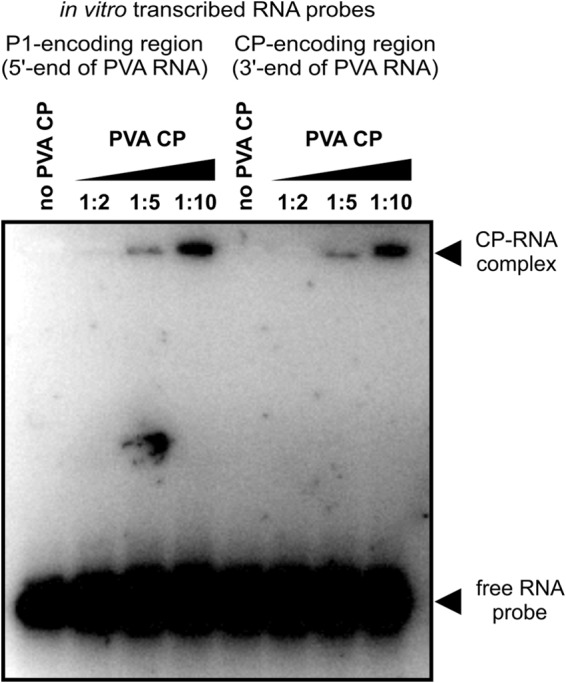
PVA CP does not have a binding preference for the CP-encoding region of PVA RNA in vitro. The figure shows results of a representative EMSA using purified PVA CP incubated with RNAs corresponding to ∼800-nucleotide regions at opposite ends of the viral genome. The RNA on the left corresponds to the 5′-terminal P1-encoding region, and the RNA on the right to the 3′-terminal CP-encoding region. RNA transcripts were incubated with increasing molar ratios of PVA CP, as indicated by numerals. The theoretical RNA/CP molar ratio required for binding saturation is 1:160. The positions of cooperatively bound CP-RNA complexes (∼5 MDa) and free RNA are indicated by arrowheads. Note the lack of a pronounced difference in PVA CP binding between the two RNA transcripts.
CP-mediated inhibition of viral gene expression occurs cotranslationally.
The absence of high-affinity CPwt binding sites in the CP-encoding region suggested that the selective CP-mediated inhibition of PVA RNA translation likely did not occur via a direct CP-RNA interaction. Therefore, another mechanism, possibly initiated by CP-CP interactions, was considered. To test whether such a mechanism could come into play, the RLuc-CP-3′UTR construct was modified by placing an in-frame stop codon upstream of the CP gene, generating the RLucSTOP-CP-3′UTR construct. The stop codon prevented CP translation from this construct, which also enabled us to investigate if CP needs to be translated from the reporter construct for this inhibition to occur. When RLuc levels were measured for RLuc-CP-3′UTR and RLucSTOP-CP-3′UTR (Fig. 5A, left panel), there was no significant difference in expression of both constructs. This demonstrated that the stop codon did not render the construct unstable. On the other hand, dramatic differences in RLuc expression levels from both constructs were observed in the presence of exogenous CP. When CPwt was transiently coexpressed in planta with the RLuc-CP-3′UTR construct, the reporter activity was inhibited >300-fold (Fig. 5A, middle panel). In contrast, CPwt coexpression with RLucSTOP-CP-3′UTR produced only a moderate inhibition of RLuc activity (∼3-fold) (Fig. 5A, right panel). We concluded that the strong CP-mediated inhibition of RNA translation required active CP translation from the RLuc-CP-3′UTR RNA. The moderate inhibition observed with the RLucSTOP-CP-3′UTR RNA was likely caused by the cooperative, sequence-nonspecific binding of CPwt to the corresponding mRNA.
FIG 5.

CP-mediated inhibition of PVA gene expression is dependent on translation of the CP cistron. We studied the effects of CPwt on the transcription and translation of viral and nonviral RNAs. Agrobacterium containing CPwt was introduced to plants by infiltration 24 h before the viral and nonviral constructs were introduced. Leaf samples were collected at 3 and 10 dpi for RLuc activity measurements as well as for RNA quantification via qRT-PCR. Firefly luciferase (FLuc) was used as an internal control to normalize RLuc activity. For qRT-PCR, six biological replicates from each sample set were pooled, and eight technical replicates were produced from them. The protein phosphatase 2A gene (a housekeeping gene) was used as a reference gene, and RLuc was used as the target during quantitative PCR. Normalized gene expression was calculated according to equation 2 of Q-Gene (32). **, P < 0.01; *, P < 0.05. (A) Expression of CPwt and GUS with RLuc-CP-3′UTR (middle panel) and RLucSTOP-CP-3′UTR (right panel). The left panel shows the expression of bicistronic constructs RLuc-CP-3′UTR and RLucSTOP-CP-3′UTR in Nicotiana benthamiana plants at 3 dpi, with no significant difference in the translation of both constructs. (B) Expression of CPwt and GUS with PVAwt (left and middle panels). The right panel shows the fold difference in RNA expression from plants expressing PVAwt and GUS and those expressing PVAwt and CPwt, collected at 10 dpi. (C) Expression of CPwt and GUS with PVASTOPCP (left and middle panels). The right panel shows the fold difference in RNA expression from plants expressing PVASTOPCP and GUS and those expressing PVASTOPCP and CPwt, collected at 10 dpi. **, P < 0.01; *, P < 0.05.
With this new hypothesis in place, we went on to investigate the CP-mediated inhibitory effect in the context of PVAwt infection. An in-frame stop codon was inserted upstream of the CP gene in PVAwt, creating PVASTOPCP, to impair CP translation. Expression of CPwt in trans with PVASTOPCP resulted in the same effect as that with RLucSTOP-CP-3′UTR. We observed a 100-fold inhibition when CPwt was coexpressed with PVAwt (Fig. 5B, left panel), as opposed to an about 4-fold inhibition of viral RNA translation when CPwt was coexpressed with PVASTOPCP (Fig. 5C, left panel). This inhibition persisted until 10 days postinfiltration (dpi), being 30-fold at this time point for PVAwt (Fig. 5B, middle panel) and 4-fold for PVASTOPCP (Fig. 5C, middle panel). Because the CP effect was significantly reduced in the presence of the stop codon, we propose that the interaction causing this inhibition occurs between CP produced in trans and CP expressed in cis from the virus and is cotranslational.
The stop codon in front of the CP cistron rendered PVAwt noninfectious and also impaired viral RNA translation by a factor of 10, as evidenced by comparing RLuc activities derived from PVAΔGDDSTOPCP (a nonreplicating PVA mutant carrying an in-frame stop codon before the CP cistron) and PVAΔGDD (data not shown). Quantification of PVAwt RNA amounts in leaves coexpressing either GUS or CPwt at 10 dpi, after which RNA encapsidation has occurred, revealed that there was 45 times less PVA RNA in the latter (Fig. 5B, right panel). The 45-fold reduction in RNA level in plants expressing PVAwt and CPwt was reflected in the 30-fold reduction in reporter activity observed above (Fig. 5B, middle panel). These results showed that CPwt coexpression interfered with the entire PVA translation/replication cycle. When the RNA amounts were similarly compared in leaves coexpressing PVASTOPCP and GUS or CP, no significant difference was observed (Fig. 5C, right panel). The substantially reduced transcript level of PVASTOPCP RNA was attributed to the absence of genome replication.
CP blocks expression of the entire PVA genome.
In line with RLuc activity measurements, immunoblot analysis using anti-CP and anti-RLuc antibodies confirmed that CP blocks translation of the 3′ end of the PVA genome. These results showed much higher PVA CP and RLuc protein levels in control plants expressing PVAwt and GUS than in experimental plants expressing PVAwt and CPwt at 10 dpi (Fig. 6A and B). Cylindrical inclusion protein (CI) is expressed from the middle of the PVA genome. It was used to verify if the CP-mediated inhibition was targeted more toward the 3′ end of the viral genome or if it affected the entire viral genome. Interestingly, CI accumulation was affected similarly to that of CP and RLuc (Fig. 6C), indicating that translation of the entire viral genome was interrupted in the presence of exogenous CP.
FIG 6.
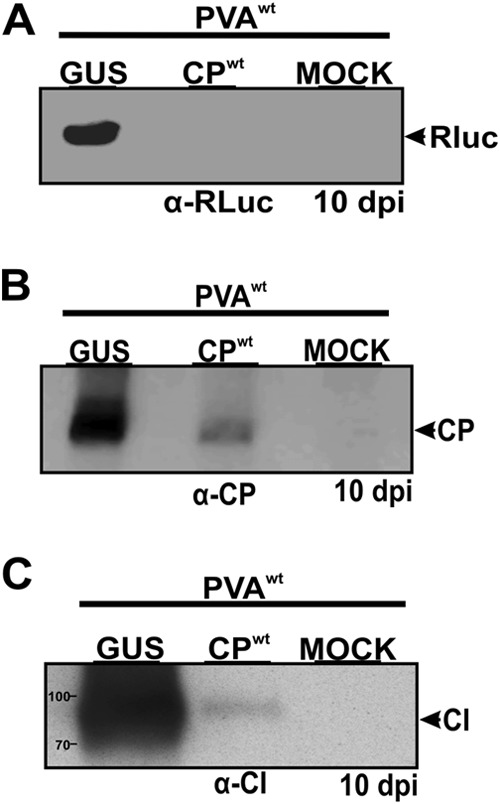
CPwt blocks gene expression from both the middle and the 3′ terminus of the viral genome. CPwt and β-glucuronidase (GUS) were expressed in trans with PVAwt. GUS was used as a positive control, while mock-treated plants (uninfected plants) were used as a negative control. Plant samples were collected at 10 dpi. Total proteins were extracted from these samples, and Western blotting was performed using specific antibodies to detect viral proteins. RLuc, CP, and CI protein levels were drastically reduced in plants coexpressing CPwt and PVAwt (experimental plants) compared to those in plants expressing GUS and PVAwt (control plants). The mock-treated negative control gave no signal with all three antibodies, showing that the protein signals came from PVA infection.
Viral RNA is encapsidated during CP-mediated inhibition.
As a next step, we investigated the implications of CP-mediated inhibition on the virus life cycle. The aim was to investigate if viral RNA was excluded from translation for encapsidation purposes. To this end, samples were collected from control plants coexpressing GUS and PVAwt and experimental plants coexpressing CPwt and PVAwt, and the formation of virus particles was monitored from 3 dpi. Virions were absent in both leaf samples at 3 dpi but became visible in the control plants from 7 dpi and in the experimental plants from 10 dpi. Viral particles from both plants were similar in length and diameter (Fig. 7A).
FIG 7.

Transmission electron microscopy (TEM) and immunocapture RT-PCR. Virion formation was assessed in Nicotiana benthamiana plants coexpressing PVAwt with GUS (control plants) or CPwt (experimental plants) at 10 dpi. (A) Virions contained in both control and experimental plants were captured on carbon-coated grids, followed by negative staining with 3% uranyl acetate for visualization on a JEOL 1400 TEM. Virions similar in shape and size were detected in both plants. (B) Immunocapture RT-PCR was used to investigate the RNA content of the virus particles. Control and experimental plants both contained the full-length RNA molecule.
Next, we verified if the particles formed in plants coexpressing CPwt and PVAwt encapsidated viral RNA. These particles were pulled down and their contents amplified via RT-PCR. The same PCR products (the CP gene and an area from the 5′ end of the viral genome) were amplified from the wild-type virions and these particles (Fig. 7B). Hence, virions were formed by the wild-type virus in the presence and absence of ectopic CP, and in both cases, viral RNA was encapsidated. This did not, however, rule out the possibility that part of the encapsidated RNAs could represent the ectopically expressed CP mRNA.
DISCUSSION
Previously, CP has been thought to function exclusively in virion assembly and viral cell-to-cell movement. More recent studies have shown that CP is a multifunctional protein involved in several processes during viral infection (see references 1 to 3 for reviews). For most RNA viruses, CP functions rely on RNA-protein as well as protein-protein interactions, and in several cases, the amount of CP determines the kind of role it will play at different stages of infection. Earlier work from our laboratory demonstrated a role for PVA CP in the downregulation of viral gene expression (9). We showed that exogenous CP interferes with PVA RNA translation and suggested that this phenomenon may be attributed to virion assembly. As a continuation of this study, here we attempted to decipher the molecular mechanism underlying the CP-mediated inhibition of viral gene expression. Based on our findings and previous literature, we propose a hypothetical model for the assembly-mediated inhibition of viral gene expression, as illustrated in Fig. 8.
FIG 8.

Proposed model for CP-mediated inhibition of translation/assembly initiation. (A) In the early stages of infection, CPIP is present in large amounts in the cell. It competes with CP expressed in trans for CP expressed in cis. Because it is more abundant in the cell, it binds CP and delivers it to HSP70, which tags it for degradation via the proteasomal pathway. CP is out of the way, so translation proceeds without interruption. (B) In the late stages of infection, CPIP is depleted and CP becomes abundant in the cells, as it is needed for assembly. CP expressed in trans interacts with CP expressed in cis, and the resulting complex binds to the CP cistron in the viral RNA and probably initiates the assembly process. In the course of assembly, ribosomes in the process of translation are removed from the viral RNA by CPs, altering translation.
CP-mediated inhibition of PVA RNA translation depends on translation of the CP cistron.
The fact that PVA CP inhibited viral gene expression by as much as 2 orders of magnitude, while the expression of nonviral control mRNA remained unaffected (9), suggested that a specific CP-binding structural element through which CP may exert its inhibitory effect may exist in viral RNA and that this structural element might play a role in RNA encapsidation. In agreement with this possibility, an encapsidation signal has been suggested to reside at the 5′ end of the potyviral genome (35). However, no further evidence to support this suggestion was obtained over the following years. Instead, it was proposed that if there is indeed an encapsidation signal, it should be located within the CP-encoding region of the viral RNA (37). At first, our results seemed to support the latter suggestion, because deletion of this region abolished the CP-mediated inhibition of viral RNA translation (Fig. 3F). Nonetheless, subsequent in vitro experiments showed that CP had no binding preference for its coding RNA (Fig. 4). This result raised the question of whether CP-RNA interactions were involved in the initiation of the CP-mediated inhibition of viral RNA translation. CP-CP interactions have been reported to have various inhibitory effects on other viruses. In the case of Tobacco mosaic virus (TMV; genus Tobamovirus), transgenic expression of the CP mutant T42W, with an enhanced ability to form CP-CP complexes, considerably reduced TMV infectivity compared to that of another mutant which failed to aggregate (38). Formation of VLPs, but not infectious virions, has been observed in transgenic plants expressing the T42W mutant. Subsequent studies have suggested that the mutant inhibits viral infection by interfering with the formation of replication complexes (39). In the case of the RNA bacteriophage MS2, several CP mutants have been found to associate with viral RNA, predominantly as dimers, repressing its translation better than wild-type CP does (40).
To shed light on the role of the CP cistron in the inhibition of PVA RNA translation, we incorporated an in-frame stop codon upstream of the CP sequence in a nonviral reporter construct and demonstrated that CP had a markedly smaller effect on reporter gene expression than the same construct without the stop codon (Fig. 5A). Furthermore, we observed a similar effect after a stop codon was inserted immediately upstream of the CP-encoding sequence in the full-length PVA genome (Fig. 5B). This finding brought up the possibility that initiation of the process leading to CP-mediated inhibition of viral RNA translation may revolve around CP-CP rather than CP-RNA interactions. However, an alternative explanation is also possible: CP might recognize a transient hairpin structure formed during translation of the CP-encoding RNA. Interestingly, a CP mutant defective in viral assembly and movement (41), CPmut, had a much smaller inhibitory effect on the expression of the RLuc-CP fusion protein than did wild-type CP when the proteins were expressed in trans (Fig. 3E). Nonetheless, when CPwt was expressed in trans with a mutant virus, PVACPmut, in a similar assay (9), viral gene expression was severely reduced. This suggests that the assembly properties of CP produced in trans need to be fully functional for inhibition to occur, while this is not a prerequisite for CP produced in cis.
Regulation of the infection process by CP.
In several RNA viruses, CPs coordinate the switch between viral RNA translation, replication, and encapsidation in a concentration-dependent manner. At low concentrations, CP may promote RNA replication or translation, whereas at higher concentrations, it may inhibit these processes in favor of RNA encapsidation (4, 42, 43). One example of such regulation is the case of Rubella virus (RUB; genus Rubivirus), in which small amounts of CP stabilize viral RNA, while larger amounts inhibit replication to promote virion assembly (43, 44). In Hepatitis C virus (HCV; genus Hepacivirus), small amounts of CP stimulate internal ribosome entry site (IRES)-dependent translation, while larger amounts of CP block translation, probably in favor of replication (5, 42). In line with the aforementioned examples, we suggest that the CP-mediated inhibition of PVA RNA translation may occur to initiate the next step in the infectious cycle. Our experiments with overexpressed CP may resemble the situation encountered in the late stages of infection, when the CP level is saturating in the cell. Therefore, it would be logical to assume a switch from translation to virion assembly. Virions similar in length to the wild-type virions formed in the presence of overexpressed CP were detected in infected cells under CP-mediated translational inhibition (Fig. 7). These particles were detected in small amounts and appeared later than in normal infection. This made it difficult to conclude beyond a reasonable doubt that RNA excluded from translation was taken directly into assembly. It was recently shown that PPV HC-Pro has the capacity to stabilize its cognate CP and enhance the yield of infectious viral particles (45). The authors of that study proposed that the increase in the HC-Pro concentration toward the later stages of infection would enhance virion formation at the expense of translation and replication. It was thought that HC-Pro either assisted the assembly of CP subunits into particles or stabilized the formed virions. It is worth noting that the system used in this study to explore the role of CP in the inhibition of translation contained only trace amounts of HC-Pro. Consequently, virion formation may have been delayed and/or disturbed, rendering viral RNA unstable, and this may explain the late appearance of virions. Further experiments are required to investigate the role of HC-Pro in virion formation following interruption of translation by CP.
There is extensive experimental evidence showing that potyviral CP-CP interactions occur both in vitro and in vivo. Importantly, self-interaction of PVA CPs with the same amino acid sequence as that used in the present study has been confirmed using the yeast two-hybrid system (46, 47). Furthermore, PPV CP-CP interactions have been confirmed in planta by bimolecular fluorescence complementation (48), using the same Agrobacterium-mediated transient-expression method as that used in this study. In that report, the encapsidation of PPV RNA was proposed to involve self-association of CP through the N-terminal domain. Moreover, self-assembly of Pepper vein banding virus (PVBV; genus Potyvirus) CP into VLPs requires the presence of N- and C-terminal surface-exposed residues in the CP. These residues are essential for the formation of octameric ring-like structures which serve as VLP assembly intermediates in E. coli (20). Although the involvement of these structures in potyvirus assembly in vivo remains to be shown, the above results, together with the results of the present study, suggest a role for CP-CP interactions in the inhibition of viral RNA translation and the initiation of virion assembly. This may also suggest that the viral RNA acts to stabilize virions, possibly via electrostatic interactions, rather than as a nucleating agent for assembly initiation. Transition of viral RNA directly from translation to assembly would provide a mechanism for the specific selection of viral RNA for encapsidation.
Regulation of CP functions by CPIP.
Potyviruses utilize a gene expression strategy producing a large polyprotein which is then proteolytically processed by viral proteases into 10 individual proteins (49). In such an expression strategy, all viral proteins, except those synthesized from the separate P3N-PIPO open reading frame (50), are theoretically produced in an equimolar ratio. Because different stages of infection require different amounts of CP and nonstructural proteins, the ratio between them must be regulated temporally. It has been shown that the amount of functionally active CP can be regulated by posttranslational modifications (11) or through targeted proteasomal degradation (9). The ubiquitin-proteasome system is used by different RNA viruses to continuously maintain optimal levels of their proteins throughout the infectious cycle (reviewed in references 51 and 52). Early in potyviral infection, excess CP is removed by means of HSP70/CPIP-mediated proteasomal degradation (9), allowing viral RNA translation and replication to proceed efficiently in the absence of premature particle assembly. The J-domain protein CPIP, a cochaperone of HSP70, is essential for potyvirus infection (13) and has been shown to interact with CP, relieving the CP-mediated inhibition of viral RNA translation (9). A possible mechanism for this effect may involve competition between CP-CPIP and CP-CP interactions. It is worth noting that CPmut does not bind CPIP (13) and therefore is not affected by this mechanism. As CP levels rise toward the later stages of infection, CP eventually exhausts all available CPIP, overpowering the HSP70/CPIP-mediated degradation machinery. This results in the cessation of viral RNA translation and replication and ultimately may lead to the initiation of viral RNA encapsidation at the CP cistron through cotranslational CP-CP interactions. This process can be regulated further by HC-Pro, which is thought to play a role in coordinating translation, replication, and encapsidation (45).
Conclusions.
In summary, the results of this study suggest that the CP-mediated inhibition of PVA RNA translation occurs in a cotranslational manner and that CP-CP interactions play an important role in this process. According to the model presented in Fig. 8, when CP levels rise in the late stages of infection, the chaperone-assisted degradation machinery becomes overpowered and unable to prevent CP-CP interactions. Under these conditions, individual CP molecules or CP assembly intermediates are targeted to the CP-encoding region of viral RNA via their interaction with the newly synthesized CP polypeptide chain, thereby releasing ribosomes from the viral RNA and immediately recruiting this RNA into the encapsidation process.
ACKNOWLEDGMENTS
We thank Minna Pöllänen for maintaining the plants for this study and Andres Lõhmus for his help in cloning and for a critical reading of the manuscript. We thank George P. Lomonossoff at the John Innes Center for the pEAQ-HT vector supplied by Plant Bioscience Limited.
Financial support from the Academy of Finland (grants 1134684 and 1138329) is gratefully acknowledged.
REFERENCES
- 1.Ivanov KI, Mäkinen K. 2012. Coat proteins, host factors and plant viral replication. Curr Opin Virol 2:712–718. doi: 10.1016/j.coviro.2012.10.001. [DOI] [PubMed] [Google Scholar]
- 2.Callaway A, Giesman-Cookmeyer D, Gillock ET, Sit TL, Lommel SA. 2001. The multifunctional capsid proteins of plant RNA viruses. Annu Rev Phytopathol 39:419–460. doi: 10.1146/annurev.phyto.39.1.419. [DOI] [PubMed] [Google Scholar]
- 3.Ni P, Cheng Kao C. 2013. Non-encapsidation activities of the capsid proteins of positive-strand RNA viruses. Virology 446:123–132. doi: 10.1016/j.virol.2013.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yi G, Letteney E, Kim CH, Kao CC. 2009. Brome mosaic virus capsid protein regulates accumulation of viral replication proteins by binding to the replicase assembly RNA element. RNA 15:615–626. doi: 10.1261/rna.1375509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boni S, Lavergne JP, Boulant S, Cahour A. 2005. Hepatitis C virus core protein acts as a trans-modulating factor on internal translation initiation of the viral RNA. J Biol Chem 280:17737–17748. doi: 10.1074/jbc.M501826200. [DOI] [PubMed] [Google Scholar]
- 6.Neeleman L, Linthorst HJ, Bol JF. 2004. Efficient translation of alfamovirus RNAs requires the binding of coat protein dimers to the 3′ termini of the viral RNAs. J Gen Virol 85:231–240. doi: 10.1099/vir.0.19581-0. [DOI] [PubMed] [Google Scholar]
- 7.Karpova OV, Arkhipenko MV, Zayakina OV, Nikitin NA, Kiselyova OI, Kozlovsky SV, Rodionova NP, Atabekov JG. 2006. Regulation of RNA translation in potato virus X RNA-coat protein complexes: the key role of the N-terminal segment of the protein. Mol Biol 40:628–634. doi: 10.1134/S0026893306040157. [DOI] [PubMed] [Google Scholar]
- 8.Mahajan S, Dolja VV, Carrington JC. 1996. Roles of the sequence encoding tobacco etch virus capsid protein in genome amplification: requirements for the translation process and a cis-active element. J Virol 70:4370–4379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hafrén A, Hofius D, Rönnholm G, Sonnewald U, Mäkinen K. 2010. HSP70 and its cochaperone CPIP promote potyvirus infection in Nicotiana benthamiana by regulating viral coat protein functions. Plant Cell 22:523–535. doi: 10.1105/tpc.109.072413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ivanov KI, Puustinen P, Gabrenaite R, Vihinen H, Ronnstrand L, Valmu L, Kalkkinen N, Makinen K. 2003. Phosphorylation of the potyvirus capsid protein by protein kinase CK2 and its relevance for virus infection. Plant Cell 15:2124–2139. doi: 10.1105/tpc.012567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ivanov KI, Puustinen P, Merits A, Saarma M, Makinen K. 2001. Phosphorylation down-regulates the RNA binding function of the coat protein of potato virus A. J Biol Chem 276:13530–13540. doi: 10.1074/jbc.M009551200. [DOI] [PubMed] [Google Scholar]
- 12.Baratova LA, Efimov AV, Dobrov EN, Fedorova NV, Hunt R, Badun GA, Ksenofontov AL, Torrance L, Jarvekulg L. 2001. In situ spatial organization of potato virus A coat protein subunits as assessed by tritium bombardment. J Virol 75:9696–9702. doi: 10.1128/JVI.75.20.9696-9702.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hofius D, Maier AT, Dietrich C, Jungkunz I, Börnke F, Maiss E, Sonnewald U. 2007. Capsid protein-mediated recruitment of host DnaJ-like proteins is required for potato virus Y infection in tobacco plants. J Virol 81:11870–11880. doi: 10.1128/JVI.01525-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nagy PD, Wang RY, Pogany J, Hafren A, Makinen K. 2011. Emerging picture of host chaperone and cyclophilin roles in RNA virus replication. Virology 411:374–382. doi: 10.1016/j.virol.2010.12.061. [DOI] [PubMed] [Google Scholar]
- 15.Cardinale D, Carette N, Michon T. 2012. Virus scaffolds as enzyme nano-carriers. Trends Biotechnol 30:369–376. doi: 10.1016/j.tibtech.2012.04.001. [DOI] [PubMed] [Google Scholar]
- 16.Pille J, Cardinale D, Carette N, Di Primo C, Besong-Ndika J, Walter J, Lecoq H, van Eldijk MB, Smits FC, Schoffelen S, van Hest JC, Makinen K, Michon T. 2013. General strategy for ordered noncovalent protein assembly on well-defined nanoscaffolds. Biomacromolecules 14:4351–4359. doi: 10.1021/bm401291u. [DOI] [PubMed] [Google Scholar]
- 17.Annamalai P, Rao AL. 2005. Replication-independent expression of genome components and capsid protein of brome mosaic virus in planta: a functional role for viral replicase in RNA packaging. Virology 338:96–111. doi: 10.1016/j.virol.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 18.Annamalai P, Rao AL. 2005. Dispensability of 3′ tRNA-like sequence for packaging cowpea chlorotic mottle virus genomic RNAs. Virology 332:650–658. doi: 10.1016/j.virol.2004.12.009. [DOI] [PubMed] [Google Scholar]
- 19.Jagadish MN, Hamilton RC, Fernandez CS, Schoofs P, Davern KM, Kalnins H, Ward CW, Nisbet IT. 1993. High level production of hybrid potyvirus-like particles carrying repetitive copies of foreign antigens in Escherichia coli. Biotechnology (N Y) 11:1166–1170. doi: 10.1038/nbt1093-1166. [DOI] [PubMed] [Google Scholar]
- 20.Anindya R, Savithri HS. 2003. Surface-exposed amino- and carboxy-terminal residues are crucial for the initiation of assembly in Pepper vein banding virus: a flexuous rod-shaped virus. Virology 316:325–336. doi: 10.1016/S0042-6822(03)00593-2. [DOI] [PubMed] [Google Scholar]
- 21.Jagadish MN, Ward CW, Gough KH, Tulloch PA, Whittaker LA, Shukla DD. 1991. Expression of potyvirus coat protein in Escherichia coli and yeast and its assembly into virus-like particles. J Gen Virol 72:1543–1550. doi: 10.1099/0022-1317-72-7-1543. [DOI] [PubMed] [Google Scholar]
- 22.Dolja VV, Haldeman-Cahill R, Montgomery AE, Vandenbosch KA, Carrington JC. 1995. Capsid protein determinants involved in cell-to-cell and long distance movement of tobacco etch potyvirus. Virology 206:1007–1016. doi: 10.1006/viro.1995.1023. [DOI] [PubMed] [Google Scholar]
- 23.Varrelmann M, Maiss E. 2000. Mutations in the coat protein gene of plum pox virus suppress particle assembly, heterologous encapsidation and complementation in transgenic plants of Nicotiana benthamiana. J Gen Virol 81:567–576. [DOI] [PubMed] [Google Scholar]
- 24.Gabrenaite-Verkhovskaya R, Andreev IA, Kalinina NO, Torrance L, Taliansky ME, Makinen K. 2008. Cylindrical inclusion protein of potato virus A is associated with a subpopulation of particles isolated from infected plants. J Gen Virol 89:829–838. doi: 10.1099/vir.0.83406-0. [DOI] [PubMed] [Google Scholar]
- 25.Eskelin K, Suntio T, Hyvärinen S, Hafren A, Mäkinen K. 2010. Renilla luciferase-based quantitation of Potato virus A infection initiated with Agrobacterium infiltration of N. benthamiana leaves. J Virol Methods 164:101–110. doi: 10.1016/j.jviromet.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 26.Frisch DA, Harris-Haller LW, Yokubaitis NT, Thomas TL, Hardin SH, Hall TC. 1995. Complete sequence of the binary vector Bin 19. Plant Mol Biol 27:405–409. doi: 10.1007/BF00020193. [DOI] [PubMed] [Google Scholar]
- 27.Datla RS, Hammerlindl JK, Panchuk B, Pelcher LE, Keller W. 1992. Modified binary plant transformation vectors with the wild-type gene encoding NPTII. Gene 122:383–384. doi: 10.1016/0378-1119(92)90232-E. [DOI] [PubMed] [Google Scholar]
- 28.Sainsbury F, Thuenemann EC, Lomonossoff GP. 2009. pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol J 7:682–693. doi: 10.1111/j.1467-7652.2009.00434.x. [DOI] [PubMed] [Google Scholar]
- 29.Van Larebeke N, Engler G, Holsters M, Van den Elsacker S, Zaenen I, Schilperoort RA, Schell J. 1974. Large plasmid in Agrobacterium tumefaciens essential for crown gall-inducing ability. Nature 252:169–170. doi: 10.1038/252169a0. [DOI] [PubMed] [Google Scholar]
- 30.Eskelin K, Hafren A, Rantalainen KI, Makinen K. 2011. Potyviral VPg enhances viral RNA translation and inhibits reporter mRNA translation in planta. J Virol 85:9210–9221. doi: 10.1128/JVI.00052-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu D, Shi L, Han C, Yu J, Li D, Zhang Y. 2012. Validation of reference genes for gene expression studies in virus-infected Nicotiana benthamiana using quantitative real-time PCR. PLoS One 7:e46451. doi: 10.1371/journal.pone.0046451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Simon P. 2003. Q-Gene: processing quantitative real-time RT-PCR data. Bioinformatics 19:1439–1440. doi: 10.1093/bioinformatics/btg157. [DOI] [PubMed] [Google Scholar]
- 33.Clark MF, Adams AN. 1977. Characteristics of the microplate method of enzyme-linked immunosorbent assay for the detection of plant viruses. J Gen Virol 34:475–483. doi: 10.1099/0022-1317-34-3-475. [DOI] [PubMed] [Google Scholar]
- 34.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Wu X, Shaw JG. 1998. Evidence that assembly of a potyvirus begins near the 5′ terminus of the viral RNA. J Gen Virol 79:1525–1529. [DOI] [PubMed] [Google Scholar]
- 36.Hafren A, Eskelin K, Makinen K. 2013. Ribosomal protein P0 promotes Potato virus A infection and functions in viral translation together with VPg and eIF(iso)4E. J Virol 87:4302–4312. doi: 10.1128/JVI.03198-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hema M, Subba Reddy C, Savithri HS, Sreenivasulu P. 2008. Assembly of recombinant coat protein of sugarcane streak mosaic virus into potyvirus-like particles. Indian J Exp Biol 46:793–796. [PubMed] [Google Scholar]
- 38.Bendahmane M, Fitchen JH, Zhang G, Beachy RN. 1997. Studies of coat protein-mediated resistance to tobacco mosaic tobamovirus: correlation between assembly of mutant coat proteins and resistance. J Virol 71:7942–7950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bendahmane M, Chen I, Asurmendi S, Bazzini AA, Szecsi J, Beachy RN. 2007. Coat protein-mediated resistance to TMV infection of Nicotiana tabacum involves multiple modes of interference by coat protein. Virology 366:107–116. doi: 10.1016/j.virol.2007.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peabody DS, Ely KR. 1992. Control of translational repression by protein-protein interactions. Nucleic Acids Res 20:1649–1655. doi: 10.1093/nar/20.7.1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolja VV, Haldeman R, Robertson NL, Dougherty WG, Carrington JC. 1994. Distinct functions of capsid protein in assembly and movement of tobacco etch potyvirus in plants. EMBO J 13:1482–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang J, Yamada O, Yoshida H, Iwai T, Araki H. 2002. Autogenous translational inhibition of core protein: implication for switch from translation to RNA replication in hepatitis C virus. Virology 293:141–150. doi: 10.1006/viro.2001.1270. [DOI] [PubMed] [Google Scholar]
- 43.Chen MH, Icenogle JP. 2004. Rubella virus capsid protein modulates viral genome replication and virus infectivity. J Virol 78:4314–4322. doi: 10.1128/JVI.78.8.4314-4322.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ilkow CS, Mancinelli V, Beatch MD, Hobman TC. 2008. Rubella virus capsid protein interacts with poly(A)-binding protein and inhibits translation. J Virol 82:4284–4294. doi: 10.1128/JVI.02732-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Valli A, Gallo A, Calvo M, de Jesus Perez J, Garcia JA. 2014. A novel role of the potyviral helper component proteinase contributes to enhance the yield of viral particles. J Virol 88:9808–9818. doi: 10.1128/JVI.01010-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo D, Merits A, Saarma M. 1999. Self-association and mapping of interaction domains of helper component-proteinase of potato A potyvirus. J Gen Virol 80:1127–1131. [DOI] [PubMed] [Google Scholar]
- 47.Guo D, Rajamaki ML, Saarma M, Valkonen JP. 2001. Towards a protein interaction map of potyviruses: protein interaction matrixes of two potyviruses based on the yeast two-hybrid system. J Gen Virol 82:935–939. [DOI] [PubMed] [Google Scholar]
- 48.Zilian E, Maiss E. 2011. Detection of plum pox potyviral protein-protein interactions in planta using an optimized mRFP-based bimolecular fluorescence complementation system. J Gen Virol 92:2711–2723. doi: 10.1099/vir.0.033811-0. [DOI] [PubMed] [Google Scholar]
- 49.Riechmann JL, Lain S, Garcia JA. 1992. Highlights and prospects of potyvirus molecular biology. J Gen Virol 73:1–16. doi: 10.1099/0022-1317-73-1-1. [DOI] [PubMed] [Google Scholar]
- 50.Chung BY, Miller WA, Atkins JF, Firth AE. 2008. An overlapping essential gene in the Potyviridae. Proc Natl Acad Sci U S A 105:5897–5902. doi: 10.1073/pnas.0800468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alcaide-Loridan C, Jupin I. 2012. Ubiquitin and plant viruses, let's play together! Plant Physiol 160:72–82. doi: 10.1104/pp.112.201905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi AG, Wong J, Marchant D, Luo H. 2013. The ubiquitin-proteasome system in positive-strand RNA virus infection. Rev Med Virol 23:85–96. doi: 10.1002/rmv.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]